

Abstract
Gene expression is governed by chromatin mainly through posttranslational modifications at the N-terminal tails of nucleosomal histone proteins. According to the histone code theory, peculiar sets of such modifications (marks) give rise to reproducible final effects on transcription and, very recently, a further level of complexity has been highlighted in binary switches between specific marks at adjacent residues. In particular, disappearance of dimethyl-lysine 9 in histone H3 is faced by phosphorylation of the following serine during activation of gene expression. Demethylation of lysine 9 by the lysine-specific demethylase 1 (LSD1) is a pre-requisite for addition of the phosphoryl mark to serine 10 and an essential step in the transcriptional control by estrogens. It generates a local burst of oxygen reactive species (ROS) that induce oxidation of nearby nucleotides and recruitment of repair enzymes with a consequent formation of single or double stranded nicks on DNA that modify chromatin flexibility in order to allow correct assembly of the transcriptional machinery.
We describe here the molecular mechanism by which members of the family of nuclear receptors prevent the potential damage to DNA during transcription of target genes elicited by the use of ROS to shape chromatin. The mechanism is based on the presence of phosphorylated serine 10 in histone H3 to prevent unbalanced DNA oxidation waves. We also discuss the opportunities raised by the use of voluntary derangement of this servo system to induce selective death in hormone-responsive transformed cells.
Keywords: Akt, DNA oxidation, DNA repair, IKKα, apoptosis, epigenetic marks, estrogen, nuclear receptors, retinoic acid, transcription
Introduction
Eukaryotic chromatin, made up by DNA tightly wrapped around octameric nucleosomes, is a dynamic structure that controls assembly of multiple factors involved in transcription, replication, and repair. This task is mainly realized through modifications at the N-terminal tails of core histones, today referred to as epigenetic.1 Such modifications (i.e., lysine acetylation, lysine and arginine methylation, and serine and threonine phosphorylation) are mostly reversible and conform to a precise code, with many choices open at any time to govern transcriptional patterns.2 Interestingly, in agreement with the “binary switch” concept, several clusters of adjacent modifiable residues are used indeed in a combinatorial fashion;3 in fact, activation of gene expression is marked, among others, by phosphorylation of serine 10 in histone H3 (H3S10),4 concurrent with loss of di- and trimethylated lysine 9 in the same histone (H3K9me2 and -me3).5,6
Estrogen, 17β-estradiol (E2), directs a variety of biological processes among which regulation of developmental growth in the reproductive tract is particularly relevant, and alterations in response to the hormone associate to estrogen-dependent diseases such as breast and endometrial cancer.7 The primary effects of estrogen are mediated by hormone control on transcription of target genes following its binding to cognate receptors (ERα and ERβ). ERs belong to the nuclear receptor superfamily and, in combination with specific coactivators, join responsive sequences on DNA (EREs) present at strategic sites of hormone-dependent genes,8,9 including those that stimulate cell proliferation and survival.10,11 In addition, estrogen triggers rapid actions through ERs located at the plasma membrane that induce signal-transduction pathways involving MAP kinases and the PI-3K/protein kinase B (Akt) cascade.12,13 Accordingly, the regulatory subunit of class IA PI-3Ks, p85α, has been shown to interact with several members of the nuclear receptor superfamily, such as the estrogen receptor, the thyroid hormone receptor and the retinoic acid receptor.14,15 However, the mechanism of rapid nuclear receptor signaling is still not well established.
We have previously shown that E2-induced transcription is governed by ERα-triggered demethylation of H3K9me2 catalyzed by LSD1, with consequent production of reactive oxygen species (ROS) and oxidation of guanines (8-oxo-Gs) in flanking DNA, followed by formation of breaks produced by base-excision repair (BER) enzymes that allow bridging between distant sites.16,17 Hormone treatment of estrogen-responsive breast cancer cells also enhances phosphorylation of ERα (serine 118) and of serine 10 in histone H3 by the NFkB-inducer IkB kinase α (IKKα) in order to stimulate transcription of estrogen-sensitive genes, labeling the receptor as the crossroads between hormone and inflammatory signaling pathways.18
In contrast to E2, retinoic acid (RA) exerts a growth suppressor function that is dependent on its transcriptional control of genes that inhibit proliferation and induce apoptosis of target cells.19 RA effect is mediated by its binding to members of the same nuclear receptor superfamily (RARs and RXRs) assembled to retinoic acid responsive elements (RAREs) on DNA where they repress or induce transcription according, respectively, to the absence or presence of RA.20-22 Interestingly, RAREs exhibit, on a genomic scale, a large co-localization with EREs in the proximity of genes antagonistically regulated by both nuclear receptors.23 On the other hand, RARα cooperates with the estrogen receptor on EREs in the transcriptional control of E2-responsive genes,24 from where it disappears after addition of RA.25 Till now, rapid activation of the PI-3K/Akt pathway initiated by interaction between p85α and RARs has been evidenced in the transcriptional control of target genes by RA in human breast cancer cells.26
Despite of their very different phenotypical effects, we highlight here the similarities between E2- and RA-dependent pathways in the generation of chromatin and DNA modifications that drive the expression of responsive genes. We also report that the extra-genomic effects elicited by both ligands, leading to recruitment of active Akt to chromatin, are essential for phosphorylation of serine 10 in histone H3 by IKKα. Finally, we elucidate the molecular consequences of interference with presence of H3S10ph on balanced production of ROS throughout the transcriptional process triggered by E2 and RA, with involvement of the DNA damage response (DDR) pathway that, if overthrown, induces programmed cell death (PCD).
