

Abstract
Secretory phospholipase A2 (sPLA2) cleave phospholipids at sn-2 ester bonds, releasing lysophospholipids and fatty acids, and are over expressed in several pathologies, including inflammation, arthritis, sepsis and breast and prostate cancers. Herein we evaluated the therapeutic activity of liposomes engineered to be responsive to different sPLA2 isoforms compared to clinically used long-circulating (pegylated) sterically stabilized liposomes (SSL) in vitro and in vivo, and assess differences in role of sPLA2 in the mechanism of uptake and delivery of these nanoparticles. Exposing sPLA2 responsive liposomes (SPRL) to sPLA2 increased the release of intraluminal entrapped contents in a time-dependent manner that was inhibited by the sPLA2 inhibitor LY3117272. Treatment of prostate cancer cells with doxorubicin encapsulated in SSL and SPRL resulted in cytotoxicity in LNCaP, DU-145 and PC-3 cells lines comparable to free drug. Interestingly, cytotoxicity was not altered by sPLA2 inhibition. Tracking of drug and liposome delivery using fluorescence microscopy and flow cytometry, we demonstrated that drug uptake was liposome-dependent, as encapsulation of doxorubicin in SPRL resulted in 1.5 to 2-fold greater intracellular drug levels compared to SSL. Liposome uptake was cell-dependent and did not correlate to doxorubicin uptake; however, doxorubicin uptake was generally greatest in PC-3 cells, followed by DU-145 cells and then LNCaP cells. In almost all cases, uptake of one of our formulations, SPRL-E, was greater than SSL. The therapeutic activity of SPRL in vivo was demonstrated using a mouse xenograft model of human prostate cancer, which showed that doxorubicin entrapped within SPRL decreased tumor growth compared to SSL, suggesting that SPRL are more effective at slowing tumor growth than a SSL formulation similar to the FDA approved DOXIL™. Collectively, these data show the therapeutic activity of SPRL compared to SSL, yield insights into the mechanisms of action of these nanoparticles and suggest that SPRL could be useful for treatment of other pathologies that over express sPLA2.
Keywords: Secretory phospholipase A2, drug delivery, nanoparticle, liposome, targeting
Introduction
Pathological changes in physiology can be exploited to enhance the delivery and efficacy of drugs encapsulated in nanoparticles. Long-circulating nanoparticulate drug carriers, such as pegylated, sterically-stabilized liposomes (SSL), can stably entrap drug, alter drug disposition, improve activity and minimize toxicity [1]. However, the inability to accurately control drug-release kinetics has limited their clinical potential [2]. Secretory phospholipase A2 (sPLA2) degrade phospholipids at the sn-2 ester position to release a fatty acid and a lysophospholipid [3]. They require calcium for their enzymatic activity, and play diverse roles in several physiological functions, such as degradation of dietary phospholipids, defense against bacterial infections and arachidonic acid production [3, 4]. sPLA2 are also hypothesized to promote inflammatory diseases, such arthritis, atherosclerosis, sepsis, and cancers [5-10]. Recent evidence, including that from our laboratory, has hypothesized that the over expression of sPLA2 in these pathologies makes them good targets to control drug release from lipid-based nanoparticles, such as liposomes [11-13].
sPLA2 are becoming of note in cancer biology because recent studies show that these enzymes are over expressed in prostate [14-16] and several other cancers [17-22]. Moreover, it is usually the more aggressive, high-grade metastatic prostate tumors that over express sPLA2 and increased expression is inversely correlated to 5-year survival [5, 16, 23]. This suggests the hypothesis that targeting liposomes to interact with sPLA2 would be most beneficial in those patients with the worst clinical prognosis.
We recently hypothesized that the increased expression of sPLA2 in prostate cancer could be exploited by designing nanoparticle-based therapies that contain phospholipids targeted to sPLA2 (i.e., those with short sn-2 acyl chain and anionic polar head groups). In studying this hypothesis we tested 17 different formulations that differed in terms of the lengths of the fatty acyl chain present in the phospholipids, types of polar head groups and the presence and absence of polythethylene glycol and cholesterol [12, 13]. Our goal was to identify formulations that were selectively degraded by sPLA2 compared to clinically standard SSL. We used electrospray ionization-mass spectrometry to assess liposome degradation and release of 6-carboxyfluorescein (6-CF) as a surrogate marker of drug release. These studies resulted in the identification of sPLA2 responsive liposomes (SPRL), whose degradation and release of payload was increased significantly in the presence of exogenously added sPLA2. Two of these SPRL, termed E and G for the presence of ethanolamine and glycerol head groups, had significantly greater levels of lipid degradation and payload release compared to the all the others studied. Unfortunately, these studies were limited in that experiments were not performed in cells or animals, and that release was not correlated to any marker of therapeutic potential.
With the limitations of the previous study in mind, the goal of this study was to determine the therapeutic potential of SPRL in vitro and in vivo. We also identified sPLA2- and cell-dependent differences in the cytotoxicity, uptake and delivery of SRPL and SSL to investigate the mechanisms mediating the distinct behavior of these formulations. In particular, the ability, or lack thereof, of each formulation to be taken up by cells in culture, the correspondence to this uptake to drug delivery, and the translation of these data in vivo was assessed. These data were used to propose molecular interaction and distinct delivery mechanisms. It is our hope that understanding of these mechanisms gained will provide a basis for novel targeting strategies for cancers and other pathologies where sPLA2 are over expressed.
Materials and Methods
Materials
Distearoylphosphatidylcholine (DSPC), distearoylphosphatidylglycerol (DSPG), distearoylphosphatidylethanolamine (DSPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol) 2000 (DSPE-PEG) were purchased from Avanti Polar Lipids Inc (Alabaster, USA). 6-Carboxyfluorescein (6-CF) was purchased from Acros Organics (Geel, Belgium). Group III sPLA2 (bee venom sPLA2) was purchased from Cayman Chemical Company (Ann Arbor, MI). Sephadex G-75 was purchased from Pharmacia (Uppsala, Sweden). LY311727 was purchased from Tocris Bioscience (Minneapolis, MN). LNCaP, DU-145 and PC-3 cells were purchased from ATCC (Manassas, VA) and maintained in RPMI 1640, Eagle's Minimum Essential Medium, and F-12K medium supplemented with 10% (v/v) fetal bovine serum and antibiotics all purchased from ATCC as well. Doxorubicin was purchased from Toronto Research Chemicals (North York, ONT, Canada). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DiO) was purchased from Invitrogen (Grand Island, NY). All other reagents were of analytical quality.
Formulation of SSL and SPRL
SSL and SPRL were formulated as described previously [12, 13]. Based on our previous study we only used two types of SPRL, those containing 10% DSPE (SPRL-E) or 10% DSPG (SPRL-G). These SPRL were chosen as they had the greatest level of 6-CF release in the presence of sPLA2 and the greatest increase in lipid degradation in our previous studies [12, 13]. The individual formulations used in this study are described in Table 1.
