

Abstract
Bacterial infections play an important role in the multifactorial etiology of rheumatoid arthritis. The arthropathic properties of Gram-positive bacteria have been associated with peptidoglycan–polysaccharide complexes (PG-PS), which are major structural components of bacterial cell walls. There is little agreement as to the identity of cellular receptors that mediate innate immune responses to PG-PS. A glycosylphosphatidylinositol-linked cell surface protein, CD14, the lipopolysaccharide receptor, has been proposed as a PG-PS receptor, but contradictory data have been reported. Here, we examined the inflammatory and pathogenic responses to PG-PS in CD14 knockout mice in order to examine the role for CD14 in PG-PS-induced signaling. We found that PG-PS-induced responses in vitro, including transient increase in intracellular calcium, activation of nuclear factor-κB, and secretion of the cytokines tumor necrosis factor-α and interleukin-6, were all strongly inhibited in CD14 knockout macrophages. In vivo, the incidence and severity of PG-PS induced acute polyarthritis were significantly reduced in CD14 knockout mice as compared with their wild-type counterparts. Consistent with these findings, CD14 knockout mice had significantly inhibited inflammatory cell infiltration and synovial hyperplasia, and reduced expression of inflammatory cytokines in PG-PS arthritic joints. These results support an essential role for CD14 in the innate immune responses to PG-PS and indicate an important role for CD14 in PG-PS induced arthropathy.
Keywords: bacterial, cell surface molecules, rheumatoid arthritis, transcription factors
Introduction
Rheumatoid arthritis (RA) is characterized by a chronic, erosive inflammation of peripheral joints. The etiology of RA is not known, but both genetic and environmental factors contribute to the disease. Gram-positive bacterial infection is one environmental factor that has been linked with RA pathology [1]. The link between systemic bacterial infection and RA is based on observations that bowel-related diseases such as Crohn's disease and ulcerative colitis are frequently complicated by arthritis, that RA patients have significantly elevated levels of antibodies to bacterial products (e.g. antipeptidoglycan antibodies), and that bacterial products can frequently be found in RA synovial fluids and tissues [2-5]. The arthropathogenic properties have been attributed to poorly degradable peptidoglycan–polysaccharide complexes (PG-PS) – covalently bound polymers that comprise cell walls of Gram-positive bacteria [2,6]. When injected in susceptible strains of animals, crude preparations of bacterial cell walls or purified PG-PS produce disease with clinical features resembling those of human RA [6].
Injected PG-PS accumulate in macrophages of the spleen, liver, and mesenteric lymph nodes [7], resulting in persistent cell activation and secretion of inflammatory cytokines. The activation of macrophages by PG-PS represents an example of innate immune response, a defensive reaction based on recognition of conserved pathogen-associated molecular patterns (PAMPs) shared by large groups of micro-organisms (for review [8]). PAMP recognition in mammals is mediated by the Toll-like receptor (TLR) family of pattern recognition receptors. Engagement of TLRs triggers signal transduction pathways that culminate in the activation of transcription of defensive genes. Individual TLRs produce overlapping but distinct patterns of gene expression, permitting tailoring of host responses to different pathogens. The precise molecular mechanisms whereby distinct PAMPs activate individual TLRs are not well defined. Recognition of lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria, is the best studied model of innate immunity [9]. In serum, LPS molecules are opsonized by LPS-binding proteins that recruit LPS to CD14 – a glycosylphosphatidylinositol-anchored cell surface protein that is expressed by phagocytic cells. The tertiary LPS–LPS binding protein–CD14 complex generates the transmembrane signal of cell activation through an association with the TLR-4 receptor. A principal target and mediator of the TLR-4-induced signaling is the transcription factor nuclear factor-κB (NF-κB), which controls the expression of a variety of proinflammatory cytokines, including IL-1, IL-6, and tumor necrosis factor (TNF)-α [8,10].
It is less clear which primary events and cellular receptors mediate the innate immune responses to and the arthritopathogenic properties of PG-PS. The intracellular signaling that occurs in response to stimulation with peptidoglycans of Gram-positive bacteria is triggered by TLR-2 [11]. A number of in vitro studies have suggested that, similar to the involvement of CD14 in LPS signaling, CD14 plays an essential role in the activation of PG-PS induced innate immune responses [12-16]. However, there are reports that peptidoglycans can directly bind TLR-2 [17] and activate cells independently of CD14 [18]. Studies conducted in vivo have also yielded contradictory results. Whereas CD14 knockout mice were resistant to Gram-negative bacteria-induced and LPS-induced shock [19], CD14-deficient mice, when challenged with live or killed Gram-positive bacteria, had survival rates similar to those in wild-type animals [20].
In order to elucidate the role played by CD14 in the innate immune response to PG-PS in vitro and in vivo, we assessed the inflammatory responses to PG-PS in CD14 knockout primary macrophages and examined the development of PG-PS induced arthritis in CD14 knockout mice. Our findings demonstrate that, in CD14 knockout macrophages, PG-PS failed to activate inflammatory responses in vitro, and that the development of PG-PS induced arthropathy was significantly inhibited, albeit not abolished, in CD14 knockout mice.
Materials and methods
Purified PG-PS (fraction 100P) from group A Streptococcus pyogenes was purchased from Lee Labs (Garrison, GA, USA). Endotoxin contamination was under 0.35 pg endotoxin/μg PG-PS, as determined using a Limulus amebocyte lysate assay system (Pyrogent®plus; Biowhittaker, Walkersville, MD, USA). Therefore, the concentration of endotoxin in in vitro experiments was under 35 pg/ml, even at the highest (100 μg/ml) concentrations of PG-PS. All reagents, unless otherwise noted, were from Sigma (St. Louis, MO, USA) and the culture medium was from GIBCO (Grand Island, NY, USA).
