

Abstract
Peripheral artery disease (PAD) is characterized by vessel occlusion and ischemia in the limbs. Treatment for PAD with surgical interventions has been showing limited success. Moreover, recent clinical trials with treatment of angiogenic growth factors proved ineffective as increased angiogenesis triggered severe inflammation in a proportionally coupled fashion. Hence, the overarching goal of this research was to address this issue by developing a biomaterial system that enables controlled, dual delivery of pro-angiogenic C16 and anti-inflammatory Ac-SDKP peptides in a minimally-invasive way. To achieve the goal, a peptide-loaded injectable microgel system was developed and tested in a mouse model of PAD. When delivered through multiple, low volume injections, the combination of C16 and Ac-SDKP peptides promoted angiogenesis, muscle regeneration, and perfusion recovery, while minimizing detrimental inflammation. Additionally, this peptide combination regulated inflammatory TNF-α pathways independently of MMP-9 mediated pathways of angiogenesis in vitro, suggesting a potential mechanism by which angiogenic and inflammatory responses can be uncoupled in the context of PAD. This study demonstrates a translatable potential of the dual peptide-loaded injectable microgel system for PAD treatment.
Keywords: polymer, polycaprolactone, polyethylene glycol, angiogenesis, inflammation, peptide
Introduction
Peripheral artery disease (PAD) develops as arteries leading to the extremities become activated by inflammatory signals, followed by accumulation of plaques which limit blood flow to distal tissues. Eighty million people in the United States suffer from PAD- ranging from symptoms of intermittent pain when walking (claudication) to critical limb ischemia (CLI).[1, 2] Although surgical interventions can alleviate symptoms, these measures are not an option for over 50% of patients due to age, diabetes, or widespread blockages.[2] Several pre-clinical treatment strategies have focused on maximizing angiogenesis and arteriogenesis to restore blood flow to ischemic tissues- including gene transfer and cell delivery approaches.[2] However, these strategies overlook a causative role of inflammation in PAD progression.[3-5] Inflammatory cues activate the vascular endothelium, enabling the diapedesis of macrophages into the intima where they accumulate cholesterol to form plaques. For this reason inflammation should be minimized in PAD treatment.
Nonetheless, some level of inflammation is needed for the initiation of angiogenesis to promote collateral vessel formation and restore blood flow to ischemic tissues. On the other hand, angiogenesis can promote inflammation, meaning that therapeutics designed to promote angiogenesis can trigger an inflammatory response as well.[6-9] [10, 11] Therefore, independent control of inflammation and angiogenesis would be an ideal treatment for PAD. In our previous study, we demonstrated successful independent control of angiogenesis and inflammation by delivering pro-angiogenic C16 and anti-inflammatory Ac-SDKP peptides via an implantable polymer scaffold.[12] C16 is a pro-angiogenic peptide derived from laminin-1[13] which is known to bind to the αvβ3 and α5β1 integrins and increase VEGFR2 and FGFR2 production.[14, 15] The tetrapeptide Ac-SDKP is derived from thymosin β-4 which can be found in platelets and wound fluid. Ac-SDKP has been identified as an anti-inflammatory and anti-fibrotic molecule which decreases macrophage infiltration and TGF-β expression.[12, 16]
Microgels were fabricated from a combination of polyethylene glycol (PEG) and poly-ε-caprolactone (PCL). PEG is a hydrophilic polymer which causes repulsion of proteins and cells.[17] PCL is a hydrophobic, slowly degrading polymer which is well-known for favorable cellular and interactions when used as tissue engineering constructs.[18] Hence, the co-polymer microgels can serve as a synthetic ECM for cell attachment and growth.[19, 20] As these injectable polymers are thermo-responsive, they exist as a solution at room temperature but become a hydrogel at body temperature. This solution-to gel (sol-gel) transition allows for easy mixing of therapeutics, including peptides with the polymer solution at room temperature before injecting into the body. Upon injection of polymers containing peptides, a stable hydrogel forms and serves as a drug depot in a site-specific manner, thereby avoiding systemic side effects.[21, 22] The injectable polymer microgel system was evaluated for gelation time and biocompatibility in vitro before transitioning to in vivo experiments to evaluate the regulation of angiogenesis and inflammation in a murine model of PAD. As a comparison to these injectable microgels, solid implantable scaffolds were also loaded with peptides and used in in vivo experiments.