Table 1. Liposome Composition.
| Formulation | Lipid Components | Cholesterol | PEG | Charged Lipid |
|---|---|---|---|---|
| SSL | 1,2-distearoyl-sn-glycero-3-phosphocholine 9 μmol/ml | 5 μmol/ml | 1 μmol/ml | N/A |
|
| ||||
| SPRL-E |
1,2-distearoyl-sn-glycero-3-phosphocholine 8 μmol/ml 1,2-distearoyl-sn-glycero-3-phosphoethanolamine 1 μmol/ml |
5 μmol/ml | 1 μmol/ml | DSPE
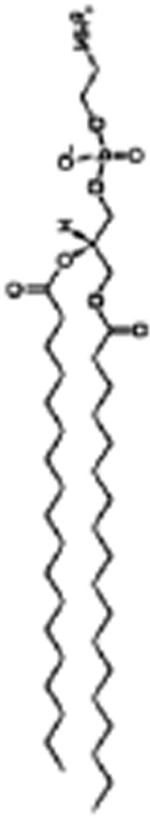
|
|
| ||||
| SPRL-G |
1,2-distearoyl-sn-glycero-3-phosphocholine 8 μmol/ml 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) 1 μmol/ml |
5 μmol/ml | 1 μmol/ml | DSPG
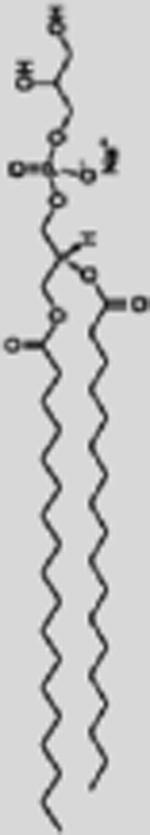
|
Preparation of 6-CF Liposomes
6-Carboxyfluorescein (6-CF, 100 mM) solution was prepared by dissolving 6-carboxyfluorescein in 5 mM Tris-HCl buffer (pH 7.4). Liposomal vesicles were prepared by hydration of thin-film followed by freeze-thawing and extrusion. Briefly, phospholipids, cholesterol or DSPE-PEG (total phospholipid: 10 μmol) in chloroform were mixed together, and dried under vacuum conditions for 25 minutes using a rotary evaporator to form a thin-film. The phospholipid film was rehydrated using the previously prepared 100 mM 6-CF solution. The dispersion then underwent 7 freeze–thaw cycles using liquid nitrogen and a 65°C water bath prior to at least five extrusions through double-stacked polycarbonate membranes (80 nm, Osmonics Inc., Minnetonka, MN) using a Lipex extruder (Northern Lipids Inc., Vancouver, BC, Canada) at 65°C. The final liposome sample was stored at 4°C and protected from light under a nitrogen atmosphere until use. Prior to use, total phospholipid was quantified using the Bartlett inorganic phosphate assay [12, 24]. Free 6-CF was removed by size exclusion chromatography using a Sephadex G-75 column. The mobile phase for these separations consisted of 5 mM Tris-HCl buffer (pH 7.4).
Dox-Loaded and DiO-Labeled Liposomes
Dox-loaded liposomes were prepared by remote loading using an ammonium sulfate gradient as described previously [1, 25]. Briefly, lipids and cholesterol in chloroform were mixed and subsequently dried using a rotary evaporator. The resulting lipid film was rehydrated in 250 mM ammonium sulfate. This dispersion then underwent 7 freeze-thaw cycles and at least five extrusions as described above. Following extrusion the liposomes were immediately placed on ice for 10 minutes then dialyzed overnight in an isotonic 10% (w/v) sucrose solution to remove excess, unencapsulated ammonium sulfate. Drug loading was performed the following day by adding doxorubicin to the dialyzed liposomes at a 0.2:1 molar ratio. The suspension was mixed and incubated for 1 hour at 65°C with periodic mixing and then immediately put on ice for 15 minutes. The loaded liposomes were then dialyzed overnight in a 10% (w/v) sucrose solution. Doxorubicin loading was quantified spectroscopically in acidified ethanol and lipid concentration determined using the Bartlett assay as described above [12, 24].
Fluorescent DiO-labeled liposomes were prepared according to the method of Kamps et. al., with slight alterations [26]. Briefly, lipids and cholesterol in chloroform were mixed and 1 mol% DiO was added before the solution was evaporated to a lipid film. The resulting film was then rehydrated in PBS or ammonium sulfate depending on whether doxorubicin would subsequently be loaded. The rest of the procedure was the same as above.
6-CF Release Assay
6-CF release from liposomes was determined as previously described [12]. Briefly, liposomal samples (0.05 μmol/ml) were incubated at 37°C in the presence and absence of Group III sPLA2 (2.5 μg/ml) and 100 μM LY311727 in F-12K medium supplemented with 10% FBS. Fluorescent intensity of 6-CF was measured using a Bio-Tec synergy HT spectrofluorometer (BIO-TEK Instruments Inc, Winooski, VT) at excitation and emission wavelengths of 480 and 510 nm, respectively. The time points used to determinate fluorescence were 1, 2, 4, 8, 12, 24, 36, 48, 72 and 108 hours. After determining initial fluorescence at each time point, 10% (v/v) of Triton X-100 was added to the samples to disrupt liposomes and permit calculation of total 6-CF. Percentage of 6-CF leakage was calculated by the equation:
where Ft represents the fluorescent intensity (FI) at a specific time point and F0 represents FI at time zero. FTriton represents FI after addition of Triton x-100.
Measurement of MTT Staining
The staining of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was used as an indicator of toxicity and corroborated with phase contrast microscopy. Cells were seeded in 96 well plates at 20-30,000 cells per well depending on cell type. After 24 hours cells were incubated with 2.5 μM doxorubicin or the liposomal equivalent and either 0 or 100 μM LY311727. A concentration of 2.5 μM was used as this was the dose that resulted in ∼50 to 80% cell kill after 72 hours in these cell lines (supplemental data). At 24, 48 and 72 hours 0.25 mg/ml of MTT was added to each well. The plates were then incubated for 2 hours before media was aspirated and replaced with DMSO. Plates were shaken vigorously for 15 minutes to dissolve all precipitates and absorbance was determined at 544 nm with a FLUOstar OPTIMA plate reader (BMG Lab technologies, Inc., Durham, NC).
