

Abstract
Interconnected functional strategies govern chromatin dynamics in eukaryotic cells. In this context, A and B type lamins, the nuclear intermediate filaments, act on diverse platforms involved in tissue homeostasis. On the nuclear side, lamins elicit large scale or fine chromatin conformational changes, affect DNA damage response factors and transcription factor shuttling. On the cytoplasmic side, bridging-molecules, the LINC complex, associate with lamins to coordinate chromatin dynamics with cytoskeleton and extra-cellular signals.
Consistent with such a fine tuning, lamin mutations and/or defects in their expression or post-translational processing, as well as mutations in lamin partner genes, cause a heterogeneous group of diseases known as laminopathies. They include muscular dystrophies, cardiomyopathy, lipodystrophies, neuropathies, and progeroid syndromes. The study of chromatin dynamics under pathological conditions, which is summarized in this review, is shedding light on the complex and fascinating role of the nuclear lamina in chromatin regulation.
Keywords: laminopathies, lamins, chromatin, LADs, nuclear envelope proteins, Emery-Dreifuss muscular dystrophy, familial partial lipodystrophy, Hutchinson-Gilford progeria, mandibuloacral dysplasia
Lamins and Their Post-Translational Modifications
Lamins are type V intermediate filament proteins located in the nuclear lamina, beneath the inner nuclear membrane. Lamin A and C are the major splicing products of the LMNA gene, which also encodes lamin C2 and lamin A delta 10 and it is located on chromosome 1.1 Lamin B1 is encoded by LMNB1 gene located on chromosome 5, while lamin B2 and B3 are alternative splicing products of LMNB2 gene on chromosome 19.1-3 Lamin A and B type lamins are transcribed as precursor proteins that undergo cysteine farnesylation at their C-terminal CaaX box, aaX cleavage by the endoprotease ZMPSTE24 (prelamin A) or RCE1 (prelamin B and in some instances prelamin A), carboxy-methylation by the methyltransferase Icmt. Prelamin A is also subjected to a second cleavage step by ZMPSTE24, leading to release of a 15 aminoacids C-terminal peptide and mature lamin A (Fig. 1). Thus, B type lamins remain permanently farnesylated, while lamin A is devoid of the farnesylated and carboxymethylated C-terminus.4,5 Lamin A processing (Fig. 1) is very efficient, so that prelamin A is almost undetectable in normal cells, but it is accumulated under particular conditions including stress,6,7 organism aging,7 and senescence6 or differentiation of some cell types.8,9 The role of lamin post-translational processing is not completely elucidated. It has been proposed that the farnesylation state avoids formation of mixed networks of A and B type lamins10 and ensures lamin B1 anchorage to the nuclear envelope. Although prelamin A and lamin B maintain localization at the nuclear rim even in their non-farnesylated form,11 farnesylation clearly influences the interaction of lamins with chromatin and chromatin-associated proteins12-18 and protein localization.11,13 For instance, farnesylated prelamin A selectively interacts with NARF and SUN1,9 while fails to bind HP1alpha.12 Moreover, non-farnesylated prelamin A recruits LAP2alpha,12 HP1alpha, and BAF into nuclear foci,13,15 thus potentially influencing chromatin remodeling. Defects in prelamin A processing and accumulation of prelamin A forms are the cause of progeroid laminopathies and LMNA-linked lipodystrophies16 (Fig. 1). Of note, Hutchinson-Gilford Progeria (HGPS) cells accumulate an alternatively spliced form of prelamin A known as progerin (Fig. 1), which lacks 50 aminoacids at its C-terminus, including the second ZMPSTE24 cleavage site, and is permanently farnesylated (Fig. 1).

Figure 1.LMNA splicing products involved in diseases and prelamin A forms accumulated in laminopathies. (A) Genomic organization and protein domain structure of the LMNA splicing products involved in laminopathies: prelamin A, progerin, and lamin C. (B) Prelamin A processing steps. The four different prelamin A forms are represented. Below each prelamin A processing intermediate structure, the disease(s) showing accumulation of that form are indicated in parentheses. The processing steps of progerin seem to be the same as for prelamin A and are not represented in this scheme. Question marks indicate possible accumulation in laminopathies.
Lamin A/C and B type lamins are also phosphorylated at diverse serine and threonine residues in both interphase19 and mitotic cells.1,20-22 Lamins phosphorylation in mitosis favors nuclear envelope breakdown, while complex and not fully elucidated roles of lamin phosphorylation are observed in interphase cells.19 Importantly, lamin B phosphorylation has been proposed to regulate lamina attachment to MARs (matrix attachment regions, DNA sequences that help generate an open chromatin domain),23 while phosphorylation of lamin A and prelamin A in interphase cells19 regulates several pathways, including protein degradation21 and chromatin binding. Interaction of lamins with chromatin regulated through phosphorylation at specific sites has been demonstrated for lamin C and lamin Dm0,10,24 as well as for LBR and lamin B.1 Specific interplay between lamins and gene promoter sequences depending on the phosphorylation status has been further reported21 and implicated in tissue differentiation. Finally, phosphorylation by the kinase AKT1 targets non-farnesylated prelamin A to lysosomal degradation mostly in G2, thus providing a fine tool to modulate prelamin A levels during the cell cycle.21
Lamins undergo sumoylation at known residues25 and this post-translational modification is disrupted by laminopathy-causing mutations. Although the relevance of lamin A sumoylation has been elusive, it has been proposed that loss of sumoylated residues26 as in cardiomyopathy25 and Familial Partial Lipodystrophy (FPLD2) might impair binding of partner proteins, such as the transcription factor SREBP1 that should regulate chromatin activity.27
Importantly, all of the known post-translational modifications of A and B type lamins are dynamically regulated during cell cycle, cellular differentiation, and aging1,9,21 and confer to the nuclear lamina an impressive plasticity and an unexpected role of “signal transmitter” from the cytoskeleton to chromatin and back.28,29
Mechanisms of Chromatin Regulation
Chromatin regulation in eukaryotes occurs through complex and interconnected mechanisms that ensure hetereochromatin maintenance and compartimentalization of chromosome domains, DNA damage response and genome stability, chromatin conformational changes before and after mitosis, gene silencing and transcriptional activation, and chromatin remodeling at specific promoters. In this paper, we refer to these events as a whole using the term “chromatin dynamics.” Chromatin dynamics involves a number of protein families including epigenetic enzymes, DNA repair factors, heterochromatin proteins, ATPases, and even nuclear actin. Moreover, transcription factors and transcriptional regulators need to be specifically targeted to sequences after nuclear import to elicit remodeling of chromatin. Although lamins have been involved in almost all the above-mentioned processes, three main functions of lamins in chromatin regulation have been described: modulation and maintenance of heterochromatin domains, recruitment of the DNA damage response machinery, and transcription factor binding. These and new proposed functions of lamins in chromatin dynamics will be discussed in the next paragraph.
Why Chromatin Needs Lamins
Lamins regulate heterochromatin30 through interaction with diverse chromatin-binding factors (Fig. 2) including histone methyltransferases such as Suv39H1, histones such as H2, H3, and H4,34 the heterochromatin proteins HP1alpha31 and β, bridging proteins such as LAP2alpha and BAF, which in turn bind nucleosomal proteins. The lamin A/C and prelamin A binding partner LAP2alpha associates with the nucleosomal protein HMGN5 within a protein platform that should regulate chromatin mobility and the degree of heterochromatin condensation.32,33 HP1 interaction with lamins is required for heterochromatin anchorage at the end of mitosis,31 another key role of lamins in chromatin dynamics.

Figure 2. Known and Predicted nuclear lamina-chromatin networks. (A) The group of nuclear envelope proteins is shown in the left end corner. Chromatin-associated proteins are shown in the right upper corner. (B) The group of nuclear envelope proteins is shown in the left end corner. Transcription factors and transcriptional regulators are shown in the right upper corner. The gene name is reported in most cases. Light gray lines indicate co-occurrence. The graphs have been obtained using String 9.1 application (http://string-db.org/).
The interaction between lamins and chromatin is complex and also involves lamin interplay with proteins of the nuclear membrane, including the lamin B receptor (LBR), LAP2beta,30 emerin,14 BAF,15 and SUN proteins,1,9 which in turn interact with chromatin partners.34-36 The interplay between lamins and their nuclear envelope partners is relevant to the timing and specificity of chromatin regulation processes. A key role has been described for emerin, which interacts with histone deacetylase 3 (HDAC3) and influences its activity, specifically targeting muscle genes during myogenesis.37 An extensive study30 has recently reported that lamin A/C and LBR, through interaction with their respective nuclear envelope and nuclear matrix partners, regulate location of heterochromatin domains depending on extracellular stimuli and differentially affect expression of the same group of genes at different stages of muscle precursors differentiation.
In the same paper30 a major role of lamin A/C and LBR in heterochromatin tethering is unraveled. The authors show that in cells that do not express LBR or lamin A/C, heterochromatin is located at the nuclear interior and euchromatin is tethered at the nuclear periphery. This unconventional pattern of chromatin organization is found in the retina rods of nocturnal mammals and can be reproduced by experimental knockdown of both LBR and lamin A/C in other cell types. Importantly, expression of LBR is sufficient to tether heterochromatin at the nuclear periphery, while lamin A/C expression does not ensure heterochromatin anchorage unless LBR or other LMNA partners are expressed.30 Thus, a hierarchical function of nuclear envelope components to regulate heterochromatin domains is being unraveled, yet not completely understood.
