

Abstract
Blockade of chemokines or chemokine receptors is emerging as a new potential treatment for various immune-mediated conditions. This review focuses on the therapeutic potential in rheumatoid arthritis, based on studies in animal models and patients. Several knockout models as well as in vivo use of chemokine antagonists are discussed. Review of these data suggests that this approach might lead to novel therapeutic strategies in rheumatoid arthritis and other chronic inflammatory disorders.
Keywords: chemokines, rheumatoid arthritis, synovial tissue
Introduction
Chemokines form a large superfamily of small (8–14 kDa) cytokines that play crucial roles in cell migration. They interact with G-protein-coupled receptors, which possess a seven transmembrane domain. To date about 50 chemokines have been identified that signal through some 20 distinct receptors [1].
A subset of the chemokine family is active under normal physiological conditions. These so-called homeostatic chemokines are involved in maintaining normal leucocyte traffic and cell compartmentalization in lymphoid tissues under non-inflammatory conditions [2].
Most chemokines play a role in inflammatory conditions by inducing integrin activation, chemotaxis, and angiogenesis. Apart from modulating migration directly, chemokines can stimulate cells to release (pro)inflammatory mediators such as cytokines and matrix metalloproteinases [3]. Increased expression of inflammatory chemokines has been found in many inflammatory disorders, including hepatic disease, multiple sclerosis, transplant rejection and inflammatory bowel disease [4]. Analysis of synovial tissue, synovial fluid and peripheral blood from patients with rheumatoid arthritis (RA) revealed abundant expression of a variety of inflammatory chemokines and their receptors [5,6]. In vitro studies have suggested that both so-called homeostatic chemokines and inflammatory chemokines, including CC chemokine receptor (CCR)1, CCR2, CCR5, CC chemokine ligand (CCL)2/monocyte chemoattractant protein (MCP)-1, CCL5/RANTES (regulated on activation, normal T-cell expressed and secreted) and CXCL8/IL-8, are intimately involved in cell migration toward the synovial compartment in RA [7-10].
Although these studies might suggest therapeutic potential for chemokine and chemokine receptor blockade in inhibiting chronic synovial inflammation, there are some possible pitfalls that could hamper the clinical use of this approach. Of particular importance is the redundancy of the system, based on in vitro studies. Because one receptor can usually bind multiple ligands and vice versa, one may anticipate that blockade of one ligand or receptor may be compensated for by other members of the superfamily. In addition, some ligands may be agonists at one receptor and antagonists at others. Another issue is that one should not interfere with the role played by these molecules in normal homeostasis.
Recently, there has been an enormous upsurge in investigations on the potential of chemokine blockade as a novel therapeutic strategy to inhibit inflammation because of the advent of new biotechnology-derived antagonists. Many biological agents as well as small molecules that target chemokines and chemokine receptors are currently in clinical development [11].
This review focuses on the available in vivo data, which may provide more insight into the chances that disrupting one single factor of the complicated chemokine network could be clinically effective in chronic inflammatory disorders such as RA.
Lessons from knockout models
Because of apparent overlapping biological activities in vitro, it is difficult to determine the precise role of specific chemokine–chemokine receptor interactions in vivo. Gene deletion approaches have proved particularly useful in dissecting the physiological role played by specific chemokines and chemokine receptors. To date various models of receptor and ligand deletion have been reported [12].
Only one (homeostatic) chemokine receptor knockout mouse model was shown to lead to perinatal death, namely the CXC chemokine receptor (CXCR)4 knockout mouse [13]. Deletion of its only known ligand, CXC chemokine ligand (CXCL)12/stromal cell derived factor (SDF)-1α, yielded a phenotype similar to that in the CXCR4 knockout mouse. Although under normal, unchallenged circumstances most chemokine receptor knockout mice are healthy, suggesting compensation by chemokine receptor family members, it is clear that they have an altered immune system. Chemokine receptor knockout mice are more susceptible to infections, for instance with Aspergillus fumigatus and Listeria monocytogenes, than are their wild-type counterparts [14,15]. Moreover, in some disease models deleting chemokine receptor genes appears to have a protective effect; for example, CCR2 knockout mice are resistant to experimental autoimmune encephalitis, and CCR1 knockout mice had prolonged allograft survival in a cardiac transplant model [16,17].