Results and Discussion
Inhibition of IKKα activity during estrogen-induced transcription enlarges the wave of oxidized guanines in surrounding DNA and engages DNA damage-related kinases
A major mechanism by which estrogen exposure contributes to the initiation and/or progression of breast cancer is the induction of DNA damage due to double stranded DNA breaks as also revealed in rat models.27,28 DNA damage is recognized and processed by several DDR pathways that check for its severity and drive cell cycle arrest, DNA repair, or, in extreme cases, apoptosis.29 Small lesions of damaged or modified nucleotides are eliminated by BER, nucleotide excision repair, or mismatch repair enzymes. These lesions are extremely common and the protective pathways are activated by constant genome surveillance also coupled to DNA replication and transcription. The DDR prevents irreversible damage through engagement of several effector kinases among which the Ataxia telangiectasia mutated (ATM) and the ATM Rad3-related (ATR) play a pivotal role, the former recognizing double stranded breaks and the latter responding to single stranded overhangs. However, DNA damage may also accumulate as a consequence of insufficient timely repair mechanisms activated by the cell that possibly result into double stranded breaks.30
Estrogen control on transcription in hormone-responsive cells needs addition of the phosphoryl mark by the NFkB-inducer IKKα to serine 10 in histone H3 near promoters of target genes, in order to prevent an excess of ROS normally produced throughout the transcriptional output after demethylation of H3K9me2 catalyzed by LSD1: in fact, upon interfering with this servo system, we induced apoptosis in hormone-stimulated cells.31 However, the mechanism raised by derangement of the control on ROS generation still awaits to be elucidated at the molecular level. Therefore, to explore the effects of ROS overproduction, we first assessed by ChIP the involvement of the DNA repair-related kinases (ATM and ATR) at the promoter of the paradigmatic estrogen-responsive pS2 gene in MCF-7 cells where transcription of target genes can be selectively enhanced (high H3S10ph, and low H3K9me2 and -me3) or silenced (vice versa) by presence or absence of E2 in the culture medium (Fig. 1A).32 The analyzed recruitment was selective, as an intronic pS2 fragment from the same ChIP samples shown in Figure 1A failed to be amplified (data not shown). Of note, even though both kinases joined chromatin after hormone addition, inhibition of IKKα activity obtained by treating cells with the specific inhibitor BAY11-7082 (BAY) further increased their presence on the promoter, with an effect especially evident for ATR, testifying for creation of nearby single stranded nicks that gave rise to DNA sticky ends. Therefore, we analyzed the boundaries of the oxidation wave in hormone challenged cells under normal or inhibited IKKα activity. Amplification of neighboring fragments along with the estrogen-responsive bcl-2 coding region starting from the EREs,11 and captured in ChIP experiments by anti-8-oxo-G antibodies revealed that DNA oxidation was undetectable in wild-type cells at a distance of roughly 1 kilobase downstream from the enhancer locus, where it was still clearly visible in cells with silent IKKα (Fig. 1B). This appears an interesting observation on the basis of our previous data showing that at the distance explored in Figure 1B no epigenetic changes involving H3 lysine 9 had been evidenced, definitely labeling the enhancers as the sites where ROS were generated.16

