

Abstract
Collagen-induced arthritis (CIA) in mice is accompanied by splenomegaly due to the selective expansion of immature CD11b+ myeloblasts. Both disease manifestations are more pronounced in interferon-γ receptor knock-out (IFN-γR KO) mice. We have taken advantage of this difference to test the hypothesis that the expanding CD11b+ splenic cell population constitutes a source from which osteoclast precursors are recruited to the joint synovia. We found larger numbers of osteoclasts and more severe bone destruction in joints of IFN-γR KO mice than in joints of wild-type mice. Osteoclast-like multinucleated cells appeared in splenocyte cultures established in the presence of macrophage colony-stimulating factor (M-CSF) and stimulated with the osteoclast-differentiating factor receptor activator of NF-κB ligand (RANKL) or with tumour necrosis factor-α (TNF-α). Significantly larger numbers of such cells could be generated from splenocytes of IFN-γR KO mice than from those of wild-type mice. This was not accompanied, as might have been expected, by increased concentrations of the intracellular adaptor protein TRAF6, known to be involved in signalling of RANKL- and TNF-α-induced osteoclast formation. Splenocyte cultures of IFN-γR KO mice also produced more TNF-α and more RANKL than those of wild-type mice. Finally, splenocytes isolated from immunised IFN-γR KO mice contained comparatively low levels of pro-interleukin-1β (pro-IL-1β) and pro-caspase-1, indicating more extensive conversion of pro-IL-1β into secreted active IL-1β. These observations provide evidence that all conditions are fulfilled for the expanding CD11b+ splenocytes to act as a source of osteoclasts and to be indirectly responsible for bone destruction in CIA. They also provide a plausible explanation for the higher susceptibility of IFN-γR KO mice to CIA.
Keywords: osteoclast differentiation factor, osteoprotegerin ligand, receptor activator of NF-κB ligand, tumour necrosis factor receptor-associated factor
Introduction
Collagen-induced arthritis (CIA) is a well-characterised experimental model for rheumatoid arthritis in humans. One common aspect of the two conditions is the occurrence of bone destruction in the joints caused by osteoclast activation in the synovium. Mice lacking a functional interferon-γ (IFN-γ) receptor (interferon-γ receptor knockout [IFN-γR KO] mice) are more susceptible to CIA than wild-type mice [1,2]: the median day of disease onset is reduced from 43 to 21 days and both the severity and the cumulative incidence of arthritis are higher. Similarly, in wild-type mice, disease onset is accelerated and scores of arthritis are increased by treatment with neutralising monoclonal antibodies against IFN-γ [2]. Accelerated disease onset in both experimental settings is associated with an increased expansion of CD11b+ myeloid cells in the spleen [3]. In this study we investigated the possibility that these CD11b+ cells can differentiate into osteoclasts and therefore that their expansion in IFN-γR KO mice can in part account for the higher susceptibility of such mice to CIA. In addition we analysed the molecular signals for osteoclastogenesis in IFN-γR KO and wild-type mice.
Osteoclasts and osteoblasts are essential for bone homeostasis and remodelling, a process that continues throughout life [4-6]. In a healthy organism the activities of both cell types are in balance. Generalised imbalance causes either osteoporosis or osteopetrosis. Localised impairment of the equilibrium can cause local damage of the bone tissue. This is considered to be a major pathogenic process in rheumatoid arthritis and similarly in CIA [7,8], as articular lesions evolve in parallel with increased numbers of osteoclasts in the inflamed synovium [8]. Osteoclast precursors belong to the monocyte/macrophage lineage [5,9,10]. They can be recruited from the bone marrow and, in mice, from the spleen [11]. Their differentiation into active osteoclasts is regulated by several cytokines: receptor activator of NF-κB ligand (RANKL); osteoprotegerin (OPG); tumour necrosis factor-α (TNF-α); interleukin (IL)-1β; and macrophage colony-stimulating factor (M-CSF).
RANKL is the most commonly used denomination of the cytokine also known as osteoclast differentiation factor (ODF) [11], TRANCE (TNF-related activation-induced cytokine) [12] and OPGL (osteoprotegerin ligand) [13]. RANKL belongs to the TNF superfamily [13]. It exists in both a membrane-bound and a soluble form [14] and is expressed by several cell types, including activated T cells [15], osteoblasts and stromal cells of the bone marrow [13] and fibroblast-like synoviocytes [16]. RANK (receptor activator of NF-κB) is the essential signalling receptor for RANKL in osteoclastogenesis [17]. In CIA, RANK+ cells are abundantly present in inflamed synovia and their numbers are correlated with disease severity [18]. RANKL can also bind a soluble protein, OPG [19], also called OCIF (osteoclastogenesis inhibiting factor) [20], which is a secreted member of the TNF receptor superfamily and acts as a decoy receptor for RANKL.
TNF-α can have a dual role in osteoclast formation. Through the activation of one of its receptors, TNFR1, it can promote osteoclastogenesis, whereas via TNFR2 it exerts an inhibitory effect [21]. Although TNF-α can act in synergy with RANKL [21], there is also evidence that it directly stimulates osteoclastogenesis in the absence of RANKL [22].
In contrast to TNF-α and RANKL, IL-1β is not able to trigger osteoclastogenesis but can activate preformed osteoclasts [16]. IL-1β is synthesised as pro-IL-1β, which remains in the cytosol until it is cleaved by caspase-1 and can be transported out of the cell. Caspase-1 is similarly produced as an inactive 45 kDa precursor protein that requires two internal cleavages before becoming the enzymatically active heterodimer comprising a 10 kDa and a 20 kDa subunit, cleaving pro-IL-1β into mature secreted IL-1β [23].
RANKL, TNF-α and IL-1β rely for signalling on intracellular adaptor proteins called TRAF (TNF receptor associated factor). One of these, TRAF6, is known to be involved in osteoclastogenesis induced by RANKL as well as by TNF-α [24,25] and in IL-1β signalling [26]. Moreover, a link between IFN-γ and RANKL signalling via TRAF6 has also been demonstrated in bone marrow cultures in which IFN-γ was shown to accelerate the degradation of TRAF6 [27].
Finally, osteoclasts can develop only in an environment in which M-CSF is present [28].
The purpose of the experiments described here was to investigate whether endogenous IFN-γ could be protective against CIA by inhibiting osteoclastogenesis in vivo. We investigated whether the accelerated CIA in IFN-γR KO mice coincides with earlier appearance and higher numbers of osteoclasts in the joints. We examined the capacity of splenocytes of IFN-γR KO and wild-type mice to differentiate into osteoclasts in vitro, and we investigated whether the extramedullar splenic CD11b+ cell population, expanded after immunisation with collagen type II in complete Freund's adjuvant (CII/CFA) in the IFN-γR KO mice, can be regarded as possible osteoclast precursors. We further analysed the capacity of the splenocytes of both mouse strains to express cytokines, receptors and intracellular key proteins regulating osteoclast differentiation in IFN-γR KO and wild-type mice.
