

Abstract
Leflunomide is an immunomodulatory agent used for the treatment of rheumatoid arthritis. In this study, we investigated the effect of A77 1726 – the active metabolite of leflunomide – on the production of IL-1 receptor antagonist (IL-1Ra) by human synovial fibroblasts and articular chondrocytes. Cells were incubated with A77 1726 alone or in combination with proinflammatory cytokines. IL-1Ra production was determined by ELISA. A77 1726 alone had no effect, but in the presence of IL-1β or tumour necrosis factor-α it markedly enhanced the secretion of IL-1Ra in synovial fibroblasts and chondrocytes. The effect of A77 1726 was greatest at 100 μmol/l. In synovial fibroblasts and de-differentiated chondrocytes, A77 1726 also increased IL-1β-induced IL-1Ra production in cell lysates. Freshly isolated chondrocytes contained no significant amounts of intracellular IL-1Ra. A77 1726 is a known inhibitor of pyrimidine synthesis and cyclo-oxygenase (COX)-2 activity. Addition of exogenous uridine did not significantly modify the effect of A77 1726 on IL-1Ra production, suggesting that it was not mediated by inhibition of pyrimidine synthesis. Indomethacin increased IL-1β-induced IL-1Ra secretion in synovial fibroblasts and de-differentiated chondrocytes, suggesting that inhibition of COX-2 may indeed enhance IL-1β-induced IL-1Ra production. However, the stimulatory effect of indomethacin was consistently less effective than that of A77 1726. A77 1726 increases IL-1Ra production by synovial fibroblasts and chondrocytes in the presence of proinflammatory cytokines, and thus it may possess chondroprotective effects. The effect of A77 1726 may be partially mediated by inhibition of COX-2, but other mechanisms likely concur to stimulate IL-1Ra production.
Keywords: articular cartilage, IL-1 receptor antagonist, leflunomide, synovium
Introduction
Leflunomide is an oral immunomodulatory agent, which is considered effective for the treatment of rheumatoid arthritis (RA). Leflunomide is a disease-modifying antirheumatic drug that is approved for treatment of RA, and radiographical findings indicate that it delays joint damage [1-4]. Its therapeutic profile closely resembles that of methotrexate. The latter drug is the most widely used disease-modifying antirheumatic drug but, despite a favourable efficiency–toxicity profile, in numerous patients it is either insufficient or associated with unacceptable side effects.
In vivo, leflunomide is rapidly converted into its pharmacologically active metabolite A77 1726 [5]. The recommended dose of leflunomide for the treatment of RA patients is 20 mg/day, which produces steady-state serum levels of A77 1726 of approximately 25–45 μg/ml (75–115 μmol/l) [6]. Although the precise mode of action of leflunomide in vivo remains elusive, A77 1726 has been shown in vitro to inhibit reversibly dihydro-orotate dehydrogenase (DHODH), which catalyzes a rate-limiting step in the de novo synthesis of pyrimidines [7,8]. The inhibition of DHODH activity by A77 1726 might explain part of its mechanism of action in suppressing inflammation. Indeed, many effects of A77 1726 can be reversed by exposing target cells to the product of DHODH activity, namely uridine. Leflunomide is a potent noncytotoxic inhibitor of the proliferation of stimulated B and T lymphocytes, which depend on de novo pyrimidine synthesis to fulfill their metabolic needs [4,5]. Furthermore, leflunomide blocks tumour necrosis factor (TNF)-α-mediated cellular responses in T cells by inhibiting nuclear factor-κB – a mechanism that also depends on pyrimidine biosynthesis [9,10]. In addition, A77 1726 exerts a direct inhibitory effect on cyclo-oxygenase (COX)-2 activity, both in vitro and in vivo [11,12]. Finally, it has been reported that, at higher concentrations, A77 1726 inhibits different types of receptor and nonreceptor tyrosine kinases that are involved in cytokine and growth factor signalling [13-15].
RA is characterized by synoviocyte proliferation and infiltration of inflammatory cells, such as lymphocytes and macrophages, into the joint. Local release of proinflammatory mediators and metalloproteinases causes joint cartilage destruction and leads to the perpetuation of joint inflammation. Potential direct anti-inflammatory effects of A77 1726 on joint cells are thus of interest because of their relevance to the effectiveness of leflunomide in treating RA and other cartilage-damaging diseases. In a previous study, A77 1726 was found to inhibit the expression of monocyte-activating factor at the surface of T lymphocytes, which in turn decreased the activation of monocyte/macrophages, and thus their production of IL-1β and matrix metalloproteinase (MMP)-1 [16]. A further study showed that A77 1726 inhibits the production of prostaglandin E2 (PGE2), MMP-1 and IL-6 in human synovial fibroblasts [12]. The inhibition of MMP-1 and IL-6 production was due to the well known inhibitory effect of A77 1726 on pyrimidine synthesis, because it was reversed by the addition of uridine. PGE2 production appeared to be inhibited by the direct action of A77 1726 on COX-2. More recently, A77 1726 was reported to decrease TNF-α, intercellular adhesion molecule-1 and COX-2 expression in synovial macrophages [17]. A77 1726 also inhibited IL-1β, TNF-α, nitric oxide and MMP-3 production in activated human synovial tissue cultures [18]. Thus, several studies indicate that A77 1726 inhibits the production of proinflammatory mediators by synovial fibroblasts.
Methotrexate also exhibits many of these effects, and in addition it has been shown to stimulate the synthesis of the anticatabolic factor IL-1 receptor antagonist (IL-1Ra) [19]. Increased production of IL-1Ra by joint cells in response to A77 1726 might potentially be beneficial by contributing to prevent joint damage in inflammatory arthropathies such as RA. However, it has not been determined whether A77 1726 has direct effects on the production of this anti-inflammatory molecule in synovial fibroblasts. Furthermore, potential direct effects of A77 1726 on articular cartilage and chondrocytes have not yet been examined.
In the present study we investigated the effect of A77 1726 on the production of IL-1Ra in human synovial fibroblasts, as well as in freshly isolated and in subcultured human articular chondrocytes.