Only a few knockouts have been used in arthritis models. CXCR2 was shown to be important for neutrophil migration in a model of acute gout [18]. In that study urate crystals were injected into subcutaneous air pouches. In mice that lacked the murine CXCR2 homologue urate crystals induced a leucocyte-poor exudate. The same receptor also proved to be important in neutrophil recruitment in Lyme arthritis. Infection of CXCR2-/- mice with Borrelia burgdorferi resulted in a significant decrease in severity of arthritis but had little effect on spirochete loads in joint tissue [19]. In contrast, infection of CCR2-/- mice in the same model had little effect on the development of arthritis or on spirochete clearance. The notion that this might be accounted for by redundant recruitment mechanisms is supported by the observation that monocytes were still present within the inflammatory infiltrates in the joints of the CCR2-/- mice [19].
Data from the knockout mice suggest that at least some individual inflammatory chemokine receptors are pivotal in the inflammatory process in both infections and immune-mediated disorders.
Chemokine blockade in animal models
In addition to knockout models, which may help to identify potential targets, blocking studies with neutralizing antibodies or small molecules could provide insight into the overlapping and distinct effects of chemokines and their receptors. Furthermore, animal models can be used to assess the role of several pathogenic factors at various stages of disease, and thus may serve as a sophisticated tool with which to study the relevance of individual chemokines and chemokine receptors in vivo.
Despite the availability of multiple highly specific compounds, species specificity of small molecules and neutralizing antibodies complicates the use of these compounds in animal models. Nevertheless, various chemokines and chemokine receptors have been targeted successfully in animal models of arthritis. Studies using this approach suggest that redundant recruitment mechanisms do not necessarily exclude the possibility that biological and clinical effects may occur after specific chemokine blockade.
For instance, a study using specific blockade of CXCL8/IL-8, an important stimulant of neutrophil accumulation in acute inflammation, showed that it is possible to block neutrophil migration selectively [20]. In that study a highly specific neutralizing antibody against IL-8 was administered in several types of acute inflammatory disease, including lipopolysaccharide/IL-1 induced arthritis. Anti-IL-8 treatment prevented neutrophil infiltration and resulting tissue damage, despite the fact that CXCL8/IL-8 is also a known ligand for CXCR1, which may be present at high concentrations in the synovial compartment.
Similarly, injection of a specific neutralizing monoclonal antibody against rat CCL2/MCP-1 in rats with collagen-induced arthritis resulted in reduced ankle swelling, in association with decreased macrophage numbers in the joints [21]. Paw swelling of the hindfeet in the antirat MCP-1 treated rats was decreased to about 70% of that in untreated rats. Moreover, destruction of the joints was significantly reduced. This was confirmed in the MRL/lpr mouse model of RA. In MRL/lpr mice that spontaneously develop chronic inflammatory arthritis, daily injection of the antagonist MCP-1(9–76) prevented the onset of arthritis whereas controls treated with native MCP-1 had enhanced arthritis symptoms [22]. Of importance for clinical use, there was also a marked reduction in symptoms and histopathology if the antagonist was given only after the disease had already developed. The protective effect on cartilage and bone destruction might be explained in part by the fact that CCL2/MCP-1 is able to stimulate matrix metalloprotease-3 [23].
The CXCR4–CXCL12/SDF-1α complex appears to be another interesting target. Because CXCL12/SDF-1α only recognizes a single receptor (i.e. CXCR4), which itself is only recognized by CXCL12/SDF-1α, and because the deletion of CXCR4 has dramatic effects on the phenotype, targeting this molecule in animal models is expected to have significant effects. Blockade of CXCR4 with a synthetic, nonpeptide antagonist that does not crossreact with other chemokine receptors exerted clear beneficial effects, both histopathologically and clinically, in murine collagen-induced arthritis [24]. Clinical improvement was also achieved when treatment was initiated at the time of disease onset. Apparently, this effect was solely due to inhibition of migration of CXCR4+MAC-1+ cells through the interference with the chemotactic activity of CXCL12/SDF-1α.