The bulk of chromatin associated with lamins has been defined as LADs, after Lamina-Associated chromatin Domains.38 LADs are highly conserved gene poor chromosome domains, which include around 40 percent of the human genome and associate with the nuclear periphery in a stochastic way.39 These domains have been identified by a specific labeling technique to mark and trace chromatin that contacts the nuclear lamina.40 In LADs, lamin B and lamin A play similar roles, yet not fully elucidated.38 However, a major remodeling of LADs has been recently reported in cellular senescence and it has been linked to downregulation of lamin B1,41 suggesting a key role of lamins in the recruitment and release of those chromatin domains during aging. Perinucleolar heterochromatin has been defined as NADs,42 after Nucleolus-Associated Domains. The LADs and NADs sequences are mobile and are relocated upon appropriate stimuli,42 depending on levels of lamin B1, lamin B2, and/or BAF, although location of genes in the perinucleolar area appears to be mostly dependent on BAF and lamin A.43 Importantly, LADs and NADs are flanked by insulator sequences such as CTCF42 and D4Z4,44 which tethers chromatin domains at the nuclear periphery in a lamin A-dependent way.45 Noteworthy, D4Z4 macrosatellite repeats number is affected in facioscapulohumeral muscular dystrophy (FSHD),44 leading to derepression of transcriptional regulators through epigenetic modifications of DNA and histones.46 An interesting hypothesis suggests that LADs and NADs are not merely structured chromosome domains, but are dynamic domains aimed at maintenance of a default setting in nuclei. Such a default setting may vary from one cell type to another and even under different developmental or metabolic conditions42 and disruption of the mechanism is likely to be involved in several diseases.
Prelamin A plays a major role in chromatin dynamics.12 The role of prelamin A in heterochromatin anchorage is associated with its ability to bind the heterochromatin protein HP1, LAP2alpha,12 and BAF. BAF is recruited to the nuclear envelope in a prelamin A-dependent way15 and, in turn, influences specific histone modifications leading to different degree of chromatin condensation.34 Importantly, prelamin A binding to HP1alpha involves LAP2alpha and it is lost when prelamin A undergoes farnesylation.12 This specificity might have a functional role in normal human muscle cells that accumulate farnesylated prelamin A upon myogenic commitment.9 In fact, release of HP1alpha from the prelamin A-LAP2alpha complex, which has been detected in differentiating myoblasts,8 is expected to influence chromatin arrangement and gene transcription.8
However, it is widely accepted that accumulation of prelamin A causes reorganization of heterochromatin domains under both physiological12,13,15 and pathological conditions47-51 and it is becoming increasingly evident a major role of prelamin A in nuclear events regulating normal aging.6,7
Moreover, lamins have been shown to play a role in the anchorage of factors involved in the DNA damage response. In particular, a major role is exerted by lamins in the recruitment of 53BP1 in the nucleus and proper localization of 53BP1 foci in cells subjected to oxidative stress. Recruitment of 53BP1 in the nucleus and protein stability rely on lamin A52 and prelamin A7 and favors DNA double-strand breaks repair by non-homologous end joining. Consistent with this role, loss of A-type lamins hinders the processing of dysfunctional telomeres by non-homologous end joining.53 It has been further reported that lamin B1 anchors proteins involved in nucleotide excision repair (NER), including PCNA.54 PCNA recruitment in nuclei occurs through lamin A Ig-fold interaction,55 so that both LMNA and LMNB1 integrity appear to be required for proper anchorage of the DNA processivity factor. Importantly, a group of DNA repair proteins in UV damaged cells are disrupted following lamin B1 loss,54 indicating that lamins maintain a complex platform required for genome integrity.
In addition, lamins play an indirect role in chromatin dynamics due to their ability to anchor transcription factors at the nuclear periphery thus regulating their nuclear import. This has been shown for SREBP1, Sp1, and Oct-1, which interact with prelamin A56-58 and for pRb, Mok2, and cFos, which bind mature lamin A or lamin C.59,60 Also lamin B1 is able to recruit the transcription factor Oct-1 to the nuclear envelope.61,62 Most of these interactions elicit an inhibitory effect on gene activity, indicating the nuclear envelope as a gate or a resting place for transcriptional regulators.56,63,64 As expected, disruption or accumulation of nuclear lamina proteins leads to mislocalization and altered dynamics of these transcriptional regulators,56,58,65 as detailed below.
However, although a sequence-specific effect of lamin mutations has been recently demonstrated,66 the bulk of available data suggest that lamins act as wide-scale regulators of chromatin dynamics and organization.13,32,67
Lamin-Dependent Chromatin Dynamics during Stem Cell Differentiation
The LMNA gene is not expressed at the early stages of embryonic development, while B type lamins appear to be constitutively expressed.1 Although recent studies have detected a small amount of lamin A in pluripotent mouse embryonic stem cells68 and lamin B has been shown to be dispensable for development of some cell types,69 this general rule can still be accepted. However, it has been demonstrated that no lamin type is required for self-renewal and differentiation of embryonic stem cells (ESCs),70 while lamin B binding to promoters of developmentally regulated genes increases after ESCs differentiation.71 This observation and the finding that both lamin B1 and B2 are required for neuronal migration during mouse brain development support the view that B type lamins have evolved to facilitate the integration of different cell types into the complex tissues found in animals.71,72 In this context, both A and B type lamins seem to coordinate the chromatin transcriptional status with the nuclear and cell movement, an intriguing hypothesis that is gaining evidence from these71,72 and other studies.18 Despite these considerations, lamins are involved in chromatin dynamics even in ESCs.71,73 Stem cells that lack lamin A/C are characterized by chromatin fluctuations74 and the epigenetic markers of heterochromatin such as trimethylated H3K9 are generally reduced. Transient transfection of lamins into stem cells, providing stiffness to the nuclear envelope and interaction with specific chromatin domains, elicits reduction of the fluctuations.74 Recent studies have confirmed that lamins are physiologically required for the rearrangement of chromatin in the initial steps of development in order to induce proper cell maturation.73 In particular, ectopic expression of LMNA gene influences histone dynamics and contributes to formation of heterochromatin domains, while absence of lamin A/C causes hypermobility of histones and loss of heterochromatin with final impairment of differentiation marker expression.73 Along this line, non-functional lamins, such as progerin, alter chromatin dynamics in fibroblasts, but do not affect iPS. For instance, HGPS-derived iPS reacquired expression of HP1alpha and HDAC1 and trimethylation of H3K9, which were dramatically reduced in skin fibroblasts.75-77
In agreement with the latter observation, a major role of lamins and particularly of lamin A has been restricted to differentiation of adult stem cells.78 In that context, chromatin remodeling occurs to activate cell type-specific genes and silence genes linked to proliferation and multipotency. This process is governed by lamin-triggered epigenetic modifications through recruitment of histone deacetylases and methyltransferases and interaction with LEM domain proteins such as LAP2alpha, BAF, and emerin.
A key feature of differentiating cells is cytoskeleton reorganization. Mechano-transduction requires lamin function to propagate extracellular stimuli to the nuclear compartment as demonstrated by lamin knockdown and impairment of gene transcriptional activation.79,80 This process is likely mediated by the activity of histone deacetylases (HDACs) that, following lamin signaling, induce the modification of chromatin organization to promote gene expression.80
An additional mechanism of chromatin regulation through lamins during cellular differentiation is transcription factor anchorage. In fact, SREBP1 translocation in differentiating pre-adipocytes is regulated through lamin A or prelamin A binding at the nuclear periphery, while β-catenin export from the nucleus during adipogenesis is regulated by lamin-emerin interaction.81 Moreover, interaction of lamin A/C with pRb regulates cell cycle exit required to initiate cellular differentiation.78
Of note, lamin-dependent regulation of chromatin activity relies also on the fact that the expression of lamins and their functional partners is regulated in differentiating cells. For instance, lamin A/C, prelamin A, emerin, SUN1, and LAP2alpha expression and nuclear recruitment are modulated in differentiating muscle cells and/or during regeneration,8,9,82-84 thus representing a dynamic platform for chromatin interaction.
The whole evaluation of the available data suggests that lamin-mediated modulation of chromatin is primarily involved in the tissue homeostasis.
Lamin-Dependent Chromatin Dynamics during Aging
Heterochromatin remodeling has been reported to occur during cellular senescence. Partially contrasting data have been published showing either formation of senescence associated heterochromatin foci or loss of heterochromatin during the aging process.85 A recent paper recapitulates both proposed mechanisms by showing that peripheral heterochromatin domains are redistributed in aging cells leading to global loss of condensed chromatin and formation of some heterochromatic foci at the nuclear interior. Downregulation of lamin B1 has been involved in this phenomenon, since reduction of lamin B1 levels in senescent cells leads to LADs release and re-localization.41 More recently, it has been shown that, in cells from very old individuals (aged 95–105 years), accumulation of prelamin A at sub-toxic levels elicits heterochromatin decondensation and recruitment of DNA repair factors ensuring prompt response to stress.7 The main factor recruited by prelamin A in centenarian fibroblasts is the DNA repair protein 53BP1, which is involved in non-homologous end joining (NHEJ).7 It is suggested that a low energy mechanism such as NHEJ allows cells from very old individuals to efficiently repair damaged DNA under conditions of caloric restriction. This condition is mimicked by rapamycin, a drug known to increase lifespan in mouse models.7,21 Importantly, transient accumulation of prelamin A has been observed under oxidative stress conditions. Prelamin A accumulation facilitates recruitment of 53BP1 and elicits an open chromatin conformation, ultimately accelerating DNA repair.7 The overall evaluation of these data suggests that very old individuals have developed a prelamin A-mediated mechanism to counteract repeated stress insults during aging and preserve genome integrity. Thus, at least lamin B1 downregulation86,87 and prelamin A accumulation7,88 play a role in chromatin remodeling linked to stress response and aging. However, lamin B1 accumulation has been also observed in cells subjected to oxidative stress and entering senescence.89 Further, SUN1 recruitment at the nuclear envelope in centenarian cells has been observed.7 As a whole, nuclear envelope/lamina remodeling appears to play a major role in stress response and cellular and organism senescence. In this context, the nuclear envelope acts as a sensor of stress conditions and favors chromatin dynamics (heterochromatin decondensation, recruitment of 53BP1, rapid repair of damaged DNA) aimed at cell survival and genome maintenance. Nevertheless, exacerbation of lamina remodeling as it occurs in progeroid laminopathies elicits opposite and deleterious effects, as detailed below.