Methods
Materials
Cell culture reagents were obtained from Gibco (Life Technologies AG, Basel, Switzerland). A77 1726 (HMR1726) – the active metabolite of leflunomide – was generously provided by Aventis Pharma (Frankfurt am Main, Germany). A77 1726 was dissolved as a 10 mmol/l stock solution in phosphate-buffered saline. Recombinant human IL-1β and TNF-α were purchased from R&D Systems (Abingdon, UK). Indomethacin was obtained from Sigma Fine Chemicals (St. Louis, MO, USA).
Cell isolation and culture
Synovium was obtained from patients undergoing joint replacement (knee or hip prosthetic surgery) for osteoarthritis (OA) or RA. Cartilage was obtained from patients undergoing joint replacement (knee or hip prosthetic surgery) for OA or broken femoral neck (normal adult cartilage), or from children undergoing spinal surgery for scoliosis (vertebral posterior joints, normal paediatric cartilage). Synovial fibroblasts and chondrocytes were isolated by collagenase digestion as reported previously [20,21] and cultured in Dulbecco's modified eagle medium supplemented with L-glutamine, streptomycin, penicillin and 10% foetal calf serum (FCS). Synovial fibroblasts were used between passages 1 and 7, as indicated. Primary chondrocytes were used directly after isolation from cartilage and de-differentiated chondrocytes were used between passages 1 and 7, as indicated. In total, the effect of A77 1726 was evaluated in 15 independent cultures of synovial fibroblast, 18 independent cultures of freshly isolated primary chondrocytes and 13 independent cultures of de-differentiated chondrocytes. Cells were plated in 96-well plates (40,000 cells per well) and used 24 hours after plating. To reduce nonspecific effects of FCS, the cells were incubated with the various agents in low-serum (0.5% FCS) medium. The cells were preincubated with A77 1726 or indomethacin for 2 hours before stimulation with IL-1β or TNF-α for 48 hours. In some experiments, uridine (50 or 200 μmol/l) was added 30 min before A77 1726.
Assessment of IL-1 receptor antagonist and prostaglandin E2 production
Culture supernatants were collected and stored at -20°C. Cell lysates were obtained by adding fresh medium containing 1% NP-40 to the cells. The concentrations of IL-1Ra in supernatants and lysates were determined by sandwich ELISA, as previously described [22]. The sensitivity of this assay is 78 pg/ml. Concentrations of PGE2 in supernatants were determined as previously described [12].
Statistical analysis
The statistical significance of differences was calculated using analysis of variance. P < 0.05 was considered statistically significant.
Results
Impact of A77 1726 on production of IL-1 receptor antagonist
We first investigated the effect of A77 1726 on IL-1Ra production in human synovial fibroblasts. Although A77 1726 alone had no significant effect, it markedly stimulated IL-1Ra secretion in the presence of IL-1β (Fig. 1a). The effect of A77 1726 was dose-dependent and highest at 100 μmol/l. Higher doses of A77 1726 (200 or 300 μmol/l) decreased cell viability (data not shown).
Figure 1.
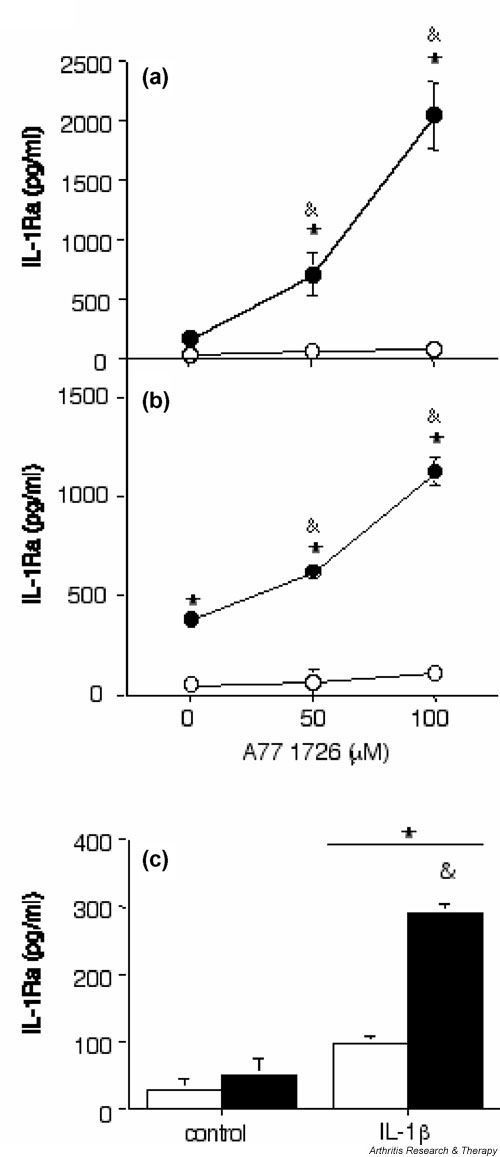
A77 1726 increases IL-1 receptor antagonist (IL-1Ra) secretion in human synovial fibroblasts and articular chondrocytes in the presence of IL-1β. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 2) and (b) de-differentiated human OA articular chondrocytes (passage 7) were stimulated (closed symbols) or not (open symbols) with 1 ng/ml IL-1β alone or in combination with 50 μmol/l or 100 μmol/l A77 1726 for 48 hours. (c) Freshly isolated normal human articular chondrocytes were stimulated (black columns) or not (white columns) with 100 μmol/l A77 1726 alone or in combination with 1 ng/ml IL-1β for 48 hours. A77 1726 was added 2 hours before stimulation with IL-1β. IL-1Ra concentrations in culture supernatants were measured using ELISA. Results are expressed as means ± SEM for three determinations in a representative experiment. Similar results were obtained with cells from three different donors (three OA samples) for synovial fibroblasts and three different donors (three OA samples) for de-differentiated chondrocytes. On freshly isolated chondrocytes, a stimulatory effect of A77 1726 was observed in cells from 12 (five normal adult, six OA and one normal paediatric sample) out of 18 different donors. *P < 0.05 versus control; &P < 0.05 versus IL-1β alone.