Materials and methods
Induction of CIA and assessment of the symptoms
IFN-γR KO mice were generated by crossing wild-type DBA/1 mice with a mutant mouse strain (129/Sv/Ev) in which the gene coding for the α-chain of the IFN-γ receptor was disrupted by insertion of a neo gene into exon V. Functional inactivation of the IFN-γR gene was verified [29]. These IFN-γR KO mice were backcrossed with wild-type DBA/1 mice for 10 generations to obtain a DBA/1 IFN-γR KO mouse strain. To identify homozygous IFN-γR KO mice, genomic tail-skin DNA was amplified by polymerase chain reaction (PCR), with 5'-CCCATTTAG-ATCCTACATACGAAACATACGG-3' as a sense primer and 5'-TTTCTGTCATCATGGAAAGGAGGGATACAG-3' as an antisense primer. On the wild-type allele these primers amplified a 189 base pair fragment; on the disrupted allele the amplification encompassed the inserted neo gene and therefore resulted in a 1282 base pair fragment. Pure DBA/1 strain mice were used as wild-type controls. The experiments were performed in mice 8–12 weeks old that were matched for age and sex within each experiment. Both the wild-type and the IFN-γR KO mice were bred in the Experimental Animal Centre of the Katholieke Universiteit Leuven.
CII from chicken sternal cartilage (Sigma-Aldrich, St Louis, MO, USA) was dissolved at 2 mg/ml in phosphate-buffered saline (PBS) containing 0.1 M acetic acid by stirring overnight at 6°C, and emulsified in an equal volume of CFA with added heat-killed Mycobacterium butyricum (Difco Laboratories, Detroit, MI, USA), reaching a final Mycobacterium content of 750 μg/ml emulsion. Mice were injected intradermally with 100 μl of emulsion at the base of the tail on day 0.
From day 10 onwards, mice were scored for symptoms of arthritis; the disease severity was recorded on a scoring system for each limb: 0, normal; 1, redness and/or swelling in one joint; 2, redness and/or swelling in more than one joint; 3, redness and/or swelling in the entire paw, 4; deformity and/or ankylosis [2].
In vitro induction of osteoclast formation by splenocytes
Spleens were isolated, cut into small pieces and passed through cell strainers (Becton Dickinson Labware, Franklin Lakes, NJ, USA), to obtain single-cell suspensions. Red blood cells were lysed by two incubations (5 and 3 min at 37°C) of the splenocyte suspension with NH4Cl solution (0.083% in 0.01 M Tris-HCl, pH 7.2). Remaining cells were washed twice with ice-cold PBS and resuspended in α-minimal essential medium containing 10% fetal calf serum (Gibco, Invitrogen Corporation, Paisley, UK). Cells (2.5 × 104) in a total volume of 400 μl were seeded in chamber slides (Lab-Tek Brand Products, Nalge Nunc International, Naperville, IL, USA). Cells were incubated for 6 days with 20 ng/ml M-CSF alone or with M-CSF and 100 ng/ml RANKL or with M-CSF and 20 ng/ml TNF-α. All cytokines were obtained from R&D Systems Europe (Abingdon, UK). On day 7, media were removed and cells were stained for the presence of tartrate-resistant acid phosphatase (TRAP).
TRAP staining, histology and immunohistochemistry
All reagents were obtained from Sigma-Aldrich. Cells from the in vitro cultures were fixed with 3.7% formaldehyde in Ca2+- and Mg2+-free PBS for 10 min and subsequently for 1 min with a 50/50 (v/v) solution of ethanol/acetone. Cells were incubated for 10 min with the staining solution (0.01% naphthol AS-MX phosphate [Sigma-Aldrich N4875], 50 mM tartrate, 0.06% fast red violet LB salt in 0.1 M acetate buffer, pH 5.0), washed with distilled water and kept in water for 20 min. Staining solutions were freshly prepared before use.
For the detection of TRAP+ cells in histological slides of joints, amputated limbs were fixed in 1% paraformaldehyde for 16 hours and washed with PBS. The tissues were decalcified by incubation in 0.5 M EDTA/PBS, pH 7.4, for 10 days, in which the EDTA solution was changed every day. Tissues were embedded in paraffin and 6 μm sections were made. Deparaffinised, rehydrated sections were either stained with haematoxylin and eosin or preincubated for 2.5 hours at 37°C in a 12.5 mM sodium tartrate solution in 100 mM acetate buffer, pH 5.5. Subsequently, sections were incubated for 1 hour at 37°C in acid phosphatase substrate solution (0.05% naphthol AS-BI phosphate [Sigma-Aldrich N2905], 50 mM sodium tartrate, 0.16% p-rosanilin, 0.16% NaNO2, 25% Michaelis' 0.14 M acetate/barbital buffer, pH 5.0, in distilled water). Sections were washed with distilled water, counterstained with 0.15% Lightgreen SF Yellowish in 0.2% acetic acid, incubated for 10 s in 1% acetic acid and dried at 37°C. Red-staining cells were considered to contain TRAP, and TRAP+ multinucleated cells (three or more nuclei) were regarded as osteoclasts.
Immunohistochemistry for CD11b+ cells was performed with biotinylated rat anti-mouse CD11b monoclonal antibody (IgG2b) or biotinylated isotype control immunoglobulin (BD Biosciences Pharmingen, San Diego, CA, USA). Paraffin-embedded sections were dewaxed, quenched in 3% H2O2 in water to limit endogenous peroxidase activity, and washed with Tris-buffered saline containing 0.1% Triton (TBS/Triton). For antigen retrieval, sections were microwaved twice for 10 min in 10 mM sodium citrate buffer. After washing in TBS/Triton, nonspecific binding was blocked by preincubation for 30 min with a 1:5 dilution of donkey serum (Dako, Glostrup, Denmark) in TBS/Triton. Sections were incubated overnight at 4°C with either anti-CD11b antibody (1:100 dilution) or control antibody in a humidified chamber. The antibody–biotin conjugates were detected with a streptavidin–biotin–horseradish peroxidase complex (StreptABComplex/HRP; Dako) applied for 30 min at 21°C, using diaminobenzidine (Dako) as a substrate. Nuclei were counterstained with haematoxylin. Slides were mounted in Pertex mounting medium (Histolab Products, Göteborg, Sweden).
Detection of RANKL, TNF-α and IFN-γ
RANKL was detected by an enzyme-linked immunosorbent assay (ELISA) developed in our laboratory. Wells of 96-well plates were coated overnight at 4°C with 100 μl per well containing 0.5 μg/ml purified polyclonal goat anti-mouse RANKL IgG (R&D Systems Europe) in PBS, subsequently washed with 0.05% Tween 20/PBS buffer and blocked with 300 μl PBS containing 1% BSA (Sigma-Aldrich) and 5% sucrose. After incubation for 2 hours, wells were washed with 0.05% Tween 20/PBS. Serial dilutions of RANKL standards and samples were prepared in 0.05% Tween 20/PBS buffer containing 0.1% BSA and then incubated in duplicate in the coated wells. After being washed with 0.05% Tween 20/PBS, each well received 100 μl of detection antibody; that is, 500 ng/ml biotin-conjugated purified polyclonal goat anti-mouse RANKL (R&D Systems Europe). After incubation for 2 hours, the wells were washed and replenished with 100 μl of streptavidin–HRP (Jackson Immunoresearch Laboratories Inc., West Grove, PA, USA) was incubated for 20 min. Wells were washed with 0.05% Tween 20/PBS. Finally, 3.3 μl of the chromogen 3,3',5,5'-tetra-methylbenzidine (Sigma-Aldrich), dissolved in 250 μl of dimethyl sulphoxide, and 3.3 μl of the substrate H2O2 were added to 25 ml of the reaction buffer (100 mM sodium acetate/citric acid, pH 4.9); 100 μl of this reaction buffer was added to the wells for a 10 min incubation. Reactions were stopped by adding 50 μl of 4 M H2SO4, and colour intensity was measured at 450 nm.
TNF-α levels were measured with the DuoSet ELISA Development System (R&D Systems Europe).
IFN-γ concentrations were determined by sandwich ELISA as described previously [30].