Liposome and Doxorubicin Uptake
Cells were seeded in 12 well plates at 7.0-8.0×104 cells per well and allowed to attach for 24 hours. Cells were then treated with PBS, free doxorubicin (Dox), empty liposomes, empty DiO-labeled liposomes, Dox-loaded liposomes or DiO-labeled Dox-loaded liposomes. Free drug and formulations containing drug were dosed at 2.5 μM doxorubicin equivalents. Phosphate assays were performed to determine lipid concentrations, and empty and DiO-labeled liposomes were dosed at equal lipid concentrations compared to doxorubicin loaded equivalents, ∼10 nmol lipid/mL. At 24, 48 and 72 hours post dosing, cells were washed 3 times with ice cold PBS, released from the plate using trypsin/EDTA and pelleted. Pellets were washed again with PBS and suspended in PBS supplemented with 1 mg/ml glucose. Samples were analyzed immediately using a CyAn flow cytometer (Beckman Coulter, Brea, CA). Samples were excited with a 488 nm argon laser and emission was determined at 575 and 613 nm. Only whole cells were analyzed, as determined by forward and side scatter, and at least 5000 events were counted per run. Data was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Activity of Liposomes In Vivo
The activity of SSL and SPRL liposomes in vivo was determined by implanting PC-3 cells subcutaneously in 7-8 week old male athymic nude (NCr, nu/nu) mice that were acclimated two weeks after receipt from Taconic Farms, Inc., (Germantown, NY). Animals were housed and maintained in accordance with an approved Institutional Animal Care and Use Committee (IACUC) protocol at the University of Georgia and in accordance with the U.S. Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals. Animals were housed in pathogen-free barrier cages within a light and temperature controlled isolated room and provided with autoclaved rodent chow and autoclaved water ad libitum. For tumor implantation, sub-confluent PC-3 cells grown in 10% fetal bovine serum supplemented F-12K were harvested using 0.25% (v/v) trypsin. Cells were counted and suspended in serum free media to a final concentration of 1×107 cells/mL. Media was mixed with ice-cold Matrigel (1:1, v/v), and 200 μL of the mixture was injected subcutaneously into the mouse flank. Tumors were allowed to grow and mice were monitored every other day. Tumor diameters were measured using digital calipers, recorded and tumor volumes were calculated according to the following formula: (larger dimension) × (smaller dimension)2 × 0.5 [27, 28]. When tumors reached ∼400 mm3 mice were randomly selected to be treated with 5 mg/kg of doxorubicin or liposomal equivalent via tail vein injection once a week [28]. Individual tumor volumes were normalized to their tumor volume on the day treatment was initiated. Treatment continued for 4 weeks (i.e., 5 total doses), tumor dimensions and animals weights were measured every other day. Animals were euthanized roughly 2 weeks after the last treatment.
Statistical Analysis
All in vitro experiments were completed at least three times (n = 3) in triplicate. In vivo studies were performed with four to five mice per treatment group. Results are shown as the average of all replicates ± SEM. Results were compared using Student's T-test or one-way ANOVA, where applicable, and considered significant if p ≤ 0.05.
Results
sPLA2-dependent release from SPRL and SSL
Our lab previously designed liposomes whose degradation and release of intraluminal contents was increased in the presence of sPLA2 isoforms compared to SSL formulations [12]. These liposomes were called sPLA2 responsive liposomes (SPRL) to denote their preference, as opposed to selectivity, for sPLA2. Unfortunately, our previous study was limited in that it did not test the effect of sPLA2 inhibition on the release of intraluminal contents, nor did it assess the therapeutic activity of SPRL in a model of disease. To address these limitations we tested the hypothesis that LY311727, a broad-spectrum sPLA2 inhibitor, prevented the release of 6-CF, which was used as a drug marker, from SSL and two different formulations of SPRL (SPRL-E and SPRL-G). As mentioned above, SPRL-E and SPRL-G were used based on our recent publication showing that these formulations yielded the highest responsiveness to sPLA2 compared to SSL [12]. The differences between these liposome formulations are shown in Table 1. As shown in Figure 1, 6-CF release in the absence of sPLA2 was minimal up to 108 hours in media containing 10% FBS. Exposure of formulations to 2.5 μg/ml sPLA2 increased 6-CF release in all formulations. sPLA2-mediated release from SSL was significantly greater than that from untreated liposomes starting at 48 hours. In contrast, significant increases compared to control were seen in SPRL-E and G treated liposomes at time points as early as 24-36 hours. The overall release of 6-CF was less than 20%, which is consistent with the non-burst slow release profile reported in our previous study [12].
Figure 1. Effect of sPLA2 inhibition on carboxyfluorescein release from SPRL and SSL.
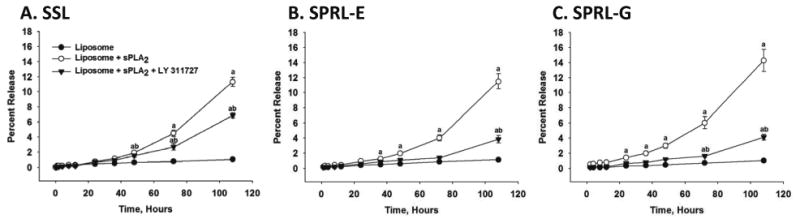
6-CF was loaded into liposomes that were then incubated in F-12K medium containing 10% FBS for 108 hours at 37°C in the presence and absence of 100 mM LY311727 (sPLA2 inhibitor). At the specified time points, samples were removed and analyzed for fluorescence. Data are presented as the mean +/- SEM of 5-6 different experiments. “a” Denotes a significant (P < 0.05) difference compared to control and “b” represents a significant difference compared to liposomes + sPLA2.
Treatment of SSL with LY311727 decreased 6-CF release roughly 50% compared to SSL treated with sPLA2 alone (Figure 1A). In contrast, exposure of SPRL-E and G to LY311727 reduced 6-CF release almost to control levels. These data support our previous observations that 6-CF release from SPRL is more dependent on sPLA2 than SSL.
Anti-tumor activity of SPRL and conventional liposomes in human prostate cancer cell lines
The anti-tumor activity of SPRL was initially determined in vitro using multiple prostate cancer cell lines. Prostate cancer cell lines used were chosen because this cancer is reported to over express sPLA2 at levels 5-20 fold higher than normal prostate tissue and because sPLA2 expression correlates to poor prognosis and decreased survival [16]. Thus, they represent excellent models to assess the therapeutic activity of these liposomes.
Therapeutic activity was first assessed by measuring the cytotoxicity of free doxorubicin, a commonly used anti-cancer drug, or doxorubicin encapsulated in SSL or SPRL-E or G (Figure 2). Free doxorubicin induced a concentration-dependent decrease in MTT staining with an IC50 of to 2.5 μM depending on the cell line (supplemental data). As expected, MTT staining and potency (IC50) of doxorubicin was also cell (LNCaP, DU-145 and PC-3) dependent. For example, doxorubicin induced roughly 60, 70 and 80% decreases in MTT staining in LNCaP cells at 24, 48 and 72 hours, respectively (Figure 2A). In contrast, MTT levels were significantly greater in DU-145 cells at these same time points (Figure 2B), and similar to controls in PC-3 cells after 72 hours of exposure to free doxorubicin (Figure 2C).
