

Abstract
In regenerative medicine, clinical imaging is indispensable for characterizing damaged tissue and for measuring the safety and efficacy of therapy. However, the ability to track the fate and function of transplanted cells with current technologies is limited. Exogenous contrast labels such as nanoparticles give a strong signal in the short term but are unreliable long term. Genetically encoded labels are good both short- and long-term in animals, but in the human setting they raise regulatory issues related to the safety of genomic integration and potential immunogenicity of reporter proteins. Imaging studies in brain, heart and islets share a common set of challenges, including developing novel labeling approaches to improve detection thresholds and early delineation of toxicity and function. Key areas for future research include addressing safety concerns associated with genetic labels and developing methods to follow cell survival, differentiation and integration with host tissue. Imaging may bridge the gap between cell therapies and health outcomes by elucidating mechanisms of action through longitudinal monitoring.
Introduction
Many tissues and organs in the human body, such as the heart, brain and spinal cord, cannot regenerate in response to disease or trauma; damage leads not to restoration of structure and function but to an inflammatory response and scar formation. Regenerative medicine aims to achieve functional recovery of damaged tissues by providing specific cell populations, alone or incorporated in biomaterial scaffolds, that enhance the body’s intrinsic healing capacity1. The field has seen considerable progress in several areas, including development of new sources of transplantable cells and improved approaches to test the safety and efficacy of experimental therapies. However, many difficult challenges remain. Transplantation into diseased tissues is a stressful experience for cells. Most cells leak out from the injection site or die through multiple mechanisms2. The hardy survivors have to migrate, proliferate and self-organize into a tissue, integrate functionally with the host parenchyma and recruit a vascular supply to support their long-term survival and function. Transplanted cells are often immature and are required to mature in situ. All of these processes must occur in a damaged tissue environment that is hostile to transplanted cells. After excessive cell loss, most tissues produce scars, such as a fibro-collagenous scar in the heart or a glial scar in the central nervous system. The scarring response is one of the banes of regenerative medicine because it blocks integration of stem cell grafts with surrounding host tissue. Other hostile factors in damaged tissues include an ischemic environment that limits oxygen and nutrient delivery, and acute and chronic inflammation, which generates reactive oxygen species and injurious cytokines.
With a few notable exceptions, such as pluripotent stem cell–derived cardiomyocytes, dopaminergic neurons and pancreatic islet cells, most cell therapies have not acted like the ‘bricks and mortar’ repair cells we originally imagined. The limited functional restoration that has been achieved may be elicited by paracrine mechanisms that are poorly understood. Rational improvement of the design of cell therapies and of clinical protocols for administering them will require a deeper understanding both of the cells and of their fate after transplantation as they interact with an inhospitable environment. Some of these questions can be elucidated through imaging. Each organ in the body has unique imaging characteristics that make it possible to differentiate pathological and regenerative responses to therapy. Many aspects of impaired organ function can be readily identified by clinical imaging, including gross morphological changes (e.g., ruptures, cystic cavities, geometric remodeling); volume loss (e.g., atrophy); hemorrhage or iron by-products; thrombosis; hypoperfusion; increased vascular permeability and resulting edema; metabolic shifts compared to surrounding parenchyma; oxygenation; fibrosis; and loss of function. Such data can provide an indirect measure of graft function and shed light on therapeutic mechanisms. However, direct tracking of the fate and function of transplanted cells in humans is at an early, experimental stage and is subject to substantial limitations. Imaging approaches for cell therapies based on the action of secreted paracrine factors is fundamentally different from imaging of cell replacement and requires the ability to track the structural and functional changes within the host organ.
This review explores the benefits and limitations of clinical imaging technologies for applications in regenerative medicine. We discuss imaging modalities that may have a role in clinical trials within a decade rather than early-stage, experimental technologies or approaches that are limited to animal research. We apologize to colleagues whose work is not included, and we refer interested readers to recent reviews3, 4, 5, 6, 7. We begin with a short primer on the capabilities of clinical imaging, which is subject to more constraints than the same technologies used in animal models. Next, we discuss cell-labeling approaches that are in the clinic or in development, and their applications for tracking transplanted cells and understanding the host environment and the response to therapy. These issues are then explored in detail for three tissues—the heart, pancreatic islet cells and the brain—chosen because they are among the most advanced in human translation and because, at least in some instances, they include cell grafts that survive long term in the host environment. We propose recommendations to researchers interested in incorporating imaging in their studies and conclude with a discussion of important areas for future research.
Capabilities and limitations of clinical imaging in regenerative medicine
Imaging is used routinely in clinical practice both to provide secondary endpoints that complement primary health outcomes and to investigate the mechanisms of therapeutic successes and failures. In early-stage clinical trials that are not powered for endpoints such as mortality or rehospitalization, imaging is often the primary ‘surrogate’ endpoint—for example, to assess structural and functional responses of the tissue or to monitor adverse reactions such as immunogenicity and tumorigenicity8. The main clinical imaging modalities in use today are magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET), single photon emission tomography (SPECT) and ultrasound, as well as multimodality methods that rely on co-registration of images (Box 1). These technologies have improved considerably in recent years and now provide sophisticated multimodality imaging, shorter acquisition times and lower radiation doses. The latest clinical scanners offer new opportunities in regenerative medicine for assessing the tissue composition of organs, monitoring transplanted cells and evaluating the effects of therapy on tissue function. They are beginning to be applied to non-invasively assess the survival, migration, biodistribution, and differentiation of transplanted cells and the underlying mechanisms of cell therapy9. However, imaging in humans is inherently more constrained than imaging in animals owing to limits on radiation, image acquisition times and, more importantly, limits on the use of genetically modified cells for long-term cell tracking. These limits translate into reduced sensitivity, specificity and ability to monitor changes over time. The benefits and challenges of clinical imaging in regenerative medicine are summarized in Box 2.
Box 1. Clinical imaging modalities.
A limited number of medical imaging technologies are currently approved for clinical use. We provide brief descriptions of them below.
Magnetic resonance imaging
MRI uses magnets (from 0.5–7 Tesla magnetic field strength) to polarize the hydrogen nuclei in water molecules in human tissues. Combinations of time-varying gradient magnetic fields and pulse sequences of radio frequency waves provide the spatial distribution of signals emitted from protons, which are displayed as high-resolution, multidimensional images. MRI does not use ionizing radiation and can be performed serially over time. It is the most versatile clinical imaging modality, with superior sensitivity in detecting morphology, pathology and function.
X-ray computed tomography
CT uses computer-processed X-rays to produce tomographic images. The disadvantage is the use of ionizing radiation, which damages DNA. Two new approaches under development rely on more exotic forms of radiation: proton beams and synchrotron radiation. Proton CT records the position, direction and energy loss from a proton beam as it traverses a patient’s body, providing a more detailed image of the body’s density and requiring less radiation exposure. CT images based on synchrotron X-rays have much higher photon energies than conventional X-rays, reducing radiation dose.
Positron emission tomography
PET is a tomographic technique that produces images of functional processes in the body through detection of biologically active positron-emitting radio-tracer, such as fluorine-18, attached to a small molecule. The short lifetime of radioligands requires fast image acquisition the same day as tracer synthesis. Sensitivity is very high and there is no limit on tissue penetration depth, so PET can be used to track cells expressing reporter proteins. PET scans are usually co-registered with MRI or CT images.
Single photon emission tomography
SPECT is a tomographic technique that uses gamma rays to detect radioactive probes, such as technetium-99m (99mTc) and indium-111 (111In), injected into the bloodstream. Sensitivity is very high and there is no limit on tissue penetration depth. There is only one FDA-approved SPECT agent (111In oxine). SPECT can report whole-body biodistribution of injected radiolabeled cells but does not provide high-resolution anatomical information that allows a precise delineation of cellular location. The main limitation of this method is signal decay; labels cannot be tracked over weeks, the signal diminishes as cells divide and the radio-labeled components are metabolized. Ionizing radiation may cause DNA damage, so exposure to radioactivity must be closely monitored.
Ultrasound
Images are generated by the reflections and echos of an oscillating sound wave on tissues. Ultrasound provides images in real time (no processing delay after an acquisition), can be brought to a sick patient’s bedside, is substantially lower in cost than other imaging modalities and does not use ionizing radiation. Ultrasound is widely used in the clinic and in research to evaluate the structure, function and blood flow of organs. Sufficient image resolution for cell tracking requires high frequencies, but the depth penetration of high-frequency ultrasound waves is limited. Therefore, ultrasound is not widely used to track transplanted cells.
Multimodality imaging
Multimodality imaging involves co-registration of images obtained from different scanners. It combines high-sensitivity but low-resolution methods, such as PET and SPECT, with high-resolution anatomical images acquired with MRI or CT scanners. Examples include MRI/PET, PET/CT, SPECT/CT and ultrasound/CT.
Box 2. Benefits and challenges of imaging in regenerative medicine.
Diagnostic and interventional imaging can be incorporated into all aspects of cell therapy research and clinical trials. The main benefits and challenges are summarized below.
Subject selection
Imaging is essential for determining whether subjects meet inclusion or exclusion criteria for entry into clinical trials. Imaging results should be part of inclusion and exclusion criteria for subjects entering into the study. Investigators should verify that accepted subjects would tolerate imaging studies.
Subject evaluation
Diagnostic imaging is essential for defining the extent of pathology and organ dysfunction to be treated. Interventional imaging can aid evaluation of organ vascular anatomy and flow.
Route of administration
Imaging is essential for selecting the best route of administration (e.g., direct implantation, intravascular, intranasal, endoscopic, surface application).
Patient safety
Imaging of cell homing and dispersion is useful for evaluating potential toxicity, graft-versus-host disease, tumor formation and undesirable changes in the host tissue.
Host microenvironment
Multimodality imaging is valuable for assessing whether the targeted region can support the cell graft.
Cell tracking
A challenge is to develop labeling approaches that uniquely identify cells over the desired temporal and spatial scales.
Cell dosing
A challenge is to use clinical imaging to determine the potency, volume of distribution and lifespan of the cell product, and to develop a protocol for administration to patients. Novel labels to track the fate and function of cell products will bring mechanistic insights into clinical trials.
Label dilution and transfer
Loss of exogenous labels by cell division limits long-term tracking. A challenge is to develop agents that remain within cells, turn off upon cell death or are not transferred to host cells such as macrophages. A change in the imaging signature of the label may indicate metabolism or cell engraftment. A decrease in the signal volume may indicate cell death or migration away from implantation site.
Mechanism of action
A challenge is to develop imaging strategies that can reveal the mechanisms of action of transplanted cells (e.g., cell replacement or paracrine effects).
Cell dose
The possibility of imaging transplanted cells depends in part on their number. The optimal cell dose required for a therapy is determined empirically, guided by the normal cell content of the tissue or organ (Fig. 1) and the cell deficit in individual patients. It is important to remember that all organs have reserve capacity and that disease ensues only when cells are depleted below a critical threshold. This means that full regeneration is not necessary for substantial improvement in organ function or symptoms.
Figure 1.
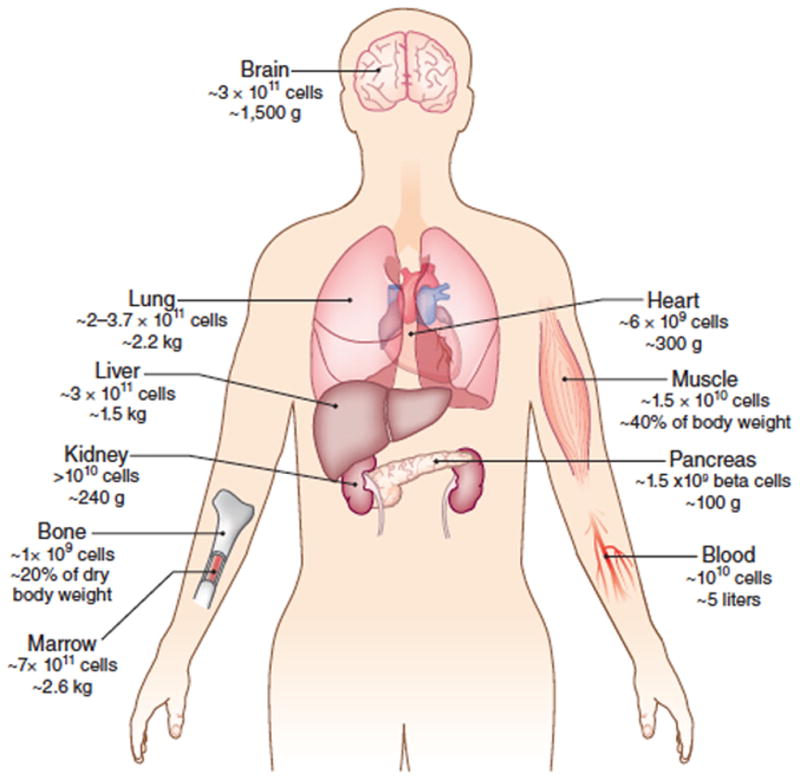
The human body contains ~3.7 × 1013 cells10.