In the current work we demonstrate the ability of C16 and Ac-SDKP-loaded, injectable polymer microgel systems to successfully increase collateral vessel formation without inflammatory exacerbation in a PAD model of murine hind limb ischemia. The minimally-invasive, injectable polymer microgel approach used in this study reduces surgical injuries expected from scaffold implantation, and this is a significant advantage in the clinic as a number of PAD patients have comorbidities which prevent them from being eligible for surgical interventions.
To elucidate a mechanism of peptide-mediated decoupling of angiogenesis and inflammation, we investigated the roles of MMP-9 and TNF-α in modulating angiogenesis and inflammation. MMP-9 promotes angiogenesis by degrading collagen in the basement membrane of vasculature, enabling blood vessel remodeling and growth. Transcriptional regulation of MMP-9 is mediated by the transcription factors nuclear factor kappa beta (NF-κβ), forkhead box protein O4 (FoxO4), and activator protein (AP-1).[23, 24], [25] Binding of these transcription factors to the promoter regions of the MMP-9 gene is stimulated by inflammatory cytokines and growth factors, such as TNF-α, IL-1α, IL-1β, PDGF and bFGF.[26] Through these inflammatory cytokine-mediated pathways of MMP regulation, angiogenesis and inflammation are highly interconnected. TNF-α is an inflammatory cytokine that is produced primarily by activated macrophages early in the acute phase of inflammatory response, and results in recruiting neutrophils to the site of inflammation.[27-29] TNF-α binds to TNF receptor 1 (TNFR1) on the cell membrane, which activates Nf-κβ, MAPK pathways, or death signaling.[30] In particular, NF-κβ signaling in macrophages stimulates phagocytosis.[31] In this study we elucidated the role of TNF-α in controlling inflammation independently of MMP-9 mediated regulation of angiogenesis.
Materials and Methods
Chemicals and Reagents for Injectable Polymer Microgel
Tin (II) ethyl hexanoate (Sn(Oct)2), ε-caprolactone, monomethoxypoly(ethylene glycol) (mPEG) (Mn= 750Da), anhydrous tetrahydrofuran (THF), anhydrous toluene, dichloromethane, and diethyl ether were purchased from Sigma- Aldrich (St. Louis, MO, USA). ε-caprolactone was dried and distilled over CaH2 (Alfa Aesar, Ward Hill, MA, USA) immediately before polymerization. Tin (II) ethyl hexanoate was distilled under high vacuum.
Synthesis and Characterization of Injectable Polymer Microgels
The injectable polymer is presented as 21%PEG-79%PCL (individual mole percentage) (Fig. 1A). Our previous studies have showed that this format of combinatorial polymers provides tunable degradation, mechanical, and thermal properties by changing the mole percentages.[20, 32] PEG-PCL was synthesized by ring opening polymerization of ε-caprolactone according to previously published methods.[20, 33] The structure and the number average molecular weight (Mn) calculated by the molar ratio of PEG and PCL was verified by NMR (Fig. S1). Injectable polymers were dissolved in H2O to form a 13% polymer by weight solution at 25°C and then incubated at 37° C and observed every 10 seconds until a stable gel formed to determine the gelation time (Fig. 1B).
Figure 1. Injectable polymer microgel and in vivo peptide release.