Figure 2. Time-dependent effects of SPRL and SSL on MTT staining in human prostate cancer cells.
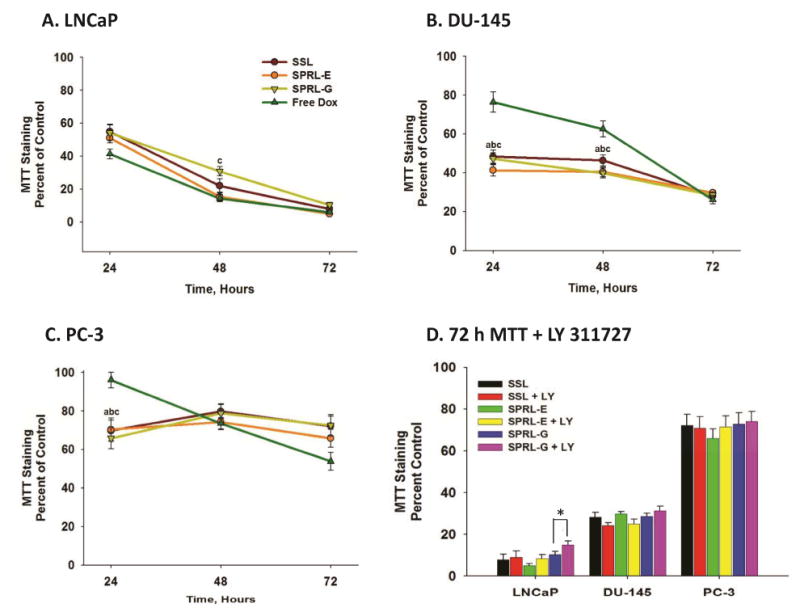
LNCaP (A), DU-145 (B), and PC-3 (C) cells were dosed with 2.5 μM doxorubicin or liposomal equivalents and MTT staining was assessed at 24, 48 and 72 hours. Panel D shows the effect of LY311727 on MTT staining in the presence and absence of SSL and SPRL-E and G after 72 hours. Data are presented as the mean +/- SEM of at least 3 different experiments. In panels A-C “a” denotes a significant difference (P < 0.05) between Free Dox and SSL, “b” denotes a significant difference between Free Dox and SPRL-E, and “c” denotes a significant difference between Free Dox and SPRL-G. In panel D “*” denotes a significant difference between the presence and absence of LY311727.
Similar to their responses to free doxorubicin, each cell line responded differently to SSL and SPRL-E and -G. For the most part, exposure of LNCaP cells to doxorubicin encapsulated in SSL or SPRL-E or G resulted in decreases similar to that seen with free drug (Figure 2A). In contrast to LNCaP cells, exposure of DU-145 cells to doxorubicin encapsulated in SSL, SPRL-E and -G resulted in significantly lower levels of MTT staining compared to free doxorubicin at 24 and 48 hours (Figure 2B). A similar trend was seen in PC-3 cells, but only at 24 hours. Interestingly, there did not appear to be a formulation-dependent difference in cytotoxicity.
LY311727 was used to assess the role of sPLA2 activity in cytotoxicity. Treatment of cells with 100 mM LY311727 prior to exposure to doxorubicin alone or doxorubicin encapsulated in SSL or SPRL did not alter MTT staining compared to cells exposed to these compounds alone, with the exception of SPRL-G in LNCaP cells (Figure 2D). LY311727 did not decrease MTT staining alone, and its ability to inhibit sPLA2 activity was verified in separate experiments (data not shown).
Tandem tracking of liposomes and doxorubicin in human prostate cancer cell lines
Flow cytometry and fluorescent microscopy were used to simultaneously track carrier and payload delivery. Figure 3 represents scatter plots and microscopy images demonstrating the fluorescence of doxorubicin and DiO labeled liposomes in PC-3 cells after 72 hours of exposure. Figure 3A represents cells treated with empty (unlabeled) liposomes demonstrating a lack of both doxorubicin fluorescence on the X-axis and DiO fluorescence on the Y-axis. Figure 3B represents the change in fluorescence on the Y-axis in PC-3 cells treated with DiO labeled liposomes, as indicated by an increase in staining in the upper left hand quadrant compared to Figure 3A. Figure 3C represents PC-3 cells treated with liposomes containing only doxorubicin. An increase in fluorescence can be observed in the lower right hand quadrant compared to control cells. Figure 3D represents cells treated with DiO labeled liposomes containing doxorubicin. As expected, increased fluorescence was seen in the upper right hand quadrant compared to control. Fluorescent intensities of both DiO and doxorubicin were time- and concentration-dependent and linear over the dose ranges tested (data not shown). These data demonstrate that drug and nanoparticle delivery to cells could be tracked simultaneously.
Figure 3. Tandem tracking of doxorubicin, SPRL and SSL in PC-3 cells using flow cytometry and fluorescence microscopy.
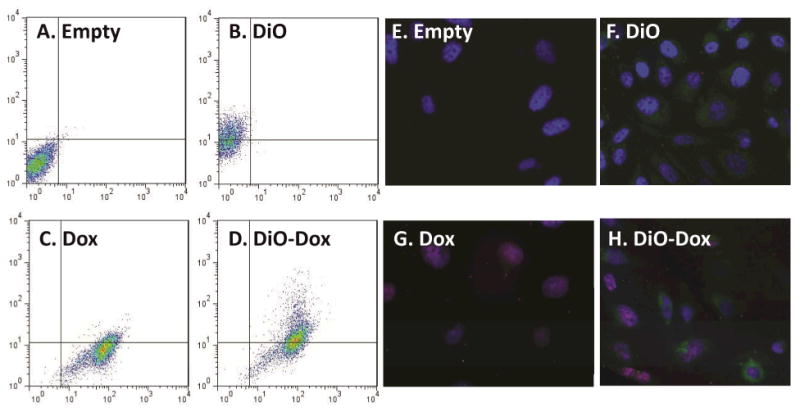
PC-3 cells were treated with DiO-labeled, doxorubicin loaded (Dox) liposomes and liposomes labeled with both doxorubicin and DiO (Dio-Dox). Both liposomes and doxorubicin were tracked concurrently using flow cytometry (A-D) and fluorescent microscopy (E-H). Panels A-D represent scatter plots showing fluorescence for empty liposomes (A), DiO-labeled liposomes (B) Dox-loaded liposomes (C) and doxorubicin-loaded liposomes labeled with DiO (D). Panels E-F represent fluorescence microscopy of these cells stained with DAPI and treated with empty liposomes (E), DiO-labeled liposomes (F), doxorubicin-loaded liposomes (G) and doxorubicin loaded liposomes labeled with DiO (H). Data are representative of at least 3 separate experiments.