In human articular chondrocytes A77 1726 also enhanced IL-1Ra secretion in the presence of IL-1β but had no effect on its own (Fig. 1b,1c). In subcultured de-differentiated chondrocytes, A77 1726 exhibited a marked dose-dependent effect on IL-1β induced IL-1Ra production, which was greatest at 100 μmol/l (Fig. 1b). In freshly isolated primary chondrocytes, A77 1726 used at 100 μmol/l also enhanced secretion of IL-1Ra induced by IL-1β (Fig. 1c). However, the levels of IL-1Ra secreted by freshly isolated chondrocytes on stimulation with A77 1726 and IL-1β were consistently much lower than those produced by stimulated synovial fibroblasts or subcultured de-differentiated chondrocytes. Furthermore, in freshly isolated chondrocytes, the stimulatory effect of A77 1726 on IL-1β-induced IL-1Ra production was more difficult to reproduce than in the two other cell types, and was significant only in about two-thirds of all experiments performed. Finally, when using A77 1726 at 50 μmol/l, we were unable to observe a consistent effect on IL-1Ra production in primary chondrocytes.
In parallel to enhancing the production of secreted IL-1Ra, A77 1726 also increased the amount of IL-1Ra present in cell lysates of synovial fibroblasts stimulated with IL-1β (Fig. 2a). IL-1β alone stimulated the production of intracellular IL-1Ra in these cells and this effect was markedly enhanced by A77 1726. Cell lysates of primary human chondrocytes contained undetectable or very low levels of IL-1Ra, even after stimulation with A77 1726 and IL-1β (data not shown). However, the levels of IL-1Ra in chondrocyte lysates increased with the number of passages; in subcultured, de-differentiated chondrocytes, IL-1β stimulated slightly the production of intracellular IL-1Ra, and A77 1726 considerably enhanced this effect (Fig. 2b).
Figure 2.
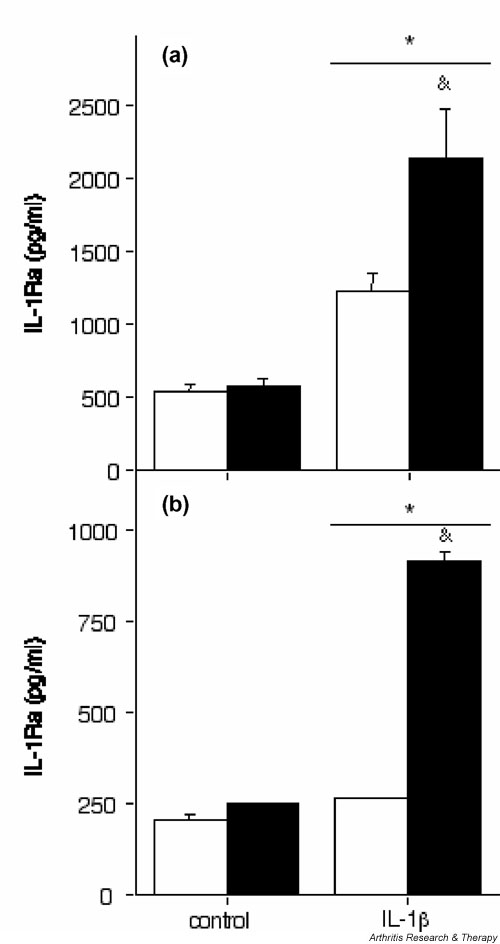
A77 1726 increases the production of intracellular IL-1 receptor antagonist (IL-1Ra) in human synovial fibroblasts and de-differentiated articular chondrocytes. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 1) and (b) de-differentiated human OA articular chondrocytes (passage 5) were stimulated (black columns) or not (white columns) with 100 μmol/l A77 1726 alone or in combination with 1 ng/ml IL-1β for 48 hours. A77 1726 was added 2 hours before stimulation with IL-1β. IL-1Ra concentrations in cell lysates were measured by ELISA. Results are represented as means ± SEM for three determinations in a representative experiment. Similar results were obtained with cells from five different donors (four OA and one rheumatoid arthritis sample) for synovial fibroblasts and two different donors (two OA samples) for de-differentiated chondrocytes. *P < 0.05 versus control; &P < 0.01 versus IL-1β alone.
We next investigated whether the stimulatory effect of A77 1726 was restricted to IL-1Ra production induced by IL-1β, or whether it could also be observed in the presence of other proinflammatory cytokines, such as TNF-α. As in the case for IL-1β, A77 1726 markedly and dose-dependently increased IL-1Ra secretion in synovial fibroblasts and chondrocytes in the presence of TNF-α (Fig. 3). The effect of A77 1726 was highest at 100 μmol/l. Furthermore, stimulation of IL-1Ra secretion by A77 1726 in human synovial fibroblasts was also dependent on the dose of IL-1β or TNF-α added (Fig. 4).
Figure 3.
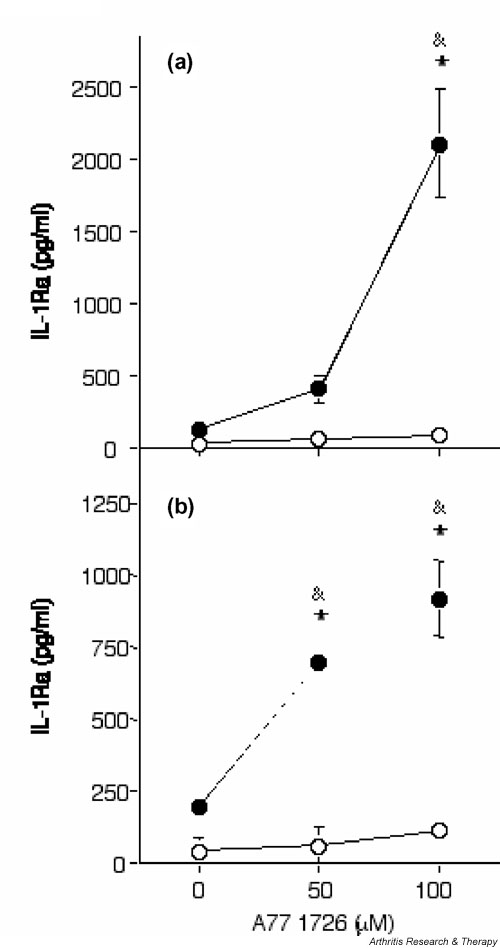
A77 1726 increases IL-1 receptor antagonist (IL-1Ra) secretion in human synovial fibroblasts and articular chondrocytes in the presence of tumour necrosis factor (TNF)-α. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 2) and (b) de-differentiated human OA articular chondrocytes (passage 7) were stimulated (closed symbols) or not (open symbols) with 10 ng/ml TNF-α alone or in combination with 50 μmol/l or 100 μmol/l A77 1726 for 48 hours. A77 1726 was added 2 hours before stimulation with TNF-α. IL-1Ra concentrations in culture supernatants were measured by ELISA. Results are represented as means ± SEM of three determinations in a representative experiment. A similar dose dependency was observed in cells obtained from three different donors (three OA samples) for synovial fibroblasts and three different donors (three OA samples) for de-differentiated chondrocytes. *P < 0.001 versus control; &P < 0.001 versus TNF-α alone.