Animals and arthritis model
Female CD14 knockout mice [19] were backcrossed 10 times onto the BALB/c background, as described previously [20,21]. The expression of CD14 mRNA was evaluated by using RNase protection assay, as described previously [21]. Age-matched wild-type female BALB/c mice were used as controls. To induce acute polyarthritis, animals were injected intravenously with PG-PS (3 mg rhamnose/kg). At this dose, the maximal amount of co-injected contaminating endotoxin was less than 30 pg/mouse. The development of arthritis was evaluated by using a scoring system based on a scale from 0 to 4 for each of four paws, as described elsewhere [22]: 0 = normal paw; 1 = swelling of individual digits; 2 = moderate swelling and redness of ankle or wrist joints; 3 = swelling and redness of at least two joints; and 4 = swelling of the whole paw. According to this system, the total arthritis score of a mouse varied in the range 0–16. To compare the frequencies of arthritis in wild-type and CD14 knockout animals, each mouse that had any signs of arthritis (i.e. total score ≥ 1) was considered positive. To minimize bias, animals were coded and assessed randomly.
Histologic analysis
Following the intravenous PG-PS injection on day 0, mice were killed on the next day after the joint inflammation had reached a peak (usually, on day 4). The hind ankle joint and paws were fixed in 10% formalin, decalcified, paraffinized, cut into 5 μm sections, deparaffinized, stained with hematoxylin–eosin, and evaluated histologically. Morphologic assessments of arthritis were performed in a blinded manner using a scoring system with a scale from 0 (no damage) to 4 (severe damage), based on the overall infiltration of inflammatory cells, synovial hyperplasia, and deposition of fibrinous exudates in the joints and surrounding tissue [4].
Cell culture
Splenocytes were harvested as described elsewhere [23]. Cells were resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, HEPES (10 mmol/l) and antibiotics (100 U/ml penicillin G and 100 μg/ml streptomycin sulfate), and incubated at 37°C in 5% carbon dioxide for 1 hour. The nonadherent cells were washed out gently twice with phosphate-buffered saline and once with DMEM. Cell viability was typically in excess of 95%, as assessed by trypan blue exclusion. Peritoneal macrophages were collected as previously described [20]. Cells were recruited by an intraperitoneal injection of a sterile solution of 3% thioglycollate. Three days after intraperitoneal injection, the peritoneal exudates were collected and washed thee times, and cells were allowed to adhere to plastic for 30 min, followed by removal of nonadherent cells. Unless otherwise indicated, cells were maintained for 24 hours before use in a DMEM media at 37°C in 5% carbon dioxide.
Assessments of intracellular calcium
The intracellular calcium concentration ([Ca2+]i) in macrophages was measured using fura-2, a fluorescent [Ca2+]i indicator, as described elsewhere [23]. Macrophages were plated on coverslips in a DMEM media supplemented with 10% fetal bovine serum at 24 hours before measurement. Growth media was replaced with a modified Hanks' balanced buffer (115 mmol/l NaCl, 5 mmol/l KCl, 0.3 mmol/l Na2HPO4, 0.4 mmol/l KH2PO4, 5.6 mmol/l glucose, 0.8 mmol/l MgSO4, 1.26 mmol/l CaCl2, and 15 mmol/l HEPES; pH = 7.4) containing 5 μmol/l acetoxymethyl ester of fura-2 (Molecular Probes, Eugene, OR, USA). Cells were incubated at room temperature for 30 min and stimulated with indicated concentrations of PG-PS. The [Ca2+]i measurements were performed in individual cells by using a microspectrofluorometer (InCyt Im2™ Imaging, Cincinnati, OH, USA) interfaced with an inverted microscope TMS-F (Nikon, Kanagawa, Japan) [23]. The fluorescence intensity was monitored at excitation wavelengths 340 and 380 nm, and emission wavelength 510 nm. Each value was corrected by subtracting the system dark noise and autofluorescence. The [Ca2+]i was calculated as [Ca2+]i = Kd([R - Rmin]/[Rmax - R]) × (F0/Fs) as previously described [23]. Cells were selected at random and analyzed using InCyt Im2™ image acquisition and analysis software.
Assessments of nuclear factor-κB activation
Cellular localization of the RelA (p65) subunit of NF-κB was detected by immunocytochemistry. Splenic macrophages were stimulated with PG-PS for 30 min, fixed for 10 min with ice cold methanol, and blocked for 30 min in a 10% normal goat serum (Sigma). Cells were immunostained using a primary polyclonal rabbit anti-RelA antibody (Rockland Immunochemicals, Gilbertsville, PA, USA) diluted at 1:200 in 10% normal goat serum, and visualized with the secondary rhodamine isothiocyanate-conjugated antirabbit antibody. Nuclei were counterstained with Hoechst 33342 (Molecular Probes).
RNA isolation and reverse transcription polymerase chain reaction
Total RNA was extracted from homogenized tissues by using Trizol reagent (Sigma). One nanogram of RNA was subjected to reverse transcription by using MLV reverse transcriptase (Promega, Madison, WI, USA). The resulting cDNA was PCR amplified with the following primers: TNF-α: 5'-TAC TGA ACT TCG GGG TGA TTG GTC C-3' (forward) and 5'-CAG CCT TGT CCC TTG AAG AGA ACC-3' (reverse); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH): 5'-TGA AGG TCG GTG TCA ACG GAT TTG-3' (forward) and 5'-GTA CAT CCG TAC TCC AGG TGG TG-3' (reverse). In a typical PCR reaction, 50 ng cDNA template was added to a mixture containing 75 μmol/l of primers, 2.5 mmol/l dNTPs, and 1 U Taq polymerase. For detection of TNF-α message, the template was amplified by 26 cycles of PCR (at the annealing temperature 56°C). GAPDH was amplified by using 24 cycles (annealing temperature 63°C). The PCR products were visualized by agarose gel electrophoresis.