The cell number and weights of individual organs160,161 provide a baseline for understanding the numbers of cells that may be needed for replacement therapies and the associated challenges for imaging.
When cell products are injected intravenously, a typical dose is 1–5 × 106 cells/kg (or 70–350 × 106cells in a 70 kg person). In a 5-liter blood volume containing ~1010 nucleated cells10, this amounts to an initial dilution of 1 cell in 103–105 blood cells, making initial tracking of transplanted cells akin to finding a needle in a haystack. As the cells distribute throughout the body, they localize primarily to the lungs, liver, spleen or marrow spaces, with relatively few cells homing to diseased sites11, 12,13. The alternative to intravenous injection is direct injection into tissues. In these therapies, doses may be smaller than ~105 (e.g., for repair of the cornea or the retinal pigment epithelium14) or larger than ~109 cells (e.g., to rebuild a heart). To be detectable by clinical imaging, at least ~104–106 cells must engraft in a specific location in the body.
Below we discuss in detail the prospects for imaging transplanted cardiomyocytes, pancreatic islets and neural cells. The left ventricle of the heart has ~20 million cardiomyocytes per gram of tissue15, or ~4 billion in total. A myocardial infarction that causes heart failure can kill ~25% of these cells16, or ~1 billion cardiomyocytes. Cell therapy for diabetes involves similar numbers of cells. A healthy human pancreas has about ~1 million islets of Langerhans, each containing ~2,000 cells, of which 65–80% are insulin-producing beta cells, for a total of 1.3–1.6 × 109 beta cells. The dose of beta cells needed to dispense with insulin therapy can be estimated from current protocols for transplantation of cadaveric islets into the liver. Patients generally receive two infusions of 5,000–11,000 islet equivalents (IEQ) per kilogram (or 0.7–0.9 × 109 beta cells IEQ/75 kg human) per infusion17. After transplantation through the portal vein, islets are lodged as single islets or islet clusters in the capillaries of the liver, where they start secreting insulin. However, the majority of these cells die soon after transplantation, and optimized therapies may require fewer cells.
The projected cell dose needed to treat neurological conditions varies widely, mostly depending on whether the aim is to replace cells or to deliver therapeutic factors. For instance, the putamen region of a healthy brain has ~7.61 ng/mg of dopamine18 supplied by ~500,000 dopaminergic neurons from the substantia nigra. In a cell therapy for Parkinson’s disease, transplantation of only ~138,000 fetal neural cells showed efficacy, increasing dopamine release from 25% to 62% of normal levels19. However, replacement or regeneration of an anatomical region, such as the putamen, may require ~7 × 106 cells20, 21.
Whether one is transplanting cardiomyocytes, islet cells, neural cells or other cell types, the ability to detect them ultimately depends on the cell density in the target tissue and the sensitivity and specificity of the imaging technique (Table 1).
Table 1.
Characteristics of clinical imaging modalities
| Modality | Applications | Labels (exogenous, endogenous reporter gene) |
Main characteristics |
Depth of penetra- tion |
Sensitivity | Image resolution (voxel size) |
Acquisition time |
Injected reagents |
Clearance of label after cell death |
Disadvantages |
|---|---|---|---|---|---|---|---|---|---|---|
| MRI | Anatomy Pathology Metabollsm Chemical exchange Physiology Function Intervention Cellular Molecular |
Iron oxide particles Gd chelates Microcapsules with fluorine sodium carbon Ferritin Lysine-rich protein protamine |
Versatile High soft tissue contrast Metabolite concentrations Perfusion Characterization of the microenvironment Short- and medium-term cell tracking Graft size |
No limit | Moderate | ~<1–3 mm3 | Microseconds to minutes | Stable Commercially available | Slow | Not compatible for patients with implants Acoustic noise Specialized coils (19F 23Na, 13C) |
| PET | Metabolism Physiology Function Cellular Molecular |
18FDG 18FHBG 18FDOPA |
Graft proliferation Tissue viability Inducible cell death Longitudinal serial imaging Differentiation possibilities |
No limit | High | ~3–5 mm3 | Minutes to hours | Labile Commercially available, Custom synthesized | Rapid Depends on T1/2 Isotope |
Ionizing radiation Biohazardous labels |
| SPECT | Metabolism Physiology Cellular Molecular |
Radioisotopes: Tc-99m In-111 NIS Dopamine receptor |
Distribution of injected cells | No limit | High | ~5 mm3 | Minutes to hours | Labile Commercially available Custom synthesized | Rapid | Ionizing radiation Limited short-term cell tracking |
| X-ray, CT | Anatomy Pathology Intervention Vascular delivery Biopsy |
Microcapsules Barium Gold nanoparticles |
Multimodal pairing with PET, SPECT Bone, lung |
No limit | High | <1 mm3 | Seconds | Stable Commercially available | Slow | Ionizing radiation Not suitable for soft tissue imaging |
| Ultrasound | Anatomy Pathology Intervention Vascular delivery Biopsy |
Microbubbles Microcapsules Liposomes |
Real-time fast imaging | Trade-off with spatial resolution | Moderate | 1 mm | Seconds to minutes | Stable Commercially available | Rapid | Tradeoff between depth of penetration and spatial resolution Limited field of view |
Limits of detection
Clinical imaging is a macroscopic technology and has limitations when applied to the detection of transplanted cells. The cell density in the target tissue corresponds to the number of labeled cells per voxel, the size of which varies with the imaging technique. The sensitivity and specificity of the imaging technique is a function of the concentration of the labeling agent in the cells and the contrast-to-noise ratio on the acquired images22. Uptake or expression of labels and their retention over time varies for different cell types and labels. Imaging contrast can be lost or diluted with cell division. Transplanted labeled cells, or free label released by dying cells, can be engulfed by macrophages, confounding interpretation of the imaging signal. Phagocytosis by macrophages may begin minutes to hours after transplantation and can continue for months, depending on the lifespan of the graft. Imaging of single cells23, 24 or small clusters (~50 cells)22,25, 26, 27 of labeled cells has been reported in animal models (using 7 T23 and 11.7 T24 MRI scanners) but in clinical imaging, in the best-case scenario, at least 600 labeled cells per voxel are needed to produce a detectable signal28. However, tracking of single or small numbers of cells is unnecessary and, for the foreseeable future, unattainable by clinical imaging.
Cell labeling
Cells transplanted in humans cannot be imaged with current technologies unless the cells are first labeled in vitro. There are two main labeling strategies: direct labeling, by nanoparticles or chemical agents, and indirect labeling, by genetic incorporation of reporter genes8, 29, 30, 31, 32 (Fig. 2). The ideal cell label for clinical applications would be nontoxic, would be retained by target cells over sufficiently long time periods at high concentration, would correlate stoichiometrically with cell number, would be cleared rapidly after cell death and would raise no long-term safety concerns. Unfortunately, none of the available labeling agents satisfy all of these criteria.
Figure 2.

Tracking cell fate by noninvasive imaging requires either direct or indirect labeling.
(a) Direct labeling. Exogenous labeling with either MRI-based contrast agents or radioprobes for PET or SPECT imaging. Cells take up SPIO or 19F nanoparticles (Fnp) primarily through endocytosis, whereas 99mTcHMPAO or 111Indium oxine are lipophilic and pass through cell membranes by passive diffusion. FDG is a glucose analog and is taken up though glucose transporter channels on cells. Small molecules can attach to cell surface markers or enter cells through channels. (b) Indirect labeling. Reporter genes introduced into the genome express surface proteins, channels, storage proteins or enzymes that are detectable or that bind detectable probes. HSV1-tk, herpes simplex virus thymidine kinase; NIS, sodium iodide symporter; LRP, lysine-rich protein; SPIO, superparamagnetic iron oxide nanoparticles.
Direct labeling
The most common contrast agents used for direct labeling are nanoparticles or radionuclides33, 34, 35. Nanoparticles suitable for MRI include superparamagnetic iron oxide (SPIO) nanoparticles (i.e., ferumoxides, ferucarbotran-1, ferumoxytol), gadolinium-filled microcapsules and liposomes, perfluorocarbon nanoparticles36 and manganese-based particles37 (Table 1). Advantages of nanoparticle-based MRI cell tracking are a strong signal, allowing high-resolution visualization of the migration and homing of injected cells, as well as relative ease of cell labeling. SPIO nanoparticles can also be detected by ultrasound, although this approach is not widely used38. SPIO-based labeling has been used clinically to track transplanted cells in the brain for up to 7 weeks by MRI39, 40. Labeling cells with perfluorocarbon nanoparticles is a promising approach that is being tested in clinical trials.
Direct labeling with radionuclides for SPECT and PET imaging has excellent sensitivity compared with MRI, CT and ultrasound because the background signal is low, allowing detection of as few as ~104–106 cells/voxel4. Radionuclide labeling is readily quantified and can offer even greater sensitivity than MRI, but half-lives are relatively short, and the limited spatial resolution of clinical PET and SPECT requires image co-localization with CT or MRI. Moreover, concerns regarding radiation exposure may limit these approaches41. Direct labeling with radioisotopes such as111Indium (In)-oxine or 99mTc chelates is used in the clinic for SPECT imaging of inflammation42. The relatively long half-life (67 h) and high in vivo stability of 111In-oxine provides the option to acquire images after 24 h or more, whereas 99mTc chelated agents have a relatively short half-life (6 h). 18F-fluorodeoxyglucose (FDG) also can be taken up and metabolically trapped by the cells and subsequently tracked in vivo43, 44.
Although direct labeling gives strong signals in the first days after cell transplantation, it has several drawbacks for long-term imaging. First, it cannot distinguish live and dead cells34, 45, 46. An ideal contrast agent would dissipate or be metabolized after the death of a labeled cell, but nanoparticles released from dead cells are phagocytosed by macrophages47, 48, which produce imaging signals identical to those of labeled cells. Soluble radioisotopes may be less prone to this artifact; to our knowledge, their uptake by macrophages has not been studied. Second, contrast agents are diluted as cells divide, resulting in a gradual disappearance of the signal49, 50, 51. Third, imaging signals can be difficult to distinguish from background. For example, SPIO nanoparticles are detectable in MRI as areas of decreased signal intensity, and similar signal voids can be caused by iron depositions in the form of hemosiderin from old hemorrhage52. Finally, all contrast agents raise concerns of cellular toxicity, particularly at high concentrations. High doses of SPIOs have been shown to inhibit mesenchymal stem cell (MSC) migration, colony formation ability53 and chondrogenesis54, although these results are controversial55. 111In-oxine labeling may impair cell proliferation42.
Indirect labeling
In indirect labeling, cells are genetically modified to express reporter genes encoding proteins that generate imaging signals, often upon interaction with a molecular probe or substrate that is taken up by cells. Reporter genes incorporated into the genome are propagated by daughter cells, and the imaging signal is stoichiometrically related to live cell mass. Episomal expression of reporter genes is also possible, but dilution of episomal vectors during cell division makes them unsuitable for long-term tracking of mitotically active cells. Reporter-gene imaging is widely used in animal research, but clinical applications for use in tracking cells to treat cardiovascular or central nervous system diseases have been limited by concerns about the safety of genomic integration and about potential immune responses to some of the foreign reporter proteins.
A variety of reporter proteins detectable by different imaging modalities have been developed. Firefly luciferase, detectable by bioluminescence imaging, is often used in small-animal research3; however, absorption of the emitted light precludes its use in most large-animal and human studies. For large-animal and clinical applications, there are several potential options.
The iron-storage protein ferritin is detectable by MRI as voxels with reduced signal intensity56, 57,58. Because ferritin is ubiquitous in most organisms, it may be possible to use it clinically. However, MRI sensitivity to ferritin is lower than to SPIO nanoparticles (work of A.V.N., C.E.M. and colleagues)46.
Herpes simplex virus thymidine kinase type 1 (HSV1-tk) is frequently used for PET imaging in large animals32, 59, and it has been incorporated into sensitized, cytolytic T-cells to image metastases in patients with glioblastoma using the ganciclovir analog 18F-9-(4-[18F]fluoro-3-hydroxymethylbutyl) guanine (18FHBG)60. HSV1-tk phosphorylates 18FHBG, trapping it within cells. Because mammalian thymidine kinases have low affinity for this reagent, it is possible to distinguish transplanted HSV1-tk cells from surrounding tissues by PET32, 59, 61, 62.