CCR5 attracted much attention as a potential therapeutic target for treatment of HIV infection. A nonpeptide antagonist of this chemokine receptor, namely TAK-779, has also been tested in murine collagen-induced arthritis [25]. Subcutaneous treatment with the CCR5 antagonist initiated a few days before clinical signs of arthritis developed markedly reduced the incidence and severity of the disease, in association with significantly decreased leucocyte migration to the joints.
Taken together, these studies in animal models of arthritis suggest that specific chemokine (receptor) blockade may result in clinically meaningful effects, despite the large number of chemokine family members and their existing overlapping functions. It should be stated, however, that the data are still limited. It remains to be shown whether long lasting effects can be achieved, because it is conceivable that compensatory feedback systems need more time to become effective.
Experience in patients
Data on the effects of chemokine blockade in patients are still very limited. The area is still relatively new, and it is difficult to assess the effectiveness of some of the compounds in animal models because of species selectivity, which could delay development. In addition, development may be hampered by low oral bioavailability of some of the compounds [26,27].
It has recently been suggested that treatment with a monoclonal antibody against CXCL8/IL-8 was not effective in a phase II study in RA patients [28]. It is difficult to interpret the results of that study because the full dataset has not yet been disclosed.
The only published study on chemokine receptor blockade in patients with chronic inflammatory disease to date is a relatively small phase Ib study in RA patients using a CCR1 antagonist [29]. CCR1-positive cells are scattered throughout the rheumatoid synovium, and most of the CCR1-positive cells are macrophages, which play a key role in synovial inflammation. The rationale for the clinical study was supported by interesting properties of the novel small-molecular-weight CCR1 antagonist CP-481,715, which was shown to inhibit 90% of the monocyte chemotactic activity present in the synovial fluid of the majority of RA patients [30]. In a randomized study patients with active RA were treated for 2 weeks with a highly specific CCR1 antagonist or placebo [29]. Synovial tissue analysis revealed a marked decrease in the total number of cells, especially in the number of macrophages and CCR1+ cells after treatment (Fig. 1). Because only cells capable of expressing CCR1 were affected, the results confirmed the specificity of the antagonist and showed the potential of selective chemokine receptor blockade in RA. Although the study was not designed to evaluate clinical efficacy, initial data were promising because one-third of the patients fulfilled the ACR20% criteria after active treatment.
Figure 1.
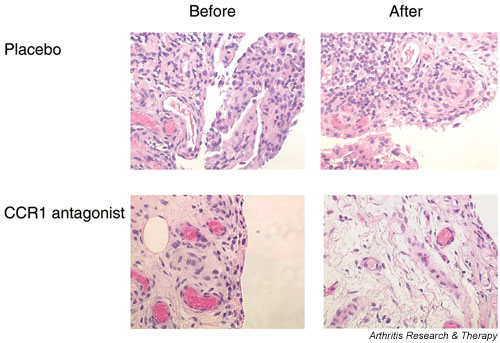
Representative synovial tissue before and after specific CC chemokine receptor (CCR)1 blockade for 14 days in a patient with rheumatoid arthritis (haematoxylin–eosin staining; original magnification × 400). After active treatment there was a marked reduction in synovial cellularity, which was not observed in patients who received placebo. The reduction in cell infiltration was due to a specific decrease in CCR1-positive cells [29].
The data from studies with chemokine antagonists in humans are at present not very comprehensive. However, the initial data are promising. It can be anticipated that several clinical trials exploring this approach will be reported in the near future.
Conclusion
The available data in animal models and initial data in human disease suggest that chemokine family members might be attractive targets for therapeutic intervention. Targeting one specific chemokine (receptor) could be sufficient to reduce inflammation, despite the apparent redundancy of the system. Theoretical advantages of the use of small molecules serving as chemokine receptor antagonists include oral delivery, controllable safety issues during infection in light of the short half-life (the drug could be discontinued during infection, allowing inflammatory cells to migrate to the site of infection), and the potential of inhibiting the migration of cells that are able to produce an array of proinflammatory cytokines at the site of inflammation.