Statistical analysis
Unless otherwise indicated, all results are expressed as means ± standard error of the mean. Statistical differences between groups were determined using analysis of variance with Tukey's or Scheffe's post hoc test. Scores were compared using the Mann–Whitney rank sum test. P < 0.05 was considered statistically significant.
Results
CD14 mediates the PG-PS-induced increase in intracellular calcium concentration
PG-PS stimulation induces a rapid, transient increase in [Ca2+]i, which is a prerequisite for induction of inflammatory gene expression by PG-PS and LPS [23-25]. Here, we compared the increase in [Ca2+]i in primary spleen macrophages from wild-type and CD14 knockout mice. Stimulation of cells with PG-PS in a serum-free medium did not induce an increase in [Ca2+]i (data not shown), which is consistent with previously published data [14]. In the presence of serum, PG-PS caused a rapid and transient increase in [Ca2+]i (Fig. 1a), which peaked at approximately 2 min after stimulation. This response was dose dependent and reached a plateau at concentrations above 20 μg/ml (Fig. 1b). The increase in [Ca+2]i was significantly reduced in CD14 knockout cells. At the saturating concentrations of PG-PS (20 μg/ml), the maximal increase in [Ca 2+]i in the wild-type cells was 47 ± 5 nmol/l as compared with 12 ± 7 nmol/l in CD14 knockout cells (Fig. 1b), indicating that the PG-PS-induced transient increase in [Ca2+]i was mediated by CD14.
Figure 1.
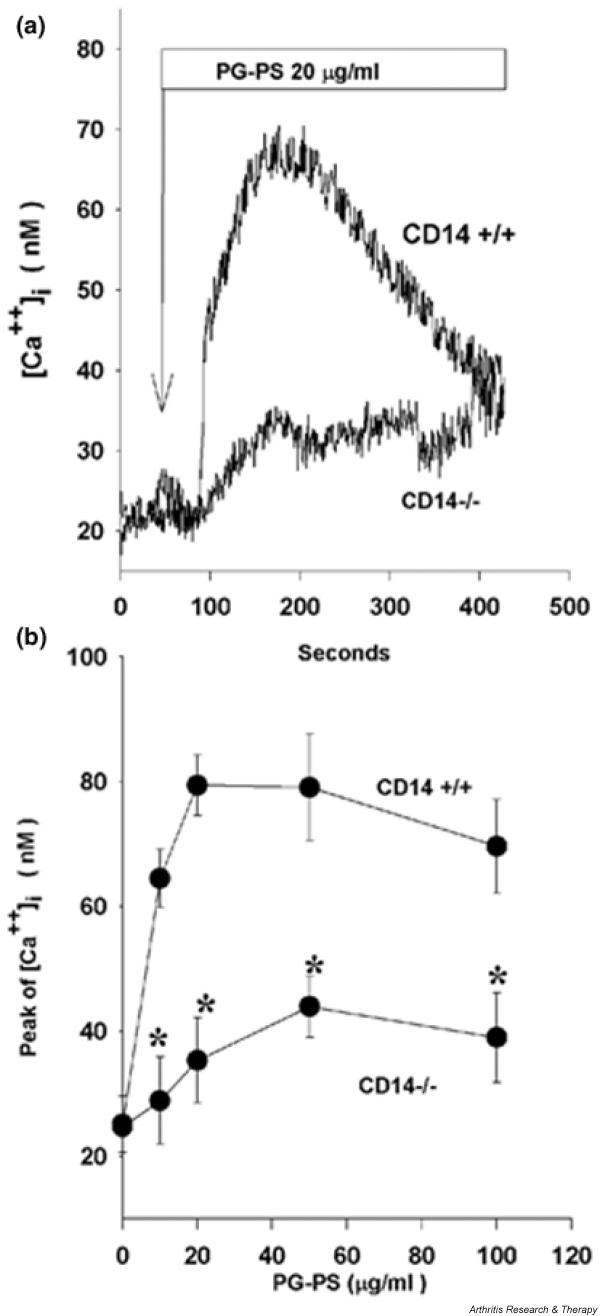
Peptidoglycan–polysaccharide (PG-PS)-induced increase in intracellular calcium concentration ([Ca2+]i) is inhibited in CD14 knockout cells. The levels of [Ca2+]i in spleen macrophages were evaluated by using fura-2, a fluorescent [Ca2+] indicator. (a) Kinetics of PG-PS-induced [Ca2+]i (average from six to eight cells). Representative data from two experiments are shown. (b) The average maximal increase in [Ca2+]i shown as a function of PG-PS concentration. Data represent means ± standard error of the mean for 12–34 cells from two independent experiments. The significance of the difference between the groups was calculated using one-way analysis of variance with Scheffe's post-hoc test. *P < 0.05 versus wild-type.
PG-PS-induced nuclear factor-κB activation is mediated by CD14
The transcription factor NF-κB is a pivotal regulator of genes that are involved in inflammation and immunity [26]. NF-κB is an essential component of TLR signaling [8]. The activity of NF-κB is controlled by interaction an inhibitory molecule known as IκB. Cell stimulation induces degradation of IκB, thereby allowing NF-κB to enter the nucleus and initiate the transcription of target genes [27]. Thus, the nuclear localization of NF-κB is indicative of NF-κB activation.
To assess NF-κB activation, wild-type and CD14 knockout spleen macrophages were stimulated with PG-PS and immunostained with antibodies against the RelA (p65) subunit of NF-κB. As shown in Fig. 2, NF-κB was largely cytoplasmic in resting cells (top panels) whereas PG-PS stimulation induced nuclear translocation of NF-κB in the majority of wild-type cells (middle panels). In contrast, only few, if any, of the PG-PS-stimulated CD14 knockout cells had nuclear NF-κB (lower panels), indicating that PG-PS-induced NF-κB activation is CD14-dependent.