Another approach is to express reporter proteins on the cell surface. Biotinylated peptides expressed on the plasma membrane were imaged with a contrast agent conjugated to streptavidin in rodent models63, 64. The human membrane-protein sodium-iodide symporter (NIS), responsible for iodide uptake in the thyroid, stomach, salivary gland and choroid plexus but minimally expressed elsewhere, was used to follow the survival and distribution of transplanted cells in pigs by hybrid SPECT/CT32. The cells were pre-labeled with iodine-123, permitting immediate tracking of the grafts, and they could be imaged long term by systemic administration of iodine-123. This approach is especially promising for clinical applications because a human protein should not be immunogenic. Radioactive iodine scans are common in clinical medicine and could be adapted to cell therapies.
The chemical exchange saturation transfer (CEST) technique uses radiofrequency saturation pulses to detect molecules containing protons that exchange rapidly with water. The exchanging intracellular protons have a unique offset resonance that distinguishes them from extracellular water protons. Exchange rate and CEST contrast depend strongly on the pH65, 66, 67. Because cell death and inflammation are associated with extracellular acidification, CEST probes can be used as sensors of cell viability68. For example, CEST contrast based on amine proton transfer has identified heterogenous regions within gliomas in patients; this technique might be helpful in identifying the infiltration of tumors into surrounding parenchyma and tumor grade69. Unique, genetically encoded, CEST-detectable amino acids and proteins (e.g., L-arginine, lysine-rich protein and protamine) enabled imaging of transplanted cells by MRI in experimental models31. The threshold for detecting CEST protein–tagged cells by clinical MRI field strengths may be 104–106 cells/voxel22. Recently, a novel reporter probe, 5-methyl-5,6-dihydrothymidine (5-MDHT), was described for detection of HSV1-tk expression with CEST imaging70; this opens up the possibility of using MRI for HSV1-tk imaging. The main limitation of CEST imaging is difficulty in distinguishing the reporter from intrinsic macromolecules.
The major disadvantage of indirect labeling for clinical studies relates to safety concerns about the effects of genetic manipulation. Insertion of reporter genes in the genome may alter cell potency, immunogenicity71, differentiation potential and tumorigenicity72. Another issue is reporter-gene silencing, which can be misinterpreted as cell death. As with direct labels, reporter proteins in dead or live cells can be phagocytosed by macrophages, resulting in short-term, false-positive signals47,48, but this is not an issue for long-term imaging.
Choosing a labeling strategy
In evaluating whether to use direct or indirect labeling, it is important to assess cell viability, cell proliferation and contrast-agent retention30. The choice of label may also depend on the culture conditions for maintaining and expanding the cells and on whether the therapy is autologous or allogeneic. In our view, direct labels are best suited to study events in the first few days rather than months to years after transplantation. A potentially important application of direct labels is real-time MRI guidance of cell delivery, as no other imaging modality provides as precise anatomical information about cell migration and short-term biodistribution in vivo39. In our view, indirect labeling approaches are best used when it is necessary to monitor the fate of implanted cells over weeks, months or years in order to ensure that engraftment has occurred in the targeted tissue. An additional advantage of indirect labeling is the potential to turn a reporter gene on or off and to monitor differentiation of implanted cells into desired functional phenotypes. It should also be noted that genetic tagging of cells requires additional clearance through the Recombinant DNA Advisory Committee, which may delay approval of clinical trials.
Imaging of cell fate and host environment
In general, very little is understood about what happens to cells after they are transplanted and how they interact with the host environment, which makes it difficult to design improvements to the therapy. Recent imaging studies show potential for addressing these questions, but much requires further investigation.
Imaging cell fate
It would be useful to be able to characterize basic phenomena such as cell retention, survival, migration, proliferation, differentiation and integration into the host. The most progress has been made in measuring retention, or biodistribution, of cells soon after transplantation. For example, in patients with dilated cardiomyopathy, biodistribution of ~108 99mTc–labeled CD34+ hematopoietic stem cells delivered by intracoronary or transendocardial injection was monitored by SPECT. The cells cleared the lungs and localized primarily to the liver and spleen, with relatively few cells homing to the heart73 (Fig. 3).
Figure 3.
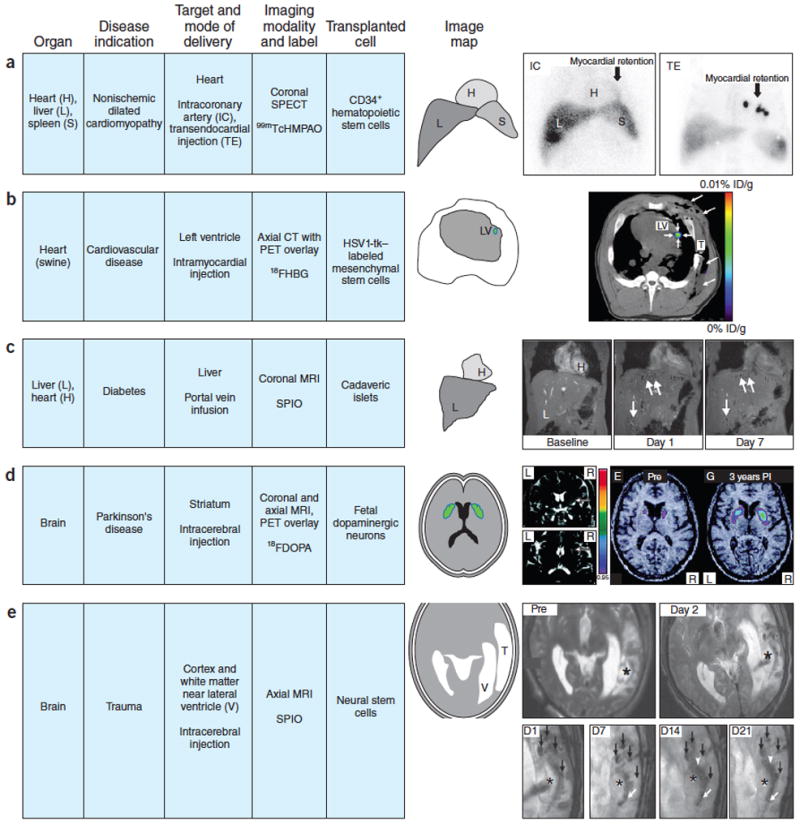
Examples of clinical imaging used to identify and track labeled cells in the body.
(a) Coronal SPECT images of chest and upper abdomen obtained 18 h after intracoronary (IC) versus transendocardial (TE) delivery of about 108 99mTcHMPAO CD34+ hematopoietic stem cells in patients with nonischemic dilated cardiomyopathy. IC route of administration clearly shows less retention of cells in the myocardium compared to TE route with both delivery techniques showing the distribution of CD34+ cells in liver and spleen (IC > TE). This comparison demonstrates the value of short-term labeling of a cell product and the value in assessing the cell delivery route. Figure from ref. 73 reprinted with permission. (b) PET/CT fused axial multimodal imaging of pig chest performed on clinical scanner. MSC transduced by adenovirus containing cytomegalovirus promoter driving HSV1-tk reporter gene implanted with matrigel into porcine left ventricle (LV) myocardium after thoracotomy (long arrows). T is the chest tube inserted during surgery. 108 human MSCs injected into the myocardium were visualized (short arrows) 4 h after intravenous infusion of 9-(4-18F-3-[hydroxymethyl]butyl)-guanine, a thymidine analog that is phosphorylated by HSV1-tk reporter following uptake by cells. % ID/g is percentage uptake per gram of tissue. Figure from ref. 32 reprinted with permission. (c) Coronal MRI performed at 3 T of liver from a diabetic patient baseline, day 1 and day 7 following portal vein infusion of 4.81 × 105SPIO-labeled islet equivalents demonstrating hypointense voxels throughout the liver (white arrows) that cleared rapidly over 24 weeks of imaging follow-up. The number of hypointense areas in liver decreased by about 50–60% in the first week after infusion, representing rapid clearance of the transplanted islets despite immunosuppression of the patient. Figure adapted from ref. 104 with permission. (d) Coronal and axial, T2-weighted, 1.0 T MRI following implantation of fetal neurons in a patient with Parkinson’s disease. Coronal MRI displays the cell transplantation needle track (arrowheads) following injection of ~3.2 × 106 cells. 18F fluorodopa PET parametric map fused with MRI performed before (PRE) and 3 years post implantation (PI) of fetal cell neurons that matured with time that resulted in increased uptake of the dopamine analog by the innervated cells in the putamen. Figure adapted from ref. 76 with permission. (e) T2-weighted, axial, 3 T MRI before (PRE) and day 1 after implantation in the left temporal lobe of SPIO-labeled autologous neural stem cells in patient with traumatic brain injury (* is site of injury). Magnified serial T2*-weighted, axial MRI performed on days (D) 1,7,14,21 post implantation (PI) of labeled cells are shown. Four hypointense areas (black arrows) were observed on PI days 1,7,14, 21 around lesion site (*) followed by migration of cells (white arrowhead and arrows) along the border of the damaged area. Hypointense areas where labeled cells were injected cleared over time (D14, D21), and by week 7 PI, areas were no longer visible on MRI. Figure adapted from ref. 40 with permission.
Cell survival in the target organ is often monitored in preclinical studies, for example, using PET imaging with reporter-gene labels62. However, few labeled cell–tracking studies have been carried out in humans. The problem of cell death after transplantation remains a major roadblock to regenerative therapies. Here, researchers could contribute by moving reporter-gene studies into large animals and by working with regulators to establish safety criteria for clinical trials of these labels.
Proliferation of transplanted cells can also be assessed using reporter genes because the imaging signal is passed to daughter cells8, 74. The most common approach, demonstrated in animals, is to image serially and to estimate growth kinetics from the rate of change in population size. Repetitive, long-term (up to 5 months) PET/CT studies of MSCs expressing HSV1-tk were conducted with a porcine model of myocardial infarction. Cell proliferation peaked at 33–35 days after injection in peri-infarct regions, but surprisingly, most of the transplanted cells were found in the major cardiac lymphatic vessels and lymph nodes75.
Imaging differentiation of transplanted cells has received relatively little attention, and there are interesting opportunities in this area. Promising approaches include genetically integrated stage-specific promoters and labeled small molecules that interact with stage-specific cellular targets. The latter approach is well suited to PET and SPECT if the small molecule can be slightly modified by addition of a radiolabel. For example, the PET agent 18F-DOPA was used to monitor maturation of fetal neurons transplanted in the brain of Parkinson’s disease patients (Fig. 3) as maturation increased uptake of the dopamine analog76. Approximately 104 functioning graft cells were detected with PET images overlaid on MRI scans. However, the use of small-molecule probes requires agents that can cross cellular barriers and vasculature (i.e., the blood-brain barrier) and interact specifically with their targets. The development of small-molecule agents and protocols will help guide clinical trials and should be a priority.
Imaging the host environment
Despite its many limitations for tracking transplanted cells, clinical imaging is indispensable in regenerative medicine for understanding the host environment. Imaging can answer many specific questions about tissues both for pretreatment planning and for longitudinal evaluation of response to therapy (Table 2). For example, what is the status of the targeted region with regard to various pathophysiological processes—inflammation, residual stunned or dormant native parenchyma, cellular and morphological alterations, and poor perfusion? Is the targeted region metabolically active or hypoxic, and can it support transplanted cells? Multimodal imaging of cell proliferation, tissue structure, perfusion, vascularity, permeability and metabolism can help delineate the feasibility of cell therapy and measure therapeutic outcomes. Initial and post-transplant assessment of the target tissue may include evaluation of structural integrity, tissue loss (e.g., atrophy, infarction), cellular composition (e.g., fibrosis, gliosis, percentage of viable cells) and inflammation, as well as underlying tissue infrastructure (extracellular matrix, vascularity), perfusion (e.g., blood flow and volume), oxygenation, acidosis (pH), and metabolic state (ATP, lactate, phosphocreatine content). Measuring hormone and neurotransmitter production, contractility and responses to external stimuli can report on the recovery of organ function. Imaging should also be useful for ensuring that cells are delivered to the optimal site (i.e., non-necrotic regions) at the optimal time and dose.
Table 2.
Interrogating the host microenvironment by clinical imaging
| Tissue characteristics | Imaging techniques | ||||||
|---|---|---|---|---|---|---|---|
| MRI | MRSI | X-ray angiography | CT | SPECT/CT | PET/CT | Ultrasound | |
| Anatomy (structure, volume loss, vasculature) | ++++ | − | ++++ | +++ | + | + | +++ |
| Vascular permeability | +++ | − | − | ++ | + | + | ++ |
| Pathology (e.g., mass) | ++++ | − | − | ++ | + | ++ | + |
| Compositions (e.g., fibrosis) | +++ | − | − | ++ | ++ | ++ | ++ |
| Inflammation (acute, subacute, chronic) | +++ | ++ | − | − | ++ | ++ | − |
| Physiology and function | ++++ | − | − | ++ | +++ | +++ | ++ |
| Perfusion | ++++ | − | − | − | − | ++ | |
| Metabolism | − | +++ | − | − | − | ++++ | − |
| Oxygenation | +++ | − | − | − | − | ++ | − |
| Cell viability | − | + | − | − | − | ++++ | − |
| Cell identity | − | − | − | − | +++ | ++++ | − |
MRSI, magnetic resonance spectroscopy imaging. MRI techniques include blood oxygenation level dependent contrast, diffusion tensor imaging, MR elastography, dynamic contrast enhance MRI. − not useful; + limited to ++++ most useful.