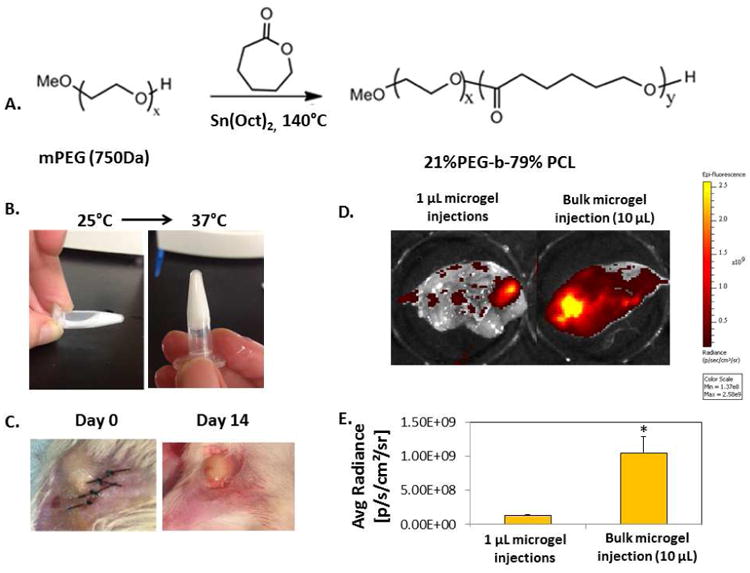
A) A co-polymer of 21% polyetheylene glycol (PEG) and 79% poly-ε-caprolactone (PCL) (%: molar ratio) was synthesized by ring opening polymerization with tin (II) ethyl hexanoate (Sn(Oct)2) catalyst. B) The injectable polymer microgel was a liquid at room temperature (25°C, left) and formed a stable gel at body temperature (37°C, right). C) Injectable polymer microgel immediately after surgery (day 0, left), or after 14 days (right). D) FITC-tagged Ac-SDKP was mixed with polymer microgel at 25°C before injecting either a single bulk 10 μL injection (right), or ten individual 1 μL injections into the muscle around the site of femoral artery ligations. After 7 days, the skin was removed from the thigh muscle and imaged using a fluorescence in vivo imaging system (IVIS). Areas with high fluorescent intensity are colored yellowed in the images. E) Fluorescence intensity of peptides that remained in the tissue after 7 days was quantified using IVIS software. As evidenced by the lower fluorescence intensity of peptides retained in the tissue, more of the peptides were released with multiple 1 μL injections than with the bulk injection over the course of 7 days. D-E) n=4; Data are means ± SEM from four or five mice. *p<0.05 vs. 1 μL polymer microgel injections (Tukeys' Range Test).
In Vitro Biocompatibility Assay
HUVECs (ATCC) were seeded at a density of 1×105 cells/mL in MesoEndo media (Cell Applications) on top of pre-gelled injectable polymer microgels and cultured for 1 or 3 days at 37°C with 5% CO2 and stained with LIVE/DEAD® Viability/Cytotoxicity Kit (Invitrogen) according to supplier's protocol (n=4 per condition).
Mouse Model of Hind Limb Ischemia
All animal experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee (IACUC) in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Wild type A/J mice were used to develop a model of PAD as described previously,[34] by ligating the femoral artery and vein at one ligation below the epigastric artery and a second ligation around the artery and vein at a distal location just proximal to the deep femoral branch. The femoral artery and vein were then cut between these two sutures. A 13% by weight solution of injectable polymers in H2O was mixed with 75 μg Ac-SDKP, C16, or the combination of Ac-SDKP and C16 at 25°C. In order to control the hydrogel size considering the possibility that the hydrogel size may change peptide release and therefore inflammatory responses,[35] a single, 10 μL bulk injection or ten, 1 μL injections of peptide-loaded polymer were made into the thigh muscle adjacent to the femoral artery ligations. The surgical incision was then closed with non-degradable sutures. As controls, femoral artery ligation surgery was performed on animals without any microgels or peptide treatment or with peptide in PBS injections into the subcutaneous tissue adjacent to femoral artery ligations. The left hind limb (unoperated) was also used as a surgical control.
In Vivo Peptide Release from Injectable Microgels
A 13% by weight solution of injectable polymers in H2O was mixed with 75 μg of FITC-labeled SDKP (GenScript) at 25°C. A single, 10 μL bulk injection or ten, 1 μL injections of peptide-loaded polymer were made into the thigh muscle adjacent to the femoral artery ligations. After 7 days, mice were sacrificed by CO2 inhalation and death was verified by cervical dislocation. The skin on the ischemic hind limb was removed and the adductor muscle was imaged on an IVIS 200 preclinical in vivo imaging system (Perkin Elmer, Waltham, MA) to visualize peptide retention in the tissue (n=4 mice per treatment).
Non-invasive Imaging of Ischemia
LDPI
LDPI was performed on the footpad region of the hind limb of the mice using a Periscan PIM II device. This technique images surface perfusion by measuring Doppler changes in the reflectance of light due to blood flow. During imaging, ambient light and temperature were carefully controlled to avoid background variations in LDPI measurements.[36] Three scans were performed per mouse at each time point: day 0, 3, 7 and 14 after femoral artery ligation and microgel injection (n=6 mice per treatment). The perfusion ratio was calculated by normalizing the average perfusion value of the ischemic footpad (right) to the average perfusion value of the control, un-operated footpad (left) using Image J (NIH).