Lamin-Dependent Chromatin Dynamics under Pathological Conditions
Laminopathies include three main groups of diseases: muscular laminopathies, lipodystrophies, and developmental-progeroid syndromes. Although laminopathies may be caused by LMNA mutations or by mutations in functional partners of lamin A/C, all the so far investigated diseases share defects in chromatin organization and/or dynamics pointing to a major pathogenetic mechanism caused by loss of chromatin functionality.48-50,90-93
An early observation in laminopathic cells was the presence of a percentage of misshapen nuclei (Fig. 3) and the so-called honeycomb structures.93-95 These structures are areas devoid of B type lamins and accumulating lamin A/C93 and LAP2alpha. Lamin A/C and emerin assume a honeycomb appearance at these areas, which are often located at one pole of the nucleus. Underneath the nuclear lamina, chromatin is dispersed, again assuming a honeycomb appearance. It is suggested that formation of these structures is related to altered protein-protein interplay at the nuclear envelope during post-mitotic reconstitution of the nucleus.96,97 In support of this, we demonstrated that lack of emerin as in EDMD1 fibroblasts causes honeycomb formation and blebbing in nuclei upon overexpression of prelamin A, thus showing that altered prelamin A-emerin interplay either due to lamin mutations or to emerin absence is the cause of nuclear dimorphism.14 The hypothesis that these events derive from failure of mutated lamins to interact at the beginning of G1 phase is supported by the observed persistence of mid-body and its altered composition in laminopathic cells.90,98
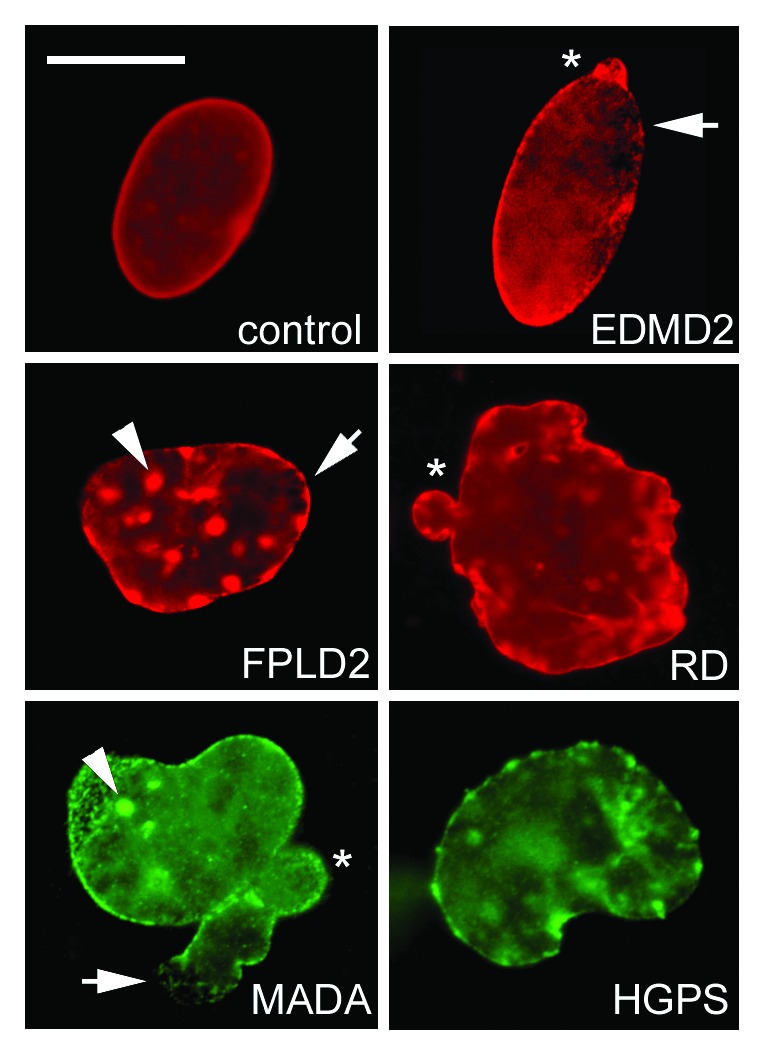
Figure 3. Altered nuclear shape in laminopathies. Representative control, EDMD2, FPLD2, and RD nuclei labeled using anti-lamin A/C antibody (Santa Cruz Sc-6215) are shown in the upper rows, OCT-1 (Santa-Cruz) and farnesylated prelamin A staining (Diatheva 1188–2) of a MADA and HGPS nucleus, respectively, are shown in the lower row. Honeycomb structures, characteristic of EDMD2 (and EDMD1) nuclei are also observed in FPLD2 and MADA (arrows). Lamin A/C aggregates (also labeled by SUN1 and prelamin A) are observed in FPLD2 and MADA (arrowheads), nucler envelope blebs (asterisk) are found in most laminopathies. Bar, 10 μm.
The first involvement of lamin-chromatin interplay in the pathogenesis of laminopathies99 came from observation that mutations in lamins or their functional partners were linked to abnormal chromatin organization in Emery-Dreifuss muscular dystrophy muscle fibers92 and Dunningan type familial partial lipodystrophy skin fibroblasts.93,100 These findings were strikingly supported by the study of progeroid laminopathies. In fact, complete loss of peripheral heterochromatin and dispersion of euchromatin were observed in HGPS and MADA cells, starting from the early ultrastructural analyses.47,48,50 Large-scale defects in heterochromatin were associated with altered genome organization,101 mislocalization of HP1,50 altered methylation pattern of H3K9,102 loss of LAP2 α, and defective histone deacetylase activity.
Muscular Laminopathies
Laminopathies affecting skeletal and/or cardiac muscle are Emery-Dreifuss muscular dystrophy (EDMD1 and EDMD2), Limb-Girdle muscular dystrophy type 1B (LGMD1B), and dilated cardiomyopathy with conduction system defects (CMD-CD). These disorders are caused by mutations in LMNA, EMD, or SYNE1/2, encoding lamin A/C, emerin, or nesprin1/2 respectively, all of which are nuclear envelope constituents. Mutations in LAP2alpha,103 FHL-1,104 and more recently SUN1/2 (Meinke et al., 2014) have been also associated with EDMD-like or cardiomyopathy phenotypes, suggesting a complex picture of the possible pathogenetic pathways, also involving lamina-associated nucleoplasmic proteins (LAP2alpha), transcriptional regulators (FHL-1), and strengthening the role of the LINC complex (SUN1/2).
In EDMD1 and EDMD2 detachment of peripheral heterochromatin from the nuclear lamina and focal loss of heterochromatin domains are observed in a significant percentage of nuclei both in fibroblasts94 and mature muscle fibers.91,92 Similar chromatin defects have been reported in Lmna −/− muscle fiber nuclei,105 supporting the view that altered chromatin organization in EDMD2 is due to loss of function of mutated lamins. Consistent with loss of heterochromatin domains, but possibly associated with more complex defects occurring in LMNA-mutated muscle fibers, myonuclei appear elongated both in mouse105 and human laminopathic muscle.9,106 We suggest that these enlarged nuclei observed in EDMD2 skeletal muscle and in the myocardium of patients affected by dilated cardiomyopathy (DCM-CD)106 are in fact derived from clustered nuclei, which fail to properly position during muscle stem cell differentiation and regeneration.9 Several implications might derive from the presence of elongated and clustered nuclei in EDMD2 muscle, including the altered regulation of myonuclear domains, i.e., the cytoplasmic regions regulated by each myonucleus. In agreement with this hypothesis, altered sarcomere structure has been observed in close proximity of altered myonuclei in Lmna −/− mice105 and in a new EDMD-like phenotype associated with SUN1 mutations (Meinke et al., 2014). Several lines of evidence show in fact that transcriptional regulation is defective in EDMD muscle. Histone H3 trimethylated at lysine 27 (H3K27) and phosphorylated RNA polymerase II, markers of inactive and active chromatin domains, respectively, are altered in EDMD2 myoblasts.107 In addition, acetylation of histone H3 on lysine 9 is defective in Lmna-null muscle fibers, indicating deregulated activation of gene expression.105 Such an altered mechanism has been also described at the neuromuscular junctions in EDMD2 muscle.108 Moreover, in a C. elegans model of EDMD1, lacking emerin expression, histone deacetylases are downregulated,109 suggesting that enzymes involved in epigenetic modifications of histones could play a role in EDMD pathogenesis. This is consistent with the occurrence of a specific interaction between emerin and HDAC3.37 However, although the only tissue apparently affected in EDMD is striated muscle, the reason why altered lamin-dependent heterochromatin dynamics affects tissue-specific pathways is not completely elucidated. To address this issue, we can consider the following reported data. (1) Muscle represents the only tissue where prelamin A increase has been demonstrated during normal differentiation.8,9 Since prelamin A has been shown to regulate large scale heterochromatin organization,12,13 it appears likely that altered prelamin A modulation occurring in EDMD29 might affect heterochromatin dynamics related to differentiation-linked positioning of chromatin domains and specific gene expression.8,107 (2) Along this line, it has been shown that EDMD2-linked point mutations in lamin impair reorganization of heterochromatic arrays during muscle-specific promoter activation,66 including the myogenin promoter,110 indicating that lamin mutations also affect gene expression at specific sites. (3) A recent study using the DamID technology has shown enrichment of muscle and neuronal genes in association with emerin in C. elegans, pointing to an upstream role of the nuclear membrane protein with respect to lamins in the regulation of those sequences.109 Interestingly, lack of emerin, a common situation in EDMD1, shifted those sequences to lamin association, a phenomenon that might explain the much lower frequency of deleterious emerin mutations causing EDMD1, with respect to the huge number of pathogenetic LMNA mutations.109 (4) The interaction of lamin A/C and emerin with their binding partners including SUN1, SUN2, BAF, and LAP2alpha could be also relevant to the effects of lamin mutations on chromatin specifically in muscle. Published data show that SUN1 levels are reduced in EDMD2 myotubes and mature muscle due to failure of interaction with mutated prelamin A. Moreover, SUN2 is not polarized in EDMD2 myonuclei, a defect associated with myonuclear clustering both in myotubes and mature muscle fibers9 (Meinke et al., 2014). Moreover, based on the reported association of LAP2alpha with prelamin A,8,12 the heterochromatin protein HP1,12 DNA, and the nucleosomal protein HMGN5,32 it is likely that redistribution of LAP2alpha in EDMD muscle stem cells or even in mature muscle might drive chromatin alterations. Altered localization and/or lamin interplay with the DNA-binding protein BAF15 is also possible in EDMD2 and it is most likely to occur in EDMD1, where the BAF-binding protein emerin is not expressed in most cases.