Pit-forming assay
Splenocyte suspensions were obtained as described above, washed twice with ice-cold PBS, and resuspended in α-minimal essential medium containing 10% fetal calf serum (Gibco). Cells (106) were cultured for 7 days with M-CSF (20 ng/ml) and RANKL (100 ng/ml) (both from R&D Systems Europe) on transparent quartz slides coated with a calcium phosphate film (BioCoat Osteologic Discs; BD Biosciences Pharmingen, San Diego, CA, USA). Cells were removed and resorption of the film was assessed by light microscopy.
Flow cytometric analysis
Single-cell suspensions (5 × 105 cells) were incubated with the Fc-receptor-blocking antibodies anti-CD16/anti-CD32 (BD Biosciences Pharmingen) and then stained for 30 min with biotin-conjugated purified polyclonal goat anti-mouse RANK antibody (R&D Systems Europe) or biotinylated pre-immune goat IgG (Jackson Immunoresearch Laboratories Inc.). Cells were washed and incubated for 20 min with fluorescein isothiocyanate-conjugated streptavidin (BD Biosciences Pharmingen). Subsequently, after being washed, cells were incubated with phycoerythrin-conjugated anti-CD11b antibody (BD Biosciences Pharmingen). Cells were washed, fixed with 0.37% formaldehyde in PBS, and analysed with a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA).
Western blotting
Spleens were isolated and single-cell suspensions were prepared as described above. Splenocytes were lysed in RIPA buffer (PBS containing 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulphonyl fluoride, 30 μl/ml aprotinin and 1 mM sodium orthovanadate; all products from Sigma-Aldrich). The lysate was subjected to SDS–polyacrylamide-gel electrophoresis and proteins were transferred to Hybond nitrocellulose membranes (Amersham Pharmacia Biotech, Little Chalfont, UK). The blot was probed with polyclonal antibodies against mouse IL-1β (R&D Systems Europe), mouse caspase-1 (a gift from Dr P Vandenabeele, University of Ghent, Belgium) or against mouse TRAF6 (MBL International, Woburn, MA, USA). Immunoreactivity was revealed with an enhanced-chemiluminescence method (NEN Renaissance Products, Perkin Elmer, Boston, MA, USA).
PCR
Synovial tissues from the ankle joints were carefully isolated under a stereomicroscope. Total RNA was extracted with Trizol reagent (Invitrogen), in accordance with the manufacturer's instructions. Complementary DNA (cDNA) was obtained by reverse transcription with a commercially available kit (Thermoscript; Invitrogen Corporation) with oligo(dT)20 as primer. PCRs were performed as previously described [31]. Complementary DNA was mixed with 0.5 U Taq polymerase (Eurogentec, Seraing, Belgium), 0.2 mM dNTP, 0.5 μM specific primers and 1.5 mM MgCl2. Primer pairs were designed with Vector NTI software (Informax, North Bethesda, MD, USA). Primer sequences were as follows: RANKL sense, 5'-CTCTGCTCTGATGTGCTGTG-3'; RANKL antisense, 5'-TCGCCCTGTTCTTCTATTTC-3'; M-CSF sense, 5'-TGA-CGGGTCACCCACACACTGTGCCCATCTA-3'; M-CSF antisense, 5'-CTAGAAGCATTTGCGGTGGACGATGGA-GGG-3'; β-actin sense, 5'-TGACGGGGTCACCCACAC-TGTGCCCATCTA-3'; β-actin antisense, 5'-CTAGAA-GCATTTGCGGTGGACGATGGAGGG-3'.
All PCRs were performed in a Perkin Elmer Thermal Cycler 9600 (Applied Biosystems; Lennik, Belgium). After denaturation at 95°C for 2 min, cycles were 10 s at 94°C, 10 s at 60°C, and 30 s at 72°C. Cycling was followed by 10 min of elongation at 72°C. PCR products were subjected to electrophoresis in 1.2% agarose gels in Tris–borate–EDTA electrophoresis buffer, stained with ethidium bromide and detected by ultraviolet transillumination. Complementary DNA samples were normalised for the housekeeping gene β-actin.
For real-time quantitative PCR, cDNA was synthesised with Superscript II RT (Gibco-BRL). Real-time quantitative PCR was performed as described by Maes and colleagues [32]. Specific forward (5'-AACCGAACCTGG-TCCAACTATACT-3') and reverse (5'-TCAGCATGG-AAGCAACCAAA-3') primers, and probe (5'-AAATGC-GTACGTTCTTTATTACCTGGCTCTTGTG-3') with fluorescent dye (5-carboxyfluorescein; FAM) and quencher (5(6)-carboxy-tetramethylrhodamine; TAMRA) were designed for the mouse calcitonin receptor. The sequence of the amplicon was verified. Expression levels of the gene were normalised for the hypoxanthine transferase gene.
Results
Accelerated CIA in IFN-γR KO mice coincides with earlier appearance of osteoclasts in the joints
In a first experiment, IFN-γR KO or wild-type mice were immunised with CII in CFA and were observed for symptoms of arthritis (Fig. 1a). As in previously reported experiments [2,3], IFN-γR KO mice developed CIA more readily: symptoms appeared from day 21 onwards, as opposed to day 31 in wild-type mice. A similar experiment (Fig. 1b) was done to confirm increased expansion of the CD11b+ splenic cell population in IFN-γR KO mice [3], but also to document the appearance of osteoclasts in affected joints. To that end the mice were killed on day 27 for histological examination of joints and for flow-cytometric analysis of splenocytes. As can be seen in Fig. 1b, spleens of IFN-γR KO mice showed an increased proportion of CD11b+ haematopoietic cells but unchanged proportions of CD4+ and CD8+ T cells and B cells. Haematoxylin staining of joint sections revealed the presence of many multinucleated cells dispersed in the synovia and lining the calcified bone tissue of IFN-γR KO mice (Fig. 1c), whereas such cells were as yet (on day 27) absent from joints of wild-type mice (Fig. 1d). Staining with TRAP allowed us to confirm the osteoclast-like nature of these cells (Fig. 1e). The numbers of TRAP+ osteoclasts in two IFN-γR KO mice were 61 and 53 (means of three sections). No TRAP+ cells could be seen in wild-type sections. Interestingly, the multinucleated cells lining the calcified bone tissue also stained positive for CD11b (Fig. 1f).
Figure 1.
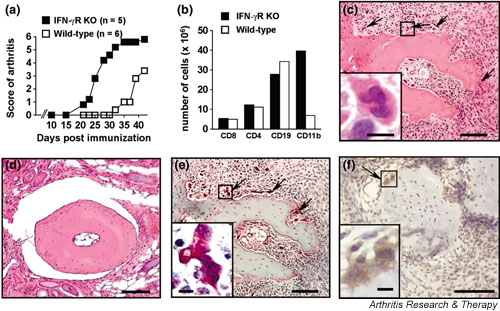
Accelerated collagen-induced arthritis (CIA) in interferon-γ receptor knock-out (IFN-γR KO) mice is accompanied by CD11b+ splenocyte expansion and osteoclast formation in the joints. Mice were immunised with collagen type II in complete Freund's adjuvant. (a) Accelerated disease onset and more severe arthritic scores in IFN-γR KO mice than in wild-type mice. (b) Expansion of the CD11b+ splenocyte population in IFN-γR KO mice 27 days after immunisation. Splenocytes were obtained from three mice, counted and then pooled for flow cytometric analysis; numbers indicated are per spleen. (c,d) Haematoxylin staining on paraffin sections of the joints on day 27 after immunisation, showing bone erosion and multinucleated giant cells in IFN-γR KO mice (arrows in (c) and detail in inset). (d) Section of wild-type mouse joint, showing normal histological appearance. (e) Tartrate-resistant acid phosphatase (TRAP) staining on paraffin sections of the joint of IFN-γR KO mice, showing multinucleated giant cells (arrows; detail in inset) staining positive for TRAP. TRAP+ multinucleated cells (three or more nuclei) can be considered to be osteoclasts. (f) CD11b+ cells in CIA. Staining with anti-mouse CD11b antibody demonstrating the presence of both CD11b+ (brown) mononuclear cells and multinuclear osteoclast-like cells (arrow; enlarged in inset). The section was counterstained with haematoxylin. Sections that were stained with an isotype control antibody revealed no positive staining (not shown). Bars in the pictures and insets represent, respectively, 100 μm and 10 μm.