Imaging in selected organs
Here we discuss three organ systems in more depth to illustrate the advances and limitations of noninvasive imaging in regenerative medicine. Studies in heart, islets and brain share common challenges, including cell-seeding efficiency, graft cell death and achieving host integration. In the three settings, combined imaging approaches are useful in delineating organ morphology and function before and after cell therapy. Differences in routes of administration, cell labeling approaches and detection limits, which depend on organ characteristics, underscore challenges specific to each system.
Imaging myocardial regeneration
The heart is one of the least regenerative organs in the body. Whole-heart transplantation is currently the only definitive treatment for end-stage heart failure, and it is limited by organ availability to fewer than 0.1% of heart failure patients. Infarction causes significant loss of cardiomyocytes (often 25% of the left ventricle), which cannot be restored by current pharmaceutical therapies. Stem cells offer the possibility of rebuilding the damaged heart from its component parts. We submit that the following three conditions must be achieved to prove true myocardial regeneration: (i) an increase in the volume of viable myocardium within the infarct zone; (ii) structural integration of the new myocardium with the host tissue, including restoration of myocardial fiber architecture; and (iii) functional integration of the new myocardium with host myocardium—that is, synchronous contraction of new and old host myocardium without conduction delay or arrhythmias. Medical imaging is essential in the evaluation of cardiac repair by regenerative therapy. Measureable end points include heart contractile function, morphology, vascularity, inflammation, infarct size, tissue viability and metabolism. In animal studies with genetically labeled cells, it is possible to demonstrate formation of even small amounts of new myocardium by MRI or PET. In humans receiving genetically unmodified cells, it should be possible to identify new myocardium by MRI if it is large enough (i.e., >2% of the left-ventricle mass) and reasonably localized. Structural alignment of grafted cells with host myofibers can be visualized in humans with diffusion tensor MRI77. However, demonstrating functional integration currently requires more-invasive techniques.
Clinical imaging studies have shown that cell therapy in the human heart to date is hampered by low retention of transplanted cells. In patients with acute myocardial infarction, low retention (1.5%) was observed 2 h after intracoronary administration of 18F-FDG–labeled peripheral hematopoietic stem cells78. Intracoronary delivery of 111In-labeled peripheral blood–derived progenitor cells led to retention of ~6.9% in acute infarct patients and ~2.5% in a subgroup of chronic infarct patients79. Low viability of the infarcted myocardium and reduced coronary flow reserve were significant predictors of pro-angiogenic progenitor cell homing79.
Imaging studies have also revealed that retention in the ischemic myocardium depends on the delivery method. In the hematopoietic stem cell trial discussed above73, at 18 h after the procedure, myocardial retention was higher in the transendocardial group than in the intracoronary group. Transendocardial delivery also correlated with greater improvement in ventricular function 6 months after infusion. In a 5-year follow-up study80, cell therapy was associated with increased left ventricular ejection fraction, increased 6-minute-walk distance, and decreased N-terminal, B-type natriuretic peptide (a biomarker of heart failure). Left-ventricular ejection fraction improvement was greater in patients with higher myocardial homing of injected cells. These studies demonstrate the advantages of direct implantation compared with intravascular injection for cardiac cell therapies. However, direct implantation has several challenges, including minimizing invasiveness, increasing graft viability and promoting vascularization. Imaging researchers can contribute by developing guidance systems for cell delivery and by designing systems that integrate blood flow dynamics with tissue mechanics.
Systematic comparisons of different routes of administration have been carried out in animal models. MRI and PET imaging of acutely infarcted rats showed that direct injection afforded 14% retention, substantially greater than rates with injection into the left ventricular cavity (3.5%) or intravenous delivery (1.2%)81. Another biodistribution study found that a substantial number of bone marrow mononuclear cells injected into the rodent myocardium actually migrated to the bone marrow, liver and spleen82.
The use of imaging to reveal the low retention of transplanted cells provides a possible explanation for poor clinical results and suggests strategies for improving therapeutic efficacy. Retention has been increased by injecting cells in a hydrogel made of natural or synthetic materials. This approach has been tested in pigs using human MSCs expressing HSV1-tk and PET/CT imaging32(Fig. 3). Surprisingly, myocardial radiotracer (18FHBG) uptake was not elevated when 200 × 106human MSCs alone were directly injected into the myocardium, despite the grafts being readily detectable by histology. When the same number of cells was injected in Matrigel, signal-to-background ratio increased to 1.87, and increased further to 8.02 when 600 × 106 cells were administered. The relatively low sensitivity for PET detection in this study may relate to timing: imaging was done the same day as cell transplantation, so delivery of radiotracer required its diffusion into avascular cell clumps. Presumably, a vascularized graft would be more easily detected.
Once cells are successfully retained in the heart, the next challenge is the wave of cell death that is initiated by ischemia, loss of survival signals due to matrix detachment, and the toxicity of reactive oxygen species and cytokines2. In practice, one can consider cell loss over the first few hours after injection to be a retention problem, whereas loss after that is likely due to death. Imaging of long-term survival of transplanted cells in the heart has not been tested in humans, but animal studies using indirect labels show the feasibility of monitoring cells for as long as 15 weeks83. For example, cell tracking in the infarcted mouse heart with the MRI reporter gene ferritin showed that the signal attenuation by ferritin-containing grafts persists for at least one month and correlates well with histologically determined graft size57, 58 (work of A.V.N., C.E.M. and colleagues). One drawback of ferritin is a relatively low sensitivity, requiring development of complicated, time-consuming imaging protocols. A more sensitive MRI reporter gene, and particularly one that yielded a gain of signal rather than signal attenuation, would be a significant advance in this field.
Assessment of the host environment and response to various therapies is conducted routinely in cardiology. Myocardial perfusion and infarct size are measured using US Food and Drug Administration (FDA)-approved gadolinium-chelate contrast agents and MRI, and this approach has been adapted for clinical trials of cell therapies. Clinical trials of autologous c-kit+ cardiac cells or cardiosphere-derived cells84, 85 transplanted into patients with ischemic heart failure86 showed a reduction in the zone of delayed gadolinium enhancement and an increase in viable tissue volume, which were described as possible improvements in heart contractility and shrinkage of the infarct scar. However, the reduction in gadolinium accumulation in the interstitial space and contrast enhancement could also have been the consequence of reduced vessel permeability leading to reduced extravasation or of enhanced lymphatic drainage of injured tissue. Although the increased mass of viable tissue is consistent with regeneration, it is also consistent with hypertrophy of pre-existing cardiomyocytes.
Quantitative imaging, such as diffusion-tensor imaging (DTI), can be used to assess the fiber architecture in remodeled myocardium in animals and humans77, 87, 88. Myocardial fibers have a complicated helical structure that is critical for efficient contractile and conductive function of the heart. DTI tractography has shown a smooth transition in fiber orientation from epicardium to endocardium in the healthy heart and severe disruption of myofiber architecture after infarction in different species (human, sheep, rat)88. Preliminary DTI results in a murine infarct model77 suggest that this technique has potential for assessing structural integration and alignment of transplanted cells with host myocardium in humans.
Finally, some aspects of functional integration are accessible by imaging. Global heart function, regional contractility and wall motion were imaged by high-resolution MRI after transplantation of human ESC-derived cardiomyocytes to the rat heart89 (work of A.V.N., C.E.M. and colleagues). It is important to determine whether the graft is electrically connected with the host tissue and whether transplanted cells create areas of arrhythmia. Recently, a genetically encoded Ca2+-indicator fused with green fluorescent protein was used to demonstrate that human ESC–derived cardiomyocyte grafts are electrically coupled with host myocardium of the guinea pig90 and of a non-human primate91 (work of C.E.M. and colleagues), indicating that this cell type meets physiological criteria for true heart regeneration. However, new imaging techniques for evaluating electro-mechanical coupling of grafted cells in patients’ hearts are needed.
In summary, imaging has played an important role in advancing cell-based cardiac repair, providing information on transplanted cell retention, viability, proliferation, graft size, integration with the host tissue and restoration of myofiber architecture, in addition to providing evidence of enhanced cardiac structure and function. What is needed are new imaging probes capable of detecting dynamic changes in grafted cells (i.e., that label live transplanted cells and quickly disappear after cell death) and techniques for genetically labeling cells for long-term tracking in humans.
Imaging pancreatic islets
Diabetes mellitus, a metabolic disease caused by insufficient insulin signaling, is traditionally divided into types I and II, with type I resulting from autoimmune destruction of pancreatic beta cells92 and type II resulting from resistance to insulin93. In its later stages, type II diabetes is thought to result in beta-cell death, adding insulin deficiency to the problem of insulin resistance. Transplantation of pancreatic islets or beta cells emerged as a promising treatment for diabetes with the development of the Edmonton Protocol, which allowed patients to attain insulin independence through transplantation of cadaveric islets into the liver94. However, insulin independence proved to be transient in most recipients95. The decline is attributed primarily to drastic graft loss—up to 60% during the early post-transplant period, as shown by histology in animal models96, and nearly 100% in patients with long duration of type 1 diabetes (determined at autopsy)97. A noninvasive approach to assess cell viability and function over time is urgently needed to investigate the immunological and nonimmunological factors underlying the loss of transplanted cells98, whether the cells are cadaveric islets94 or beta cells derived from human pluripotent stem cells99, 100, 101, 102.
In the first clinical trial of labeled islets, no correlation was seen between the number of injected islets and the SPIO signal on MRI images103. However, a subsequent trial using ferucarbotran-labeled islets demonstrated a 60% decrease in graft volume one week after transplantation in eight patients by MRI104 (Fig. 3). Although no independent method was used to confirm this finding, significant C-peptide levels and near-normal HbA1c values were achieved in these patients with a substantial reduction of insulin dose, suggesting that the remaining grafts were functional104. Two clinical trials using PET imaging of 18F-FDG–labeled islets showed early engraftment in the liver105, 106. However, only 50–70% of the expected signal was observed, suggesting substantial islet loss during the procedure. Importantly, no side effects attributed to islet labeling by 18F-FDG were detected.
These clinical trial results, although promising, leave much to be desired. An ideal imaging modality would detect retention in the liver by a quantitative correlation between the number of islets present and the number of hyper/hypointense voxels or ‘hot spots’. This has been demonstrated in small animals107 (work of A.M. and colleagues) but not yet in humans103. The detection limit of intrahepatically transplanted islets using clinical magnetic resonance scanners is not known, although studies in rodents have detected ~75 SPIO-labeled islets scattered throughout the liver using a 4.7 T scanner107, 108 (work of A.M. and colleagues) and 200 islets under the kidney capsule using a 1.5 T clinical magnet109. Long-term survival of islets in the human liver is also difficult to assess. Although the greatest clinical need is for monitoring during the early engraftment period (2–5 wks) when graft loss is the highest, long-term monitoring would provide valuable information about recurrent autoimmunity and rejection and/or the options in modulating the regimen of immunosuppression in response to such recurring events. This is not possible with 18F-FDG–labeled islets owing to the short half-life of 18F and rapid washout. Finally, islets transplanted in the liver suffer from hypoxia, which could result in impaired insulin secretion. There are no established clinical methods to evaluate islet graft function noninvasively.
The limitations of current islet imaging approaches are due in part to the unique, complex structure of pancreatic islets, which consist of four cell types (beta cells, alpha cells, delta cells and pancreatic polypeptide-producing cells)110. Labeling of this rather large cluster of cells is nonspecific and occurs only by diffusion. In the case of SPIO nanoparticles, 12–48 h are required to achieve a concentration of 2–12 pg/cell103, 108, 111 (work of A.M. and colleagues). In vivo, quantification of SPIO-labeled grafts is complicated by the presence of susceptibility artifacts, the inability to distinguish single islets from islet clusters and low signal-to-noise ratios between the grafts and liver parenchyma.
Several emerging strategies, which have been demonstrated in animals, may help to address the remaining challenges in clinical islet imaging. The issue of islet quantification is being explored using ‘off-resonance’ techniques, such as automatic quantitative ultrashort echo time imaging, which provide positive contrast. A study in rats showed high positive contrast from SPIO-labeled islets on three-dimensional dual echo ultrashort echo time images and suppression of the signal from liver and small vessels, allowing for automatic quantification. Quantification of hyperintense pixels correlated with the number of injected IEQs due to the uniform suppressed background and high contrast and appears to be superior to standard imaging and manual counting methods112.