Figure 4.
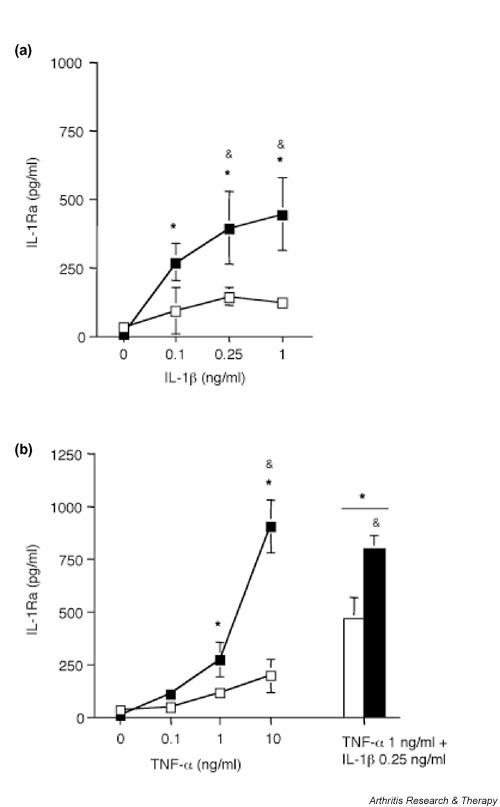
Dose dependent and additive effects of IL-1β and tumour necrosis factor (TNF)-α on IL-1 receptor antagonist (IL-1Ra) secretion in the presence of A77 1726 in human synovial fibroblasts. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 4) were stimulated with various doses of IL-1β, or (b) various doses of TNF-α or with 0.25 ng/ml IL-1β and 1 ng/ml TNF-α together, in the absence (open symbols) or presence (closed symbols) of 100 μmol/l A77 1726, for 48 hours. A77 1726 was added 2 hours before stimulation with IL-1β. IL-1Ra concentrations in culture supernatants were measured using ELISA. Results are represented as means ± SEM of three determinations in a representative experiment. Similar results were obtained with cells from three different donors (three OA samples). *P < 0.05 versus control; &P < 0.05 versus cytokines without A77 1726.
Finally, suboptimal doses of IL-1β and TNF-α had additive effects on IL-1Ra production, both in the presence and in the absence of A77 1726 (Fig. 4).
Mechanisms underlying the stimulatory effect of A77 1726 on IL-1 receptor antagonist production
In order to determine whether stimulation of IL-1Ra production by A77 1726 might be related to its well known inhibition of pyrimidine synthesis, we tested whether uridine could reverse the induction of IL-1Ra expression by A77 1726. However, uridine (50 and 200 μmol/l) had no impact on the enhancement of IL-1Ra secretion by A77 1726 in IL-1β-treated synovial fibroblasts, or de-differentiated or primary chondrocytes (data not shown).
We previously observed inhibition of COX-2 activity and of PGE2 production by A77 1726 in human synovial fibroblasts, and we therefore investigated whether A77 1726 could also inhibit PGE2 production in human articular chondrocytes. A77 1726 at 100 μmol/l completely blocked the production of PGE2 induced by IL-1β in human synovial fibroblasts, as well as in de-differentiated and in freshly isolated chondrocytes (Fig. 5). A similar inhibition of PGE2 synthesis was observed in the presence of 5 μg/ml indomethacin in the three cell types. We repeatedly observed much lower levels of IL-1β-induced PGE2 production in freshly isolated primary chondrocytes than in de-differentiated chondrocytes and synovial fibroblasts (Fig. 5).
Figure 5.
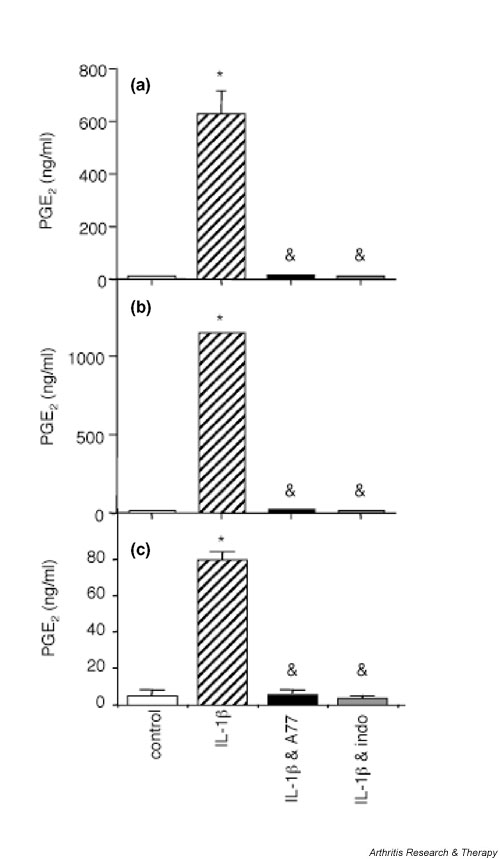
A77 1726 inhibits IL-1β-induced prostaglandin E2 (PGE2) productionin human synovial fibroblasts and articular chondrocytes. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 1), (b) de-differentiated human OA articular chondrocytes (passage 4), and (c) freshly isolated human OA articular chondrocytes were stimulated or not (control; white bars) with 1 ng/ml IL-1β (hatched bars), the combination of 1 ng/ml IL-1β and 100 μmol/l A77 1726 (black bars), or the combination of 1 ng/ml IL-1β and 5 μg/ml indomethacin (grey bars) for 48 hours. A77 1726 or indomethacin was added 2 hours before stimulation with IL-1β. PGE2 concentrations in culture supernatants were measured as described elsewhere [12]. Results are represented as means ± SEM of three determinations in one representative experiment. Similar results were obtained with cells from two different donors (two OA samples) for synovial fibroblasts, three different donors (three OA samples) for de-differentiated chondrocytes and five different donors (three normal and two OA samples) for freshly isolated chondrocytes. *P < 0.01 versus control; &P < 0.01 versus IL-1β.