These observations supported the hypothesis that the osteoclast-like cells were in part derived from the CD11b+ myeloblasts in the spleen, and hence that the earlier expansion of CD11b+ cells in IFN-γR KO mice had a role in the accelerated CIA process in these mice.
Increased RANKL/TNF-α-induced osteoclastogenesis in splenocyte cultures from IFN-γR KO mice
To test the hypothesis that facilitated CIA in IFN-γR KO mice might be due to an inhibitory effect of endogenous IFN-γ on osteoclastogenesis, we studied RANKL/TNF-α-induced osteoclast formation by culturing splenocytes of IFN-γR KO and wild-type mice. Whole splenocyte suspensions were cultured for 6 days in an environment containing M-CSF alone, M-CSF plus RANKL, or M-CSF plus TNF-α. Splenocytes were derived from either naive mice or mice immunised 21 days previously with CII in CFA. We verified whether IFN-γ was produced during splenocyte osteoclastogenesis. IFN-γ levels were similar after stimulation with RANKL or TNF-α, but were higher in IFN-γR KO-derived than in wild-type-derived cultures. Thus, the average IFN-γ levels in supernatant of splenocytes stimulated with M-CSF plus RANKL (n = 3) were 273 ± 27 pg/ml (for IFN-γR KO) compared with 114 ± 41 pg/ml (for wild-type); the levels in the supernatant of cultures stimulated with M-CSF plus TNF-α were 241 ± 25 pg/ml (for IFN-γR KO) compared with 135 ± 5 pg/ml (for wild-type). Similar data were obtained with RANKL-stimulated and TNF-α-stimulated splenocytes from naive mice (264 ± 49 and 105 ± 6 pg/ml for IFN-γR KO and wild-type cells, respectively, stimulated with RANKL). The higher levels in IFN-γR KO-derived cultures can be explained by the failure of the mutant cells to internalise IFN-γ, owing to the absence of the IFN-γ receptor [33]. Osteoclasts were identified by their multinucleate aspect combined with TRAP staining, and by testing their activity in a pit-forming assay, which proved their capacity to resorb a calcium phosphate film. No osteoclast differentiation was observed in cultures stimulated with M-CSF only. The results (Fig. 2a,2b) show that more osteoclasts were generated in cultures stimulated with M-CSF plus RANKL (Fig. 2a) than in those stimulated with M-CSF plus TNF-α (Fig. 2b). Whatever the osteoclast differentiating stimulus, and whatever the immunisation status of the mice, osteoclastogenesis was more pronounced in splenocyte cultures from IFN-γR KO mice than in those from wild-type mice (Fig. 2c,2d). This was not associated with higher levels of mortality or apoptosis in the wild-type-derived cultures (data not shown). In addition, when M-CSF plus RANKL was used as the stimulus, significantly more osteoclasts were generated out of cells taken from immunised mice than out of those taken from naive mice (Fig. 2a). When TNF-α was used instead of RANKL, the immunisation status of the mice did not affect the capacity of the splenocytes to differentiate into osteoclasts (Fig. 2b). The ratio between the numbers of osteoclasts that were generated and the surface of calcium phosphate that was resorbed in the pit-forming assay was the same in IFN-γR KO cultures as in wild-type cultures, indicating that in vitro generated osteoclasts derived from either mouse strain are equally active (Fig. 2e,2f).
Figure 2.
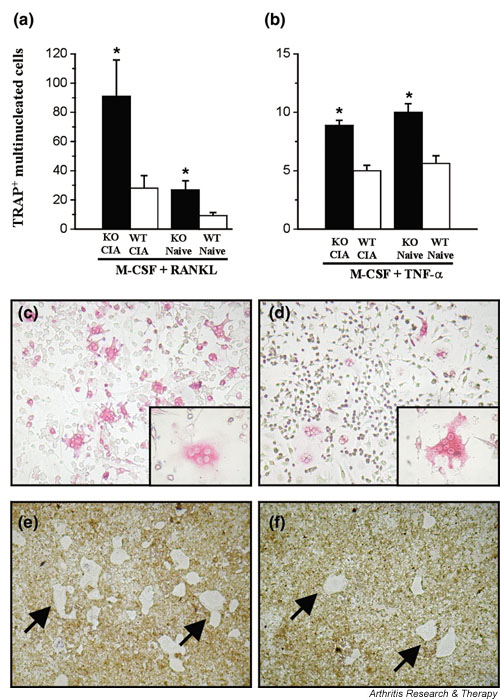
Increased osteoclast formation from splenocytes of interferon-γ receptor knock-out (IFN-γR KO) mice. Splenocytes of IFN-γR KO (KO) mice and wild-type (WT) mice were isolated. Mice were either naive or had been immunised with collagen type II in complete Freund's adjuvant 21 days previously. Cells were stimulated for 6 days in chamber slide cups with 20 ng/ml macrophage colony-stimulating factor (M-CSF) and 100 ng/ml receptor activator of NF-κB ligand (RANKL) (a) or 20 ng/ml M-CSF and 20 ng/ml tumour necrosis factor-α (TNF-α) (b). After stimulation, cultures were fixed and stained for the presence of tartrate-resistant acid phosphatase (TRAP). (a,b) TRAP+ multinucleated (three or more nuclei) cells were counted within each cup. In each group, bars represent averages ± standard error of the mean for five mice. *P < 0.05 compared with wild–type mice (Mann-Whitney U-test). (c,d) Representative pictures of TRAP-stained cultures of RANKL-stimulated IFN-γR KO (c) and wild-type (d) splenocytes. Insets show details of multinucleated TRAP+ cells. (e,f) Osteoclast activity after stimulation by RANKL of IFN-γR KO (e) and wild-type (f) splenocyte cultures, as analysed by their ability to resorb a calcium phosphate film coated on a quartz substrate. Sites of resorption are indicated by arrows. CIA, collagen-induced arthritis.
Expression of RANK and production of RANKL and/or TNF-α by splenocytes
Increased osteoclast formation in IFN-γR KO mice, in vivo in arthritic joints and in vitro in stimulated splenocyte cultures, might be due to an increased production of RANKL and/or TNF-α or to an increased expression of RANK, a receptor for RANKL. To test this possibility, IFN-γR KO and wild-type mice were given the CII/CFA immunisation schedule for the induction of CIA. On day 21, spleens were removed and the splenocyte population was analysed by flow cytometry for the expression of RANK. Because we were interested in the possibility that the increased number of osteoclasts in the joints of IFN-γR KO mice could be derived from the expanding CD11b+ haematopoietic cell population, we focused on both the CD11b+ and the CD11b- splenocyte populations. As can be seen in Fig. 3, RANK+ cells were found only within the CD11b+ population, indicating that at least those cells can differentiate into osteoclasts. Moreover, the larger numbers of CD11b+ cells in the spleens of IFN-γR KO mice as opposed to those of wild-type mice was associated with even larger portions of RANK+ cells. This selective effect of the IFN-γR KO mutation on emergence of the double-positive CD11b+/RANK+ population is a further argument for the hypothesis that the larger numbers of osteoclasts in immunised IFN-γR KO mice originate in the expanding CD11b+ haematopoietic population.