Gadolinium chelates used in MRI have potential disadvantages, including risk of nephrogenic systemic fibrosis in patients with renal insufficiency113 and obscuring of the imaging signal in patients with hemosiderosis114. Instead, fluorine-19 MRI of perfluorocarbon-labeled islets appears to be a more promising approach because of lack of toxicity and the absence of background fluorine-19 signal in tissues115, 116, 117. Human islets have been incorporated into microcapsules that contain perfluorocarbon, and the microcapsules were transplanted in mice and visualized by19F MRI and CT118, 119. Perfluorocarbon labeling of the microcapsules did not interfere with long-term insulin secretion in vivo.
Long-term tracking of human SPIO-labeled islet grafts for up to 188 days has been shown in diabetic immune-compromised mice108, 111, non-human primates120 (work of A.M. and colleagues) and swine107. The feasibility of this approach for long-term tracking of transplanted islets in humans has already been demonstrated with data going out to 24 weeks104, although quantitative correlation between the number of infused and remaining islets was lacking and should be addressed in future trials.
The possibility of evaluating graft function by MRI has also been suggested in preclinical studies by quantifying zinc co-released with insulin in response to glucose challenge from endogenous islets using the Zn(2+)-responsive T1 agent, GdDOTA-diBPEN121. This approach offers the exciting potential for deep-tissue monitoring of beta cell function after islet transplantation in patients.
Finally, combination of several imaging modalities could provide considerably more information regarding graft fate than a single approach. To that end, a trimodal microcapsule for islet labeling containing gold nanoparticles functionalized with DTDTPA (dithiolated diethylenetriaminepentaacetic acid) and gadolinium chelates was developed for co-encapsulation with islets using protamine sulfate as a clinical-grade alginate cross-linker. Microencapsulation decreased islet rejection due to the presence of a semi-permeable membrane that prevents the passage of antibodies but allows the passage of cell-derived factors, including insulin. Intra-abdominally engrafted islets were detectable by multimodal imaging (i.e., MRI, CT and ultrasound)114, 122. Inclusion of perfluorocarbon emulsions in alginate constructs for islet encapsulation118, 119 also allows for their detection by 19F MRI, ultrasound and CT (in the case of perfluorooctyl-bromide) and does not alter the permeability of the capsules or affect islet function. These new approaches not only improved islet protection but also allowed for MRI of transplanted islets with positive contrast and for islet infusions under fluoroscopic and ultrasound guidance, which are common techniques in clinical practice and could ease clinical translation. However, wide clinical applicability of these methods would require the use of approved materials for islet labeling and further optimization.
In summary, the ability to image transplanted pancreatic islets or islet cells provides a unique opportunity for clinicians to monitor their distribution, survival and function in the short and long term. However, algorithms for precise quantification of infused and engrafted islets are still under development, as are probes that are quantitative, nontoxic and long retained by islet cells. Furthermore, the described methods label islet cells and are not specific for beta cells.
Imaging brain repair
Acute brain injury from stroke and chronic neurodegenerative disease are prevalent in our aging societies and increasingly burden healthcare systems. The human brain has little or no regenerative capacity. Although endogenous neurogenesis has been described in the injured human brain123, it is insufficient to promote significant repair. The possibility of cell therapy in the brain arose with the development of methods for MRI-guided stereotactic delivery directly into the parenchyma124, 125, 126.
The most striking results in clinical cell therapy for the brain came in the mid-1990s in trials for Parkinson’s disease127. The disease is caused by degeneration of dopaminergic neurons, which have their cell bodies in the substantia nigra and project axons to the caudate and putamen. Implanting fetal neural tissue grafts ‘ectopically’ in the caudate and putamen improved outcomes in patients, whereas implants in the substantia nigra had no benefit128, 129. In cell therapy for stroke, minor improvements in patient outcomes were achieved by brain transplantation of neuroteratocarcinoma-derived neurons130, 131. This approach aimed both to replace lost neurons and to provide trophic factors that promote angiogenesis and plasticity in existing neural networks. Re-myelination was achieved in patients with Pelizaeus-Merzbacher disease through implantation of neural stem cells into the brain132. In patients with multisystem atrophy, infusion of unlabeled autologous MSCs into the carotid artery led to increased uptake of 18F-FDG in various areas of the brain compared to the control group and improvement in functional neurological outcome over a period of 1 year133. Of concern, however, diffusion-weighted MRI detected areas of microinfarction, presumably due to MSC clumping in vasculature. Longitudinal imaging studies are an essential part of cell therapy clinical trials to monitor damage and toxicity.
Efficacious cell therapy requires delivery to the appropriate region of the brain. Thus, imaging of the retention and biodistribution of transplanted cells in the human and animal brain is important for interpreting therapeutic outcomes. In one early-phase clinical study, SPIO-labeled, autologous brain-derived cells were transplanted into a patient with an open brain trauma40. MRI at 3T could delineate the damaged parenchyma, the implantation site of the labeled cells, and movement of the label through the parenchyma for 21 days (Fig. 3); the hypointense voxels disappeared at seven weeks after implantation40. Imaging has also revealed that intracerebral retention of transplanted cells requires delivery into the parachyma rather than into the circulation134. In stroke patients who received 99mTc-labeled bone marrow mononuclear cells, only a very small proportion of intravenously or intra-arterially injected cells invaded the central nervous system, and most cells were cleared within 24 h135. Nevertheless, intravenous cell injection is the most common route of delivery for acute brain injury, with some evidence of therapeutic efficacy134, 136. The lack of intracerebral penetration and retention of MSCs after intravenous injection indicates that any observed efficacy is not mediated by cell replacement.
Imaging of cell proliferation and differentiation has not been demonstrated in the human brain. A major obstacle has been the development of suitable probes that can cross the blood-brain barrier. Potential indicators of integration, such as brain plasticity and cell differentiation, can be assessed by functional MRI137 or MR spectroscopy138, 139, respectively. Re-myelination after neural stem cell implantation in patients with Pelizaeus-Merzbacher disease was evident in the restoration of the tissue microenvironment, as indicated by an increase in fractional anisotropy and reduced radial diffusivity on diffusion MRI132. The restoration of neurochemical tone, as in the case of Parkinson’s disease, can be determined reliably and quantitatively by PET using the dopamine D2 receptor-binding agent, raclopride76, 140. Reduction of raclopride binding correlates to the presence of endogenous dopamine, and this approach has revealed the function of transplants even 15 years after transplantation141. Indeed, PET imaging of neurotransmitter activity also revealed that graft-induced dyskinesia is mediated by serotonergic neurons contained within the graft128, 142. Paracrine and juxtacrine pathways, such as those discussed for the heart, may also play a role in the clinical benefits. PET ligands targeting receptors of paracrine factors, such as the TrkB receptor, are being developed for brain imaging in rodents143, and should eventually allow investigation of paracrine pathways in patients.
As with cell therapies for many organs, imaging of the brain is essential for evaluating the tissue environment before and after treatment. MRI and PET scans can reveal regional damage, structural and functional connectivity, and metabolic and vascular changes131, 144, 145. Incorporation of non-invasive imaging into clinical trials will further benefit the safety assessment (e.g., small bleeds, edema) and quality control (e.g., location of cells) of cell delivery. The host inflammatory response in patients can be monitored using 11C-PK11195 PET, which binds to the peripheral benzodiazepine receptor on activated microglia146, as well as ultrasmall SPIO uptake into peripheral macrophages detectable by MRI147. In rodents, an endogenous neural stem cell response to damage and cell therapy can potentially be monitored using the proliferation marker 3′-deoxy-3′-18F-fluoro-L-thymidine (18F-FLT)148, 149, whereas regional changes in metabolic activity indicating an increase in cellular density can be assessed in the human brain using FDG-PET150.
Although systemic cell administration or intracerebral implantation of cell suspensions can potentially improve behavior, repair of large cavities in damaged tissue is still problematic151. In these cases a scaffolding support is required152; various biomaterials can serve this purpose, and future studies are needed to determine the clinical efficacy of this approach153. The most appropriate cell types may be fetal neural stem cells, which are positionally specified to develop into a particular type of tissue, or pluripotent stem cell–derived neural stem cells, which require positional specification to differentiate into cells of the appropriate brain region. However, the skull restricts delivery of large constructs to the brain. To avoid tissue damage, presurgical planning for each subject based on imaging is essential to determine both the volume and location of injection154. In one study in rats, imaging enabled visualization of scaffolding material in the brain, although the survival and distribution of implanted cells was not determined155. Monitoring of scaffold biodegradation and formation of new tissue are other unsolved problems. More sophisticated imaging approaches are needed to visualize the cell and biomaterial components independently156.
In summary, readily available clinical imaging tools used to diagnose or monitor disease can in many cases be applied to measure the therapeutic efficacy of cell therapy in the brain. However, before such therapies could be widely adopted, it will be essential to devise new, robust imaging technologies. Current techniques, such as 99Tc SPECT imaging, are adequate to determine short-term, labeled-cell infiltration in the human brain but not to assess precise anatomical distribution or long-term survival (with the exception of dopaminergic neurons). A reasonable goal would be the capability to measure cell location at <1 mm accuracy, cell survival for >1 month, and the interaction of a few hundred transplanted cells with the microenvironment.
Future directions
The path to clinical translation of regenerative therapies is difficult. For researchers intrepid enough to consider jumping into this area, Box 3 offers suggestions on developing programs that include imaging. Incorporating imaging in a cell therapy requires preparation of additional data for Investigational New Drug (IND) submissions. Specific recommendations are presented in Box 4.
Box 3. A framework for implementing imaging in regenerative medicine.
Some specific steps for researchers wishing to incorporate imaging into cell therapies on a path to clinical translation are provided below.
Teamwork
Establish a team that includes stem cell biologists, physiologists, chemists, specialty clinicians, radiologists and imaging scientists to determine the most appropriate imaging strategy.
Regulatory requirements
Learn the requirements by communicating early and often with overseeing regulatory agencies. Clearance from an institutional review board may be required. A consultant familiar with imaging in IND submissions may be helpful.
Standard operating procedures
These are required for production, labeling and handling of cells, data collection, in vivo imaging and analysis, histological verification and clinical outcome measures. Document all results and do not deviate from standard operating procedures once established.
In vivo imaging
Ensure that labeled cells are detectable in vivo with high sensitivity and specificity using clinical scanners. Understanding the number of cells/voxel that migrate to targeted regions is important in determining preclinical doses, schedules and administration routes. It is impossible and unnecessary to track single cells.
Scale up
The ability to move a labeling technique from the bench to a clinical Good Manufacturing Practice facility may be difficult, but it is essential for producing large numbers of cells for clinical trials. Phase 1 trials that include dose escalation should also be used to determine delivery approaches, safety, toxicity and maximum tolerated dose of the labeled cells.
Box 4. Recommendations for preclinical evaluation of imaging methods.
If imaging is to be used in a cell-therapy clinical trial, it is necessary to compile appropriate data for presentation during pre-IND discussions with the regulatory agency. These data should include the following.
In vitro tests of direct or indirect labeling methods should be compared to unlabeled cells and include determination of labeling efficiency; label concentration; rate of cell death; short and longer-term proliferation capacity; differentiation capacity; migration capacity; immunogenicity in a mixed lymphocyte reaction; and surface markers.
It is important to document that labeling does not change cell potency. For cells whose function is secretory, potency can be defined as hormone, neurotransmitter, cytokine/chemokine or growth factor release. For mechanically active cells, such as cardiomyocytes, potency includes electrical and mechanical activity. Gene expression profiling has not been required or routinely recommended to evaluate direct labeling methods because of potential variability between donor cells and uncertainty in how these data correlate with potency.
For indirect labels, it may be necessary to determine the chromosomal location of the label to avoid proximity to oncogenes. If a suicide or therapeutic gene is inserted with the reporter gene, expression of both genes must be documented, along with efficacy of the suicide gene. Long-term passaging of cells is needed to ensure stability of transgenes and lack of malignant potential.
An estimate of the dilution rate of the label over multiple cell divisions can be provided by pulse-chase experiments (i.e., cell labeling followed by expansion).
Sensitivity to the minimal number of cells that can be detected by clinical scanners can be estimated by in vitro serial dilutions of labeled cells if these studies are carried out with clinically relevant voxel sizes and acquisition times.
Preliminary in vivo imaging studies for detecting and tracking cell fate are useful to determine the biodistribution of the cells in normal and experimental models. It is necessary to determine whether preclinical studies use xenogeneic (i.e., human cells into mouse), allogeneic or autologous transplantation. Histological analysis of multiple organs should be performed to assess inflammatory responses and to identify labeled cells in targeted and nontargeted tissues.