Fluorescence microscopy was used to verify the flow cytometry results (Figure 3E-H). For these experiments, all cells were fixed and stained with DAPI after 72 hours of exposure to various treatments. Figure 3E represents control PC-3 cells. Blue stained, normal nuclei are clearly discernible as a result of the DAPI staining. Figure 3F represents PC-3 cells exposed to DiO only labeled SPRL-E, demonstrating increased green fluorescent staining compared to control cells. Figure 3G represents cells exposed to SPRL-E loaded with doxorubicin. While doxorubicin fluorescence is usually red, in this case it appears purple when overlaid with the DAPI stain as both signals co-localize to the nucleus. Figure 3H represents cells exposed to DiO labeled SPRL-E liposomes containing doxorubicin. Evidence can be seen of cells staining for green fluorescence representing SPRL-E, as well as cells staining for doxorubicin. Additionally evidence of altered nuclear morphology can be seen, supporting the conclusion that these liposomes induced cell death.
Having demonstrated the presence of nanoparticle and drug uptake in cells, we used flow cytometry to compare cell- and nanoparticle-dependent differences in fluorescence at 24, 48, and 72 hours (Figure 4). Doxorubicin fluorescence was lowest in LNCaP cells and greatest in PC-3 cells, as can be seen in Figure 4A. Likewise, DiO fluorescence was generally greater in PC-3 cells, but similar in LNCaP and DU-145 cells, with the exception of SPRL-G fluorescence, which was equal in all three cell lines (Figure 4B). DiO fluorescence for SPRL-E was visibly greater in each cell line, compared to SSL and SPRL-G, especially in LNCaP and PC-3 cells. In general, the level of doxorubicin fluorescence correlated with DiO fluorescence in both SSL and SPRL-E, but this correlation was not observed for SPRL-G.
Figure 4. Quantification of fluorescence of doxorubicin and DiO in prostate cancer cells exposed to SPRL and SSL.
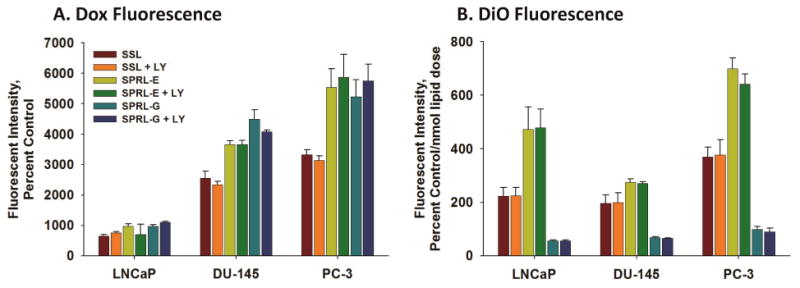
LNCaP, DU-145, and PC-3 cells were treated with dual-labeled liposomes for 72 hours and examined with flow cytometry. Panel A shows the intensity of doxorubicin fluorescence while panel B shows the intensity of DiO fluorescence normalized by the nmol dose of lipid. Data are presented as the mean +/- SEM of at least 3 different experiments. Differences were considered significant with a P < 0.05.
Cytotoxicity may alter liposome uptake. This is especially relevant as LNCaP cells had increased levels of cytotoxicity compared to DU-145 and PC-3 cells. To assess liposome uptake in cells in the absence of cytotoxicity we determined time-dependence differences in fluorescence in cells exposed to liposomes labeled only with DiO (Figure 5). Exposure of LNCaP cells to DiO-labeled SSL resulted in comparable levels of fluorescence at 24, 48 and 72 hours (Figure 5A). The levels of fluorescence were similar to cells exposed to SSL containing both DiO and doxorubicin (Figure 4B). DiO fluorescence in LNCaP cells was also similar to that seen in DU-145 and PC-3 cells exposed to SSL (Figure 5B and C), with the exception of slightly greater levels in PC-3 cells at 72 hours. DiO fluorescence was similar in all cells lines exposed to SPRL-G at all time points measured, which agrees with data reported in Figure 4. In contrast, DiO fluorescence was significantly (p<0.05) greater in cells exposed to SPRL-E, compared to SSL and SPRL-G. Interestingly, DiO fluorescence was higher in LNCaP cells exposed to SPRL-E at all time points, compared to DU-145 or PC-3 cells.
Figure 5. Time-dependence of DiO uptake in prostate cancer cells exposed to SPRL and SSL.
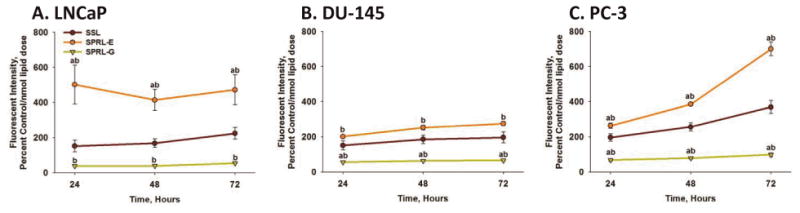
LNCaP (A), DU-145 (B) and PC-3 (C) cells were exposed to DiO-labeled liposomes for 24, 48 and 72 hours, after which cells were detached, washed, and assessed for fluorescence using flow cytometry. Data are presented as the mean +/- SEM of at least 3 different experiments. One-way ANOVA test was performed to determine differences between groups. Differences between SPRL formulations and SSL are denoted by “a” and differences between SPRL-E and G are denoted by “b”.
It is also possible that labeling liposomes with DiO may alter doxorubicin fluorescence. Thus, the fluorescence of doxorubicin was determined in cells after exposure to doxorubicin alone, or doxorubicin encapsulated in SSL, SPRL-E and SPRL-G (Figure 6). Doxorubicin fluorescence was lower in LNCaP cells exposed to SSL, compared to cells exposed to doxorubicin alone, or to SPRL formulations (Figure 6A). There was a time-dependent decrease in doxorubicin fluorescence in LNCaP cells, with significantly (p<0.05) lower levels being observed at 48 and 72 hours, compared to 24 hours. There were no differences in fluorescence between the formulations tested at 48 and 72 hours.
Figure 6. Time-dependence of doxorubicin uptake in prostate cancer cells exposed to SPRL and SSL.
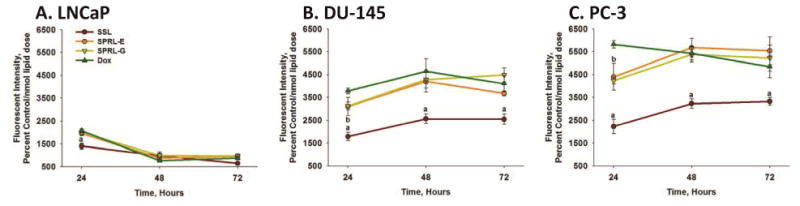
LNCaP (A), DU-145 (B) and PC-3 (C) cells were exposed to doxorubicin alone (Dox) or encapsulated in SPRL-E, G and SSL for 24, 48, and 72 hours, after which cells were detached, washed, and assessed for fluorescence using flow cytometry. Data are presented as the mean +/- SEM of at least 3 different experiments. “a” Denotes a significant (P < 0.05) difference between Dox and SSL, “b” denotes a significant difference between Dox and SPRL-G. There were no significant differences between Dox and SPRL-E.