Overall, these findings suggest that altered lamin-mediated chromatin remodeling plays a pathogenetic role in EDMD, through deregulation of chromatin dynamics and gene expression.
Familial Partial Lipodystrophy
Familial partial lipodystrophy linked to LMNA mutations (FPLD2) is the prototype of lipodystrophic laminopathies. Lipodystrophic phenotypes are also observed in Mandibuloacral dysplasia type A (partial lipodystrophy) or type B (generalized lipodystrophy) and in Hutchinson-Gilford progeria (HGPS). All those diseases share the common feature of prelamin A accumulation at diverse levels.111 Two main pathogenetic mechanisms involving altered chromatin dynamics have been described for FPLD2. The first one involves altered organization of peripheral heterochromatin associated with accumulation of prelamin A and BAF in FPLD2 cells.56,67,93 In fact, BAF recruitment to the nuclear envelope is triggered by prelamin A and its mutated R482Q form found in FPLD2 cases.67 The other mechanism implies that prelamin A elicits altered transcription factor import in the nucleus. In fact, impaired SREBP1 translocation and SREBP1-mediated transactivation of adipocyte-specific genes including PPARG have been reported not only in LMNA-mutated lipodystrophies,56,100 but also in acquired lipodystrophies caused by accumulation of prelamin A due to anti-retroviral therapy.112 Moreover, accumulation of prelamin A by anti-retroviral protease inhibitors treatment of mesenchymal stem cells has been shown to affect Sp1 import in nuclei and expression of genes related to lipid metabolic processes.113 In the same cellular model, Oct-1 activity has been shown to be impaired due to sequestering at the nuclear periphery by prelamin A.113 Impairment of Oct-1 transactivation activity has been shown to block the autophagic process,113 a mechanism involved in white adipose tissue differentiation.114 Aberrant differentiation of adipocyte precursors has been also suggested by Oldenburg et al.115 The authors found that lamin A associates with the RNA-binding protein Fragile X syndrome related protein 1 (FXR1P) and upregulation of FXR1P in FPLD2 adipogenic precursors causes conversion to the myogenic lineage. These findings point to a complex regulation of adipogenesis by lamins, involving not only control of gene expression, but also microRNA activity and miR-regulated expression of target genes.115 Interestingly, FXR1P is also altered in FSHD, a disease linked to altered expression of D4Z4 repeats, acting as insulators and regulating chromatin dynamics through lamin A interaction.
Finally, differentiation into the adipogenic lineage has been shown to reset lamin A-promoter interactions at PPARG, FABP8, FABP9, FABP4, and FABP12, leading to transcriptional activation.116 Thus, lipodystrophy-linked LMNA mutations could affect chromatin activity in terms of altered transcription factor and/or transcriptional regulators targeting and activity. The whole picture of altered gene expression in FPLD2 and in the other LMNA-linked lipodystrophic disorders needs to be defined and it is further complicated by the occurrence of two distinct and opposite clinical phenotypes (lipoatrophy and adipose tissue hypertrophy) downstream of the same lamin A mutations. Consistent with this phenotype, it has been shown that mutated lamin A downregulates adipocyte gene expression, including PPARG2, RB1, CCND3, and LPL in thigh but not in abdomen subcutaneous adipose tissue in patients with FPLD2.100 Moreover, it is conceivable that LMNA defects might affect a group of adipogenic genes, depending on the type of adipogenic precursor (white or brown pre-adipocytes, adipogenic mesenchymal stem cells, etc.). Along this line, Xiong et al.77 demonstrated that expression of truncated prelamin A (progerin) in iPS selectively impairs expression of late adipocyte differentiation genes.
Progeroid and Developmental Laminopathies
In progeroid and developmental laminopathies, including HGPS, MADA and MADB and restrictive dermopathy (RD), a prominent pathogenetic role is exerted by prelamin A (or its alternatively spliced form, progerin48), which, in most cases, elicits complete loss of heterochromatin in the nucleus of cultured cells.48,50,51 This phenomenon is directly linked to the ability of the farnesylated form of the lamin A precursor to elicit nuclear size increase, nuclear envelope misfolding and heterochromatin dispersion.13,111 The mechanism upstream of prelamin A-mediated chromatin remodeling has been elusive, but most likely involves anomalous interplay between progerin or farnesylated prelamin A and enzymes involved in epigenetic pathways. Our first study performed in HGPS fibroblasts had shown that progerin levels could be reduced by the use of trichostatin A, a drug known to inhibit class I and II mammalian histone deacetylases (HDACs).48 However, statin pretreatment of cells was required to efficiently recover the chromatin phenotype of HGPS cells (Fig. 4), suggesting that an interaction between HDACs and progerin could be strengthened by the farnesylated residue of truncated prelamin A. Further, aberrant interaction of progerin with the facultative heterochromatin-associated trimethyl-H3K27 has been reported and, noteworthy, trimethyl-H3K27-enrichement in gene-poor chromosome regions was reduced in HGPS.117,118 More recently, loss of SIRT1 from the nuclear matrix has been reported in HGPS fibroblasts and partial rescue of the chromatin phenotype and of lamin A-SIRT1 binding by SIRT1 activators has been shown.119 SIRT1 is a pleiotropic enzyme platform with HDACs activity and its role in chromatin remodeling is not fully elucidated. Moreover, the possibility that SIRT1 interaction with lamin A, prelamin A, or progerin might cause different effects on chromatin epigenetic modifications deserves further studies. For instance, the deacetylation of SUV39H1 by SIRT1 enhances SUV39H1 activity and facilitates H3K9 trimethylation and heterochromatin formation.120 However, the finding that both HDAC and DNA methyltransferases inhibitors affect prelamin A and/or progerin levels suggests that prelamin A-progerin interplay with HDACs48,119 and/or DNA methyltransferases13,48 might be involved in pathogenetic mechanisms. Moreover, these observations suggest that epigenetic enzymes and their inhibitors and/or activators may be further explored in progeria as potential therapeutic targets or pharmacological tools. In the context of chromatin epigenetic modifications occurring in progeroid laminopathies, the methylation status of histones has been widely investigated. Trimethylation of histone H3K9, a marker of constitutive heterochromatin and H3K27, a marker of facultative heterochromatin has been reported to be reduced in HGPS cells48,51 and in MADA at high passage number.48,50 Importantly, a stepwise redistribution of trimethyl-H3K9 was observed in MADA cells50 and decrease of trimethyl-H3K9 with passage number was observed both in controls and laminopathic fibroblasts,48,50,121 suggesting that cell cycle progression and population doublings influence lamin-mediated epigenetic modifications. Intriguingly, the dynamics of heterochromatin loss is less obvious in RD cells, which do not express mature lamin A,49 suggesting a more severe, yet not understood mechanism of chromatin de-regulation in the human disease, only in part recapitulated by mouse models. In HGPS and MADA cells, loss of the heterochromatin protein HP1 has been also reported in several studies.50,51 However, some controversial data can be found in the literature, showing beneficial effects of the histone methyltransferase Suv39H1 knockdown in progeroid mice.121 As in the field of normal aging, formation of heterochromatin foci or, vice versa, heterochromatin loss in senescent cells is matter of debate.7,85 The most likely interpretation of the available data are that lamin A and prelamin A forms, contribute to the default setting of chromatin in cells by “locking in” a particular transcriptional status. In this context, accumulation of anomalous amount of prelamin A as in RD or MADB or mutated prelamin A forms as in the other progeroid laminopathies impairs such a fundamental function of the lamin network causing unordered localization of chromatin domains and loss or increase of some markers depending on stochastic events. Consistent with this view, unordered distribution of heterochromatin markers is always observed in laminopathic cells.49,51 Further, an explanation for the results showing that inhibition of enzymes involved in heterochromatin organization improves the phenotype in progeria models48,121 is that the active enzymes are possibly entrapped by progerin or farnesylated prelamin A, so that their inhibition may favor their release and restore heterochromatin dynamics. We are currently testing this hypothesis in human progeroid laminopathies.