If the enhancing effect of A77 1726 on IL-1β-induced IL-1Ra production was due to inhibition of COX-2 activity, then the effect of indomethacin on IL-1Ra production should be comparable to that of A77 1726. Indeed, indomethacin increased IL-1β-induced IL-1Ra secretion in human synovial fibroblasts and in de-differentiated chondrocytes (Fig. 6a,6b), suggesting that the inhibition of COX-2 enhanced IL-1Ra production in the presence of IL-1β in these cells. However, because the stimulatory effect of indomethacin was consistently less effective than that of A77 1726, additional mechanisms account for the increased IL-1Ra production in response to the latter compound. Finally, in agreement with the low levels of PGE2 produced by freshly isolated chondrocytes, which are suggestive of low COX-2 expression or activity, indomethacin had no significant effect on IL-1β-induced IL-1Ra production in these cells (Fig. 6c).
Figure 6.
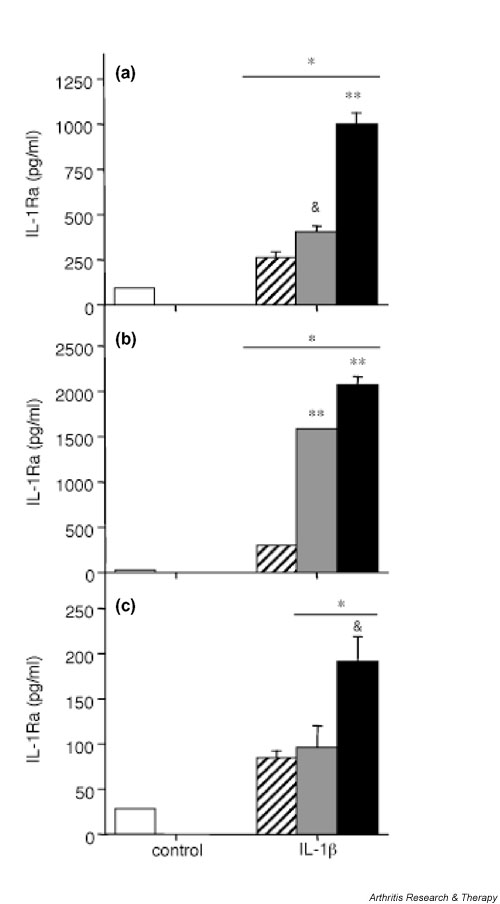
Effect of indomethacin on IL-1 receptor antagonist (IL-1Ra) secretion in human synovial fibroblasts and articular chondrocytes. (a) Human osteoarthritis (OA) synovial fibroblasts (passage 1), (b) de-differentiated human OA articular chondrocytes (passage 2), and (c) freshly isolated human OA articular chondrocytes were stimulated or not (control; white bars) with 1 ng/ml IL-1β (hatched bars), the combination of IL-1β and 5 μg/ml indomethacin (gray bars), or the combination of IL-1β and 100 μmol/l A77 1726 (black bars) for 48 hours. Indomethacin or A77 1726 was added 2 hours before stimulation with IL-1β. IL-1Ra concentrations in culture supernatants were measured by ELISA. Results are presented as means ± SEM of three determinations in a representative experiment. Similar results were obtained with cells from six different donors (six OA samples) for synovial fibroblasts, seven different donors (seven OA samples) for de-differentiated chondrocytes and six different donors (three normal and three OA samples) for freshly isolated chondrocytes. *P < 0.05 versus control; &P < 0.05 versus IL-1β alone; **P < 0.001 versus IL-1β alone.
Discussion
In the present study we investigated the effect of the active metabolite of leflunomide – A77 1726 – on the production of IL-1Ra by human joint cells. We observed that A77 1726, while having no effect alone, markedly enhanced the secretion of IL-1Ra in the presence of IL-1β or TNF-α in synovial fibroblasts and articular chondrocytes. The effect of A77 1726 was maximal at 100 μmol/l – a dose that lies within the range of plasma concentrations that may be observed in leflunomide-treated patients [6,23]. Because IL-1Ra has been shown to exert chondroprotective effects, our observations suggest that in the presence of proinflammatory cytokines, which are present in significant amounts in inflamed joints, A77 1726 might exert a beneficial effect by increasing the local production of this anti-inflammatory mediator by joint cells.
IL-1Ra, which was initially discovered for impeding the binding of IL-1 to lymphoma cells, is produced in four different isoforms, one secreted and three intracellular, which are derived from the same gene [24,25]. The exact biological functions of the different IL-1Ra isoforms are still not clear [25-27]. The major role of secreted IL-1Ra is to block the effects of IL-1 by binding competitively to IL-1 receptor I without inducing signal transduction. The intracellular isoforms may be released from cells under certain circumstances, but they have also been suggested to perform important regulatory roles within cells. Synovial fibroblasts and de-differentiated chondrocytes produce both secreted and intracellular IL-1Ra [28], and in these cells IL-1β-induced IL-1Ra production was enhanced in culture supernatants and in cell lysates in response to A77 1726. In contrast, cell lysates of freshly isolated chondrocytes contained no significant amounts of IL-1Ra, even after stimulation with IL-1β and A77 1726, which is consistent with our previous observations [28,29].