Exposure of DU-145 and PC-3 cells to doxorubicin alone resulted in greater levels of fluorescence, compared to LNCaP cells, at all time points tested (Figure 6B and C). Once again fluorescence was lower in cells exposed to SSL, and unlike LNCaP cells, this trend was maintained at both 48 and 72 hours. For the most part, fluorescence was similar in DU-145 and PC-3 cells exposed to doxorubicin alone or that encapsulated in SPRL-E or G, with the exception of 24 hours in PC-3 cells. These data suggest that the uptake of SSL and SPRL is cell- and formulation-dependent.
In vivo evaluation of SPRL and SSL
While our in vitro data was promising, in vivo evaluation is better for suggesting real clinical utility. For these studies, we used a human PC-3 xenograft model in aythmic mice, as PC-3 cells are our most aggressive cell line. Additionally, LNCaP cells do not readily form tumors in this model and DU-145 are slower growing. For the purpose of testing, we evaluated the SPRL-E formulation, given its comparatively high levels of uptake, and the SSL formulation as a clinically relevant comparison. Treatment of mice, via tail vein injection, with doxorubicin encapsulated in SSL resulted in slight decreases in tumor volume compared to controls after 21 days and 3 treatments (Figure 7A). Tumor volume continued to increases in SSL treated mice throughout the length of the study, but was decreased compared to control after 35 days. In contrast, treatment of mice with doxorubicin encapsulated in SPRL-E resulted in significantly lower tumor volumes than either control or SSL treated mice. Tumor volume was lower than controls and SSL at day 17 and remained lower than controls even after treatment was stopped at 21 days. Body weights were not significantly different between control, SSL and SPRL-E exposed mice after 21 days, and only slightly lower in treated groups after that (Figure 7B). Necropsies performed following sacrifice to look for evidence of cardiotoxicity and signs of cardiomyopathy were negative. These data suggest that SPRL-E are more effective at limiting tumor growth than the clinically utilized SSL.
Figure 7. Effect of doxorubicin containing SPRL and SSL on PC-3 xenograft tumor growth.
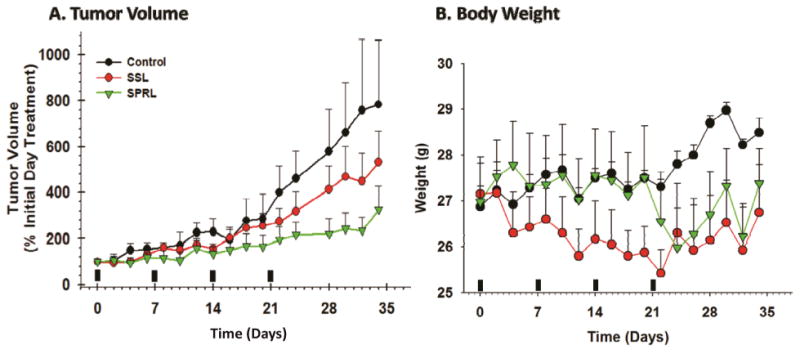
Athymic mice bearing PC-3 xenograft tumors were treated (indicated by solid bars) with SSL and SPRL-E formulations on a weekly basis for 4 weeks after tumors reached 400 mm3 and tumor volume (A) and mouse weight (B) were determined every 2 days for 34 days. Data are presented as the mean +/- SEM of at least 4 different mice.
Discussion
This study demonstrated the novel finding that SRPL formulations containing doxorubicin were effective at decreasing human prostate cancer cell growth in vitro and in vivo. The data support the hypothesis that SPRL may be used to treat cancer, as well as other diseased where sPLA2 is over expressed and may be more effective than the clinically utilized SSL formulations. The increased efficacy of SPRL-E, compared to SSL, in vivo may have been a result of increased uptake. This hypothesis is supported by our in vitro studies in all three prostate cancer cells lines. However, SPRL-G was also more effective than SSL at delivering drug inside the cell, although uptake of these particles was limited by comparison. Thus, other mechanisms, in addition to uptake, may be mediating the increased efficacy of SPRL in vivo.
The fact that these nanoparticles induced cytotoxicity at levels comparable to free drug in vitro is an unexpected finding. Free drug typically displays greater antitumor activity than liposomes in vitro, as encapsulated drugs must first be released from nanoparticles. Toxicities equal to free drug suggests that there is enhanced release and/or uptake mechanisms at work beyond the simple diffusion of particles across the membrane. The nature of this mechanism is unknown, but must be cell mediated, as the release studies in Figure 1 demonstrate that drug release is effectively zero in the absence of cells or sPLA2.
SPRL were designed to interact with sPLA2, but all of the formulations, including SSL, had similar levels of in vitro cytotoxicity in spite of having different levels of DiO fluorescence. This shows that cytotoxicity does not always correlate to cellular uptake and suggest that the mechanisms mediating cytotoxicity of these nanoparticles are distinct from those mediating uptake. One possible explanation for these differences may be a product of extracellular degradation of liposomes, resulting in the release and subsequent uptake of doxorubicin independent of the nanoparticle. It is unlikely that differences in cytotoxicity are mediated by differences in sPLA2 activity between these cells as LY311727 had little to no effect on MTT staining, even though it decreased sPLA2-mediated release of payload in earlier studies. This suggests that the mechanism of cytotoxicity or uptake does not require enzymatic activity, and again, points to the possibility of multiple mechanisms mediating cytotoxicity.
The inability of LY311727 to alter uptake or antitumor activity does not necessarily mean that these liposomes are not responsive to sPLA2. sPLA2 have functions that are independent of their lipolytic activity and several proteins exist in mammalian cells that bind sPLA2 independently of the sPLA2 active site [3]. One of these proteins is a receptor in the C-type lectin superfamily called PLA2R and it is responsible for internalizing sPLA2 back inside the cell via endocytosis after it has been secreted [29]. Studies in other cells types show that binding of sPLA2 to PLA2R does not require lipase activity [3]. Thus, an alternative mechanisms for uptake of these formulations, independent of lipase activity, is that the liposome are interacting with sPLA2, which forms a complex with the PLA2R membrane receptor that is then transported into the cell. This type of facilitated uptake has not been reported for liposomes or other drug carriers and may hold great potential in terms of developing a novel targeting strategies.
One clear finding of this study is that the mechanisms mediating SPRL uptake and drug delivery are cell-dependent. The cell lines used in this study differentially express multiple sPLA2 isoforms [30]. It is possible that these differences may account for disparities in drug delivery and SPRL uptake, as each sPLA2 isoform displays differential preferences for binding to lipid substrates, PLA2R and other extracellular features of the membrane [4, 29].