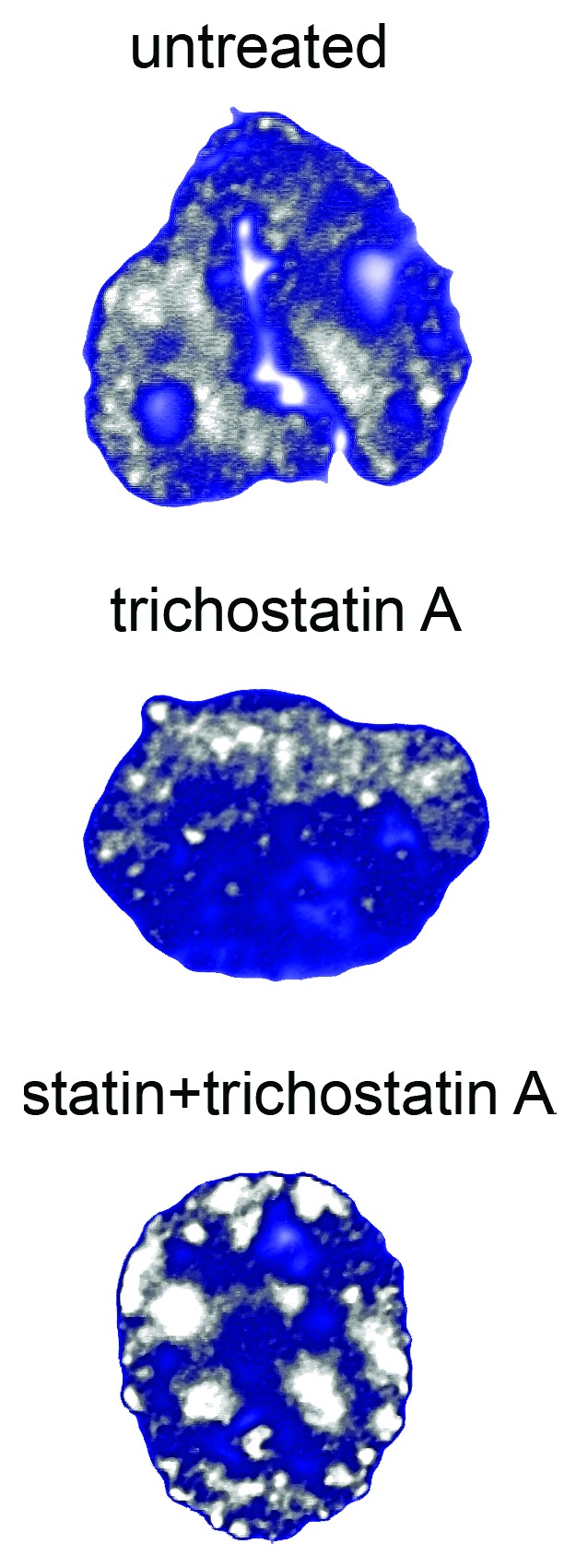
Figure 4. Chromatin remodeling in HGPS fibroblasts. The experiment was performed using the histone deacetylase inhibitor trichostatin A or combination of trichostatin A and statins (that convert progerin into its non-farnesylated form and elicit accumulation of wild-type prelamin A). The images obtained using anti-trimethyl-H3K9 antibody to label heterochromatin domains have been elaborated using Photoshop 7. Note recovery of heterochromatin domains (gray) using combined drugs, indicating that heterochromatin status in HGPS is regulated by prelamin A forms, most likely through histone deacetylase interaction.
Another feature of HGPS and MADA nuclei is that they become more susceptible to ROS- or ionizing radiation-induced DNA damage, although prelamin A accumulation does not induce per se genomic instability.7,122 The problem is that DNA damage is less efficiently repaired in HGPS and RD nuclei123 due to altered recruitment of DNA repair factors. This effect is dependent on the environment created by progeroid cells and opens a new question on the extent to which systemic effects rather than (or along with) cell intrinsic factors contribute to disease.124,125
The role of prelamin A binding partners in the chromatin remodeling that occurs in progeroid laminopathies has been better defined in the last years. A master regulator of the interplay between progerin and chromatin is BAF,15 which is recruited to the nuclear lamina upon accumulation of any wild-type or mutated prelamin A form.67 BAF is a DNA and emerin binding factor, which also causes, when mutated, a rare progeroid disorder featuring major bone involvement.126
BAF also interacts with LAP2alpha, located in the nucleoplasm, but recruited by farnesylated prelamin A at the nuclear periphery.12 LAP2alpha is mostly downregulated in the nuclear interior in HGPS cells as well as in MADA, while peripheral staining is preserved in nuclei (Cenni et al., 2014). Intriguingly, LAP2alpha interacts with HP1alpha, which is displaced from the complex by farnesylated prelamin A.13
An interesting mechanism has been proposed as the cause of altered chromatin dynamics in HGPS. The authors propose that progerin accumulation results in disruption of functions of some replication and repair factors, causing the mislocalization of XPA (xeroderma pigmentosum group A) protein to the replication forks and replication fork stalling and arresting cell-cycle progression. Further, it has been shown failure to recruit the DNA processivity factor PCNA and DNA polymerase delta due to anomalous cleavage of the replication factor C.127 These data might explain both the altered DNA damage repair and the prolonged S-phase observed in progeroid laminopathies (Cenni et al., 2014).
Finally, the relevance of pathogenetic lamin A mutations on LADs organization and function is being investigated in recent years. Lamin B1 depletion and overexpression are known to cause senescence and to affect LADs localization in nuclei as well as DNA binding.87 In cells from an atypical progeria syndrome carrying the E145K LMNA mutation lamin B1 levels are reduced.128 However, this is not the case of all progeroid laminopathies, where increased or decreased levels of lamin B1 can be found in nuclei at low passage. These apparently contradictory effects are recapitulated in the interpretation that a fixed rate of nuclear lamina constituents is required for proper chromatin modulation, while loss of this condition elicits cell cycle arrest and senescence linked to relocation and activation or inactivation of specific genes.129,130 Here, we suggest that downregulation or upregulation of lamin B1, as well as upregulation of SUN1 or recruitment of BAF and LAP2alpha at the nuclear periphery are part of the cellular response to the unstable situation caused by prelamin A/progerin accumulation. Whether this “pathological nuclear envelope remodeling” worsens the cellular phenotype or attenuates deleterious effects on chromatin remains to be established.
Other Laminopathies
The autosomal dominant leucodystrophy (ADLD) is a laminopathy affecting the nervous system. It is caused by LMNB1 duplication and features high levels of lamin B1 accumulation in nuclei at the onset of disease, which occurs around the fourth decade.61 Heterochromatin disorganization and altered import of the transcription factor Oct-1 in nuclei have been reported in ADLD.61 Of note, Oct-1 sequestering in ADLD muscle nuclei impairs transcription of Myosyn heavy chain 2B, one of the Oct-1 target genes, playing a major role in muscle function.61 The latter finding involves regulation of muscle genes in ADLD pathophysiology, consistent with clinical and morphological observations.61 Moreover, studies performed in animal models show that lamin B1 overexpression causes reduced occupancy of Yin Yan transcription factor at the promoter of proteolipid protein PLP, the most abundant protein in the central nervous system myelin sheet.131 The authors suggest that re-localization of the promoter by lamin B1 to a more heterochromatic region may reduce accessibility to the transcription factor. This hypothesis is consistent with the observed alteration of heterochromatin domains in ADLD tissues.61
The lamin B receptor LBR, when mutated or knocked down as it occurs in Pelger-Huet Anomaly and in the severe skeletal disease known as Greenberg Skeletal Dysplasia, elicits formation of heterochromatin clumps and impairs nuclear fragmentation in granulocytes,132 in agreement with the recently identified role of LBR in chromatin organization and with its ability to bind heterochromatin proteins.30
Fine mechanisms of chromatin control are probably at the basis of LMNA-linked Charcot-Marie Tooth disease (CMT2B1) or Heart Hand syndrome, both diseases caused by mutations in LMNA and presenting with relatively mild clinical phenotype.1 Due to the rarity of samples, these disorders have been poorly investigated.
Conclusions and Perspectives
In summary, at least four modes of action of lamins for chromatin regulation (Fig. 5) are affected in laminopathies. (1) Transcription factor retention at the nuclear lamina appears to be a common mechanism shared by different laminopathies, including FPLD2, MADA,56 ADLD48 and also observed in EDMD mouse models.133 (2) Altered interplay between mutated lamins and LADs is an emerging pathogenetic mechanism supporting and going in depth into the first observations that heterochromatin loss occurred in laminopathies. (3) Specific effects at promoters exerted by lamin-dependent alterations of epigenetic molecules such as HDACs, HP1, and histone methyltransferases have been shown in a limited number of cases, although lamin interplay with chromatin regulatory factors is gaining increasing evidence. (4) Impaired tethering of DDR factors has been mostly observed in progeroid laminopathies.

Figure 5. Chromatin dynamics dependent on lamins. Four major groups of events that contribute to chromatin dynamics are represented in the large ellipses. Blue arrows indicate A type lamin pathways, purple arrows B type pathways. Lamin partners involved in chromatin regulation are indicated in the respective arrows. Some targets of lamin activity (HP1alpha, HDAC3, SREBP1, OCT-1) are common to lamin A and lamin B-related pathways or are misregulated in diverse diseases (SREBP1, OCT-1, HP1alpha). Targets affected in laminopathies are in yellow boxes. See text for references.
The pathogenic effects described above are linked not only to the presence of mutated lamins, but also to their altered expression levels and defective protein processing. This apparently trivial observation must be kept in mind when thinking about therapeutic approaches, which should be aimed at reducing levels of toxic proteins and/or strength of their aberrant interactions with chromatin and chromatin regulatory factors. In this context, the use of epigenetic drugs such as HDACs inhibitors and/or activators deserves further investigation.