In a recent study we observed that over-expression of either the secreted or the type I intracellular IL-1Ra isoform similarly protected mice from collagen-induced arthritis, blocking inflammation and joint damage [30]. In RA, IL-1Ra has been shown to be one of the most potent agents available to decrease the progression of joint destruction [31-33], although its effects on inflammation and symptoms are frequently considered disappointing. It is generally considered that a 10- to 100-fold molar excess of IL-1Ra over IL-1 is required to suppress completely the biological effects of IL-1, although lower amounts of IL-1Ra can significantly inhibit IL-1-induced responses [34]. In the present study, the levels of IL-1Ra produced by synovial fibroblasts and de-differentiated chondrocytes on stimulation with A77 1726 and IL-1β usually ranged between equimolar concentrations and a twofold molar excess of IL-1Ra over IL-1. Even higher molar ratios of IL-1Ra:IL-1 were obtained when IL-1 was combined with TNF-α. Although large amounts of IL-1 are needed to obtain maximal catabolic effects in vitro, multiple lines of evidence (for example [35]) indicate that even very low levels of catabolic cytokines, including IL-1, can synergize to induce substantial effects. In vivo, it is likely that multiple cytokines present in low amounts act in synergy to induce proinflammatory and catabolic effects. Thus, the blockade of low amounts of IL-1 might be sufficient to decrease such a synergistic effect in vivo. The increased production of both secreted and intracellular IL-1Ra, which was observed in joint cells in response to A77 1726, might therefore be potentially beneficial by contributing to prevent joint damage in inflammatory arthropathies such as RA. In this regard, administration of leflunomide has been shown to limit joint destruction and improve function scores significantly, and to a greater degree than with methotrexate, according to at least two studies [1,36]. The mechanisms that result in this protection are likely to be multiple. However, a stimulatory effect on IL-1Ra synthesis might be particularly relevant, given the important role of IL-1 in joint destruction [37].
We investigated putative pathways involved in mediating the stimulatory effect of A77 1726 on IL-1Ra production, first focusing on the known effect of A77 1726 on pyrimidine synthesis. Addition of exogenous uridine did not significantly modulate the effect of A77 1726, suggesting that it was unlikely to be related to the inhibition of pyrimidine synthesis.
A77 1726 was previously reported to inhibit COX-2 activity [11]. Also, the findings reported here confirm a previous report that 100 μmol/l A77 1726 completely blocked IL-1β-induced PGE2 production in synovial fibroblasts [12]. Similarly, we observed that A77 1726 inhibited IL-1β-induced PGE2 production in chondrocytes. Interestingly, IL-1β triggered production of lower amounts of PGE2 in freshly isolated chondrocytes than in de-differentiated chondrocytes, suggesting low levels of expression and/or activity of COX-2 in these primary cells. This observation substantiates a recent report, which described a similar, differentiation stage-dependent regulation of COX-2 expression and PGE2 production in rabbit articular chondrocytes [38]. Indomethacin increased IL-1β-induced IL-1Ra production in synovial fibroblasts and de-differentiated chondrocytes, suggesting that inhibition of COX-2 may indeed enhance IL-1Ra production in the presence of IL-1β in these cells. However, the stimulatory effect of indomethacin was repeatedly less potent than that of A77 1726. In addition, indomethacin did not affect IL-1Ra production in primary chondrocytes. This observation is consistent with the low levels of PGE2 produced in these cells, which are suggestive of low COX-2 expression/activity. It is also in agreement with a previous study that described a lack of effect of indomethacin on IL-1Ra production in IL-1-stimulated OA chondrocytes [39]. Moreover, both in synovial fibroblasts and in chondrocytes, we observed that IL-1β-induced PGE2 production was strongly inhibited at relatively low concentrations (10–50 μmol/l) of A77 1726, as compared with the higher doses (50–100 μmol/l) that were required to enhance IL-1Ra production efficiently (data not shown). The dose dependency of these two effects thus appeared to be slightly different. Taken together, these observations strongly suggest that, in addition to the inhibitory effect of A77 1726 on COX-2 activity and PGE2 production, other mechanisms contribute to its stimulatory effect on IL-1Ra secretion.
Because high doses of A77 1726 have been reported to inhibit different types of receptor and nonreceptor tyrosine kinases [13-15], we assessed whether tyrosine kinase inhibitors would affect IL-1β-induced IL-1Ra production in fibroblasts and chondrocytes. Two types of inhibitors were tested: genistein, a broad range tyrosine kinase inhibitor; and PP1, a more specific inhibitor of the src family of tyrosine kinases, the members of which have been reported to mediate IL-1 signalling in various cell types [40,41]. Both inhibitors decreased IL-1β-induced IL-1Ra expression in synovial fibroblasts and chondrocytes (data not shown). These observations suggest that the increase in IL-1Ra production observed in the presence of A77 1726 is unlikely to be due to the inhibition of tyrosine kinases. Thus, although our findings suggest that part of the effect of A77 1726 on IL-1Ra production occurs through the inhibition of COX-2 activity, other unknown mechanisms, which remain to be characterized, are likely to be involved.
Conclusion
A77 1726 enhanced the production of IL-1Ra by human synovial fibroblasts and articular chondrocytes in the presence of the proinflammatory cytokines IL-1β and TNF-α. This might explain some of the protective effects exerted by leflunomide in RA and other cartilage-damaging diseases.
Competing interests
This work was supported in part by an unrestricted research fund from Aventis Pharma (Frankfurt am Main, Germany).
Abbreviations
COX = cyclo-oxygenase; DHODH = dihydro-orotate dehydrogenase; ELISA = enzyme-linked immunosorbent assay; FCS = foetal calf serum; IL = interleukin; IL-1Ra = IL-1 receptor antagonist; MMP = matrix metalloproteinase; OA = osteoarthritis; PGE2 = prostaglandin E2; RA = rheumatoid arthritis; TNF = tumour necrosis factor.
Acknowledgments
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant 3100-064123.00/1 to PAG, grants 3200-054955.98 and 3231-05454.98 to CG, and grant 3200-068286.02 to JMD) and by Aventis Pharma (Frankfurt am Main, Germany).