Figure 1. Excess of nicks on DNA finally results into apoptosis. (A) ChIP analysis showing the effect of inhibition of IKKα activity with BAY11-7082 (BAY, 20 μM) on specific recruitment of ATM and ATR kinases to pS2 promoter after E2 treatment for 45 min. The DNA region (reported on top of each gel) has been amplified by fixed PCR reactions realized within the range of cycles with exponential duplication of template sequences from sonicated chromatin before immunoprecipitation (Input, as graphically plotted on the left). The antibodies used are indicated on the left of an illustrative gel that, as in the following panels, represents one of at least three different experiments. All shown bands have been quantified and plotted in graphs whose values revealed a statistical significance where 0.05 ≥ P ≥ 0.01. Error bars represent SEM. Even though illustratively reported only in panel D of Figure 2, antibodies to α-Tubulin have been used in all ChIP experiments as negative control; in addition, an intronic fragment of the same gene has been used as negative control to assess site-specificity of protein recruitment (not shown). (B) Evaluation upon the same tool of boundaries of the DNA oxidation waves along with bcl-2 coding region that harbors the EREs, in wild type and IKKα-inhibited cells.11 Location of primer pairs used in PCRs is reported on the left. (C) Estimate of the percentage of apoptotic MCF-7 cells challenged with E2 (10 nM) for 24 h, in the absence or presence of BAY11-7082 (20 μM) added for the first 6 h. Addition of 25 mM NAC to IKKα-inhibited/E2-challenged cells (6 h as for BAY) highlighted the role of ROS in hormone effect. Evocative fields of cells stained with May Grünwald-Giemsa and photographed by light microscopy with apoptotic nuclei evidenced by black arrowheads have been reported. For each treatment, 500 cells have been counted and the apoptotic fraction (representing the mean of three different experiments) is indicated on top. (D) ChIP assays revealing the effect of OGG1 silencing on presence of oxidized Gs near pS2 promoter. Assessment of the specific knock down has been evaluated by presence of OGG1 on the same promoter. (E) The same cells have been analyzed by FACS in order to evaluate the apoptotic fraction after addition of E2 or E2/BAY. Wild-type and LSD1-knocked down cells have been used as basis for comparison. Levels of silenced LSD1 have been determined as reported in panel A of Figure 2. (F) Estimate by dedicated PCR reactions of the DNA damage induced by E2, BAY or E2/BAY added for 6 h to MCF-7 cells. DNA sonicated in order to obtain fragments of 800–1000 base pairs was quantified and equal amounts (20 ng) were amplified in PCRs with primer pairs spanning a region from bcl-2 EREs to approximately 500 bp downstream, as detailed on top of the reported gel. Graphic representation of the effect on DNA of IKKα inhibition in E2-challenged cells is reported on the left. Amplification of the coding region of the RA-responsive caspase 9 gene has been used as control.
Since the proapoptotic effect of E2 in IKKα-inhibited cells was prevented by addition of the ROS scavenger N-acetyl cysteine (NAC) (Fig. 1C),31 in order to determine whether presence of ROS per se induced apoptosis, we silenced the pivotal nick-producing enzyme 8-oxo-guanine-DNA glycosylase 1 (OGG1) in cells treated or not with E2.33 Of note, knock down of OGG1 resulted, as expected, into an increased presence of 8-oxo-Gs around pS2 promoter (Fig. 1D); however, concomitant addition of E2 and BAY to these cells revealed an apoptotic population that was half as that displayed by their wild-type complement (Fig. 1E). Therefore, presence of ROS, although necessary as confirmed by the results obtained with LSD1-silenced cells (where they were not produced in the absence of lysine 9 demethylation), was not sufficient to induce PCD (Fig. 1E).
Oxidized bases (as consequence of ROS) are mutagenic for cell progeny and must be eliminated, a task performed by BER enzymes. Therefore, we investigated the “state of health” of DNA after addition of E2 in wild-type cells and compared with that observed in their IKKα-inhibited counterpart. To this aim, we extracted the DNA from cells challenged with E2 for 6 h in the absence or presence of BAY and, after mild sonication, amplified in PCR reactions equal amounts of DNA. By use of primers spanning the bcl-2 region analyzed in Figure 1B showing an increase of oxidized Gs after E2 addition to cells with knocked down IKKα, we observed a reduced ability of that region to function as template in the same cells, evidently as a consequence of specific local DNA fragmentation: this result indicates that nicks actually represent the background for triggering PCD (Fig. 1F).