The identification of the best targets will be the subject of future research. It appears likely that redundant mechanisms may be more important for some chemokines than for others. For some pathways it might be necessary to use poly-chemokine antagonists [11] or to combine different chemokine antagonists. It can be expected that rapid developments in immunology, molecular biology and biotechnology will lead to an increase in the number of chemokine and chemokine receptor antagonists that can be tested in clinical trials. Therefore, there is a clear need for biomarkers that could be used for selection purposes during the development process. We have previously proposed examination of serial synovial samples as a method that can be used to examine the effects of targeted antirheumatic interventions [31]. This approach could be particularly helpful in studies evaluating the effects of treatment aimed at blocking cell migration to the site of inflammation. It is likely that we will also see the development of other forms of molecular imaging to assist in selection of therapeutic targets.
A possible concern is that some chemokine ligands may act as agonists rather than as antagonists [32]. Additionally, migration of cells with anti-inflammatory properties could be inhibited by some forms of chemokine blockade. This notion is supported by the observation that CCR2 knockout mice had exacerbated disease in a model of glomerulonephritis [33]. In addition, it remains to be shown whether sustained clinical efficacy can be achieved over time. Therefore, initial proof-of-principle studies followed by well controlled studies of sufficient duration will be essential to realise the potential of chemokine blockade for the treatment of RA.
Competing interests
None declared.
Abbreviations
CCL = CC chemokine ligand; CCR = CC chemokine receptor; CXCL = CXC chemokine ligand; CXCR = CXC chemokine receptor; IL = interleukin; MCP = monocyte chemoattractant protein; RA = rheumatoid arthritis; SDF = stromal cell derived factor.
Contributor Information
Jasper J Haringman, Email: j.j.haringman@amc.uva.nl.
Paul P Tak, Email: p.p.tak@amc.uva.nl.
References
- IUIS/WHO Subcommittee on Chemokine Chemokine/chemokine receptor nomenclature. J Immunol Methods. 2002;262:1–3. doi: 10.1016/S0022-1759(02)00042-X. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- Mackay CR. Chemokines: immunology's high impact factors. Nat Immunol. 2001;2:95–101. doi: 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- Ajuebor MN, Swain MG, Perretti M. Chemokines as novel therapeutic targets in inflammatory diseases. Biochem Pharmacol. 2002;63:1191–1196. doi: 10.1016/S0006-2952(02)00854-7. [DOI] [PubMed] [Google Scholar]
- Hosaka S, Akahoshi T, Wada C, Kondo H. Expression of the chemokine superfamily in rheumatoid arthritis. Clin Exp Immunol. 1994;97:451–457. doi: 10.1111/j.1365-2249.1994.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekanecz Z, Kim J, Koch AE. Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol. 2003;15:15–21. doi: 10.1016/S1044-5323(02)00124-0. [DOI] [PubMed] [Google Scholar]
- Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, Rosa R, Di Salvo J, Mudgett J, Peterson LB, Wicker LS, DeMartino JA. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol. 2003;73:771–780. doi: 10.1189/jlb.1102573. [DOI] [PubMed] [Google Scholar]
- Tylaska LA, Boring L, Weng W, Aiello R, Charo IF, Rollins BJ, Gladue RP. Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine. 2002;18:184–190. doi: 10.1006/cyto.2002.1031. [DOI] [PubMed] [Google Scholar]
- Hayashida K, Nanki T, Girschick H, Yavuz S, Ochi T, Lipsky PE. Synovial stromal cells from rheumatoid arthritis patients attract monocytes by producing MCP-1 and IL-8. Arthritis Res. 2001;3:118–126. doi: 10.1186/ar149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volin MV, Shah MR, Tokuhira M, Haines GK, Woods JM, Koch AE. RANTES expression and contribution to monocyte chemotaxis in arthritis. Clin Immunol Immunopathol. 1998;89:44–53. doi: 10.1006/clin.1998.4590. [DOI] [PubMed] [Google Scholar]
- Carter PH. Chemokine receptor antagonism as an approach to anti-inflammatory therapy: 'just right' or plain wrong? Curr Opin Chem Biol. 2002;6:510–525. doi: 10.1016/S1367-5931(02)00351-4. [DOI] [PubMed] [Google Scholar]