Depending on the disease model, dose-escalation studies over three dose levels (typically spanning at least a tenfold range) may be required to determine safety and efficacy of transplanted or infused labeled cells. Proposed cell dosing, dosing schedule, experimental model systems and required validation/toxicity outcomes (i.e., histology, serum chemistries including renal and liver function, blood counts, functional measures) should be included as part of the discussion with a regulatory agency at a pre-IND conference.
There are many examples of experimental labels for tagging cells, but few have made it into clinical trials157, 158, and fewer still are used routinely in the clinic. Researchers who synthesize novel agents do not usually pursue regulatory approval because of lack of funding158 or insufficient projected markets. However, the demand for imaging technologies to evaluate the host and to track transplanted cells is likely to grow over the coming decades. For those who wish to undertake clinical translation of novel imaging agents, some relevant considerations are summarized in Box 5. Guidelines for bringing a novel imaging agent to the clinic specify rigorous preclinical safety testing and validation for regulatory approval as drugs. Because nanoparticles or MRI contrasts agents may require nanogram to microgram concentrations in cells, their first use in humans must be a phase 1 safety study. In comparison, radiolabeled agents are used in far lower concentrations (femtograms/cell) and are dosed based on radiosensitivity rather than chemical concentration. Introduction into humans can be performed through an exploratory IND submission in a phase 0 clinical trial to determine safety, pharmacokinetics and pharmacodynamics158. The exploratory trial design is much less costly, allowing rapid screening of radioactively tagged agents developed under good laboratory practice standards.
Box 5. Considerations for the development of novel cell-tracking agents.
Before undertaking the development of a novel imaging agent for cell tracking, investigators should understand the time commitment and funding that are required for clinical translation. The following points should be considered.
Assemble a multidisciplinary team.
Confirm that the labeling method is amenable to scale-up of the cell product in a Good Manufacturing Practice facility.
Ensure high sensitivity of detection by maximizing the concentration of label in cells (consistent with safety) and minimizing the numbers of cells/voxel for detection in vivo.
Evaluate whether cellular dynamics (cell death, proliferation and interaction with the host), cellular kinetics (movement of cells in the body) and volume of distribution of the cells may affect efficacy.
Strive to limit transfer of the labeling agent from therapeutic cells to macrophages or bystander cells. Develop methods to turn off the signature of the label after engulfment.
Resist the temptation to switch to an improved agent or technique after starting preclinical experiments for an IND submission as such modifications will likely require that experiments be repeated, resulting in delays and increased costs.
Note that regulatory agencies require separate preclinical evaluation of the labeling agent and technique for each cell type and disease model.
A problem common to all contrast agents is that the imaging signal does not distinguish live and dead cells. Reporter genes represent a possible solution, although genetic modification brings additional layers of safety and regulatory concerns. With a concerted effort, it should be possible to assess the risks of knocking reporter genes into safe-harbor loci in only a few years. We encourage researchers, regulatory authorities and funding agencies to work together to solve this tractable problem. An alternative solution would be the development of novel agents that label only live cells, disappearing quickly after cell death. In addition, most cell therapies would greatly benefit from novel ligands or probes with increased imaging specificity to detect dynamic changes in the heterogeneous, injured host environment. Understanding the role of the host environment will be crucial to improving the efficacy of cell therapies.
Many results in animal models are not predictive of the human response. For example, therapeutic results in rodents do not mean that the same number of cells/kg would be safe and efficacious in patients. Similarly, imaging results demonstrated in rodent models do not necessarily translate into the clinic, primarily because of limitations in clinical scanner technology—spatial resolution or voxel size, instrumentation (i.e., radiofrequency coils and magnetic field strength), patient motion and image acquisition times. Investigating teams must be aware that the translation of regenerative therapies from bench to bedside is research, so they are advised to be patient.
Acknowledgments
The authors would like to acknowledge Kristine Evers for proofreading of the manuscript and the following grant support: M.M. was supported by the Commonwealth of Pennsylvania, Department of Health (4100061184), NINDS (R01NS082226) and NIBIB (1R01EB016629). C.E.M. was supported by US National Institutes of Health (NIH) grants P01HL094374, R01HL084642, U01HL100405 and P01GM81619. J.A.F. was supported in part by the intramural research program in the Clinical Center and National Institutes of Biomedical Imaging and Bioengineering at the US National Institutes of Health. A.M. was supported in part by NIH grant R24 DK096465.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Daar AS, Greenwood HL. A proposed definition of regenerative medicine. J Tissue Eng Regen Med. 2007;1:179–184. doi: 10.1002/term.20. [DOI] [PubMed] [Google Scholar]
- 2.Robey TE, Saiget MK, Reinecke H, Murry CE. Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol. 2008;45:567–581. doi: 10.1016/j.yjmcc.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Almeida PE, van Rappard JR, Wu JC. In vivo bioluminescence for tracking cell fate and function. Am J Physiol Heart Circ Physiol. 2011;301:H663–H671. doi: 10.1152/ajpheart.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen PK, Riegler J, Wu JC. Stem cell imaging: from bench to bedside. Cell Stem Cell. 2014;14:431–444. doi: 10.1016/j.stem.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roura S, Galvez-Monton C, Bayes-Genis A. Bioluminescence imaging: a shining future for cardiac regeneration. J Cell Mol Med. 2013;17:693–703. doi: 10.1111/jcmm.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Progatzky F, Dallman MJ, Lo Celso C. From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus. 2013;3:20130001. doi: 10.1098/rsfs.2013.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigert R, Porat-Shliom N, Amornphimoltham P. Imaging cell biology in live animals: ready for prime time. J Cell Biol. 2013;201:969–979. doi: 10.1083/jcb.201212130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu E, Chen WY, Gu J, Burridge P, Wu JC. Molecular imaging of stem cells: tracking survival, biodistribution, tumorigenicity, and immunogenicity. Theranostics. 2012;2:335–345. doi: 10.7150/thno.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagers AJ. The stem cell niche in regenerative medicine. Cell Stem Cell. 2012;10:362–369. doi: 10.1016/j.stem.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 10.Bianconi E, et al. An estimation of the number of cells in the human body. Ann Hum Biol. 2013;40:463–471. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 11.Fischer UM, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harting MT, et al. Intravenous mesenchymal stem cell therapy for traumatic brain injury. J Neurosurg. 2009;110:1189–1197. doi: 10.3171/2008.9.JNS08158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everaert BR, et al. Multimodal in vivo imaging reveals limited allograft survival, intrapulmonary cell trapping and minimal evidence for ischemia-directed BMSC homing. BMC Biotechnol. 2012;12:93. doi: 10.1186/1472-6750-12-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsden CM, et al. Stem cells in retinal regeneration: past, present and future. Development. 2013;140:2576–2585. doi: 10.1242/dev.092270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olivetti G, Capasso JM, Sonnenblick EH, Anversa P. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardialinfarction in rats. Circ Res. 1990;67:23–34. doi: 10.1161/01.res.67.1.23. [DOI] [PubMed] [Google Scholar]
- 16.Husser O, et al. Head-to-head comparison of 1 week versus 6 months CMR-derived infarct size for prediction of late events after STEMI. Int J Cardiovasc Imaging. 2013;29:1499–1509. doi: 10.1007/s10554-013-0239-1. [DOI] [PubMed] [Google Scholar]
- 17.McCall M, Shapiro AM. Update on islet transplantation. Interface Focus. 2012;3:20130001. [Google Scholar]
- 18.Moszczynska A, et al. Why is parkinsonism not a feature of human methamphetamine users? Brain. 2004;127:363–370. doi: 10.1093/brain/awh046. [DOI] [PubMed] [Google Scholar]
- 19.Kordower JH, et al. Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson’s disease. Mov Disord. 1998;13:383–393. doi: 10.1002/mds.870130303. [DOI] [PubMed] [Google Scholar]
- 20.Everall I, Barnes H, Spargo E, Lantos P. Assessment of neuronal density in the putamen in human immunodeficiency virus (HIV) infection. Application ofstereology and spatial analysis of quadrats. J Neurovirol. 1995;1:126–129. doi: 10.3109/13550289509111017. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R, et al. Global and regional putamen volume loss in patients with heart failure. Eur J Heart Fail. 2011;13:651–655. doi: 10.1093/eurjhf/hfr012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahrens ET, Bulte JW. Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol. 2013;13:755–763. doi: 10.1038/nri3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shapiro EM, Sharer K, Skrtic S, Koretsky AP. In vivo detection of single cells by MRI. Magn Reson Med. 2006;55:242–249. doi: 10.1002/mrm.20718. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro EM, et al. MRI detection of single particles for cellular imaging. Proc Natl Acad Sci USA. 2004;101:10901–10906. doi: 10.1073/pnas.0403918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahrens ET, Zhong J. In vivo MRI cell tracking using perfluorocarbon probes and fluorine-19 detection. NMR Biomed. 2013;26:860–871. doi: 10.1002/nbm.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arbab AS, Frank JA. Cellular MRI and its role in stem cell therapy. Regen Med. 2008;3:199–215. doi: 10.2217/17460751.3.2.199. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen PK, Lan F, Wang Y, Wu JC. Imaging: guiding the clinical translation of cardiac stem cell therapy. Circ Res. 2011;109:962–979. doi: 10.1161/CIRCRESAHA.111.242909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahnke H, Schaeffter T. Limits of detection of SPIO at 3.0 T using T2 relaxometry. Magn Reson Med. 2005;53:1202–1206. doi: 10.1002/mrm.20435. [DOI] [PubMed] [Google Scholar]
- 29.Rueger MA, Androutsellis-Theotokis A. Identifying endogenous neural stem cells in the adult brain in vitro and in vivo: novel approaches. Curr Pharm Des. 2013;19:6499–6506. doi: 10.2174/1381612811319360010. [DOI] [PubMed] [Google Scholar]
- 30.Arbab AS, et al. In vivo cellular imaging for translational medical research. Curr Med Imaging Rev. 2009;5:19–38. doi: 10.2174/157340509787354697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bar-Shir A, et al. Human Protamine-1 as an MRI reporter gene based on chemical exchange. ACS Chem Biol. 2013;9:134–138. doi: 10.1021/cb400617q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willmann JK, et al. Imaging gene expression in human mesenchymal stem cells: from small to large animals. Radiology. 2009;252:117–127. doi: 10.1148/radiol.2513081616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arbab AS, Liu W, Frank JA. Cellular magnetic resonance imaging: current status and future prospects. Expert Rev Med Devices. 2006;3:427–439. doi: 10.1586/17434440.3.4.427. [DOI] [PubMed] [Google Scholar]
- 34.Amsalem Y, et al. Iron-oxide labeling and outcome of transplanted mesenchymal stem cells in the infarcted myocardium. Circulation. 2007;116:I38–I45. doi: 10.1161/CIRCULATIONAHA.106.680231. [DOI] [PubMed] [Google Scholar]
- 35.de Vries IJ, et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol. 2005;23:1407–1413. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 36.Partlow KC, et al. 19F magnetic resonance imaging for stem/progenitor cell tracking with multiple unique perfluorocarbon nanobeacons. FASEB J. 2007;21:1647–1654. doi: 10.1096/fj.06-6505com. [DOI] [PubMed] [Google Scholar]