We believe that our data may also help to explain the phenotype described in IKKα knockout mice showing mammary defects previously related to impaired epithelial proliferation due to reduced cyclin D1 expression upon ERα- or NFkB-mediated activation.18,34 We may add a new tile in this puzzle: estrogen stimulation of breast tissue in the absence of active IKKα is able to induce apoptosis in a large fraction of hormone-responsive cells with consequent regression of that tissue. In fact, several studies have already evidenced paradoxical effects of estrogen in MCF-7 cells deprived of hormone for long times, where it inhibited growth and induced apoptosis.35 However, the use of high doses of estrogen to inhibit growth of hormone-responsive breast tumors has not been further developed for the serious secondary effects evidenced in women experiencing such therapy.36
Cooperation between H3K9 demethylases allows phosphorylation of H3S10 by IKKα, activated by engagement of Akt onto chromatin
We have already shown that estrogen-induced demethylation of H3K9me2 by LSD1 is a pre-requisite for phosphorylation of the following serine.31 However, since LSD1 is able to subtract the methyl group exclusively from mono- and dimethylated lysine 9 in histone H3, and activation of estrogen-target genes needs removal also of the repressive trimethyl mark from the same residue,37 we have measured recruitment to pS2 promoter of JMJC domain-containing hydroxylases that, by performing this task,38 have already been shown to cooperate with LSD1 in androgen-dependent activation of gene expression.39 After preliminary analysis, the H3K9-specific JMJD2A was selected and, to compare its role on observed H3K9me2 levels with that of LSD1, we specifically knocked down any or both enzymes. As shown in Figure 2A, silencing of each demethylase prevented reduction of H3K9me2 levels, suggesting that both are necessary to carry out this job. Interestingly, upon the same approach we found that knock down of JMJD2A was able to inhibit also the increase of H3S10ph, as already observed for its sister demethylase LSD1 confirming, in agreement with the binary switch hypothesis, that loss of methyl groups from methylated-lysine 9 (H3K9me2 and -me3) precedes addition of the phosphoryl mark to the adjacent serine by IKKα (Fig. 2B).

Figure 2. Cooperation between H3K9 demethylases controls phosphorylation of H3S10 by IKKα as triggered by promoter-recruited Akt. (A) ChIP analysis to assess how selective or concomitant silencing of LSD1 and JMJD2A demethylases influenced levels of H3K9me2 at pS2 promoter after incubation of MCF-7 cells with E2 (10 nM) for 45 min. The result of specific knock down of silenced proteins is shown on the left. (B) Assessment of the consequences of JMJD2A knock down on H3K9me3 and H3S10ph levels. The effect of demethylase silencing has been unraveled by the decrease of its assembly to chromatin, as compared with wild-type cells. (C) Western blot performed to assess the subcellular fractionation of total and active (phopshorylated at serine 473) Akt (pAkt). The PI-3K inhibitory drug LY-294002 (LY, 50 μM) has been added as negative control. α-Tubulin (Tub) and H3 histone have been evidenced as markers of the cytosolic and nuclear fractions, respectively. (D) ChIP analysis to determine recruitment to pS2 promoter of total and phosphorylated fractions of Akt and IKKα in the presence or absence of LY-294002. Antibody to α-Tubulin (as in all ChIP experiments, even if not reported) has been used as negative control. (E) Evaluation of the effect of Akt inhibition on H3K9me2 demethylation, phosphorylation of the following serine and recruitment of OGG1 to pS2 promoter. (F) Hybridization of chromatin (DPC) from quiescent or E2-challenged (10 nM for 30 min) MCF-7 cells with a probe from bcl-2 enhancer site (bcl-2 ERE, evidenced on the left) to unravel the presence of specific gene regions in retrieved DNA. Input indicates the amplifiable DNA before hybridization. Locations of primer pairs used in PCR reactions are drawn on top of each gel. On the right is graphically represented the 5′ region of bcl-2 gene where the exons have been evidenced with colored boxes. The two EREs are located in exon 2. E2-triggered changes of the spatial interactions between the promoter and enhancer sites in the presence or absence of active Akt have also been drawn. (G) RT-PCR to assess the effect of inhibition of Akt on hormone-dependent stimulation of pS2 gene expression in MCF-7 cells.
A growing body of evidence has recently assigned IKKα a novel role in activating gene expression independently from its effects on NFkB-regulated genes.40,41 On the other hand, IKKα is phosphorylated by Akt,42 that is, in turn, powered by E2 during hormone-induced entry of MCF-7 cells into the S-phase of cell cycle.43,44 Therefore, to analyze in deeper detail this pathway, we have explored whether Akt were directly involved in hormone-dependent increase of H3S10ph by IKKα at pS2 promoter. We first assessed the intracellular compartmentalization of active Akt and found that it appeared in the cytosol within 30 min of E2 addition and immediately afterwards in the nucleus, perfectly fitting with the time of H3S10ph rise at pS2 promoter (Fig. 2C).18 Therefore, we searched for direct recruitment of Akt to pS2 ERE and found that it joined, in fact, that site (Fig. 2D). Moreover, treatment of hormone-challenged cells with the PI-3K-inhibitor LY-294002 that prevents, as a consequence, activation of Akt, precluded assembly of the phosphorylated form of IKKα to the same promoter and confirmed that the Akt fraction joining chromatin was phosphorylated at its activating site (serine 473) (Fig. 2D).