Finally, we cannot rule out the possibility that systemic factors triggered by mutated lamin expression or by prelamin A accumulation might influence chromatin dynamics. Some papers report increase of cytokine expression in cells bearing EDMD-linked mutations or in progeria models.125,134 Interestingly, altered gene expression downstream of these systemic effects has been reported.134 The latter finding raises the hypothesis that systemic disorders mediated by cytokine signaling could in part cause or at least worsen (but also improve) the chromatin defects observed in laminopathic cells. The latter aspect warrants further investigation in view of possible therapeutic treatments acting on circulating factors.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The work was supported by grants from Italian FIRB MIUR 2010, AIProSaB 2013, AIDMED 2014, EU COST Action BM1002 “Nanonet” to G.L., and “5 per mille” Project 2011 to Rizzoli Orthopedic Institute. The authors acknowledge the technical assistance of S Grasso, A Valmori, and D Zini.
References
- 1.Maraldi NM, Capanni C, Cenni V, Fini M, Lattanzi G. Laminopathies and lamin-associated signaling pathways. J Cell Biochem. 2011;112:979–92. doi: 10.1002/jcb.22992. [DOI] [PubMed] [Google Scholar]
- 2.Biamonti G, Giacca M, Perini G, Contreas G, Zentilin L, Weighardt F, Guerra M, Della Valle G, Saccone S, Riva S, et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol. 1992;12:3499–506. doi: 10.1128/mcb.12.8.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schütz W, Benavente R, Alsheimer M. Dynamic properties of germ line-specific lamin B3: the role of the shortened rod domain. Eur J Cell Biol. 2005;84:649–62. doi: 10.1016/j.ejcb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Davies BS, Coffinier C, Yang SH, Barnes RH, 2nd, Jung HJ, Young SG, Fong LG. Investigating the purpose of prelamin A processing. Nucleus. 2011;2:4–9. doi: 10.4161/nucl.2.1.13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrowman J, Hamblet C, Kane MS, Michaelis S. Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS One. 2012;7:e32120. doi: 10.1371/journal.pone.0032120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200–10. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]
- 7.Lattanzi G, Ortolani M, Columbaro M, Prencipe S, Mattioli E, Lanzarini C, Maraldi NM, Cenni V, Garagnani P, Salvioli S, et al. Lamins are rapamycin targets that impact human longevity: a study in centenarians. J Cell Sci. 2014;127:147–57. doi: 10.1242/jcs.133983. [DOI] [PubMed] [Google Scholar]
- 8.Capanni C, Del Coco R, Squarzoni S, Columbaro M, Mattioli E, Camozzi D, Rocchi A, Scotlandi K, Maraldi N, Foisner R, et al. Prelamin A is involved in early steps of muscle differentiation. Exp Cell Res. 2008;314:3628–37. doi: 10.1016/j.yexcr.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 9.Mattioli E, Columbaro M, Capanni C, Maraldi NM, Cenni V, Scotlandi K, Marino MT, Merlini L, Squarzoni S, Lattanzi G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011;18:1305–15. doi: 10.1038/cdd.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaremba-Czogalla M, Piekarowicz K, Wachowicz K, Kozioł K, Dubińska-Magiera M, Rzepecki R. The different function of single phosphorylation sites of Drosophila melanogaster lamin Dm and lamin C. PLoS One. 2012;7:e32649. doi: 10.1371/journal.pone.0032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adam SA, Butin-Israeli V, Cleland MM, Shimi T, Goldman RD. Disruption of lamin B1 and lamin B2 processing and localization by farnesyltransferase inhibitors. Nucleus. 2013;4:142–50. doi: 10.4161/nucl.24089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lattanzi G, Columbaro M, Mattioli E, Cenni V, Camozzi D, Wehnert M, Santi S, Riccio M, Del Coco R, Maraldi NM, et al. Pre-Lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007;102:1149–59. doi: 10.1002/jcb.21467. [DOI] [PubMed] [Google Scholar]
- 13.Mattioli E, Columbaro M, Capanni C, Santi S, Maraldi NM, D’Apice MR, Novelli G, Riccio M, Squarzoni S, Foisner R, et al. Drugs affecting prelamin A processing: effects on heterochromatin organization. Exp Cell Res. 2008;314:453–62. doi: 10.1016/j.yexcr.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Capanni C, Del Coco R, Mattioli E, Camozzi D, Columbaro M, Schena E, Merlini L, Squarzoni S, Maraldi NM, Lattanzi G. Emerin-prelamin A interplay in human fibroblasts. Biol Cell. 2009;101:541–54. doi: 10.1042/BC20080175. [DOI] [PubMed] [Google Scholar]
- 15.Capanni C, Cenni V, Haraguchi T, Squarzoni S, Schüchner S, Ogris E, Novelli G, Maraldi N, Lattanzi G. Lamin A precursor induces barrier-to-autointegration factor nuclear localization. Cell Cycle. 2010;9:2600–10. doi: 10.4161/cc.9.13.12080. [DOI] [PubMed] [Google Scholar]
- 16.Lattanzi G. Prelamin A-mediated nuclear envelope dynamics in normal and laminopathic cells. Biochem Soc Trans. 2011;39:1698–704. doi: 10.1042/BST20110657. [DOI] [PubMed] [Google Scholar]
- 17.Lattanzi G, Marmiroli S, Facchini A, Maraldi NM. Nuclear damages and oxidative stress: new perspectives for laminopathies. Eur J Histochem. 2012;56:e45. doi: 10.4081/ejh.2012.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung HJ, Nobumori C, Goulbourne CN, Tu Y, Lee JM, Tatar A, Wu D, Yoshinaga Y, de Jong PJ, Coffinier C, et al. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc Natl Acad Sci U S A. 2013;110:E1923–32. doi: 10.1073/pnas.1303916110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack CG, Melo-Cardenas J, Imanishi SY, Goldman RD, Eriksson JE. Interphase phosphorylation of lamin A. J Cell Sci. 2014;127:2683–96. doi: 10.1242/jcs.141820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cenni V, Bertacchini J, Beretti F, Lattanzi G, Bavelloni A, Riccio M, Ruzzene M, Marin O, Arrigoni G, Parnaik V, et al. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J Proteome Res. 2008;7:4727–35. doi: 10.1021/pr800262g. [DOI] [PubMed] [Google Scholar]
- 21.Bertacchini J, Beretti F, Cenni V, Guida M, Gibellini F, Mediani L, Marin O, Maraldi NM, de Pol A, Lattanzi G, et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013;27:2145–55. doi: 10.1096/fj.12-218214. [DOI] [PubMed] [Google Scholar]
- 22.Kuga T, Nie H, Kazami T, Satoh M, Matsushita K, Nomura F, Maeshima K, Nakayama Y, Tomonaga T. Lamin B2 prevents chromosome instability by ensuring proper mitotic chromosome segregation. Oncogenesis. 2014;3:e94. doi: 10.1038/oncsis.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barboro P, Repaci E, D’Arrigo C, Balbi C. The role of nuclear matrix proteins binding to matrix attachment regions (Mars) in prostate cancer cell differentiation. PLoS One. 2012;7:e40617. doi: 10.1371/journal.pone.0040617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaremba-Czogalla M, Gagat P, Kozioł K, Dubińska-Magiera M, Sikora J, Dadlez M, Rzepecki R. Identification of new in vivo phosphosites on lamin Dm - the evidence of heterogeneity of phosphorylation sites in different Drosophila tissues. Nucleus. 2011;2:478–88. doi: 10.4161/nucl.2.5.17864. [DOI] [PubMed] [Google Scholar]
- 25.Zhang YQ, Sarge KD. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J Cell Biol. 2008;182:35–9. doi: 10.1083/jcb.200712124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boudreau É, Labib S, Bertrand AT, Decostre V, Bolongo PM, Sylvius N, Bonne G, Tesson F. Lamin A/C mutants disturb sumo1 localization and sumoylation in vitro and in vivo. PLoS One. 2012;7:e45918. doi: 10.1371/journal.pone.0045918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon DN, Domaradzki T, Hofmann WA, Wilson KL. Lamin A tail modification by SUMO1 is disrupted by familial partial lipodystrophy-causing mutations. Mol Biol Cell. 2013;24:342–50. doi: 10.1091/mbc.E12-07-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22:2335–49. doi: 10.1093/hmg/ddt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gundersen GG, Worman HJ. Nuclear positioning. Cell. 2013;152:1376–89. doi: 10.1016/j.cell.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152:584–98. doi: 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Chaturvedi P, Parnaik VK. Lamin A rod domain mutants target heterochromatin protein 1alpha and beta for proteasomal degradation by activation of F-box protein, FBXW10. PLoS One. 2010;5:e10620. doi: 10.1371/journal.pone.0010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S, Schones DE, Malicet C, Rochman M, Zhou M, Foisner R, Bustin M. High mobility group protein N5 (HMGN5) and lamina-associated polypeptide 2α (LAP2α) interact and reciprocally affect their genome-wide chromatin organization. J Biol Chem. 2013;288:18104–9. doi: 10.1074/jbc.C113.469544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rochman M, Malicet C, Bustin M. HMGN5/NSBP1: a new member of the HMGN protein family that affects chromatin structure and function. Biochim Biophys Acta. 2010;1799:86–92. doi: 10.1016/j.bbagrm.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montes de Oca R, Andreassen PR, Wilson KL. Barrier-to-Autointegration Factor influences specific histone modifications. Nucleus. 2011;2:580–90. doi: 10.4161/nucl.2.6.17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong H, Rivero F, Euteneuer U, Mondal S, Mana-Capelli S, Larochelle D, Vogel A, Gassen B, Noegel AA. Dictyostelium Sun-1 connects the centrosome to chromatin and ensures genome stability. Traffic. 2008;9:708–24. doi: 10.1111/j.1600-0854.2008.00721.x. [DOI] [PubMed] [Google Scholar]