Figure 2.
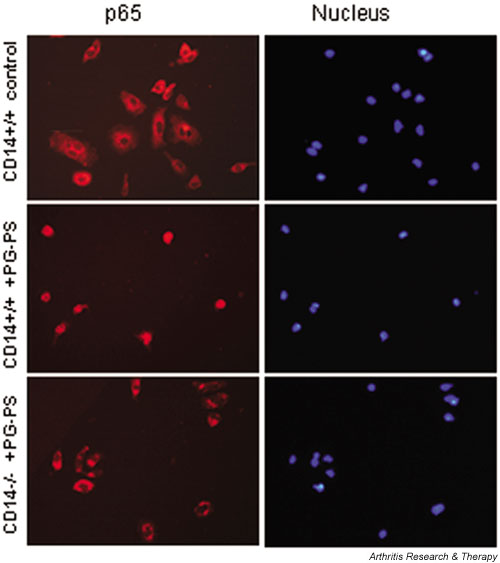
Peptidoglycan–polysaccharide (PG-PS)-induced activation of nuclear factor-κB (NF-κB) is inhibited in CD14 knockout spleen macrophages. Spleen macrophages were stimulated for 30 min with PG-PS at a concentration of 20 μg/ml. The left column shows immunostaining of the p65 (RelA) subunit of NF-κB, and the right column shows counterstaining of nuclei with Hoechst 33342. The rows show the following: top – unstimulated cells from wild-type mice; middle – PG-PS stimulated cells from wild-type mice; and bottom row – PG-PS-stimulated cells from CD14 knockout mice. Representative data from three independent experiments are shown.
CD14 mediates PG-PS-induced production of inflammatory cytokines
The signal transduction pathways activated by TLRs culminate in activation of transcription of defensive genes. The expression of many inflammatory cytokines, including IL-1, TNF-α, and IL-6, depends on NF-κB, because their promoters contain NF-κB-binding sites, and specific suppression of NF-κB blocks their production [28]. Here, we examined the production of these cytokines in response to PG-PS. Upon stimulation with PG-PS wild-type peritoneal macrophages secreted large quantities of IL-6 (Fig. 3a), whereas the IL-6 production by CD14 knockout macrophages was below the level of detection (<10 pg/ml). Similar to that finding, PG-PS-stimulated CD14 knockout macrophages produced much less TNF-α than did wild-type cells (Fig. 3b). The results in peritoneal macrophages were in good agreement with those observed in spleen macrophages (data not shown). Therefore, PG-PS-induced production of inflammatory cytokines is CD14-dependent.
Figure 3.
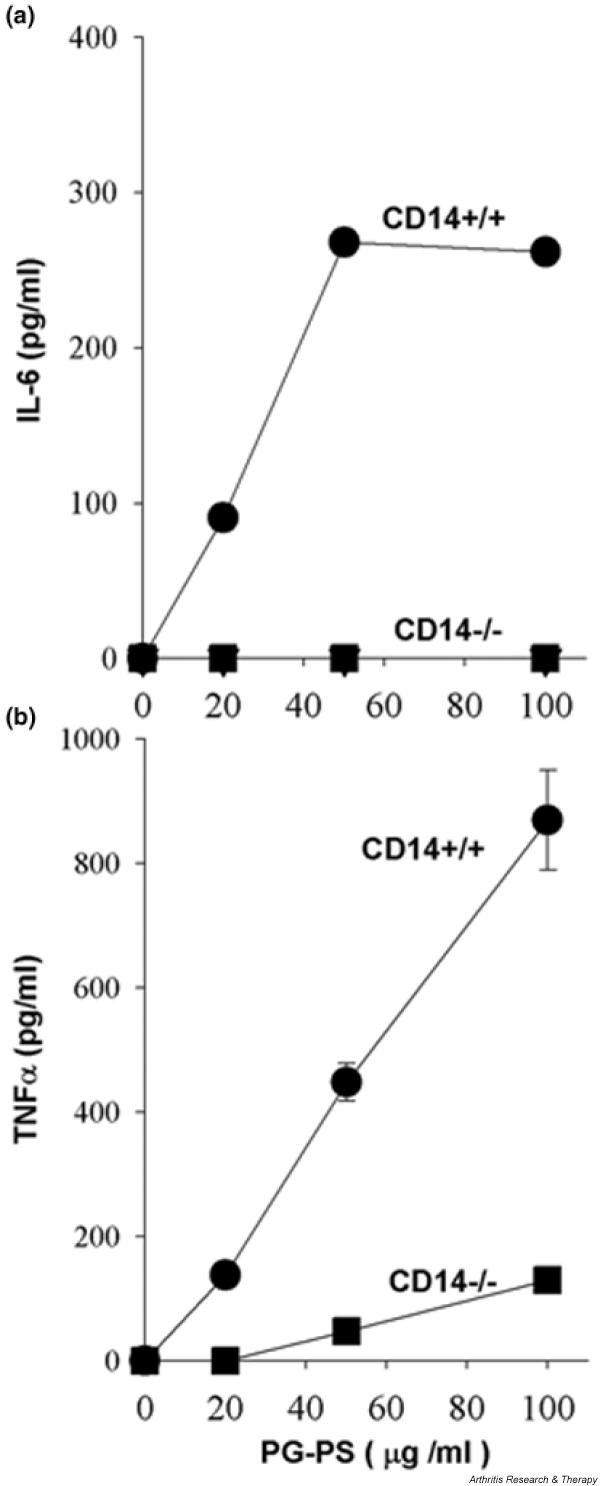
CD14 mediates peptidoglycan–polysaccharide complex (PG-PS)-induced secretion of IL-6 and tumor necrosis factor (TNF)-α. Peritoneal macrophages were stimulated for 4 hours with the indicated concentrations of PG-PS. Concentrations of TNF-α and IL-6 were determined in supernatants by ELISA. Each determination was done in duplicates. (a) TNF-α production by peritoneal macrophages. Data represent means ± standard error of the mean from three independent experiments. (b) IL-6 production by peritoneal macrophages. Data represent mean ± SEM from two independent experiments.