Notably, inhibition of Akt activity prevented either demethylation of H3K9me2 as well as phosphorylation of H3S10 together with recruitment of OGG1, indicating that assembly of the active kinase on chromatin represents a hierarchically upstream step in the transcriptional control played by estrogens (Fig. 2E). Absence of H3K9me2 demethylation means absence of nicks (due to failure of OGG1 assembly) that allow establishment of dynamic contacts between distant hormone-dependent regulatory sites. To determine whether this was the case, we used the DNA-picked chromatin (DPC) approach that unravels topographic links in the nuclear compartment between different loci that, if bridged by protein factors involved in the transcriptional output, may be co-captured by a specific DNA probe that hybridizes with one of them.17 Upon this strategy, we found that inhibition of Akt prevented hormone-induced association between the EREs of bcl-2 gene and the promoter site located more than 1.5 kb upstream on linear DNA (Fig. 2F).11,17 This condition produced negative consequences also on pS2 mRNA synthesis, as evidenced by RT-PCRs measuring running transcription (Fig. 2G).
The findings reported in Figure 2 highlight a new role of Akt in particular and, potentially, hormone-induced signal transduction pathways in general, that, besides the peculiar effects on their cytoplasmic targets, appear to be necessary to elicit ERα transcriptional activity as such.
RA-mediated signaling mirrors the route walked by E2 to control transcription-coupled production of ROS
To determine whether the role of H3S10ph in the control of balanced production of ROS and the consequences following its derangement are peculiar of ERα or can be applied to different nuclear receptors, we have analyzed the histone modifications induced by treating the RAR+ MCF-7 cells with retinoic acid, whose cognate receptors exhibit a deep crosstalk with ERα and, in contrast to E2, exerts antiproliferative and proapoptotic effects.23,24
RA challenge induced IKKα and Akt targeting to the promoter of the paradigmatic RA-responsive gene cyp26A, with appearance of the canonical modifications at lysines 4 and 9 as well as at serine 10 in histone H3, also due to recruitment of the same demethylases (LSD1 and JMJD2A) elicited by E2 (Fig. 3A and B, upper panel). Induction of cyp26A expression by RA was confirmed by the increased promoter recruitment of the actively transcribing RNA polymerase II phosphorylated at serine 5 (Fig. 3A). Therefore, IKKα should be able to form complexes also with RARs. Moreover, knock down of the same kinase produced a quicker rise of 8-oxo-Gs near cyp26A promoter (Fig. 3B). Based on this finding and similarly to what observed with E2, we then challenged cells with RA for 24 h, in the absence or presence of BAY added for the first 6 h. Measurement of the apoptotic fraction by PARP cleavage, direct cell counting, and FACS analysis all in accord revealed that addition of RA and BAY mimicked the pattern induced by estrogen, clearly indicating that both nuclear receptors elicit a very similar mechanism to control gene expression (Fig. 3C).

Figure 3. The chromatin picture induced by treatment of MCF-7 cells with RA essentially mirrors that observed with E2. (A) ChIP assays to assess the key nucleosomal modifications and transcription factors assembly to the paradigmatic RA-responsive cyp26A promoter. (B) Evaluation by ChIP of the effect of IKKα knock down on presence of H3S10 ph and 8-oxo-Gs near the same promoter. (C) PARP fragmentation (left), cell count (middle), and FACS analysis (right) to assess the role of the control on ROS production played by IKKα, whose derangement induced apoptosis in RA-treated cells. The reported fractions of apoptotic MCF-7 cells represent the mean from three different experiments.
Inhibition of IKKα influences the ability of MCF-7 cells to form colonies after addition of E2 or RA
We have finally assessed that timely restricted production of ROS is also important for establishment of the proliferative effect of estrogen. In fact, evaluation of the ability of MCF-7 cells to form colonies after hormone challenge in the absence or presence of BAY for 6 h revealed that, even though cells added with BAY formed smaller colonies, concomitant treatment with E2 further halved the number of such colonies, as assessed after two additional weeks of incubation (Fig. 4). Moreover and even more interestingly, these data were doubled by RA treatment where we observed a more than additive effect of concomitant addition with BAY, notwithstanding the fact that it controls a completely different array of genes (Fig. 4). Therefore, we can conclude that, independently from the metabolite used as trigger, the molecular mechanism we highlight in our study is directly connected with the pathway that nuclear receptors follow to induce expression of target genes.