References
- Strand V, Cohen S, Schiff M, Weaver A, Fleischmann R, Cannon G, Fox R, Moreland L, Olsen N, Furst D, Caldwell J, Kaine J, Sharp J, Hurley F, Loew-Friedrich I. Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Leflunomide Rheumatoid Arthritis Investigators Group. Arch Intern Med. 1999;159:2542–2550. doi: 10.1001/archinte.159.21.2542. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Kalden JR, Scott DL, Rozman B, Kvien TK, Larsen A, Loew-Friedrich I, Oed C, Rosenburg R. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double-blind, randomised, multi-centre trial. European Leflunomide Study Group. Lancet. 1999;353:259–266. doi: 10.1016/S0140-6736(98)09403-3. [DOI] [PubMed] [Google Scholar]
- Emery P, Breedveld FC, Lemmel EM, Kaltwasser JP, Dawes PT, Gomor B, Van Den Bosch F, Nordstrom D, Bjorneboe O, Dahl R, Horslev-Petersen K, Rodriguez De La Serna A, Molloy M, Tikly M, Oed C, Rosenburg R, Loew-Friedrich I. A comparison of the efficacy and safety of leflunomide and methotrexate for the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2000;39:655–665. doi: 10.1093/rheumatology/39.6.655. [DOI] [PubMed] [Google Scholar]
- Breedveld FC, Dayer JM. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2000;59:841–849. doi: 10.1136/ard.59.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann ML, Schleyerbach R, Kirschbaum BJ. Leflunomide: an immunomodulatory drug for the treatment of rheumatoid arthritis and other autoimmune diseases. Immunopharmacology. 2000;47:273–289. doi: 10.1016/S0162-3109(00)00191-0. [DOI] [PubMed] [Google Scholar]
- Williams JW, Mital D, Chong A, Kottayil A, Millis M, Longstreth J, Huang W, Brady L, Jensik S. Experiences with leflunomide in solid organ transplantation. Transplantation. 2002;73:358–366. doi: 10.1097/00007890-200202150-00008. [DOI] [PubMed] [Google Scholar]
- Cherwinski HM, Byars N, Ballaron SJ, Nakano GM, Young JM, Ransom JT. Leflunomide interferes with pyrimidine nucleotide biosynthesis. Inflamm Res. 1995;44:317–322. doi: 10.1007/BF01796261. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, Woodward K, Bruneau JM, Hambleton P, Spinella-Jaegle S, Morand P, Courtin O, Sautes C, Westwood R, Hercend T, Kuo EA, Ruuth E. Dihydroorotate dehydrogenase is a target for the biological effects of leflunomide. Transplant Proc. 1996;28:3088–3091. [PubMed] [Google Scholar]
- Manna SK, Aggarwal BB. Immunosuppressive leflunomide metabolite (A77 1726) blocks TNF-dependent nuclear factor-kappa B activation and gene expression. J Immunol. 1999;162:2095–2102. [PubMed] [Google Scholar]
- Manna SK, Mukhopadhyay A, Aggarwal BB. Leflunomide suppresses TNF-induced cellular responses: effects on NF-kappa B, activator protein-1, c-Jun N-terminal protein kinase, and apoptosis. J Immunol. 2000;165:5962–5969. doi: 10.4049/jimmunol.165.10.5962. [DOI] [PubMed] [Google Scholar]
- Hamilton LC, Vojnovic I, Warner TD. A77 the active metabolite of leflunomide, directly inhibits the activity of cyclo-oxygenase-2 in vitro and in vivo in a substrate-sensitive manner. Br J Pharmacol. 1999;127:1589–1596. doi: 10.1038/sj.bjp.0702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger D, Begue-Pastor N, Benavent S, Gruaz L, Kaufmann MT, Chicheportiche R, Dayer JM. The active metabolite of leflunomide, A77 inhibits the production of prostaglandin E(2), matrix metalloproteinase 1 and interleukin 6 in human fibroblast-like synoviocytes. Rheumatology (Oxford) 2003;42:89–96. doi: 10.1093/rheumatology/keg038. [DOI] [PubMed] [Google Scholar]
- Mattar T, Kochhar K, Bartlett R, Bremer EG, Finnegan A. Inhibition of the epidermal growth factor receptor tyrosine kinase activity by leflunomide. FEBS Lett. 1993;334:161–164. doi: 10.1016/0014-5793(93)81704-4. [DOI] [PubMed] [Google Scholar]
- Xu X, Williams JW, Gong H, Finnegan A, Chong AS. Two activities of the immunosuppressive metabolite of leflunomide, A77 1726. Inhibition of pyrimidine nucleotide synthesis and protein tyrosine phosphorylation. Biochem Pharmacol. 1996;52:527–534. doi: 10.1016/0006-2952(96)00303-6. [DOI] [PubMed] [Google Scholar]
- Elder RT, Xu X, Williams JW, Gong H, Finnegan A, Chong AS. The immunosuppressive metabolite of leflunomide, A77 affects murine T cells through two biochemical mechanisms. J Immunol. 1997;159:22–27. [PubMed] [Google Scholar]
- Deage V, Burger D, Dayer JM. Exposure of T lymphocytes to leflunomide but not to dexamethasone favors the production by monocytic cells of interleukin-1 receptor antagonist and the tissue-inhibitor of metalloproteinases-1 over that of interleukin-1beta and metalloproteinases. Eur Cytokine Netw. 1998;9:663–668. [PubMed] [Google Scholar]
- Cutolo M, Sulli A, Ghiorzo P, Pizzorni C, Craviotto C, Villaggio B. Anti-inflammatory effects of leflunomide on cultured synovial macrophages from patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:297–302. doi: 10.1136/ard.62.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkayam O, Yaron I, Shirazi I, Judovitch R, Caspi D, Yaron M. Active leflunomide metabolite inhibits interleukin 1beta, tumour necrosis factor alpha, nitric oxide, and metalloproteinase-3 production in activated human synovial tissue cultures. Ann Rheum Dis. 2003;62:440–443. doi: 10.1136/ard.62.5.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz M, Zwicker M, Loetscher P. Effects of methotrexate on differentiation of monocytes and production of cytokine inhibitors by monocytes. Arthritis Rheum. 1998;41:2032–2038. doi: 10.1002/1529-0131(199811)41:11<2032::AID-ART19>3.3.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Lotz M, Clark-Lewis I, Ganu V. HIV-1 transactivator protein Tat induces proliferation and TGF beta expression in human articular chondrocytes. J Cell Biol. 1994;124:365–371. doi: 10.1083/jcb.124.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L, Forsum U, Malmnas Tjernlund UK, Kabelitz D, Wigren A. Appearance of anti-HLA-DR-reactive cells in normal and rheumatoid synovial tissue. Scand J Immunol. 1981;14:183–192. doi: 10.1111/j.1365-3083.1981.tb00198.x. [DOI] [PubMed] [Google Scholar]