Figure 3.
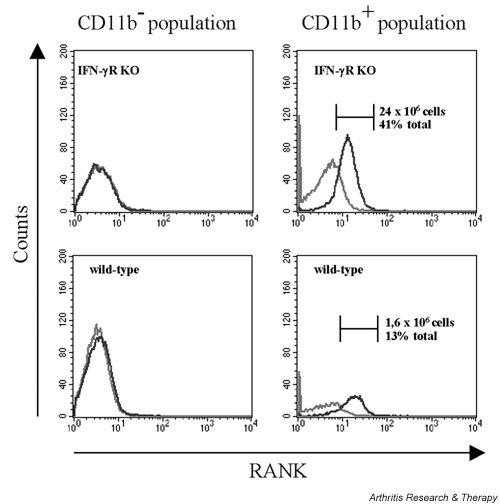
Flow cytometric analysis for receptor activator of NF-κB (RANK) on splenocytes of immunised interferon-γ receptor knock-out (IFN-γR KO) and wild-type mice. Splenocytes were isolated on day 21 after immunisation. Cells were incubated with phycoerythrin-labelled anti-CD11b antibody, biotinylated anti-RANK antibody and fluorescein isothiocyanate-labelled streptavidin. The grey line represents staining with an irrelevant biotinylated IgG. RANK expression was analysed both within the CD11b- (left panels) and within the CD11b+ (right panels) splenocyte population. The black line represents staining with anti-RANK–biotin/streptavidin–phycoerythrin. The total numbers of RANK+ cells within the CD11b+ population of the IFN-γR KO (upper right panel) and of the wild-type (lower right panel) splenocytes are indicated, together with the percentage that they represent of the total splenocyte population (each picture is representative for one mouse out of three).
The possibility that RANKL production is enhanced in immunised IFN-γR KO mice was examined by experiments both in vivo and in vitro. Attempts to detect RANKL in the serum of naive or CII/CFA-immunised IFN-γR KO or wild-type mice on day 21 were unsuccessful, as were attempts to detect RANKL in the serum of mice after administration of anti-CD3 antibody (1 or 10 μg). However, we could detect RANKL in the 6-day supernatant of splenocytes cultured in the presence of M-CSF and anti-CD3 antibody (1 μg/ml). Splenocytes of IFN-γR KO mice produced more RANKL than those of wild-type mice (Fig. 4a). Moreover, splenocytes derived from CII/CFA-immunised mice produced more RANKL than those from naive mice. Similar results were obtained when supernatant was analysed for the presence of TNF-α (Fig. 4b). Splenocyte cultures of IFN-γR KO mice produced higher levels of TNF-α than those of wild-type mice, and the level of TNF-α was higher when the cells were derived from immunised mice. These results in vitro suggest that in IFN-γR KO mice a subpopulation of splenocytes is programmed to produce more RANKL and TNF-α in response to T cell stimuli. The presence of RANKL and M-CSF mRNA in cells of the inflamed synovium, as investigated by PCR (Fig. 4c), shows that production of these osteoclast-inducing stimuli by these cells in vivo might be possible. On the assumption that osteoclasts in the joints can be derived from haematopoietic spleen cells, the question arises whether the osteoclast precursor cells first differentiate into osteoclasts and subsequently migrate to the joints, or vice versa. To distinguish between these possibilities, freshly isolated splenocytes from IFN-γR KO and wild-type mice were stained for the presence of TRAP. No TRAP+ mononucleated or multinucleated cells could be detected (data not shown). Furthermore, no mRNA for the calcitonin receptor, which is expressed on differentiated osteoclasts [5], was present within the splenocyte population. As a contrast, such mRNA was detectable in cells derived from the synovium of immunised mice but not in spleen tissue (Fig. 4d). These data indicate that osteoclast differentiation does not take place in the spleen and suggest that haematopoietic cells migrate into the joints before differentiation into osteoclasts. Interestingly, levels of calcitonin receptor mRNA were higher in IFN-γR KO than in wild-type mice, confirming the increased presence of osteoclasts in the mutant mice.
Figure 4.
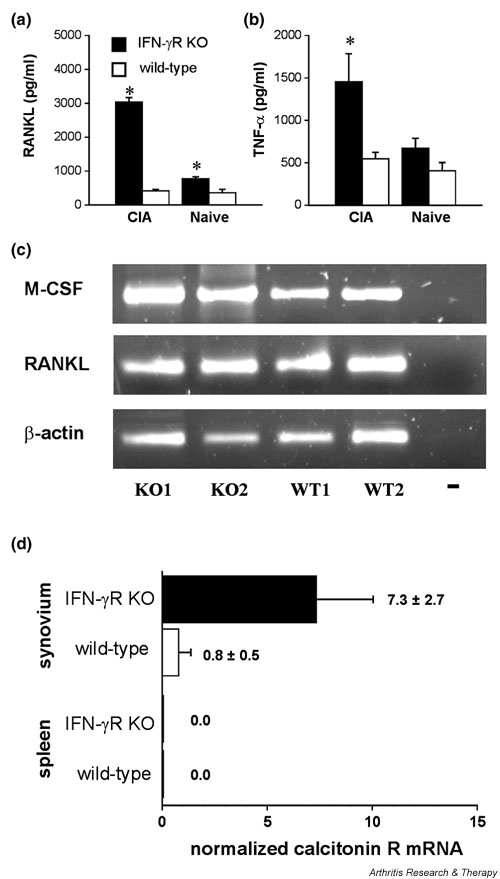
Production of receptor activator of NF-κB ligand (RANKL) and tumour necrosis factor-α (TNF-α) in vitro and expression of macrophage colony-stimulating factor (M-CSF) and RANKL in vivo. (a,b) RANKL and TNF-α concentrations measured in supernatant of anti-CD3 antibody-stimulated splenocytes. Splenocytes of naive and immunised mice (day 21 after immunisation) were cultured in the presence of M-CSF and stimulated with 1 μg/ml anti-CD3 antibody. RANKL (a) and TNF-α (b) detected in supernatants by enzyme-linked immunosorbent assay 6 days after stimulation. *P < 0.05 compared with wild-type mice (Mann–Whitney U-test). (c) Reverse transcriptase PCR performed on RNA of isolated inflamed synovia of two interferon-γ receptor knock-out (IFN-γR KO) mice (KO1, KO2) and two wild-type mice (WT1, WT2) showing transcription of RANKL and M-CSF within the inflamed synovium. The housekeeping gene β-actin was used to normalise the levels of cDNA. (d) Analysis of calcitonin receptor expression level by real-time quantitative PCR on synovium and spleen from three wild-type and three IFN-γR KO mice. Values are the numbers of calcitonin receptor mRNA copies per 1000 copies of hypoxanthine transferase. CIA, collagen-induced arthritis.