Figure 4. Ability of MCF-7 cells to form colonies after stimulation with E2 or RA under normal or inhibited IKKα activity. (A) Cells were treated as detailed in the Material and Methods section prior to be analyzed for ability to form colonies. Colonies of more than 40 cells were visible. To count also smaller colonies, a replica was obtained on transparent plastic with a black marker (as shown on the right). (B) Specific fields (two for each treatment) as examined by light microscopy (10×) revealing that addition of any or the other ligand to IKKα-inhibited cells further diminished the number of the smaller colonies induced by addition of BAY11-7082. The reported experiment has been confirmed by two different assays that originated very similar results.
In conclusion, based on our experimental results (graphically summarized in Fig. 5), we believe that physiological doses of E2, in combination with the inhibition of IKKα activity, could be used as a Trojan horse in novel therapeutic protocols developed for treatment of human hormone-responsive breast cancers.
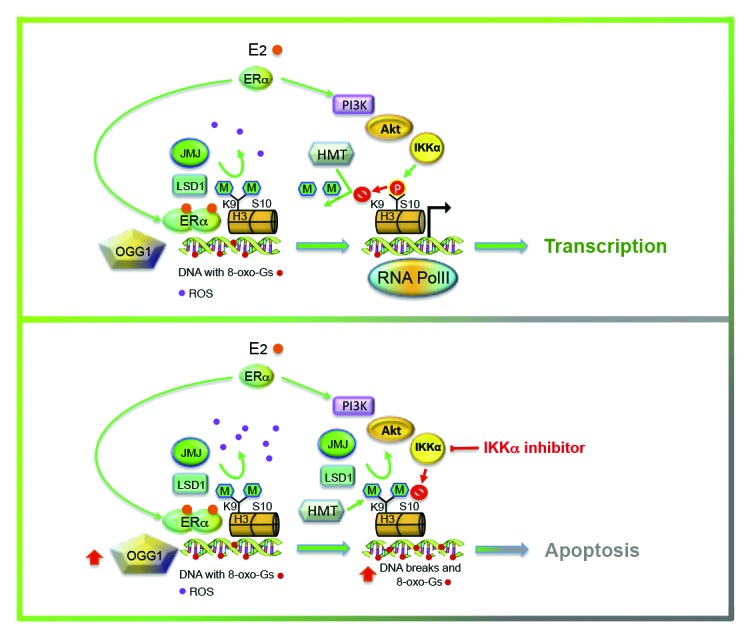
Figure 5. Graphic representation of the effect of absence of IKKα activity on productive transcription induced by E2. After hormone challenge, the transcription complex with ERα assembles on chromatin where the receptor drives demethylation of lysine 9 in histone H3 with consequent production of ROS (in violet). Following phosphorylation of serine 10, addition of methyl-marks to lysine is delayed, giving the DDR mechanisms sufficient time to eliminate the oxidized Gs (in red) in order for transcription to progress safely. Inhibition of IKKα activity increases production of ROS with disproportionate formation of nicks, DNA fragmentation and apoptosis.
Materials and Methods
Cells
Human breast cancer MCF-7 cells have been routinely grown as already described.16 To evaluate the effect of E2 or RA challenge, cells have been first incubated with phenol red-free DMEM with 0.5% dextran-charcoal-stripped FCS for 6–8 h, and then with the same medium containing 5% dextran-charcoal stripped FCS for further 3 d, prior to be challenged with 10 nM E2 or 100 nM RA according to the experimental needs. To obtain LSD1, JMJD2A, IKKα, or OGG1 knock down with siRNAs, cells have been transfected with the respective siRNAs according to manufacturers’ instructions (flexitubes, Quiagen), and incubation has been continued for 48–60 h. To inhibit IKKα activity, cells have been incubated with BAY11-7082 (20 μM). Inhibition of Akt has been obtained by LY-294002 (50 μM). To drain away reactive oxygen species, cells have been treated with NAC (25 mM).
Chromatin immunoprecipitation (ChIP)
ChIP assays have been performed as previously reported.16 Chromatin has been sonicated to obtain fragments of 300–500 base pairs. For each assay, chromatin from a total of 106 cells has been used. All bands from ethidium bromide-stained gels have been analyzed by densitometry and quantified with Total Lab ID software. Antibodies used, as well as sequences of primers and PCR conditions are available upon request.
Assessment of the apoptotic fraction by flow cytometry (FACS) or light microscopy
For FACS analysis, cells under different treatments have been incubated for 45 min in the dark with propidium iodide (50 μg/ml), RNase (1mg/ml), 0.1% NP-40, 0,1% Na Citrate, 0.1 mM EDTA in PBS. For each sample, at least 104 events have been acquired using Fluorescence Activated Cell Sorter (FACS) Calibur Flow Cytometry System (BD Biosciences), and analyzed using the ModFit LT 3.0 software (Verity Software House).