- 36.Demmerle J, Koch AJ, Holaska JM. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem. 2012;287:22080–8. doi: 10.1074/jbc.M111.325308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demmerle J, Koch AJ, Holaska JM. Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosome Res. 2013;21:765–79. doi: 10.1007/s10577-013-9381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meuleman W, Peric-Hupkes D, Kind J, Beaudry JB, Pagie L, Kellis M, Reinders M, Wessels L, van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–80. doi: 10.1101/gr.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38:1005–14. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 40.Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–92. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 41.Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, Narita M, Pérez-Mancera PA, Bennett DC, Chong H, et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013;27:1800–8. doi: 10.1101/gad.217281.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padeken J, Heun P. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr Opin Cell Biol. 2014;28:54–60. doi: 10.1016/j.ceb.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Kind J, van Steensel B. Stochastic genome-nuclear lamina interactions: modulating roles of Lamin A and BAF. Nucleus. 2014;5:124–30. doi: 10.4161/nucl.28825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ottaviani A, Schluth-Bolard C, Gilson E, Magdinier F. D4Z4 as a prototype of CTCF and lamins-dependent insulator in human cells. Nucleus. 2010;1:30–6. doi: 10.4161/nucl.1.1.10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, Desnuelle C, Gilson E, Magdinier F. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet. 2009;5:e1000394. doi: 10.1371/journal.pgen.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krom YD, Thijssen PE, Young JM, den Hamer B, Balog J, Yao Z, Maves L, Snider L, Knopp P, Zammit PS, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol Life Sci. 2005;62:2669–78. doi: 10.1007/s00018-005-5318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Columbaro M, Mattioli E, Schena E, Capanni C, Cenni V, Levy N, Navarro CL, Del Coco R, Squarzoni S, Camozzi D, et al. Prelamin A processing and functional effects in restrictive dermopathy. Cell Cycle. 2010;9:4766–8. doi: 10.4161/cc.9.23.14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filesi I, Gullotta F, Lattanzi G, D’Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23:150–8. doi: 10.1152/physiolgenomics.00060.2005. [DOI] [PubMed] [Google Scholar]
- 51.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J. 2011;30:3383–96. doi: 10.1038/emboj.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, Morgado-Palacin L, Gapud EJ, Sleckman BP, Sullivan T, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28:2414–27. doi: 10.1038/emboj.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butin-Israeli V, Adam SA, Goldman RD. Regulation of nucleotide excision repair by nuclear lamin b1. PLoS One. 2013;8:e69169. doi: 10.1371/journal.pone.0069169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shumaker DK, Solimando L, Sengupta K, Shimi T, Adam SA, Grunwald A, Strelkov SV, Aebi U, Cardoso MC, Goldman RD. The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J Cell Biol. 2008;181:269–80. doi: 10.1083/jcb.200708155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14:1489–502. doi: 10.1093/hmg/ddi158. [DOI] [PubMed] [Google Scholar]
- 57.Mahen R, Hattori H, Lee M, Sharma P, Jeyasekharan AD, Venkitaraman AR. A-type lamins maintain the positional stability of DNA damage repair foci in mammalian nuclei. PLoS One. 2013;8:e61893. doi: 10.1371/journal.pone.0061893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Infante A, Gago A, de Eguino GR, Calvo-Fernández T, Gómez-Vallejo V, Llop J, Schlangen K, Fullaondo A, Aransay AM, Martín A, et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging (Albany NY) 2014;6:264–80. doi: 10.18632/aging.100651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodríguez J, Calvo F, González JM, Casar B, Andrés V, Crespo P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J Cell Biol. 2010;191:967–79. doi: 10.1083/jcb.201004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harper M, Tillit J, Kress M, Ernoult-Lange M. Phosphorylation-dependent binding of human transcription factor MOK2 to lamin A/C. FEBS J. 2009;276:3137–47. doi: 10.1111/j.1742-4658.2009.07032.x. [DOI] [PubMed] [Google Scholar]
- 61.Columbaro M, Mattioli E, Maraldi NM, Ortolani M, Gasparini L, D’Apice MR, Postorivo D, Nardone AM, Avnet S, Cortelli P, et al. Oct-1 recruitment to the nuclear envelope in adult-onset autosomal dominant leukodystrophy. Biochim Biophys Acta. 2013;1832:411–20. doi: 10.1016/j.bbadis.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 62.Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol. 2009;184:45–55. doi: 10.1083/jcb.200804155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heessen S, Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007;8:914–9. doi: 10.1038/sj.embor.7401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcárcel J, Guigó R. Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol. 2009;16:996–1001. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 65.Dreuillet C, Harper M, Tillit J, Kress M, Ernoult-Lange M. Mislocalization of human transcription factor MOK2 in the presence of pathogenic mutations of lamin A/C. Biol Cell. 2008;100:51–61. doi: 10.1042/BC20070053. [DOI] [PubMed] [Google Scholar]
- 66.Mattout A, Pike BL, Towbin BD, Bank EM, Gonzalez-Sandoval A, Stadler MB, Meister P, Gruenbaum Y, Gasser SM. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr Biol. 2011;21:1603–14. doi: 10.1016/j.cub.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 67.Capanni C, Squarzoni S, Cenni V, D’Apice MR, Gambineri A, Novelli G, Wehnert M, Pasquali R, Maraldi NM, Lattanzi G. Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier-to-autointegration factor (BAF) nuclear redistribution. Cell Cycle. 2012;11:3568–77. doi: 10.4161/cc.21869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eckersley-Maslin MA, Bergmann JH, Lazar Z, Spector DL. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus. 2013;4:53–60. doi: 10.4161/nucl.23384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang SH, Chang SY, Yin L, Tu Y, Hu Y, Yoshinaga Y, de Jong PJ, Fong LG, Young SG. An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum Mol Genet. 2011;20:3537–44. doi: 10.1093/hmg/ddr266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim Y, Zheng X, Zheng Y. Proliferation and differentiation of mouse embryonic stem cells lacking all lamins. Cell Res. 2013;23:1420–3. doi: 10.1038/cr.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science. 2011;334:1706–10. doi: 10.1126/science.1211222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coffinier C, Chang SY, Nobumori C, Tu Y, Farber EA, Toth JI, Fong LG, Young SG. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc Natl Acad Sci U S A. 2010;107:5076–81. doi: 10.1073/pnas.0908790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Melcer S, Hezroni H, Rand E, Nissim-Rafinia M, Skoultchi A, Stewart CL, Bustin M, Meshorer E. Histone modifications and lamin A regulate chromatin protein dynamics in early embryonic stem cell differentiation. Nat Commun. 2012;3:910. doi: 10.1038/ncomms1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Talwar S, Kumar A, Rao M, Menon GI, Shivashankar GV. Correlated spatio-temporal fluctuations in chromatin compaction states characterize stem cells. Biophys J. 2013;104:553–64. doi: 10.1016/j.bpj.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Xiong ZM, LaDana C, Wu D, Cao K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging (Albany NY) 2013;5:288–303. doi: 10.18632/aging.100550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gesson K, Vidak S, Foisner R. Lamina-associated polypeptide (LAP)2α and nucleoplasmic lamins in adult stem cell regulation and disease. Semin Cell Dev Biol. 2014;29:116–24. doi: 10.1016/j.semcdb.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8. doi: 10.1172/JCI200419670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Y, Chu JS, Kurpinski K, Li X, Bautista DM, Yang L, Sung KL, Li S. Biophysical regulation of histone acetylation in mesenchymal stem cells. Biophys J. 2011;100:1902–9. doi: 10.1016/j.bpj.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tilgner K, Wojciechowicz K, Jahoda C, Hutchison C, Markiewicz E. Dynamic complexes of A-type lamins and emerin influence adipogenic capacity of the cell via nucleocytoplasmic distribution of beta-catenin. J Cell Sci. 2009;122:401–13. doi: 10.1242/jcs.026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lattanzi G, Ognibene A, Sabatelli P, Capanni C, Toniolo D, Columbaro M, Santi S, Riccio M, Merlini L, Maraldi NM, et al. Emerin expression at the early stages of myogenic differentiation. Differentiation. 2000;66:208–17. doi: 10.1111/j.1432-0436.2000.660407.x. [DOI] [PubMed] [Google Scholar]
- 83.Lattanzi G, Cenni V, Marmiroli S, Capanni C, Mattioli E, Merlini L, Squarzoni S, Maraldi NM. Association of emerin with nuclear and cytoplasmic actin is regulated in differentiating myoblasts. Biochem Biophys Res Commun. 2003;303:764–70. doi: 10.1016/S0006-291X(03)00415-7. [DOI] [PubMed] [Google Scholar]
- 84.Squarzoni S, Sabatelli P, Capanni C, Lattanzi G, Rutigliano C, Columbaro M, Mattioli E, Rocca M, Maraldi NM. Emerin increase in regenerating muscle fibers. Eur J Histochem. 2005;49:355–62. doi: 10.4081/963. [DOI] [PubMed] [Google Scholar]
- 85.Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Camps J, Wangsa D, Falke M, Brown M, Case CM, Erdos MR, Ried T. Loss of lamin B1 results in prolongation of S phase and decondensation of chromosome territories. FASEB J. 2014;28:3423–34. doi: 10.1096/fj.14-250456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–99. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013;112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543. [DOI] [PubMed] [Google Scholar]
- 89.Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, Lopez B, Bertrand P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012;31:1080–94. doi: 10.1038/emboj.2011.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Merlini L, Mattioli E, Sabatelli P, Squarzoni S, Manzoli FA. Nuclear envelope proteins and chromatin arrangement: a pathogenic mechanism for laminopathies. Eur J Histochem. 2006;50:1–8. [PubMed] [Google Scholar]
- 91.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Mattioli E, Sabatelli P, Squarzoni S, Manzoli FA. Laminopathies: a chromatin affair. Adv Enzyme Regul. 2006;46:33–49. doi: 10.1016/j.advenzreg.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 92.Sabatelli P, Lattanzi G, Ognibene A, Columbaro M, Capanni C, Merlini L, Maraldi NM, Squarzoni S. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2001;24:826–9. doi: 10.1002/mus.1076. [DOI] [PubMed] [Google Scholar]