Increased CIA in IFN-γR/KO mice coincides with depletion of TRAF6 in splenocytes
The intracellular signalling protein TRAF6 is known to be involved in osteoclastogenesis induced by RANKL as well as by TNF-α. Moreover, a link between IFN-γ and RANKL signalling via TRAF6 has also been demonstrated in bone marrow cultures, in which IFN-γ was shown to accelerate the degradation of TRAF6. We therefore investigated whether TRAF6 levels would change in splenocytes during CIA development and whether such changes might be affected by the IFN-γR KO mutation. We prepared lysates from splenocytes of CII/CFA-immunised IFN-γR KO and wild-type mice (day 21) and from corresponding naive mice, and subsequently we examined the presence of TRAF6 by Western blotting. As can be seen in Fig. 5, TRAF6 was expressed at similar levels in three of the four groups of splenocytes (those derived from naive wild-type mice, naive IFN-γR KO mice and immunised wild-type mice), but was virtually absent from splenocytes of CII/CFA-immunised IFN-γR KO mice. Similar data (not shown) were obtained from lysates of splenocyte cultures. Virtual absence of TRAF6 expression in the IFN-γR KO mice was obviously not linked to the knock-out genotype only but required immunisation. Moreover, the degradation of TRAF6 was not caused by an overall higher protease activity in the CII/CFA-immunised IFN-γR KO mice, because TRAF2 expression remained unaffected by the immunisation. Interestingly, a lack of TRAF6 expression was associated with an accelerated and severe form of arthritis with large numbers of osteoclasts in the arthritic joints. Thus, the enlarged population of splenocytes, which accompanies accelerated arthritis and osteoclastogenesis in IFN-γR KO mice, seems to be characterised by the depletion of intracellular TRAF6 stores, suggesting that the RANKL signalling pathway is (or has been) strongly solicited in these cells.
Figure 5.
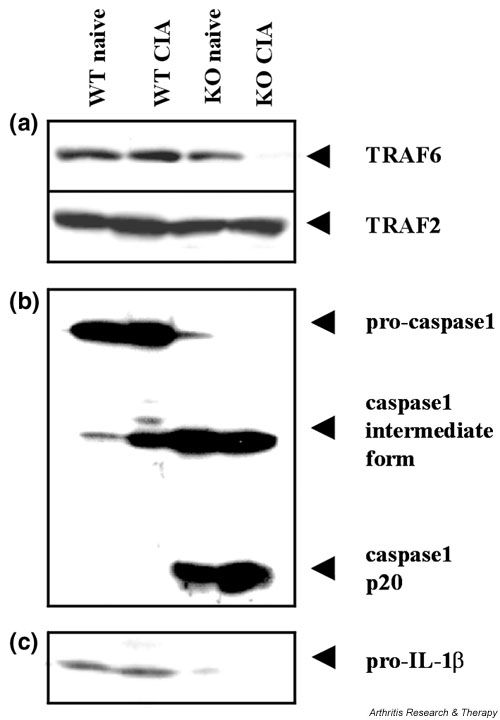
Expression of TNF receptor associated factor (TRAF)6, caspase-1 and interleukin-1β (IL-1β) in splenocytes of interferon-γ receptor knock-out (IFN-γR KO) mice (KO) and wild-type mice (WT), both naive and immunised with collagen type II in complete Freund's adjuvant (CII/CFA) (day 21 after immunisation). Splenocyte suspensions of three mice within each group were pooled and lysed. Total protein (30 μg) was loaded for electrophoresis and blotted. The blotting membrane was incubated with anti-TRAF6 or anti-TRAF2 antibody (a), anti-caspase-1 antibody (b) and anti-IL-1β antibody (c). (a) Degradation of TRAF6 in CII/CFA-immunised IFN-γR KO mice: there is unaltered expression of TRAF2, demonstrating that TRAF6 degradation is not caused by an overall higher protease activity in mutant mice. (b) Differential expression of the caspase-1 isoforms: inactive pro-caspase-1, intermediate caspase-1 form and the active 20 kDa form (p20). (c) Inactive pro-IL-1β mainly detectable only in wild-type mice. CIA, collagen-induced arthritis.
Accelerated CIA in IFN-γR KO mice is associated with activation of caspase-1-mediated IL-1 processing
To obtain evidence for the involvement of IL-1β as an additional possible stimulant for the osteoclast activation of IFN-γR KO mouse splenocytes, we also tested the splenocyte lysates for the presence of both the non-processed IL-1β and the IL-1β processing enzyme, caspase-1. Splenocytes derived from wild-type mice, irrespective of whether or not they had been immunised, contained only pro-caspase-1 and pro-IL-1β, and no detectable mature caspase-1. In contrast, splenocytes of naive IFN-γR KO mice or from CII/CFA-immunised IFN-γR KO mice hardly showed any pro-caspase-1 or pro-IL-1β levels (Fig. 5b,5c). Instead, cells of both the naive and the immunised IFN-γR KO mice contained a processed intermediate form of caspase-1 and the 20 kDa chain of its active form. Concordantly, higher active caspase-1 levels were accompanied by a depletion of pro-IL-1β, suggesting complete conversion into secreted active IL-1β. These observations demonstrate that accelerated CIA in IFN-γR KO mice is associated with the activation of caspase-1 and the proteolytic maturation of IL-1β.
Discussion
CIA develops more readily in IFN-γR KO mice than in wild-type mice: arthritic lesions appear about 2 weeks earlier and symptoms are more severe. The myelopoietic burst that accompanies the local disease manifestations and that is most evident in the spleen also occurs earlier and is more pronounced. The cell population generated by this myelopoietic burst consists mainly of CD11b+ doughnut-like cells, namely immature macrophages and neutrophils [3]. Total numbers of other investigated splenocyte subpopulations, namely CD4+ and CD8+ T cells and B cells, remain unchanged. The parallelism between systemic myelopoiesis and local lesion development has led us to postulate that the CD11b+ cells have a crucial role in CIA by invading the joint tissues and by differentiating into osteoclasts. We have already provided evidence that invasion of periarticular tissues by myeloid cells does take place and is an important element in the pathogenesis [34]. The experiments described in the present paper were conducted to provide evidence for the potential of the CD11b+ cells to differentiate into osteoclasts.
We showed that osteoclasts, identifiable by their multinucleated appearance, by their localisation close to the calcified bone material and by TRAP staining, resided in the CIA lesions of IFN-γR KO mice at the time (day 27) when macroscopic joint involvement was maximal. At that time, lesions were not yet present in wild-type mice, and osteoclasts could not yet be seen in their joint tissues. At a later time, when lesions eventually developed in these mice, osteoclasts also became visible, although their numbers were smaller. Thus, intra-articular osteoclast formation was accelerated and more pronounced in IFN-γR KO mice, in concordance with the earlier and more prolific myelopoietic burst. Immunocytochemical staining of joint sections revealed osteoclasts to be positive for CD11b+, supporting the hypothesis that mature osteoclasts in the inflamed joints tissues might be derived from extramedullar CD11b+ myelopoiesis.
Evidence for CD11b+ splenocytes being able to differentiate into osteoclasts was obtained by observations both in vivo and in vitro. Osteoclastogenesis induced by RANKL as well as by TNF-α could be demonstrated in splenocyte cultures. It was more pronounced if these cells were derived from IFN-γR KO rather than from wild-type mice in both CIA and naive conditions. IFN-γ levels were present in RANKL- and TNF-α-induced splenocyte cultures derived from immunised as well as from naive mice. Spleens of naive IFN-γR KO and wild-type mice did not significantly differ in their proportions of the splenocyte subpopulations (CD11b+ cells, T cells and B cells). This suggests that IFN-γ, aside from causing a delay in the myelopoietic response to the CII/CFA immunisation, also inhibits differentiation of immature myeloid cells into osteoclast precursors. Pit-forming assays failed to reveal any difference between osteoclasts from IFN-γR KO and wild-type mice, indicating that endogenous IFN-γ, while inhibiting differentiation of osteoclasts, does not affect their activation.