For analysis by light microscopy, MCF-7 cells have been harvested in ice-cold PBS prior to be fixed and pre-stained with ethanol solution of May Grünwald for 5 min. After washing, cells have been stained with water solution of Giemsa for 20 min and washed before being observed with light microscope (Zeiss). For each treatment, 500 cells have been counted.
Evaluation of protein levels by western blotting
Cells have been harvested and lysed in ice-cold lysis buffer (1mM EDTA, 0.2% Triton X-100, protease inhibitors mix). Twenty μg of proteins from cell lysates, or 40 μg when Poly(ADP-ribose) polymerase (PARP) was detected, have been subjected to SDS-8% PAGE. α-Tubulin, or GAPDH have been used as internal control. For evaluation of PARP fragmentation, cells have been challenged with RA for 24 h in the absence or presence of BAY11-7082 (20 μM) added for the first 6 h. Protein-antibody complexes have been revealed using western blotting detection reagents (ECL), following manufacturer’s instructions. Proteins have been assessed as indicated above.
Subcellular fractionation of MCF-7 cells
MCF-7 cells (75% confluence in 100 mm dishes) have been collected in 500 μl of ice-cold subcellular fractionation (SF) buffer (250 mM sucrose; 20 mM HEPES, pH 7.4; 10 mM KCl; 1.5 mM MgCl2; 1 mM EDTA; 1 mM EGTA; 1mM DTT; protease inhibitors) and incubated with mild shaking for 30 min at 4 °C. After centrifugation, supernatants have been transferred to new 1.5 ml tubes to be further centrifuged at 10 000 x g at 4 °C for 10 min prior to be employed as the cytosolic fraction. On the other hand, pellets have been washed with 500 μl of SF buffer and ri-centrifuged (10 min each at 4 °C) three times prior to be resuspended in 500 μl of ice-cold nuclear lysis buffer (50 mM TRIS-HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycolate; 0.1% SDS; 10% glycerol; protease inhibitors) and incubated with mild shaking at 4 °C for 15 min to be considered as the nuclear fraction. Equal amounts of proteins from cell lysates (30 μg) have been, then, subjected to SDS-12% PAGE and transferred to nitrocellulose membranes for detection. H3 histone and α-Tubulin have been utilized as markers of nuclear and cytosolic fractions, respectively. Proteins have been revealed as indicated above.
DNA-picked chromatin (DPC)
DPC has been achieved as already reported.17 MCF-7 cells have been cross-linked and collected into 100 mM TRIS-HCl (pH 9.4), 10 mM DTT [17]. Prior to sonication, cell pellets have been re-suspended into 0.5 volumes of 1x PBS-0.5% Triton X-100, with 2 μl of RNase A (20 mg/ml). After incubation at RT for 1 h and at 4 °C for 12–16 h, samples have been washed six times in 1x PBS and centrifuged. Pellet has been finally re-suspended in 300 μl of sonication buffer (50 mM TRIS-HCl, pH 8.0, 10 mM EDTA, pH 8.0, 1% SDS, protease inhibitors) and diluted in 0.4% SDS, 0.1% Sarkosyl, 100 mM NaCl, 2 mM EDTA, pH 8.0, 1 mM EGTA, pH 8.0. 100 μl aliquots (corresponding to 1.0–1.5 × 106 cells) have been added with 1 μM of biotinylated oligonucleotide probe and hybridized as follows: 25 °C for 3 min, 70 °C for 6 min, 38 °C for 60 min, 60 °C for 2 min, 38 °C for 60 min, 60 °C for 2 min, 38 °C for 120 min, and 25 °C final temperature. Eluates from samples incubated at RT for 1 h and at 65 °C for 10 min have been treated with proteinase K and DNA precipitated with ethanol prior to be amplified with PCRs. Sequences of primers and PCR conditions are available upon request.
The following 5′-biotinylated oligonucleotide has been used as probe: bcl-2 ERE: GCCAGGCCGG CGACGATTC TCC.
Reverse transcriptase (RT)-PCR
Cells have been harvested and pellets have been resuspended in 1 ml TRIZOL for 10 min at R.T. prior to proceed with RNA extraction according to manufacturer’s instructions. Two μl of RNA have been used in reactions with M-MLV reverse transcriptase (RT) according to established protocols. Primers from pS2 coding region have then been used in PCRs. Sequences of primers as well as RT and PCR conditions are available upon request.