In addition to cell-dependence, these data suggest that the mechanisms mediating the uptake of drug and nanoparticles are formulation-dependent. All of these nanoparticles were roughly identical in size (100 nm), but differed slightly in terms of phospholipid content. Our results demonstrate that incorporating as little as 10% of zwitterionic or anionic lipid into our liposome membranes can have a pronounced affect on whether or not they are taken up or release their contents extracellularly (Figures 5 and 6). This information will be invaluable for the development of future generations of SPRL, whether they are designed to treat cancer or any one of numerable other diseases.
SSL containing doxorubicin is a FDA approved treatment for some cancers, and goes under the name trade name Doxil™ [31]. SSL and SPRL-E differ only by 10 mol% DSPE, which is abundant in eukaryotic cell membranes [32]. This suggests that SPRL may be rapidly translated to clinical application with little fear of toxicity. The increased efficacy of SPRL against tumor growth in vivo suggests that SPRL may be viable for treating prostate cancers specifically, as well as other cancers that over express sPLA2.
The enhanced efficacy of SPRL-E in vivo was particularly interesting, given that all three of the formulations performed comparably in vitro. These data suggest that the the responsiveness to sPLA2 becomes more valuable in an in vivo platform. One reason for the discrepancy between the in vitro and in vivo data may be the increased uptake of both drug and in SPRL-E compared to SSL (Figures 5 and 6). This uptake mechanisms may be minimized in vivo, but play a more impoartant role in vivo. Another possible explanation for this discrepancy is that in an in vitro setting there is no means of clearance, or interaction with additional organ systems. However, in vivo such events are critical to nanoparticle efficacy. It is unlikely that differences in EPR results in increased efficacy in vivo as both particles should deposit in the tumor tissue in relatively similar amounts based on previous studies [33]. Finally, it is well established that in vitro efficacy of nanoparticles does not always translate well in vivo. Future studies focusing on differences in the mechanism of uptake and delivery in vivo will hopefully provide answers to some of these questions.
Although the experiments in this study focused on the utility of SPRL in prostate cancer, this targeting strategy may hold greater potential. As mentioned above, sPLA2 have a variety of physiological functions [3] and are commonly over expressed in a number of serious pathologies. Up regulation of sPLA2 frequently occurs in atherosclerotic plaques and in arthritic joints [6-10], as well as other inflammatory conditions [18, 19, 22]. This suggests that SPRL may be integrated, or translated, to therapies outside of just prostate cancer.
In conclusion, we showed that engineering liposomes to specifically interact with sPLA2 is a viable targeting strategy for inhibiting prostate cancer growth in vitro and in vivo. Data in this study also suggests that mechanisms independent of sPLA2 activity may, in part, mediate the toxicity and disposition of liposomes, and that the efficacy of sPLA2 targeted nanoparticles are mediated by mechanisms that are cell- and formulation-dependent. Identifying these mechanisms will be key to designing more efficacious targeting strategies for treatment of diseases that over express sPLA2.
Supplementary Material
Supplemental Figure 1. Concentration-dependence effect of doxorubicin exposure on MTT staining in PC-3, LNCaP and DU145 cells after 72 hours. Data are presented as the mean +/- SEM of at least 3-6 different experiments.
Insight Statement.
Secretory phospholipase A2 (sPLA2) are over expressed in many solid tumors, as well as inflammatory diseases such as arthritis and sepsis, and as such may be targets for treatment of numerous pathologies. We evaluated the therapeutic activity of liposomes engineered to be responsive to secretory sPLA2 in vitro and in vivo and assessed differences in their mechanism of uptake and delivery compared to long-circulating (pegylated) sterically stabilized liposomes (SSL). Treating prostate cancer cells with sPLA2 responsive liposomes (SPRL) resulted in similar cytotoxicity in vitro as that induced by free drug. Flow cytometry and fluorescent microscopy were used to track the spatial and temporal distribution of these carriers, and demonstrated how alterations in formulations affected nanoparticle behavior and induced differential patterns of uptake and delivery, compared to SSL. In vivo evaluation of SPRLs suggested that these formulations are more effective at slowing tumor growth than clinically utilized doxorubicin liposomes. These data demonstrate that SPRL may improve drug delivery in cancer and other pathologies where sPLA2 is over expressed and that differences exist in the mechanisms mediating the uptake of SPRL compared to SSL
Acknowledgments
We thank the UGA Flow Cytometry Core Facility in the Center for Tropical and Emerging Diseases, particularly Julie Nelson for all of her assistance and insight. The authors would also like to acknowledge and give special thanks to both the American Foundation of Pharmaceutical Education (AFPE) and the Achievement Rewards for College Scientists (ARCS) foundation for providing pre-doctoral fellowship assistance to JNM. This research was funded in part by Georgia Cancer Coalition Distinguished Scholar Grants and a NIH NIBIB (EB08153) to BSC/RDA.