The severity of PG-PS-induced arthritis is attenuated in CD14 knockout mice
To elucidate the role for CD14 in development of PG-PS-induced arthropathy, CD14 knockout mice were backcrossed on a BALB/c background, which is highly susceptible to PG-PS-induced acute arthritis [22]. Intravenous injection of PG-PS caused polyarthritis that reached a peak at around 2 days after injection. At that time, each animal in the wild-type group had developed arthritis (19/19 mice [100%]), with an average arthritis index of 4.1 ± 0.5 (Fig. 4). In CD14 knockout mice, both the incidence of arthritis (11/16 [68%]; P < 0.05) and the average arthritis index (2.3 ± 0.5; P < 0.05) were significantly inhibited.
Figure 4.
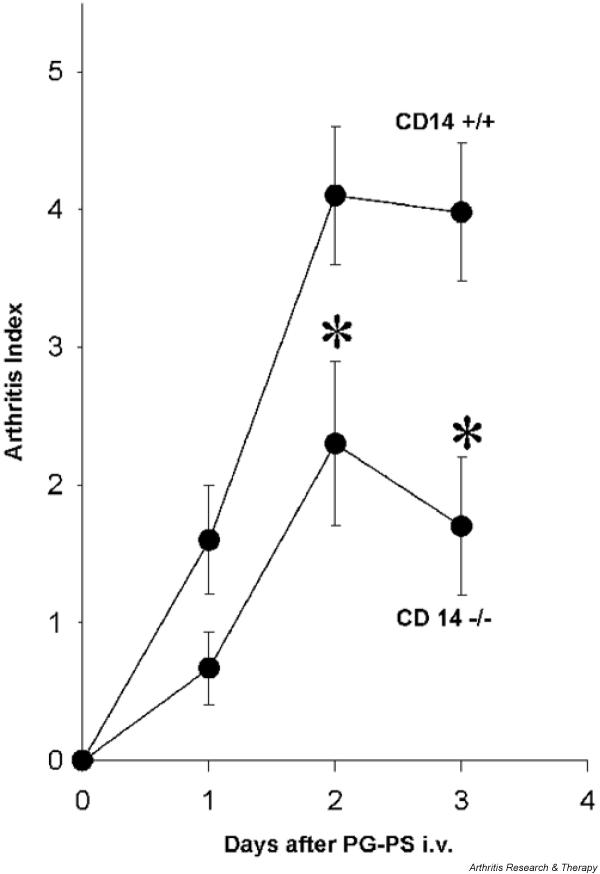
Peptidoglycan–polysaccharide (PG-PS)-induced polyarthritis is attenuated in CD14 knockout animals: gross observation score. Acute PG-PS polyarthritis was induced by intravenous injection of PG-PS (3 mg/kg). The arthritis index was scored as described in the Materials and methods section. Each point represents a mean ± standard error of the mean in 16–19 animals. The significance of the difference between the groups was evaluated by Mann–Whitney rank sum test. *P < 0.05.
The pathomorphologic changes within arthritic joints were consistent with those previously described in this model [22] (i.e. the infiltration of inflammatory cells into the synovium, synovial hyperplasia, and fibrinous deposits; Fig. 5a). Infiltration of inflammatory cells was apparent in the tibio-tarsal joint space, synovium, tendon, toes, and surrounding soft tissues. Extensive deposition of fibrinous exudate in the tibio-tarsal joint and in the bursa of the Achilles' tendon was also observed. There was neither pannus formation nor destruction of cartilage and subchondral bone. All of these changes were observed in some CD14 knockout mice as well, but the magnitude of these changes was much lower. On average, the morphologic arthritis score was 3.3 ± 0.4 in wild-type mice as compared with 1.0 ± 0.5 in CD14 knockout animals (n = 8–10 mice/group; P < 0.05; Fig. 5b). The morphologic changes correlated well with the gross observation data.
Figure 5.
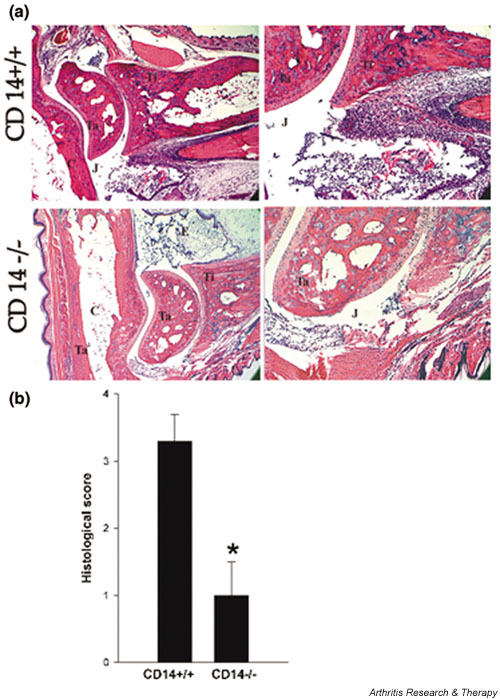
Peptidoglycan–polysaccharide (PG-PS)-induced polyarthritis is attenuated in CD14 knockout animals: morphological assessment. (a) Ankle joints at day 3 after intravenous injection of PG-PS. The upper row shows the joint of a wild-type mouse and the lower row the joint of a CD14 knockout mouse. Synovium (S), tibia (T), tarsal (Ta), joint space (J), cartilage (C), and fibrinous exudates (F) are indicated. Hematoxylin–eosin counterstaining was employed and the original magnifications are as follows: left column 12.5× and right column 40×. Representative data from 10 joints in each group are shown. (b) Histological score. The severity of arthritis was assessed as described in the Materials and methods section. Data are shown as means ± standard error of the mean (n = 10). The significance of difference between groups was calculated by using the Mann–Whitney rank sum test. *P < 0.05.