RANKL, when used in optimal doses, seemed to be a more potent osteoclast differentiating stimulus than TNF-α. Moreover, stimulation with RANKL, but not with TNF-α, revealed a facilitating effect of CII/CFA immunisation on differentiation into osteoclasts. The different RANKL sensitivities of splenocytes from wild-type versus IFN-γR KO mice and from CII/CFA-immunised versus naive ones led us to investigate whether RANKL production could also vary between these groups of mice and whether the receptor and the signalling system for RANKL (RANK and TRAF6) could be differently tuned.
Production of RANKL and TNF-α by anti-CD3-stimulated splenocyte cultures was higher if these cells were derived from IFN-γR KO (rather than wild-type) mice and from CII/CFA-immunised (rather than naive) mice, suggesting that augmented osteoclastogenesis in the immunised IFN-γR KO mice might be due in part to an increased production of RANKL and TNF-α. We found expression of RANK in the 10-fold expanded CD11b+ splenocyte population of immunised IFN-γR KO mice, whereas CD11b- cells were RANK-negative. Furthermore, the intracellular concentration of TRAF6, a RANK adapter protein, seemed to be strongly decreased in the splenocyte cultures derived from 21-day CII/CFA-immunised IFN-γR KO mice, in comparison with levels in splenocytes taken at the same time point from nonimmunised or wild-type mice. This indicates that the status of the RANKL signalling system in 21-day immunised IFN-γR KO mice is profoundly different from that in the nonimmunised mice or the wild-type controls.
Because our in vitro data prove that spleen cells are able to differentiate into osteoclasts, and that anti-CD3-stimulated splenocyte cultures can produce RANKL and TNF-α, we investigated whether osteoclast formation can occur in vivo in the spleen. No osteoclasts were detected by TRAP staining of freshly isolated splenocytes of diseased mice. Quantitative reverse transcriptase PCR revealed that mRNA of the calcitonin receptor was absent from splenocytes but present in cells residing in the inflamed synovium. These data prove that no osteoclast differentiation takes place within the spleen.
An inhibitory effect of IFN-γ on osteoclast formation via cross-talk with the RANK/RANKL system has been described in another in vivo model of bone degradation involving the injection of lipopolysaccharide into calvarial bone in mice, and, in vitro, in bone marrow macrophage (BMM) cultures exposed to RANKL [35]. Intriguingly, inhibition of osteoclast formation by IFN-γ in the BMM cultures was accompanied by decreased TRAF6 levels and by increased TRAF6 turnover. In the BMM model, IFN-γ induced degradation of TRAF6 was found to require RANK/RANKL signalling and a functional proteasome. These observations are in contrast to our findings in ex vivo lysed splenocytes (Fig. 5) and in splenocyte cultures, in which decreased TRAF6 levels occurred in association with increased osteoclastogenesis and with an absence of IFN-γ signalling in the IFN-γR KO-derived cells. The lower TRAF6 levels were not caused by decreased transcription because no differences in TRAF6 mRNA were found between IFN-γR KO and wild-type mice (data not shown). In concordance with our TRAF6 results, recent findings by Huang and colleagues [36] have shown that early exposure to IFN-γ renders osteoclast precursors resistant to the effects of RANKL and that this effect is not associated with degradation of TRAF6.
TRAF6 is a ubiquitin ligase, becoming activated by ubiquitination [37]. It has been shown that the TRAF6 protein is ubiquitinated in response to RANKL and IL-1. It has recently also been shown that ubiquitination of TRAF6 does not necessarily lead to degradation but can be followed by de-ubiquitination [38]. The ubiquitinated form of TRAF6 is not necessarily detectable with anti-TRAF6 antibody because modification with ubiquitin might alter its native epitopes. Hence, the presence of non-ubiquitinated TRAF6 can be indicative of the absence of TRAF6-activating stimuli, whereas decreasing the concentration of the non-ubiquitinated form can be indicative of a high activation state of TRAF6. It is therefore possible that the absence of non-ubiquitinated TRAF6 from the IFN-γR KO cells of our model is due to a high activation status of the RANK/RANKL system, requiring TRAF6 activation and thus resulting in a high turnover between ubiquitinated and non-ubiquitinated TRAF6.
As regards the role of TRAF6 during osteoclastogenesis, it is also important to keep in mind that, at least in vitro, RANK has been shown to associate with TRAFs 1, 2, 3, 5 and 6. Consequently, the possibility cannot be excluded that signalling through one of these occurs in the IFN-γR KO splenocytes. Moreover, TRAF6-deficient mice reportedly do produce osteoclasts within their bone, and the numbers of TRAP+ cells per square millimetre of tissue area are comparable in wild-type and knock-out mice [39]. The phenotype is nonetheless osteopetrotic owing to the inactivity of these osteoclasts. This shows that TRAF6 is not indispensable for the formation of osteoclasts but is vital for their activation. In this respect, the absence of TRAF6 should not in the first place influence the number of osteoclasts developing from splenocytes, but rather their activity. Although osteoclasts in TRAF6-deficient mice are inactive, and the splenocytes of our IFN-γR KO mice show low levels of TRAF6, the osteoclasts derived from the splenocytes of both the IFN-γR KO mice and the wild-type mice were indeed active, as proved with the pit-forming assay. This supports our first hypothesis that the lowered levels of non-ubiquitinated TRAF6 in the IFN-γR KO mice, as detected by Western blotting, are indicative of a high activity of the RANK/RANKL signalling.
Not only is the RANK/RANKL system operational in the CII/CFA-immunised IFN-γR KO mice; so also is the IL-1β system, as was evident from comparatively low levels of pro-IL-1β and pro-caspase-1 but a high level of mature caspase-1, indicating the active conversion of pro-IL-1β into secreted active IL-1β. In this respect, Guedez and colleagues [40] found that genetic ablation of IFN-γ upregulates IL-1β and enables the elicitation of CIA in the nonsusceptible C57BL/6 mouse strain. Moreover, treatment of IFN-γ KO mice with anti-IL-1β antibody reduced the incidence and severity of arthritis, indicating that in the absence of IFN-γ, IL-1β is important in the pathogenesis of CIA [40]. Important in this respect is the known osteoclast-activating property of IL-1β [41].
Together, these data indicate that, in CII/CFA-immunised mice, all the conditions are fulfilled for the expanded CD11b+ myeloid splenocytes to differentiate into osteoclasts. Our study thereby provides a link between the increased expansion of CD11b+ cells in the IFN-γR KO mouse spleens, the higher capacity of IFN-γR KO splenocytes to produce key mediators in the osteoclast-differentiating process and the higher susceptibility of the IFN-γR KO mice to CIA. Moreover, control by endogenous IFN-γ over CD11b+ myelopoiesis and osteoclastogenesis might also account for the recently reported observation that IL-10-deficient mice have an increased susceptibility to CIA in association with decreased production of IFN-γ [42]. Support for the importance of extramedullar CD11b+ myelopoiesis during the development of CIA comes from a recent study on spontaneously occurring arthritis in TNF-α-transgenic mice [43]. Higher numbers of CD11b+ osteoclast precursors were recorded in the blood and spleen of the transgenic mice. The increased numbers were correlated with the appearance of TNF-α in the circulation and with the initiation of joint inflammation. TNF-α blockade with the TNF-α antagonist etanercept did not affect enhanced RANKL-induced osteoclast formation in vitro, suggesting that TNF-α-stimulated osteoclasto-genesis in vivo was indeed due to the generation of larger numbers of osteoclast precursors rather than to accelerated differentiation beyond the precursor stage.