- 37.Zhen Z, Xie J. Development of manganese-based nanoparticles as contrast probes for magnetic resonance imaging. Theranostics. 2012;2:45–54. doi: 10.7150/thno.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bara C, et al. In vivo echocardiographic imaging of transplanted human adult stem cells in the myocardium labeled with clinically applicable CliniMACS nanoparticles. J Am Soc Echocardiogr. 2006;19:563–568. doi: 10.1016/j.echo.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Bulte JW. In vivo MRI cell tracking: clinical studies. AJR Am J Roentgenol. 2009;193:314–325. doi: 10.2214/AJR.09.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J, Zhou L, Wu FX. Tracking neural stem cells in patients with brain trauma. N Engl J Med. 2006;355:2376–2378. doi: 10.1056/NEJMc055304. [DOI] [PubMed] [Google Scholar]
- 41.Laskey WK, Feinendegen LE, Neumann RD, Dilsizian V. Low-level ionizing radiation from noninvasive cardiac imaging: can we extrapolate estimated risksfrom epidemiologic data to the clinical setting? JACC Cardiovasc Imaging. 2010;3:517–524. doi: 10.1016/j.jcmg.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Brenner W, et al. 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. J Nucl Med. 2004;45:512–518. [PubMed] [Google Scholar]
- 43.Wolfs E, et al. 18F-FDG labeling of mesenchymal stem cells and multipotent adult progenitor cells for PET imaging: effects on ultrastructure and differentiation capacity. J Nucl Med. 2013;54:447–454. doi: 10.2967/jnumed.112.108316. [DOI] [PubMed] [Google Scholar]
- 44.Wu C, et al. In vivo cell tracking via (1)(8)F-fluorodeoxyglucose labeling: a review of the preclinical and clinical applications in cell-based diagnosis and therapy. Clin Imaging. 2013;37:28–36. doi: 10.1016/j.clinimag.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 45.Terrovitis J, et al. Magnetic resonance imaging overestimates ferumoxide-labeled stem cell survival after transplantation in the heart. Circulation. 2008;117:1555–1562. doi: 10.1161/CIRCULATIONAHA.107.732073. [DOI] [PubMed] [Google Scholar]
- 46.Naumova AV, et al. Magnetic resonance imaging tracking of graft survival in the infarcted heart: iron oxide particles versus ferritin overexpression approach. J Cardiovasc Pharmacol Ther. 2014;19:358–367. doi: 10.1177/1074248414525999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pawelczyk E, et al. In vitro model of bromodeoxyuridine or iron oxide nanoparticle uptake by activated macrophages from labeled stem cells: implications forcellulartherapy. Stem Cells. 2008;26:1366–1375. doi: 10.1634/stemcells.2007-0707. [DOI] [PubMed] [Google Scholar]
- 48.Pawelczyk E, et al. In vivo transfer of intracellular labels from locally implanted bone marrow stromal cells to resident tissue macrophages. PLoS ONE. 2009;4:e6712. doi: 10.1371/journal.pone.0006712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walczak P, Kedziorek DA, Gilad AA, Barnett BP, Bulte JW. Applicability and limitations of MR tracking of neural stem cells with asymmetric cell division and rapid turnover: the case of the shiverer dysmyelinated mouse brain. Magn Reson Med. 2007;58:261–269. doi: 10.1002/mrm.21280. [DOI] [PubMed] [Google Scholar]
- 50.Arbab AS, et al. Characterization of biophysical and metabolic properties of cells labeled with superparamagnetic iron oxide nanoparticles and transfection agent for cellular MR imaging. Radiology. 2003;229:838–846. doi: 10.1148/radiol.2293021215. [DOI] [PubMed] [Google Scholar]
- 51.Thu MS, et al. Self-assembling nanocomplexes by combining ferumoxytol, heparin and protamine for cell tracking by magnetic resonance imaging. Nat Med. 2012;18:463–467. doi: 10.1038/nm.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rigol M, et al. Hemosiderin deposits confounds tracking of iron-oxide-labeled stem cells: an experimental study. Transplant Proc. 2008;40:3619–3622. doi: 10.1016/j.transproceed.2008.05.084. [DOI] [PubMed] [Google Scholar]
- 53.Schäfer R, et al. Labeling of human mesenchymal stromal cells with superparamagnetic iron oxide leads to a decrease in migration capacity and colony formation ability. Cytotherapy. 2009;11:68–78. doi: 10.1080/14653240802666043. [DOI] [PubMed] [Google Scholar]
- 54.Kostura L, Kraitchman DL, Mackay AM, Pittenger MF, Bulte JW. Feridex beling of mesenchymal stem cells inhibits chondrogenesis but not adipogenesis or osteogenesis. NMR Biomed. 2004;17:513–517. doi: 10.1002/nbm.925. [DOI] [PubMed] [Google Scholar]
- 55.Arbab AS, et al. Labeling of cells with ferumoxides-protamine sulfate complexes does not inhibit function or differentiation capacity of hematopoietic or mesenchymalstemcells. NMR Biomed. 2005;18:553–559. doi: 10.1002/nbm.991. [DOI] [PubMed] [Google Scholar]
- 56.Campan M, et al. Ferritin as a reporter gene for in vivo tracking of stem cells by 1.5-T cardiac MRI in a rat model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2011;300:H2238–H2250. doi: 10.1152/ajpheart.00935.2010. [DOI] [PubMed] [Google Scholar]
- 57.Naumova AV, et al. Ferritin overexpression for noninvasive magnetic resonance imaging-based tracking of stem cells transplanted into the heart. Mol Imaging. 2010;9:201–210. [PMC free article] [PubMed] [Google Scholar]
- 58.Naumova AV, et al. Quantification of MRI signal of transgenic grafts overexpressing ferritin in murine myocardial infarcts. NMR Biomed. 2012;25:1187–1195. doi: 10.1002/nbm.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gyöngyösi M, et al. Serial noninvasive in vivo positron emission tomographic tracking of percutaneously intramyocardially injected autologous porcine mesenchymal stem cells modified for transgene reporter gene expression. Circ Cardiovasc Imaging. 2008;1:94–103. doi: 10.1161/CIRCIMAGING.108.797449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yaghoubi SS, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6:53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, et al. Tracking stem cell therapy in the myocardium: applications of positron emission tomography. Curr Pharm Des. 2008;14:3835–3853. doi: 10.2174/138161208786898662. [DOI] [PubMed] [Google Scholar]
- 62.Yaghoubi SS, Campbell DO, Radu CG, Czernin J. Positron emission tomography reporter genes and reporter probes: gene and cell therapy applications. Theranostics. 2012;2:374–391. doi: 10.7150/thno.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tannous BA, et al. Metabolic biotinylation of cell surface receptors for in vivo imaging. Nat Methods. 2006;3:391–396. doi: 10.1038/nmeth875. [DOI] [PubMed] [Google Scholar]
- 64.So PW, et al. Efficient and rapid labeling of transplanted cell populations with superparamagnetic iron oxide nanoparticles using cell surface chemical biotinylation for in vivo monitoring by MRI. Cell Transplant. 2010;19:419–429. doi: 10.3727/096368910X498250. [DOI] [PubMed] [Google Scholar]
- 65.Ward KM, Aletras AH, Balaban RS. A new class of contrast agents for MRI based on proton chemical exchange dependent saturation transfer (CEST) J Magn Reson. 2000;143:79–87. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- 66.van Zijl PC, Yadav NN. Chemical exchange saturation transfer (CEST): what is in a name and what isn’t? Magn Reson Med. 2011;65:927–948. doi: 10.1002/mrm.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou J, Payen JF, Wilson DA, Traystman RJ, van Zijl PC. Using the amide proton signals of intracellular proteins and peptides to detect pH effects in MRI. Nat Med. 2003;9:1085–1090. doi: 10.1038/nm907. [DOI] [PubMed] [Google Scholar]
- 68.Chan KW, et al. MRI-detectable pH nanosensors incorporated into hydrogels for in vivo sensing of transplanted-cell viability. Nat Mater. 2013;12:268–275. doi: 10.1038/nmat3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou J, et al. Three-dimensional amide proton transfer MR imaging of gliomas: Initial experience and comparison with gadolinium enhancement. J Magn Reson Imaging. 2013;38:1119–1128. doi: 10.1002/jmri.24067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bar-Shir A, Liu G, Greenberg MM, Bulte JW, Gilad AA. Synthesis of a probe for monitoring HSV1-tk reporter gene expression using chemical exchange saturation transfer MRI. Nat Protoc. 2013;8:2380–2391. doi: 10.1038/nprot.2013.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TKmodified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seggewiss R, et al. Acute myeloid leukemia is associated with retroviral gene transfer to hematopoietic progenitor cells in a rhesus macaque. Blood. 2006;107:3865–3867. doi: 10.1182/blood-2005-10-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vrtovec B, et al. Comparison of transendocardial and intracoronary CD34+ cell transplantation in patients with nonischemic dilated cardiomyopathy. Circulation. 2013;128:S42–S49. doi: 10.1161/CIRCULATIONAHA.112.000230. [DOI] [PubMed] [Google Scholar]
- 74.Qiao H, et al. Death and proliferation time course of stem cells transplanted in the myocardium. Mol Imaging Biol. 2009;11:408–414. doi: 10.1007/s11307-009-0222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perin EC, et al. Imaging long-term fate of intramyocardially implanted mesenchymal stem cells in a porcine myocardial infarction model. PLoS ONE. 2011;6:e22949. doi: 10.1371/journal.pone.0022949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mendez I, et al. Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson’s disease. Brain. 2005;128:1498–1510. doi: 10.1093/brain/awh510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sosnovik DE, et al. Microstructural impact of ischemia and bone marrow-derived cell therapy revealed with diffusion tensor magnetic resonance imaging tractography of the heart in vivo. Circulation. 2014;129:1731–1741. doi: 10.1161/CIRCULATIONAHA.113.005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang WJ, et al. Tissue distribution of 18F-FDG-labeled peripheral hematopoietic stem cells after intracoronary administration in patients with myocardial infarction. J Nucl Med. 2006;47:1295–1301. [PubMed] [Google Scholar]
- 79.Schächinger V, et al. Pilot trial on determinants of progenitor cell recruitment to the infarcted human myocardium. Circulation. 2008;118:1425–1432. doi: 10.1161/CIRCULATIONAHA.108.777102. [DOI] [PubMed] [Google Scholar]
- 80.Vrtovec B, et al. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow-up. Circ Res. 2013;112:165–173. doi: 10.1161/CIRCRESAHA.112.276519. [DOI] [PubMed] [Google Scholar]
- 81.Elhami E, et al. Assessment of three techniques for delivering stem cells to the heart using PET and MR imaging. EJNMMI Research. 2013;3:72. doi: 10.1186/2191-219X-3-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van der Bogt KE, et al. Comparison of different adult stem cell types for treatment of myocardial ischemia. Circulation. 2008;118:S121–S129. doi: 10.1161/CIRCULATIONAHA.107.759480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Templin C, et al. Transplantation and tracking of human-induced pluripotent stem cells in a pig model of myocardial infarction: assessment of cell survival, engraftment, and distribution by hybrid single photon emission computed tomography/computed tomography of sodium iodide symporter transgene expression. Circulation. 2012;126:430–439. doi: 10.1161/CIRCULATIONAHA.111.087684. [DOI] [PubMed] [Google Scholar]
- 84.Malliaras K, et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction) J Am Coll Cardiol. 2014;63:110–122. doi: 10.1016/j.jacc.2013.08.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Makkar RR, et al. Intracoronary cardiosphere-derived cells for heart regenerationafter myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chugh AR, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126:S54–S64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McGill LA, et al. Reproducibility of in-vivo diffusion tensor cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2012;14:86. doi: 10.1186/1532-429X-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mekkaoui C, et al. Fiber architecture in remodeled myocardium revealed with a quantitative diffusion CMR tractography framework and histological validation. J Cardiovasc Magn Reson. 2012;14:70. doi: 10.1186/1532-429X-14-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 90.Shiba Y, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chong JJ, et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 93.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 94.Shapiro AM, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 95.Ryan EA, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 96.Barshes NR, Wyllie S, Goss JA. Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: implications for intrahepatic grafts. J Leukoc Biol. 2005;77:587–597. doi: 10.1189/jlb.1104649. [DOI] [PubMed] [Google Scholar]
- 97.Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10 (suppl 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rother KI, Harlan DM. Challenges facing islet transplantation for the treatment of type 1 diabetes mellitus. J Clin Invest. 2004;114:877–883. doi: 10.1172/JCI23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kroon E, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 100.Rezania A, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Blum B, et al. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van Hoof D, Liku ME. Directed differentiation of human pluripotent stem cells along the pancreatic endocrine lineage. Methods Mol Biol. 2013;997:127–140. doi: 10.1007/978-1-62703-348-0_10. [DOI] [PubMed] [Google Scholar]