To assess inflammation at the molecular level, we examined the expression of inflammatory cytokines TNF-α, IL-1β, and IL-6 in arthritic joints by RT-PCR. In normal joints the messages were below the level of detection, but at the peak of arthritis these cytokines were readily detectable in the wild-type mice. The expression of TNF-α mRNA was substantially lower in the arthritic joints of CD14 knockout mice (Fig. 6). Similar to that finding, IL-6 message was inhibited in the arthritic joints of CD14 knockout mice (data not shown). Combined, these data indicate that the PG-PS-induced arthropathy is significantly reduced in CD14 knockout animals.
Figure 6.

The expression of inflammatory cytokines in arthritic joints of mice with peptidoglycan–polysaccharide (PG-PS)-induced arthritis. Total RNA was extracted from ankle joints, reversely transcribed, amplified by PCR, and resolved on gel electrophoresis. The upper panel shows 24 cycles of amplification with tumor necrosis factor (TNF)-α specific primers, and the lower panel shows 22 cycles of amplification with glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific primers. The first two lanes on the left represent DNA molecular weight markers. Lane (-) represents a negative control (RT-PCR amplification in the absence of RNA sample). Lanes N1 and N2 represent RT-PCR amplified RNA from nonarthritic joints of normal (i.e. not injected with PG-PS) wild-type and CD14 knockout animals, respectively. Lanes a–d represent RT-PCR amplified RNA from four different arthritic joints of PG-PS-injected wild-type animals. Lanes e–g represent RT-PCR amplified RNA from three arthritic joints of PG-PS injected CD14 knockout animals. Representative data from two independent experiments are shown.
Discussion
The etiologic agents of RA remain to be identified. However, in spite of the inability to isolate a specific infectious organism from the joints of RA patients, there is evidence suggesting that transient exposure to indigenous bacterial products may provoke an initial response that could eventually perpetuate and amplify itself. This notion has been supported by numerous animal studies. Single intraperitoneal injection of crude bacterial cell walls or water-soluble PG-PS from Gram-positive bacteria into susceptible strains of rats induces polyarthritis that closely resembles human RA. The joint lesions have a biphasic course with an initial acute inflammation of the ankle, wrist, and small joints of extremities, followed by chronic erosive arthritis (for review [6]). The initial, acute, phase has features of the innate immune response in that inflammation is driven by neutrophils and macrophages and is T-cell independent. The chronic stage of arthritis is T-cell dependent and thus has features of the adaptive immune response [7,29]. Systemic administration of Gram-positive bacterial cell walls or PG-PS in susceptible strains of mice also produces arthritis, although, for unknown reasons, mice develop only the acute phase [22,30]. The minimal essential arthritogenic structures have been identified as PG-PS, the major structural components of Gram-positive bacterial cell walls [31]. The peptidoglycan moiety is responsible for the pathogenicity of PG-PS, because enzymatic digestion of the peptidoglycan moiety eliminates the proinflammatory and arthritopathogenic properties of PG-PS [32]. The peptidoglycan can substitute for PG-PS in the induction of the acute [22] but not chronic phase of arthritis [2,33]; it has been proposed that the polysaccharide moiety protects the peptidoglycan moiety from degradation in vivo, thereby facilitating chronic persistence of PG-PS in the host [2].
The primary events and the identity of cellular receptors that mediate the innate immune response to and the arthritogenic properties of peptidoglycan and PG-PS are incompletely characterized. There is substantial evidence that the transmembrane signal leading to cell activation in response to stimulation with Gram-positive peptidoglycans is triggered by TLR-2 [11]. CD14, has been proposed as an intermediate, connecting peptidoglycan with TLR-2-induced cell activation, but the role of CD14 is controversial. On the one hand, neutralizing antibody to CD14 prevented peptidoglycan-induced cell activation, and transfection of CD14-negative cells with exogenous CD14 conferred responsiveness to peptidoglycan [14], suggesting that CD14 plays an essential role. On the other hand, peptidoglycan was shown to bind TLR-2 directly [17] and to induce the activation of TLR-2-transfected cells regardless of CD14, although CD14 increased the affinity of PG–TLR-2 interactions [17] and potentiated cell activation [18], suggesting a facultative role for CD14. It is possible that the discrepancies between the studies can be attributed to differences in experimental conditions. In this regard, using primary cells with genetically inactivated CD14 allowed for clearer interpretation. We demonstrate that, in CD14 knockout macrophages, each step of PG-PS induced signal transduction, including the transient increase in [Ca2+]i, nuclear translocation of NF-κB, and secretion of TNF-α and IL-6, were almost completely suppressed. Thus, our data strongly support an essential role for CD14 in the innate immune responses to PG-PS.