Abbreviations
- 6-CF
6-carboxyfluorescien
- DiO- 3
3′-dioctadecyloxacarbocyanine perchlorate
- Dox
doxorubicin
- DSPC
distearoylphosphatidylcholine
- DSPE
distearoylphosphatidylethanolamine
- DSPG
distearoylphosphatidylglycerol
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PEG
polyethylene glycol
- sPLA2
secretory phospholipase A2
- SSL
sterically stabilized liposomes
- SPRL
secretory phospholipase A2 responsive liposome
References
- 1.Arnold RD, Mager DE, Slack JE, Straubinger RM. Effect of repetitive administration of Doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin Cancer Res. 2005;11:8856–65. doi: 10.1158/1078-0432.CCR-05-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drummond MF, Mason AR. European perspective on the costs and cost-effectiveness of cancer therapies. J Clin Oncol. 2007;25:191–5. doi: 10.1200/JCO.2006.07.8956. [DOI] [PubMed] [Google Scholar]
- 3.Lambeau Gr, Gelb MH. Biochemistry and Physiology of Mammalian Secreted Phospholipases A2. Annual Review of Biochemistry. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 4.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A research: from cells to animals to humans. Prog Lipid Res. 50:152–92. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Graff JR, Konicek BW, Deddens JA, Chedid M, Hurst BM, Colligan B, et al. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin Cancer Res. 2001;7:3857–61. [PubMed] [Google Scholar]
- 6.Pruzanski W, Vadas P. Secretory synovial fluid phospholipase A2 and its role in the pathogenesis of inflammation in arthritis. J Rheumatol. 1988;15:1601–3. [PubMed] [Google Scholar]
- 7.Leistad L, Feuerherm AJ, Ostensen M, Faxvaag A, Johansen B. Presence of secretory group IIa and V phospholipase A2 and cytosolic group IValpha phospholipase A2 in chondrocytes from patients with rheumatoid arthritis. Clin Chem Lab Med. 2004;42:602–10. doi: 10.1515/CCLM.2004.104. [DOI] [PubMed] [Google Scholar]
- 8.Fraser H, Hislop C, Christie RM, Rick HL, Reidy CA, Chouinard ML, et al. Varespladib (A-002), a secretory phospholipase A2 inhibitor, reduces atherosclerosis and aneurysm formation in ApoE-/- mice. J Cardiovasc Pharmacol. 2009;53:60–5. doi: 10.1097/FJC.0b013e318195bfbc. [DOI] [PubMed] [Google Scholar]
- 9.Bostrom MA, Boyanovsky BB, Jordan CT, Wadsworth MP, Taatjes DJ, de Beer FC, et al. Group v secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler Thromb Vasc Biol. 2007;27:600–6. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- 10.Rosengren B, Jonsson-Rylander AC, Peilot H, Camejo G, Hurt-Camejo E. Distinctiveness of secretory phospholipase A2 group IIA and V suggesting unique roles in atherosclerosis. Biochim Biophys Acta. 2006;1761:1301–8. doi: 10.1016/j.bbalip.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Andresen TL, Jensen SS, Kaasgaard T, Jorgensen K. Triggered activation and release of liposomal prodrugs and drugs in cancer tissue by secretory phospholipase A2. Curr Drug Deliv. 2005;2:353–62. doi: 10.2174/156720105774370203. [DOI] [PubMed] [Google Scholar]
- 12.Zhu G, Mock JN, Aljuffali I, Cummings BS, Arnold RD. Secretory phospholipase A responsive liposomes. J Pharm Sci. 100:3146–59. doi: 10.1002/jps.22530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu G, Alhamhoom Y, Cummings BS, Arnold RD. Synthesis of lipids for development of multifunctional lipid-based drug-carriers. Bioorg Med Chem Lett. 21:6370–5. doi: 10.1016/j.bmcl.2011.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong Q, Patel M, Scott KF, Graham GG, Russell PJ, Sved P. Oncogenic action of phospholipase A2 in prostate cancer. Cancer Lett. 2006;240:9–16. doi: 10.1016/j.canlet.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Kallajoki M, Alanen KA, Nevalainen M, Nevalainen TJ. Group II phospholipase A2 in human male reproductive organs and genital tumors. Prostate. 1998;35:263–72. doi: 10.1002/(sici)1097-0045(19980601)35:4<263::aid-pros5>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 16.Jiang J, Neubauer BL, Graff JR, Chedid M, Thomas JE, Roehm NW, et al. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am J Pathol. 2002;160:667–71. doi: 10.1016/S0002-9440(10)64886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamashita S, Yamashita J, Ogawa M. Overexpression of group II phospholipase A2 in human breast cancer tissues is closely associated with their malignant potency. Br J Cancer. 1994;69:1166–70. doi: 10.1038/bjc.1994.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamashita S, Ogawa M, Sakamoto K, Abe T, Arakawa H, Yamashita J. Elevation of serum group II phospholipase A2 levels in patients with advanced cancer. Clin Chim Acta. 1994;228:91–9. doi: 10.1016/0009-8981(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita S, Yamashita J, Sakamoto K, Inada K, Nakashima Y, Murata K, et al. Increased expression of membrane-associated phospholipase A2 shows malignant potential of human breast cancer cells. Cancer. 1993;71:3058–64. doi: 10.1002/1097-0142(19930515)71:10<3058::aid-cncr2820711028>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Kiyohara H, Egami H, Kako H, Shibata Y, Murata K, Ohshima S, et al. Immunohistochemical localization of group II phospholipase A2 in human pancreatic carcinomas. Int J Pancreatol. 1993;13:49–57. doi: 10.1007/BF02795199. [DOI] [PubMed] [Google Scholar]
- 21.Kuopio T, Ekfors TO, Nikkanen V, Nevalainen TJ. Acinar cell carcinoma of the pancreas. Report of three cases. APMIS. 1995;103:69–78. doi: 10.1111/j.1699-0463.1995.tb01081.x. [DOI] [PubMed] [Google Scholar]
- 22.Oka Y, Ogawa M, Matsuda Y, Murata A, Nishijima J, Miyauchi K, et al. Serum immunoreactive pancreatic phospholipase A2 in patients with various malignant tumors. Enzyme. 1990;43:80–8. doi: 10.1159/000468710. [DOI] [PubMed] [Google Scholar]
- 23.Sved P, Scott KF, McLeod D, King NJ, Singh J, Tsatralis T, et al. Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res. 2004;64:6934–40. doi: 10.1158/0008-5472.CAN-03-3018. [DOI] [PubMed] [Google Scholar]
- 24.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- 25.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201–15. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 26.Kamps JA, Morselt HW, Swart PJ, Meijer DK, Scherphof GL. Massive targeting of liposomes, surface-modified with anionized albumins, to hepatic endothelial cells. Proc Natl Acad Sci U S A. 1997;94:11681–5. doi: 10.1073/pnas.94.21.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geran RI, Schumach Am, Abbott BJ, Greenber Nh, Macdonal Mm. Protocols for Screening Chemical Agents and Natural-Products against Animal Tumors and Other Biological-Systems. Cancer Chemotherapy Reports Part 3. 1972;3:1. &. [Google Scholar]
- 28.Aljuffali IA, Mock JN, Costyn LJ, Nguyen H, Nagy T, Cummings BS, et al. Enhanced antitumor activity of low-dose continuous administration schedules of topotecan in prostate cancer. Cancer Biology & Therapy. 2011;12:407–20. doi: 10.4161/cbt.12.5.15950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanasaki K, Arita H. Phospholipase A2 receptor: a regulator of biological functions of secretory phospholipase A2. Prostaglandins Other Lipid Mediat. 2002;68-69:71–82. doi: 10.1016/s0090-6980(02)00022-9. [DOI] [PubMed] [Google Scholar]
- 30.Menschikowski M, Hagelgans A, Gussakovsky E, Kostka H, Paley EL, Siegert G. Differential expression of secretory phospholipases A2 in normal and malignant prostate cell lines: regulation by cytokines, cell signaling pathways, and epigenetic mechanisms. Neoplasia. 2008;10:279–86. doi: 10.1593/neo.07965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DOXIL approved by FDA. AIDS Patient Care. 1995;9:306. [PubMed] [Google Scholar]
- 32.Bakovic M, Fullerton MD, Michel V. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: the role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) Biochem Cell Biol. 2007;85:283–300. doi: 10.1139/o07-006. [DOI] [PubMed] [Google Scholar]
- 33.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Concentration-dependence effect of doxorubicin exposure on MTT staining in PC-3, LNCaP and DU145 cells after 72 hours. Data are presented as the mean +/- SEM of at least 3-6 different experiments.