- 103.Toso C, et al. Clinical magnetic resonance imaging of pancreatic islet grafts after iron nanoparticle labeling. Am J Transplant. 2008;8:701–706. doi: 10.1111/j.1600-6143.2007.02120.x. [DOI] [PubMed] [Google Scholar]
- 104.Saudek F, et al. Magnetic resonance imaging of pancreatic islets transplanted into the liver in humans. Transplantation. 2010;90:1602–1606. doi: 10.1097/tp.0b013e3181ffba5e. [DOI] [PubMed] [Google Scholar]
- 105.Eich T, Eriksson O, Lundgren T. Visualization of early engraftment in clinical islet transplantation by positron-emission tomography. N Engl J Med. 2007;356:2754–2755. doi: 10.1056/NEJMc070201. [DOI] [PubMed] [Google Scholar]
- 106.Eriksson O, et al. Positron emission tomography in clinical islet transplantation. Am J Transplant. 2009;9:2816–2824. doi: 10.1111/j.1600-6143.2009.02844.x. [DOI] [PubMed] [Google Scholar]
- 107.Evgenov NV, et al. In vivo imaging of immune rejection in transplanted pancreatic islets. Diabetes. 2006;55:2419–2428. doi: 10.2337/db06-0484. [DOI] [PubMed] [Google Scholar]
- 108.Evgenov NV, Medarova Z, Dai G, Bonner-Weir S, Moore A. In vivo imaging of islet transplantation. Nat Med. 2006;12:144–148. doi: 10.1038/nm1316. [DOI] [PubMed] [Google Scholar]
- 109.Tai JH, et al. Imaging islets labeled with magnetic nanoparticles at 1.5 Tesla. Diabetes. 2006;55:2931–2938. doi: 10.2337/db06-0393. [DOI] [PubMed] [Google Scholar]
- 110.Hellman B. Actual distribution of the number and volume of the islets of Langerhans in different size classes in non-diabetic humans of varying ages. Nature. 1959;184 (suppl 19):1498–1499. doi: 10.1038/1841498a0. [DOI] [PubMed] [Google Scholar]
- 111.Medarova Z, Evgenov NV, Dai G, Bonner-Weir S, Moore A. In vivo multimodal imaging of transplanted pancreatic islets. Nat Protoc. 2006;1:429–435. doi: 10.1038/nprot.2006.63. [DOI] [PubMed] [Google Scholar]
- 112.Crowe LA, et al. A novel method for quantitative monitoring of transplanted islets of langerhans by positive contrast magnetic resonance imaging. Am J Transplant. 2011;11:1158–1168. doi: 10.1111/j.1600-6143.2011.03559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grobner T. Gadolinium–a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21:1104–1108. doi: 10.1093/ndt/gfk062. [DOI] [PubMed] [Google Scholar]
- 114.Arifin DR, et al. Trimodal gadolinium-gold microcapsules containing pancreatic islet cells restore normoglycemia in diabetic mice and can be tracked by using US, CT, and positive-contrast MR imaging. Radiology. 2011;260:790–798. doi: 10.1148/radiol.11101608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Srinivas M, et al. Imaging of cellular therapies. Adv Drug Deliv Rev. 2010;62:1080–1093. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 116.Bonetto F, et al. A novel (19)F agent for detection and quantification of human dendritic cells using magnetic resonance imaging. Int J Cancer. 2011;129:365–373. doi: 10.1002/ijc.25672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bonetto F, et al. A large-scale (19) F MRI-based cell migration assay to optimize cell therapy. NMR Biomed. 2012;25:1095–1103. doi: 10.1002/nbm.2774. [DOI] [PubMed] [Google Scholar]
- 118.Barnett BP, et al. Fluorocapsules for improved function, immunoprotection, and visualization of cellular therapeutics with MR, US, and CT imaging. Radiology. 2011;258:182–191. doi: 10.1148/radiol.10092339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Barnett BP, et al. Use of perfluorocarbon nanoparticles for non-invasive multimodal cell tracking of human pancreatic islets. Contrast Media Mol Imaging. 2011;6:251–259. doi: 10.1002/cmmi.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barnett BP, et al. Magnetic resonance-guided, real-time targeted delivery and imaging of magnetocapsules immunoprotecting pancreatic islet cells. Nat Med. 2007;13:986–991. doi: 10.1038/nm1581. [DOI] [PubMed] [Google Scholar]
- 121.Lubag AJ, De Leon-Rodriguez LM, Burgess SC, Sherry AD. Noninvasive MRI of beta-cell function using a Zn2+-responsive contrast agent. Proc Natl Acad Sci USA. 2011;108:18400–18405. doi: 10.1073/pnas.1109649108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arifin DR, Bulte JW. Imaging of pancreatic islet cells. Diabetes Metab Res Rev. 2011;27:761–766. doi: 10.1002/dmrr.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Minger SL, et al. Endogenous neurogenesis in the human brain following cerebral infarction. Regen Med. 2007;2:69–74. doi: 10.2217/17460751.2.1.69. [DOI] [PubMed] [Google Scholar]
- 124.Donovan T, et al. Stereotactic MR imaging for planning neural transplantation: a reliable technique at 3 Tesla? Br J Neurosurg. 2003;17:443–449. doi: 10.1080/02688690310001613844. [DOI] [PubMed] [Google Scholar]
- 125.Kondziolka D, Steinberg GK, Cullen SB, McGrogan M. Evaluation of surgical techniques for neuronal cell transplantation used in patients with stroke. Cell Transplant. 2004;13:749–754. doi: 10.3727/000000004783983350. [DOI] [PubMed] [Google Scholar]
- 126.Muir KW, Sinden J, Miljan E, Dunn L. Intracranial delivery of stem cells. Transl Stroke Res. 2011;2:266–271. doi: 10.1007/s12975-011-0095-z. [DOI] [PubMed] [Google Scholar]
- 127.Lindvall O, et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science. 1990;247:574–577. doi: 10.1126/science.2105529. [DOI] [PubMed] [Google Scholar]
- 128.Politis M, et al. Serotonin neuron loss and nonmotor symptoms continue in Parkinson’s patients treated with dopamine grafts. Sci Transl Med. 2012;4:128ra41. doi: 10.1126/scitranslmed.3003391. [DOI] [PubMed] [Google Scholar]
- 129.Politis M, Piccini P. In vivo imaging of the integration and function of nigral grafts in clinical trials. Prog Brain Res. 2012;200:199–220. doi: 10.1016/B978-0-444-59575-1.00009-0. [DOI] [PubMed] [Google Scholar]
- 130.Kondziolka D, et al. Neurotransplantation for patients with subcortical motor stroke: a phase 2 randomized trial. J Neurosurg. 2005;103:38–45. doi: 10.3171/jns.2005.103.1.0038. [DOI] [PubMed] [Google Scholar]
- 131.Kondziolka D, et al. Transplantation of cultured human neuronal cells for patients with stroke. Neurology. 2000;55:565–569. doi: 10.1212/wnl.55.4.565. [DOI] [PubMed] [Google Scholar]
- 132.Gupta N, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee PH, et al. Autologous mesenchymal stem cell therapy delays the progression of neurological deficits in patients with multiple system atrophy. Clin Pharmacol Ther. 2008;83:723–730. doi: 10.1038/sj.clpt.6100386. [DOI] [PubMed] [Google Scholar]
- 134.Moniche F, et al. Intra-arterial bone marrow mononuclear cells in ischemic stroke: a pilot clinical trial. Stroke. 2012;43:2242–2244. doi: 10.1161/STROKEAHA.112.659409. [DOI] [PubMed] [Google Scholar]
- 135.Rosado-de-Castro PH, et al. Biodistribution of bone marrow mononuclear cells after intra-arterial or intravenous transplantation in subacute stroke patients. Regen Med. 2013;8:145–155. doi: 10.2217/rme.13.2. [DOI] [PubMed] [Google Scholar]
- 136.Battistella V, et al. Safety of autologous bone marrow mononuclear cell transplantation in patients with nonacute ischemic stroke. Regen Med. 2011;6:45–52. doi: 10.2217/rme.10.97. [DOI] [PubMed] [Google Scholar]
- 137.Lazaridou A, et al. fMRI as a molecular imaging procedure for the functional reorganization of motor systems in chronic stroke. Mol Med Rep. 2013;8:775–779. doi: 10.3892/mmr.2013.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ross BD, et al. In vivo magnetic resonance spectroscopy of human fetal neural transplants. NMR Biomed. 1999;12:221–236. doi: 10.1002/(sici)1099-1492(199906)12:4<221::aid-nbm582>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 139.Chung YL, et al. Profiling metabolite changes in the neuronal differentiation of human striatal neural stem cells using 1H-magnetic resonance spectroscopy. Neuroreport. 2013;24:1035–1040. doi: 10.1097/WNR.0000000000000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Piccini P, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat Neurosci. 1999;2:1137–1140. doi: 10.1038/16060. [DOI] [PubMed] [Google Scholar]
- 141.Kefalopoulou Z, et al. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: two case reports. JAMA Neurol. 2014;71:83–87. doi: 10.1001/jamaneurol.2013.4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Politis M, et al. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med. 2010;2:38ra46. doi: 10.1126/scitranslmed.3000976. [DOI] [PubMed] [Google Scholar]
- 143.Bernard-Gauthier V, Boudjemeline M, Rosa-Neto P, Thiel A, Schirrmacher R. Towards tropomyosin-related kinase B (TrkB) receptor ligands for brain imaging withPET: radiosynthesis and evaluation of 2-(4-[(18)F]fluorophenyl)-7,8-dihydroxy-4Hchromen-4-one and 2-(4-([N-methyl-(11)C]-dimethylamino)phenyl)-7,8-dihydroxy4H-chromen-4-one. Bioorg Med Chem. 2013;21:7816–7829. doi: 10.1016/j.bmc.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 144.Sztriha LK, et al. Monitoring brain repair in stroke using advanced magnetic resonance imaging. Stroke. 2012;43:3124–3131. doi: 10.1161/STROKEAHA.111.649244. [DOI] [PubMed] [Google Scholar]
- 145.Wardlaw JM, et al. Clinical relevance and practical implications of trials of perfusion and angiographic imaging in patients with acute ischaemic stroke: a multicentre cohort imaging study. J Neurol Neurosurg Psychiatry. 2013;84:1001–1007. doi: 10.1136/jnnp-2012-304807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gerhard A, Schwarz J, Myers R, Wise R, Banati RB. Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. Neuroimage. 2005;24:591–595. doi: 10.1016/j.neuroimage.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 147.Saleh A, et al. Iron oxide particle-enhanced MRI suggests variability of brain inflammation at early stages after ischemic stroke. Stroke. 2007;38:2733–2737. doi: 10.1161/STROKEAHA.107.481788. [DOI] [PubMed] [Google Scholar]
- 148.Rueger MA, et al. Noninvasive imaging of endogenous neural stem cell mobilization in vivo using positron emission tomography. J Neurosci. 2010;30:6454–6460. doi: 10.1523/JNEUROSCI.6092-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rueger MA, et al. Effects of minocycline on endogenous neural stem cells after experimental stroke. Neuroscience. 2012;215:174–183. doi: 10.1016/j.neuroscience.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 150.Stilley CS, et al. Changes in cognitive function after neuronal cell transplantation for basal ganglia stroke. Neurology. 2004;63:1320–1322. doi: 10.1212/01.wnl.0000140700.44904.53. [DOI] [PubMed] [Google Scholar]
- 151.Meltzer CC, et al. Serial [18F] fluorodeoxyglucose positron emission tomography after human neuronal implantation for stroke. Neurosurgery. 2001;49:586–591. doi: 10.1097/00006123-200109000-00011. [DOI] [PubMed] [Google Scholar]
- 152.Modo M, Ambrosio F, Friedlander RM, Badylak SF, Wechsler LR. Bioengineering solutions for neural repair and recovery in stroke. Curr Opin Neurol. 2013;26:626–631. doi: 10.1097/WCO.0000000000000031. [DOI] [PubMed] [Google Scholar]
- 153.Park KI, Teng YD, Snyder EY. The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat Biotechnol. 2002;20:1111–1117. doi: 10.1038/nbt751. [DOI] [PubMed] [Google Scholar]
- 154.Yannas IV. Emerging rules for inducing organ regeneration. Biomaterials. 2013;34:321– 330. doi: 10.1016/j.biomaterials.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 155.Bible E, et al. Attachment of stem cells to scaffold particles for intra-cerebral transplantation. Nat Protoc. 2009;4:1440–1453. doi: 10.1038/nprot.2009.156. [DOI] [PubMed] [Google Scholar]
- 156.Bible E, et al. The support of neural stem cells transplanted into stroke-induced brain cavities by PLGA particles. Biomaterials. 2009;30:2985–2994. [Google Scholar]
- 157.Bible E, et al. Non-invasive imaging of transplanted human neural stem cells and ECM scaffold remodeling in the stroke-damaged rat brain by (19)F- and diffusion-MRI. Biomaterials. 2012;33:2858–2871. doi: 10.1016/j.biomaterials.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Modo M, et al. Considerations for the clinical use of contrast agents for cellular MRI in regenerative medicine. Contrast Media Mol Imaging. 2013;8:439–455. doi: 10.1002/cmmi.1547. [DOI] [PubMed] [Google Scholar]
- 159.Josephson L, Rudin M. Barriers to clinical translation with diagnostic drugs. J Nucl Med. 2013;54:329–332. doi: 10.2967/jnumed.112.107615. [DOI] [PubMed] [Google Scholar]
- 160.Molina DK, DiMaio VJ. Normal organ weights in men: part II-the brain, lungs, liver, spleen, and kidneys. Am J Forensic Med Pathol. 2012;33:368–372. doi: 10.1097/PAF.0b013e31823d29ad. [DOI] [PubMed] [Google Scholar]
- 161.Molina DK, DiMaio VJ. Normal organ weights in men: part I-the heart. Am J Forensic Med Pathol. 2012;33:362–367. doi: 10.1097/PAF.0b013e31823d298b. [DOI] [PubMed] [Google Scholar]