- Gabay C, Smith MF, Eidlen D, Arend WP. Interleukin 1 receptor antagonist (IL-1Ra) is an acute-phase protein. J Clin Invest. 1997;99:2930–2940. doi: 10.1172/JCI119488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RI, Herrmann ML, Frangou CG, Wahl GM, Morris RE, Strand V, Kirschbaum BJ. Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93:198–208. doi: 10.1006/clim.1999.4777. [DOI] [PubMed] [Google Scholar]
- Seckinger P, Lowenthal JW, Williamson K, Dayer JM, MacDonald HR. A urine inhibitor of interleukin 1 activity that blocks ligand binding. J Immunol. 1987;139:1546–1549. [PubMed] [Google Scholar]
- Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- Muzio M, Polentarutti N, Facchetti F, Peri G, Doni A, Sironi M, Transidico P, Salmona M, Introna M, Mantovani A. Characterization of type II intracellular IL-1 receptor antagonist (IL-1ra3): a depot IL-1ra. Eur J Immunol. 1999;29:781–788. doi: 10.1002/(SICI)1521-4141(199903)29:03<781::AID-IMMU781>3.3.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Watson JM, Lofquist AK, Rinehart CA, Olsen JC, Makarov SS, Kaufman DG, Haskill JS. The intracellular IL-1 receptor antagonist alters IL-1-inducible gene expression without blocking exogenous signaling by IL-1 beta. J Immunol. 1995;155:4467–4475. [PubMed] [Google Scholar]
- Palmer G, Mezin F, Juge-Aubry CE, Plater-Zyberk C, Gabay C, Guerne PA. Interferon-beta stimulates interleukin-1 receptor antagonist production in human articular chondrocytes and synovial fibroblasts. Ann Rheum Dis. 2004. [DOI] [PMC free article] [PubMed]
- Palmer G, Guerne PA, Mezin F, Maret M, Guicheux J, Goldring MB, Gabay C. Production of interleukin-1 receptor antagonist by human articular chondrocytes. Arthritis Res. 2002;4:226–231. doi: 10.1186/ar411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer G, Talabot-Ayer D, Szalay-Quinodoz I, Maret M, Arend WP, Gabay C. Mice transgenic for intracellular interleukin-1 receptor antagonist type 1 are protected from collagen-induced arthritis. Eur J Immunol. 2003;33:434–440. doi: 10.1002/immu.200310018. [DOI] [PubMed] [Google Scholar]
- Bresnihan B. Effects of anakinra on clinical and radiological outcomes in rheumatoid arthritis. Ann Rheum Dis. 2002;Suppl 2:ii74–ii77. doi: 10.1136/ard.61.suppl_2.ii74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayer JM, Bresnihan B. Targeting interleukin-1 in the treatment of rheumatoid arthritis. Arthritis Rheum. 2002;46:574–578. doi: 10.1002/art.10168. [DOI] [PubMed] [Google Scholar]
- Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford) 2003;Suppl 2:ii3–ii10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- Granowitz EV, Clark BD, Vannier E, Callahan MV, Dinarello CA. Effect of interleukin-1 (IL-1) blockade on cytokine synthesis: I. IL-1 receptor antagonist inhibits IL-1-induced cytokine synthesis and blocks the binding of IL-1 to its type II receptor on human monocytes. Blood. 1992;79:2356–2363. [PubMed] [Google Scholar]
- Chabaud M, Page G, Miossec P. Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol. 2001;167:6015–6020. doi: 10.4049/jimmunol.167.10.6015. [DOI] [PubMed] [Google Scholar]
- Cohen S, Cannon GW, Schiff M, Weaver A, Fox R, Olsen N, Furst D, Sharp J, Moreland L, Caldwell J, Kaine J, Strand V. Two-year, blinded, randomized, controlled trial of treatment of active rheumatoid arthritis with leflunomide compared with methotrexate. Utilization of Leflunomide in the Treatment of Rheumatoid Arthritis Trial Investigator Group. Arthritis Rheum. 2001;44:1984–1992. doi: 10.1002/1529-0131(200109)44:9<1984::AID-ART346>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- van de Loo FA, van den Berg WB. Gene therapy for rheumatoid arthritis. Lessons from animal models, including studies on interleukin-4, interleukin-10, and interleukin-1 receptor antagonist as potential disease modulators. Rheum Dis Clin North Am. 2002;28:127–149. doi: 10.1016/s0889-857x(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Huh YH, Kim SH, Kim SJ, Chun JS. Differentiation status-dependent regulation of cyclooxygenase-2 expression and prostaglandin E2 production by epidermal growth factor via mitogen-activated protein kinase in articular chondrocytes. J Biol Chem. 2003;278:9691–9697. doi: 10.1074/jbc.M211360200. [DOI] [PubMed] [Google Scholar]
- Pelletier JP, Mineau F, Ranger P, Tardif G, Martel-Pelletier J. The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarthritis Cartilage. 1996;4:77–84. doi: 10.1016/s1063-4584(96)80009-4. [DOI] [PubMed] [Google Scholar]
- Al-Ramadi BK, Welte T, Fernandez-Cabezudo MJ, Galadari S, Dittel B, Fu XY, Bothwell AL. The Src-protein tyrosine kinase Lck is required for IL-1-mediated costimulatory signaling in Th2 cells. J Immunol. 2001;167:6827–6833. doi: 10.4049/jimmunol.167.12.6827. [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M, Tago K, Sonoda Y, Tominaga S, Kasahara T. TRAF6 and C-SRC induce synergistic AP-1 activation via PI3-kinase-AKT-JNK pathway. Eur J Biochem. 2003;270:1257–1268. doi: 10.1046/j.1432-1033.2003.03487.x. [DOI] [PubMed] [Google Scholar]