To examine the role for CD14 in PG-PS induced arthropathy, CD14 knockout mice were backcrossed to a susceptible BALB/c genetic background [22]. Because mice do not develop chronic arthritis, we were restricted to examination of acute arthritis, which is driven by the innate immune response to PG-PS. The CD14 knockout mice developed arthritis significantly less frequently (68% versus 100% in wild-type group), and the severity of arthritis was significantly reduced (average arthritis score of 2.5 versus 4.8 in the wild-type group). The gross observation data largely correlated with morphologic assessments, the most pronounced difference being in the degree of infiltration of inflammatory cells. Because CD14 knockout macrophages were largely unresponsive to PG-PS stimulation, these in vivo results were not unexpected. It was rather surprising to find that PG-PS was able to induce arthritis in a significant proportion of CD14 knockout mice. The reason for that is not clear. Our analysis of mRNA expression has shown that inflammatory gene expression in arthritic joints of CD14 knockout mice was strongly inhibited but not abolished (Fig. 6). It is possible that resident nonphagocytic cells within joints can be activated by PG-PS via CD14-independent mechanisms. In this regard, Kyburz and coworkers [34] showed that stimulation of synovial fibroblasts with staphylococcal peptidoglycan caused cell activation and elevated expression of matrix metalloproteinases and cytokines IL-6 and IL-8; it appeared that cell activation was mediated by both TLR-2-dependent and -independent pathways. It is not known whether CD14 is the major receptor for PG-PS in each cell type; alternative receptors have been proposed, including the peptidoglycan recognition proteins and nucleotide-binding oligomerization proteins (NODs) [35-37]. Nonetheless, our data indicate that CD14 is an essential receptor for activation of the innate immune response in macrophages by the arthritiogenic PG-PS, and that CD14-dependent mechanisms significantly contribute to PG-PS-induced arthropathy.
Conclusion
Bacterial infection has frequently been associated with RA pathology. The arthropathic properties have been attributed to PG-PS, which are major structural components of bacterial cell walls. The identity of receptors that mediate the arthropathic properties of PG-PS is controversial. Here, we examined the role played by CD14 in PG-PS-induced arthritis in mice. To do so, we used CD14-deficient knockout mice. We found that CD14 knockout macrophages were almost completely deficient in inflammatory responses to PG-PS stimulation in vitro. In vivo, the incidence and severity of PG-PS-induced polyarthritis in CD14 knockout mice were significantly reduced but not abolished, as compared with their wild-type counterparts. These results support an essential role for CD14 in the innate immune responses to PG-PS and indicate an important role for CD14 in PG-PS-induced arthropathy.
Competing interests
None declared.
Abbreviations
[Ca2+]i = intracellular calcium concentration; DMEM = Dulbecco's modified Eagle's medium; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; IL = interleukin; LPS = lipopolysaccharide; NF-κB = nuclear factor-κB; PAMP = pathogen-associated molecular patterns; PG-PS = peptidoglycan–polysaccharide complexes; RA = rheumatoid arthritis; RT-PCR = reverse transcription polymerase chain reaction; TLR = Toll-like receptor; TNF = tumor necrosis factor.
Acknowledgments
Acknowledgments
The authors gratefully acknowledge the technical expertise of Mrs Julie V Mitchell and Charlotte Walters at the Center for Gastrointestinal Biology and Disease (P30 KD34987). This work was supported by NIH public health grants AR/AI-44564, 5-P60 AR-30701-14, and AR/AI-44030. SM is the recipient of an Investigator Award from The Arthritis Foundation.
Contributor Information
Xiangli Li, Email: xiangli_li@med.unc.edu.
Sergei S Makarov, Email: smak@med.unc.edu.
References
- Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- Schwab JH. Phlogistic properties of peptidoglycan-polysaccharide polymers from cell walls of pathogenic and normal-flora bacteria which colonize humans. Infect Immun. 1993;61:4535–4539. doi: 10.1128/iai.61.11.4535-4539.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden IM, Wilbrink B, Tchetverikov I, Schouls LM, Hazenberg MP, Breedveld FC, Tak PP. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatioid arthritis and other arthritides. Arthritis Rheum. 2000;43:593–598. doi: 10.1002/1529-0131(200003)43:3<593::AID-ANR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Lichtman SN, Wang J, Sartor RB, Zhang C, Bender D, Dalldorf FG, Schwab JH. Reactivation of arthritis induced by small bowel bacterial overgrowth in rats: role of cytokines, bacteria, and bacterial polymers. Infect Immun. 1995;63:2295–2301. doi: 10.1128/iai.63.6.2295-2301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravallese EM, Kantrowitz FG. Arthritic manifestations of inflammatory bowel disease. Am J Gastroenterol. 1988;83:703–709. [PubMed] [Google Scholar]
- Schwab JH. Bacterial cell-wall induced arthritis: models of chronic recurrent polyarthritis and reactivation of monoarticular arthritis. In: Henderson B, Edwards JCW, Pettipher ER, editor. In Mechanisms and Models in Rheumatoid Arthritis. London: Academic Press; 1995. pp. 431–446. [Google Scholar]
- Allen JB, Malone DG, Wahl SM, Calandra GB, Wilder RL. Role of the thymus in streptococcal cell wall-induced arthritis and hepatic granuloma-formation: comparative studies of pathology and cell-wall distribution in athymic and euthymic rats. J Clin Invest. 1985;76:1042–1056. doi: 10.1172/JCI112057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Beutler B. TLR4 as the mammalian endotoxin sensor. Curr Top Microbiol Immunol. 2002;270:109–120. doi: 10.1007/978-3-642-59430-4_7. [DOI] [PubMed] [Google Scholar]