Our present and previous studies [3,44] stress the predominant role of innate immunity in the pathogenesis of CIA. Innate immunity, triggered by the mycobacterial cell wall components in CFA, might be the primary motor of the disease by stimulating myelopoiesis, causing migration and regulating osteoclast differentiation. The role of specific immunity directed at CII might consist of restricting the inflammatory response to the specific location of the joints.
Conclusions
We provide several lines of evidence strongly suggesting that the development of arthritis in CII/CFA-immunised mice is determined by the potential of an expanding CD11b+ myeloid splenic cell population to differentiate into osteoclasts, and that this process is under the downregulatory control of endogenous IFN-γ, via its effects on the production and action of several osteoclast-differentiating cytokines. This supports a pathogenesis model for CIA that assigns a predominant role to innate immunity overstimulation, most probably engendered by the mycobacterial components of CFA.
Competing interests
None declared.
Abbreviations
BMM = bone marrow macrophage; BSA = bovine serum albumin; cDNA = complementary DNA; CFA = complete Freund's adjuvant; CIA = collagen-induced arthritis; CII = collagen type II; ELISA = enzyme-linked immunosorbent assay; IFN-γ = interferon-γ; IFN-γR KO = interferon-γ receptor knock-out; IL = interleukin; M-CSF = macrophage colony-stimulating factor; ODF = osteoclast differentiation factor; OPG = osteoprotegerin; OPGL = osteoprotegerin ligand; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; RANK = receptor activator of NF-κB; RANKL = receptor activator of NF-κB ligand; TBS = Tris-buffered saline; TNF = tumour necrosis factor; TRAF = TNF receptor associated factor; TRANCE = TNF-related activation-induced cytokine; TRAP = tartrate-resistant acid phosphatase.
Acknowledgments
Acknowledgements
We thank Dr Lieve Moons for help in preparing histological slides, and Inge Derese for help in immunocytochemistry. Studies in the authors' laboratories are funded by the Concerted Research Actions (GOA) Initiative of the Regional Government of Flanders, the Interuniversity Attraction Pole Program (IUAP) of the Belgian Federal Government, and grants from the National Fund for Scientific Research of Flanders (FWO). PM holds a postdoctoral fellowship from the FWO.
References
- Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier M-C, Fournier C. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J Immunol. 1997;158:5501–5506. [PubMed] [Google Scholar]
- Vermeire K, Heremans H, Vandeputte M, Huang J, Billiau A, Matthys P. Accelerated collagen-induced arthritis in interferon-γ receptor-deficient mice. J Immunol. 1997;158:5507–5513. [PubMed] [Google Scholar]
- Matthys P, Vermeire K, Mitera T, Heremans H, Huang J, Schols D, Dewolf-Peeters C, Billiau A. Enhanced autoimmune arthritis in IFN-γ receptor-deficient mice is conditioned by mycobacteria in Freund's adjuvant and by increased expansion of Mac-1+ myeloid cells. J Immunol. 1999;163:3503–3510. [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nishikaku F, Nakatuka M, Koga Y. Osteoclast-like cells in murine collagen induced arthritis. Endocr Rev. 1998;25:1154–1160. [PubMed] [Google Scholar]
- Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13:66–80. doi: 10.1210/er.13.1.66. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Advances in bone biology: the osteoclast. Endocr Rev. 1996;17:308–332. doi: 10.1210/er.17.4.308. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, III, Frankel WN, Lee SY, Choi Y. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Caparelli C, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, Morinaga T, Higashio K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. 1998;253:395–400. doi: 10.1006/bbrc.1998.9788. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Oppers-Walgreen B, Pettit AR, Van Den BL, Joosten LA, Goldring SR, Gravallese EM, van den Berg WB. Increase in expression of receptor activator of nuclear factor κB at sites of bone erosion correlates with progression of inflammation in evolving collagen-induced arthritis. Arthritis Rheum. 2002;46:3055–3064. doi: 10.1002/art.10607. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T, Higashio K. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun. 1997;234:137–142. doi: 10.1006/bbrc.1997.6603. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-α (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem. 2001;276:563–568. doi: 10.1074/jbc.M008198200. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–265. doi: 10.1016/S1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002;277:44347–44356. doi: 10.1074/jbc.M202009200. [DOI] [PubMed] [Google Scholar]
- Kaji K, Katogi R, Azuma Y, Naito A, Inoue JI, Kudo A. Tumor necrosis factor alpha-induced osteoclastogenesis requires tumor necrosis factor receptor-associated factor 6. J Bone Miner Res. 2001;16:1593–1599. doi: 10.1359/jbmr.2001.16.9.1593. [DOI] [PubMed] [Google Scholar]
- Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. BioEssays. 2003;25:1096–1105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res Ther. 2002;4(Suppl 3):S227–S232. doi: 10.1186/ar581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-γ receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Dijkmans R, Martens E, Beuken E, Cornette F, Dillen C, Heremans H, Boraschi D, Billiau A. Murine interferon-γ/interleukin-1 fusion protein used as antigens for the generation of hybridomas producing monoclonal anti-interleukin-1 antibodies. Cytokine. 1991;3:134–140. doi: 10.1016/1043-4666(91)90034-b. [DOI] [PubMed] [Google Scholar]
- De Bari C, Dell'Accio F, Tylzanowski P, Luyten F. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44:1928–1942. doi: 10.1002/1529-0131(200108)44:8<1928::AID-ART331>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73. doi: 10.1016/S0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- Farrar MA, Schreiber RD. The molecular cell biology of interferon-γ and its receptor. Annu Rev Immunol. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson G, De Clercq E, Billiau A, Schols D. AMD 3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-γ receptor-deficient mice. J Immunol. 2001;167:4686–4692. doi: 10.4049/jimmunol.167.8.4686. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- Huang W, O'Keefe RJ, Schwarz EM. Exposure to receptor-activator of NFκB ligand renders pre-osteoclasts resistant to IFN-γ by inducing terminal differentiation. Arthritis Res Ther. 2003;5:R49–R59. doi: 10.1186/ar612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Whitehead AS. Ubiquitin activated tumor necrosis factor receptor associated factor-6 (TRAF6) is recycled via deubiquitination. FEBS Lett. 2003;553:190–194. doi: 10.1016/S0014-5793(03)00998-0. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedez YB, Whittington KB, Clayton JL, Joosten LAB, van de Loo FAJ, van den Berg WB, Rosloniec EF. Genetic ablation of interferon-γ up-regulates interleukin-1β expression and enables the elicitation of collagen-induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum. 2001;44:2413–2424. doi: 10.1002/1529-0131(200110)44:10<2413::AID-ART406>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Joosten LA, Helsen MM, Saxne T, van de Loo FA, Heinegard D, van den Berg WB. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol. 1999;163:5049–5055. [PubMed] [Google Scholar]
- Finnegan A, Kaplan CD, Cao Y, Eibel H, Glant TT, Zhang J. Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res Ther. 2003;5:R18–R24. doi: 10.1186/ar601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Schwarz EM, O'Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–276. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- Matthys P, Vermeire K, Billiau A. Mac-1+ myelopoiesis induced by complete Freund's adjuvant (CFA): a clue to the paradoxical effects of IFN-γ in autoimmune disease models. Trends Immunol. 2001;22:367–371. doi: 10.1016/S1471-4906(01)01937-8. [DOI] [PubMed] [Google Scholar]