- 93.Capanni C, Cenni V, Mattioli E, Sabatelli P, Ognibene A, Columbaro M, Parnaik VK, Wehnert M, Maraldi NM, Squarzoni S, et al. Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: altered intermolecular interaction with emerin and implications for gene transcription. Exp Cell Res. 2003;291:122–34. doi: 10.1016/S0014-4827(03)00395-1. [DOI] [PubMed] [Google Scholar]
- 94.Ognibene A, Sabatelli P, Petrini S, Squarzoni S, Riccio M, Santi S, Villanova M, Palmeri S, Merlini L, Maraldi NM. Nuclear changes in a case of X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve. 1999;22:864–9. doi: 10.1002/(SICI)1097-4598(199907)22:7<864::AID-MUS8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 95.Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22:3409–21. doi: 10.1101/gad.1735208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dechat T, Gajewski A, Korbei B, Gerlich D, Daigle N, Haraguchi T, Furukawa K, Ellenberg J, Foisner R. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J Cell Sci. 2004;117:6117–28. doi: 10.1242/jcs.01529. [DOI] [PubMed] [Google Scholar]
- 97.Kumaran RI, Thakar R, Spector DL. Chromatin dynamics and gene positioning. Cell. 2008;132:929–34. doi: 10.1016/j.cell.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Motsch I, Kaluarachchi M, Emerson LJ, Brown CA, Brown SC, Dabauvalle MC, Ellis JA. Lamins A and C are differentially dysfunctional in autosomal dominant Emery-Dreifuss muscular dystrophy. Eur J Cell Biol. 2005;84:765–81. doi: 10.1016/j.ejcb.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 99.Maraldi NM, Lattanzi G, Sabatelli P, Ognibene A, Squarzoni S. Functional domains of the nucleus: implications for Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2002;12:815–23. doi: 10.1016/S0960-8966(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 100.Araújo-Vilar D, Lattanzi G, González-Méndez B, Costa-Freitas AT, Prieto D, Columbaro M, Mattioli E, Victoria B, Martínez-Sánchez N, Ramazanova A, et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J Med Genet. 2009;46:40–8. doi: 10.1136/jmg.2008.059485. [DOI] [PubMed] [Google Scholar]
- 101.Meaburn KJ, Cabuy E, Bonne G, Levy N, Morris GE, Novelli G, Kill IR, Bridger JM. Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell. 2007;6:139–53. doi: 10.1111/j.1474-9726.2007.00270.x. [DOI] [PubMed] [Google Scholar]
- 102.Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res. 2003;291:352–62. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 103.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, et al. Familial Cardiomyopathy Registry Research Group Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–74. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 104.Gueneau L, Bertrand AT, Jais JP, Salih MA, Stojkovic T, Wehnert M, Hoeltzenbein M, Spuler S, Saitoh S, Verschueren A, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–53. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gnocchi VF, Scharner J, Huang Z, Brady K, Lee JS, White RB, Morgan JE, Sun YB, Ellis JA, Zammit PS. Uncoordinated transcription and compromised muscle function in the lmna-null mouse model of Emery- Emery-Dreyfuss muscular dystrophy. PLoS One. 2011;6:e16651. doi: 10.1371/journal.pone.0016651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. European journal of human genetics. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kandert S, Wehnert M, Müller CR, Buendia B, Dabauvalle MC. Impaired nuclear functions lead to increased senescence and inefficient differentiation in human myoblasts with a dominant p.R545C mutation in the LMNA gene. Eur J Cell Biol. 2009;88:593–608. doi: 10.1016/j.ejcb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 108.Méjat A, Decostre V, Li J, Renou L, Kesari A, Hantaï D, Stewart CL, Xiao X, Hoffman E, Bonne G, et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J Cell Biol. 2009;184:31–44. doi: 10.1083/jcb.200811035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.González-Aguilera C, Ikegami K, Ayuso C, de Luis A, Iñiguez M, Cabello J, Lieb JD, Askjaer P. Genome-wide analysis links emerin to neuromuscular junction activity in Caenorhabditis elegans. Genome Biol. 2014;15:R21. doi: 10.1186/gb-2014-15-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Håkelien AM, Delbarre E, Gaustad KG, Buendia B, Collas P. Expression of the myodystrophic R453W mutation of lamin A in C2C12 myoblasts causes promoter-specific and global epigenetic defects. Exp Cell Res. 2008;314:1869–80. doi: 10.1016/j.yexcr.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 111.Maraldi NM, Mattioli E, Lattanzi G, Columbaro M, Capanni C, Camozzi D, Squarzoni S, Manzoli FA. Prelamin A processing and heterochromatin dynamics in laminopathies. Adv Enzyme Regul. 2007;47:154–67. doi: 10.1016/j.advenzreg.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 112.Bastard JP, Caron M, Vidal H, Jan V, Auclair M, Vigouroux C, Luboinski J, Laville M, Maachi M, Girard PM, et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet. 2002;359:1026–31. doi: 10.1016/S0140-6736(02)08094-7. [DOI] [PubMed] [Google Scholar]
- 113.Ruiz de Eguino G, Infante A, Schlangen K, Aransay AM, Fullaondo A, Soriano M, García-Verdugo JM, Martín AG, Rodríguez CI. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl Med. 2012;1:309–21. doi: 10.5966/sctm.2011-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Armani A, Cinti F, Marzolla V, Morgan J, Cranston GA, Antelmi A, Carpinelli G, Canese R, Pagotto U, Quarta C, et al. Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J. 2014;28:3745–57. doi: 10.1096/fj.13-245415. [DOI] [PubMed] [Google Scholar]
- 115.Oldenburg AR, Delbarre E, Thiede B, Vigouroux C, Collas P. Deregulation of Fragile X-related protein 1 by the lipodystrophic lamin A p.R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum Mol Genet. 2014;23:1151–62. doi: 10.1093/hmg/ddt509. [DOI] [PubMed] [Google Scholar]
- 116.Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband-Goulet I, Eskeland R, Buendia B, Collas P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013;23:1580–9. doi: 10.1101/gr.159400.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–9. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bruston F, Delbarre E, Ostlund C, Worman HJ, Buendia B, Duband-Goulet I. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 2010;584:2999–3004. doi: 10.1016/j.febslet.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16:738–50. doi: 10.1016/j.cmet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 120.Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L, Reinberg D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440–4. doi: 10.1038/nature06268. [DOI] [PubMed] [Google Scholar]
- 121.Liu B, Wang Z, Zhang L, Ghosh S, Zheng H, Zhou Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun. 2013;4:1868. doi: 10.1038/ncomms2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Das A, Grotsky DA, Neumann MA, Kreienkamp R, Gonzalez-Suarez I, Redwood AB, Kennedy BK, Stewart CL, Gonzalo S. Lamin A Δexon9 mutation leads to telomere and chromatin defects but not genomic instability. Nucleus. 2013;4:410–9. doi: 10.4161/nucl.26873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet. 2011;20:3997–4004. doi: 10.1093/hmg/ddr327. [DOI] [PubMed] [Google Scholar]
- 124.de la Rosa J, Freije JM, Cabanillas R, Osorio FG, Fraga MF, Fernández-García MS, Rad R, Fanjul V, Ugalde AP, Liang Q, et al. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat Commun. 2013;4:2268. doi: 10.1038/ncomms3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Avnet S, Pallotta R, Perut F, Baldini N, Pittis MG, Saponari A, Lucarelli E, Dozza B, Greggi T, Maraldi NM, et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim Biophys Acta. 2011;1812:711–8. doi: 10.1016/j.bbadis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 126.Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadiñanos J, Fraile JM, Ordóñez GR, Puente DA, Gutiérrez-Fernández A, Fanjul-Fernández M, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–6. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tang H, Hilton B, Musich PR, Fang DZ, Zou Y. Replication factor C1, the large subunit of replication factor C, is proteolytically truncated in Hutchinson-Gilford progeria syndrome. Aging Cell. 2012;11:363–5. doi: 10.1111/j.1474-9726.2011.00779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Taimen P, Pfleghaar K, Shimi T, Möller D, Ben-Harush K, Erdos MR, Adam SA, Herrmann H, Medalia O, Collins FS, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci U S A. 2009;106:20788–93. doi: 10.1073/pnas.0911895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dreesen O, Ong PF, Chojnowski A, Colman A. The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus. 2013;4:283–90. doi: 10.4161/nucl.25808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hutchison CJ. B-type lamins and their elusive roles in metazoan cell proliferation and senescence. EMBO J. 2012;31:1058–9. doi: 10.1038/emboj.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptáček LJ, et al. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013;123:2719–29. doi: 10.1172/JCI66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clayton P, Fischer B, Mann A, Mansour S, Rossier E, Veen M, Lang C, Baasanjav S, Kieslich M, Brossuleit K, et al. Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus. 2010;1:354–66. doi: 10.4161/nucl.1.4.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bertrand AT, Renou L, Papadopoulos A, Beuvin M, Lacène E, Massart C, Ottolenghi C, Decostre V, Maron S, Schlossarek S, et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum Mol Genet. 2012;21:1037–48. doi: 10.1093/hmg/ddr534. [DOI] [PubMed] [Google Scholar]
- 134.Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, López-Otín C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–24. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]