- Zhang G, Ghosh S. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107:13–19. doi: 10.1172/JCI11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Pugin J, Heumann D, Tomasz A, Kravchenko VV, Akamatsu Y, Nishijima M, Glauser MP, Tobias PS, Ulevitch RJ. Cd14 is a pattern-recognition receptor. Immunity. 1994;1:509–516. doi: 10.1016/1074-7613(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Dziarski R, Tapping RI, Tobias PS. Binding of bacterial peptidoglycan to CD14. J Biol Chem. 1998;273:8680–8690. doi: 10.1074/jbc.273.15.8680. [DOI] [PubMed] [Google Scholar]
- Gupta D, Kirkland TN, Viriyakosol S, Dziarski R. CD14 is a cell-activating receptor for bacterial peptidoglycan. J Biol Chem. 1996;271:23310–23316. doi: 10.1074/jbc.271.38.23310. [DOI] [PubMed] [Google Scholar]
- Rietschel ET, Schletter J, Weidemann B, El-Samalouti V, Mattern T, Zahringer U, Seydel U, Brade H, Flad H-D, Kusumoto S, Gupta D, Dziarski R, Ulmer AJ. Lipopolysaccharide and peptidoglycan: CD14-dependent bacterial inducers of inflammation. Microb Drug Resist. 1998;4:37–44. doi: 10.1089/mdr.1998.4.37. [DOI] [PubMed] [Google Scholar]
- Gupta D, Wang Q, Vinson C, Dziarski R. Bacterial peptidoglycan induces CD14-dependent activation of transcription factors CREB/ATF and AP-1. J Biol Chem. 1999;274:14012–14020. doi: 10.1074/jbc.274.20.14012. [DOI] [PubMed] [Google Scholar]
- Iwaki D, Mitsuzawa H, Murakami S, Sano H, Konishi M, Akino T, Kuroki Y. The extracellular toll-like receptor 2 domain directly binds peptidoglycan derived from Staphylococcus aureus. J Biol Chem. 2002;277:24315–24320. doi: 10.1074/jbc.M107057200. [DOI] [PubMed] [Google Scholar]
- Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- Haziot A, Ferrero E, Kontgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, Goyert SM. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity. 1996;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- Haziot A, Hijiya N, Schultz K, Zhang F, Gangloff SC, Goyert SM. CD14 plays no major role in shock induced by Staphylococcus aureus but down-regulates TNF-α production. J Immunol. 1999;162:4801–4805. [PubMed] [Google Scholar]
- Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol. 2001;166:4737–4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- Koga T, Kakimoto K, Hirofuji T, Kotani S, Ohkuni H, Watanabe K, Okada N, Okada H, Sumiyoshi A, Saisho K. Acute joint inflammation in mice after systemic injection of the cell wall, its peptidoglycan, and chemically defined peptidoglycan subunits from various bacteria. Infect Immun. 1985;50:27–34. doi: 10.1128/iai.50.1.27-34.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bradford BU, Wheeler MD, Stimpson SA, Pink MH, Brodie AT, Schwab JH, Thurman RG. Dietary glycine prevents peptidoglycan polysaccharide-induced reactive arthritis in the rat: role for glycine-gated chloride channel. Infect Immun. 2001;69:5883–5891. doi: 10.1128/IAI.69.9.5883-5891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Suzuki J, Kobayashi Y. Role of calcium in tumor necrosis factor-α produced by activated macrophages. J Biochem. 1996;120:1190–1195. doi: 10.1093/oxfordjournals.jbchem.a021540. [DOI] [PubMed] [Google Scholar]
- Ikejima K, Enomoto N, Seabra V, Ikejima A, Brenner DA, Thurman RG. Pronase destroys the lipopolysaccharide receptor CD14 on Kupffer cells. Am J Physiol. 1999;276:G591–598. doi: 10.1152/ajpgi.1999.276.3.G591. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Makarov SS, Johnston WN, Olsen JC, Watson JM, Mondal K, Rinehart C, Haskill JS. NF-kappa B as a target for anti-inflammatory gene therapy: suppression of inflammatory responses in monocytic and stromal cells by stable gene transfer of I kappa B alpha cDNA. Gene Ther. 1997;4:846–852. doi: 10.1038/sj.gt.3300461. [DOI] [PubMed] [Google Scholar]
- Schrier DJ, Schimmer RC, Flory CM, Tung DKL, Ward PA. Role of chemokines and cytokines in a reactivation model of arthritis in rats induced by injection with Streptococcal cell walls. J Leukocyte Biol. 1998;63:359–363. doi: 10.1002/jlb.63.3.359. [DOI] [PubMed] [Google Scholar]
- Onta T, Sashida M, Noriyuki F, Sugawara S, Rikiishi H, Kumagai K. Induction of acute arthritis in mice by peptidoglycan derived from Gram-positive bacteria and its possible role in cytokine producion. Microbiol Immunol. 1993;37:573–582. doi: 10.1111/j.1348-0421.1993.tb01679.x. [DOI] [PubMed] [Google Scholar]
- Schwab JH, Brown RR, Anderle AK, Schlievert PM. Superantigen can reactivate bacterial cell wall-induced arthritis. J Immunol. 1993;150:4151–4159. [PubMed] [Google Scholar]
- Janusz MJ, Esser RE, Schwab JH. In vivo degradation of bacterial cell wall by the muralytic enzyme mutanolysin. Infect Immun. 1986;52:459–467. doi: 10.1128/iai.52.2.459-467.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Brown RR, Anderle SK, Chetty C, Cromartie WJ, Gooder H, Schwab JH. Arthropathic properties related to the molecular weight of peptidoglycan-polysaccharide polymers of streptococcal cell walls. Infect Immun. 1982;35:1003–1010. doi: 10.1128/iai.35.3.1003-1010.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyburz D, Rethage J, Seibl R, Lauener R, Gay RE, Carson DA, Gay S. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling. Arthritis Rheum. 2003;48:642–650. doi: 10.1002/art.10848. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- Dziarski R. Peptidoglycan recognition proteins (PGRPs) Mol Immunol. 2004;40:877–886. doi: 10.1016/j.molimm.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Hugot JP, Sansonetti PJ. Lessons from Nod2 studies: towards a link between Crohn's disease and bacterial sensing. Trends Immunol. 2003;24:652–658. doi: 10.1016/j.it.2003.10.007. [DOI] [PubMed] [Google Scholar]