Colony formation
Four × 103 cells (2 × 104 where BAY11-7082 was present) have been plated in 6-well plates and incubated for 24 h. Thereafter, they have been grown in phenol red-free DMEM containing 5% dextran-charcoal stripped FCS for further 3 d, prior to proceed with the specific treatments for 6 h, according to the experimental design. Cells have been then changed medium with normal DMEM (5% FCS) and incubated for further two weeks. Colonies have been finally fixed in methanol, stained with 0.5% crystal violet and washed with PBS. Colonies with less than 10 cells (not stained and observable with light microscopy) have been signed with a black marker pen on a translucent plastic replica.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
This work has been supported by the Associazione per la Ricerca sul Cancro (AIRC Grant IG 11520: PI Antimo Migliaccio).
References
- 1.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–71. doi: 10.1016/S0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 4.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–8. doi: 10.1016/S0959-437X(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 6.Ayyanathan K, Lechner MS, Bell P, Maul GGH, Schultz DC, Yamada Y, Tanaka K, Torigoe K, Rauscher FJ., 3rd Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17:1855–69. doi: 10.1101/gad.1102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–65. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 8.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–63. doi: 10.1016/S0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 10.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–74. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 11.Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20:2890–901. doi: 10.1128/MCB.20.8.2890-2901.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001;20:6050–9. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lösel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4:46–56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- 14.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 15.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–6. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 17.Abbondanza C, De Rosa C, Ombra MN, Aceto F, Medici N, Altucci L, Moncharmont B, Puca GA, Porcellini A, Avvedimento EV, et al. Highlighting chromosome loops in DNA-picked chromatin (DPC) Epigenetics. 2011;6:979–86. doi: 10.4161/epi.6.8.16060. [DOI] [PubMed] [Google Scholar]
- 18.Park K-J, Krishnan V, O’Malley BW, Yamamoto Y, Gaynor RB. Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–53. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 20.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–41. [PubMed] [Google Scholar]
- 21.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–54. [PubMed] [Google Scholar]
- 22.Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193–9. doi: 10.1158/0008-5472.CAN-05-1177. [DOI] [PubMed] [Google Scholar]
- 23.Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–71. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, Carroll JS. Cooperative interaction between retinoic acid receptor-α and estrogen receptor in breast cancer. Genes Dev. 2010;24:171–82. doi: 10.1101/gad.552910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ombra MN, Di Santi A, Abbondanza C, Migliaccio A, Avvedimento EV, Perillo B. Retinoic acid impairs estrogen signaling in breast cancer cells by interfering with activation of LSD1 via PKA. Biochim Biophys Acta. 2013;1829:480–6. doi: 10.1016/j.bbagrm.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi E, Kogai T, Kagechika H, Brent GA. Activation of the PI3 kinase pathway by retinoic acid mediates sodium/iodide symporter induction and iodide transport in MCF-7 breast cancer cells. Cancer Res. 2009;69:3443–50. doi: 10.1158/0008-5472.CAN-08-3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 28.Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A. 2004;101:18123–8. doi: 10.1073/pnas.0408273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pallis AG, Karamouzis MV. DNA repair pathways and their implication in cancer treatment. Cancer Metastasis Rev. 2010;29:677–85. doi: 10.1007/s10555-010-9258-8. [DOI] [PubMed] [Google Scholar]
- 30.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 31.Perillo B, Di Santi A, Cernera G, Ombra MN, Castoria G, Migliaccio A. Phosphorylation of H3 serine 10 by IKKα governs cyclical production of ROS in estrogen-induced transcription and ensures DNA wholeness. Cell Death Differ. 2014;21:1503. doi: 10.1038/cdd.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AM, Jeltsch JM, Roberts M, Chambon P. Activation of pS2 gene transcription is a primary response to estrogen in the human breast cancer cell line MCF-7. Proc Natl Acad Sci U S A. 1984;81:6344–8. doi: 10.1073/pnas.81.20.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roldán-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang RP, Augustus M, Lindahl T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94:8016–20. doi: 10.1073/pnas.94.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–75. doi: 10.1016/S0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 35.Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, Yue W, Wang J, Santen RJ. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93:1714–23. doi: 10.1093/jnci/93.22.1714. [DOI] [PubMed] [Google Scholar]
- 36.Lønning PE, Taylor PD, Anker G, Iddon J, Wie L, Jørgensen LM, Mella O, Howell A. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67:111–6. doi: 10.1023/A:1010619225209. [DOI] [PubMed] [Google Scholar]
- 37.Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–9. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 38.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–81. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 39.Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–53. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 40.Anest V, Cogswell PC, Baldwin AS., Jr. IkappaB kinase alpha and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IkappaBalpha degradation. J Biol Chem. 2004;279:31183–9. doi: 10.1074/jbc.M404380200. [DOI] [PubMed] [Google Scholar]
- 41.Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–49. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NFkappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 43.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 44.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]