- Power CA. Knock out models to dissect chemokine receptor function in vivo. J Immunol Methods. 2003;273:73–82. doi: 10.1016/S0022-1759(02)00419-2. [DOI] [PubMed] [Google Scholar]
- Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95:9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blease K, Mehrad B, Standiford TJ, Lukacs NW, Kunkel SL, Chensue SW, Lu B, Gerard CJ, Hogaboam CM. Airway remodeling is absent in CCR1-/- mice during chronic fungal allergic airway disease. J Immunol. 2000;165:1564–1572. doi: 10.4049/jimmunol.165.3.1564. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Topham PS, King JA, Smiley ST, Csizmadia V, Lu B, Gerard CJ, Hancock WW. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J Clin Invest. 2000;105:35–44. doi: 10.1172/JCI8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W. The murine homolog of the interleukin-8 receptor CXCR-2 is essential for the occurrence of neutrophilic inflammation in the air pouch model of acute urate crystal-induced gouty synovitis. Arthritis Rheum. 1998;41:900–909. doi: 10.1002/1529-0131(199805)41:5<900::AID-ART18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Brown CR, Blaho VA, Loiacono CM. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J Immunol. 2003;171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. 1994;56:559–564. [PubMed] [Google Scholar]
- Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol. 1997;182:106–114. doi: 10.1002/(SICI)1096-9896(199705)182:1<106::AID-PATH816>3.3.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J Exp Med. 1997;186:131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borzi RM, Mazzetti I, Cattini L, Uguccioni M, Baggiolini M, Facchini A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000;43:1734–1741. doi: 10.1002/1529-0131(200008)43:8<1734::AID-ANR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, De Clercq E, Billiau A, Schols D. AMD3100 a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J Immunol. 2001;167:4686–4692. doi: 10.4049/jimmunol.167.8.4686. [DOI] [PubMed] [Google Scholar]
- Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, Obika S, Imanishi T, Tsujimura T, Hamaoka T, Fujiwara H. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol. 2002;32:2124–2132. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP. AMD3100 a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4:72–77. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, Lin SW, Ying W, Smith SO, Sakmar TP, Moore JP. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci USA. 2000;97:5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keystone EC. Abandoned therapies and unpublished trials in rheumatoid arthritis. Curr Opin Rheumatol. 2003;15:253–258. doi: 10.1097/00002281-200305000-00012. [DOI] [PubMed] [Google Scholar]
- Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:715–721. doi: 10.1136/ard.62.8.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladue RP, Tylaska LA, Brissette WH, Lira PD, Kath JC, Poss CS, Brown MF, Paradis TJ, Conklyn MJ, Ogborne KT, McGlynn MA, Lillie BM, DiRico AP, Mairs EN, McElroy EB, Martin WH, Stock IA, Shepard RM, Showell HJ, Neote K. CP-481,715, a Potent and Selective CCR1 Antagonist with Potential Therapeutic Implications for Inflammatory Diseases. J Biol Chem. 2003;278:40473–40480. doi: 10.1074/jbc.M306875200. [DOI] [PubMed] [Google Scholar]
- Tak PP. Lessons learnt from the synovial tissue response to anti-rheumatic treatment. Rheumatology. 2000;39:817–820. doi: 10.1093/rheumatology/39.8.817. [DOI] [PubMed] [Google Scholar]
- Jarnagin K, Grunberger D, Mulkins M, Wong B, Hemmerich S, Paavola C, Bloom A, Bhakta S, Diehl F, Freedman R, McCarley D, Polsky I, Ping-Tsou A, Kosaka A, Handel TM. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry. 1999;38:16167–16177. doi: 10.1021/bi9912239. [DOI] [PubMed] [Google Scholar]
- Bird JE, Giancarli MR, Kurihara T, Kowala MC, Valentine MT, Gitlitz PH, Pandya DG, French MH, Durham SK. Increased severity of glomerulonephritis in C-C chemokine receptor 2 knockout mice. Kidney Int. 2000;57:129–136. doi: 10.1046/j.1523-1755.2000.00848.x. [DOI] [PubMed] [Google Scholar]