Optical Coherence Tomography
Doppler OCT and speckle-variance OCT were used to non-invasively image blood vessels in the ischemic gastrocnemius muscle of mice on days 1 and 13 after femoral artery ligation, as previously described.[37] Doppler OCT detects frequency shifts in the phase-sensitive OCT signal due to flowing blood, while speckle-variance OCT tracks variation in laser speckle over time due to red blood cell movement. Doppler OCT cross sectional scans (B-scans) were used to quantify blood flow changes over time in the hind limb, while volume intensity projections from speckle-variance OCT image volumes (C-scans) presented vessel morphology differences between groups. The OCT system uses an 860 nm center wavelength, 51 nm bandwidth laser, and has an axial resolution of 6.4 μm in air and lateral resolution of 25 μm. Prior to imaging, mice were anesthetized and hair on the hind limb was removed. To track the imaged area over time, glass microscope slides were marked with the placement of the mouse during imaging day 1 and used to correctly position the mouse leg during imaging on day 13. To avoid bulk motion artifacts, OCT scans of the calf muscle were gated between breaths of the mouse. Six Doppler OCT B-scans (4 mm, 800 A-scans, Doppler number of 9) were performed per mouse at each time point and perfusion was quantified by calculating the ratio of the number of blood vessel pixels per scan over the total imaged area per scan (n=6 mice per time point).
Angiogenesis and Phagocytosis Assays
Fourteen days after femoral artery ligation, mice were sacrificed and tissue and microgels were harvested for analysis. Immediately before sacrificing the mice, functional fluorescence microangiography was performed to visualize angiogenesis in and around peptide-loaded microgels, as described previously.[12] As a result of fluorescence microangiography, only the functional capillaries with a perfusion capacity, including those around injected microgels, show red fluorescence in the mouse body. Excised tissue from the site of microgel injections was imaged using an Olympus FV100 confocal microscope. For quantification of vessel perfusion capacity, the red fluorescence intensity was quantified using Image J software (n=6 images per mouse, n=6 mice per treatment).[38, 39]
A phagocytosis assay was performed in harvested microgels using Vybrant Phagocytosis Assay kit according to manufacturer's protocol.[40], [12] Green fluorescence from internalized E. coli particles in excised microgels and surrounding tissue was visualized through confocal imaging. The intensity of green fluorescence in each image was quantified using Image J (n=6 images per mouse, n=6 mice per treatment).
Histological Analysis of Angiogenesis and Inflammation
After sacrificing mice, ischemic muscle samples were prepared for histological analysis as described elsewhere.[41] Briefly, hind limbs were detached and placed in methanol overnight after removal of skin. Adductor muscle samples adjacent to the microgels were cut from the limb and placed in 10% phosphate buffered formalin for 24 hours, embedded in paraffin, sectioned (5 μm sections), mounted on slides, antigen retrieval, and stained with either hemotoxylin and eosin (H&E), or biotinylated rat anti-mouse F4/80 antibodies by the Vanderbilt Translational Pathology Shared Resource Core. Immunohistochemical (IHC) staining to identify activated inflammatory cells by F4/80 expression[42] was quantified by normalizing the total F4/80 positive area (indicated by brown staining) to the total cell number (determined by hematoxylin nuclear staining) using Image J.
Cell Culture
RAW 264.7 macrophages (ATCC) were cultured in DMEM (Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin. Mouse aortic endothelial cells (mAECs) were a generous gift from Dr. Ambra Pozzi at Vanderbilt University Medical Center. mAECs were cultured in EGM-2 Basal Media supplemented with BulletKit (Lonza, Allendale, NJ) and 10 units/mL IFN-γ (Sigma). RAW 264.7 mouse macrophage cells (Simga) were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin. For cell culture studies with peptides, 75 μg/mL of Ac-SDKP or C16 peptides was used. For inhibition studies, 5μM of MMP-9 inhibitor-1 (CTK8G1150; AG-L-66085, Santa Cruz Biotechnology, Dallas, TX)[43] or 5 μg/mL of LEAF Purified Mouse TNF-α antibody (BioLegend) was used.[44]
In Vitro Peptide Uptake
mAECs or RAW 264.7 cells were incubated with DilC12 (BD Biosciences) for 2 hours; washed two times with PBS; and seeded 3×105 cells/mL on pre-gelled injectable polymer microgels loaded with FITC-tagged Ac-SDKP or C16 peptides (75 μg peptide/mL media, GenScript). After 72 hours, cells were washed with PBS and imaged using Zeiss LSM 710 confocal microscope for visualization of peptide uptake (n=4 per treatment).
Gene Expression
mAECs or RAW 264.7 cells were seeded at a density of 3×105 cells/mL on TCPS with 75 μg/mL of Ac-SDKP, C16, or the combination of C16 and Ac-SDKP peptides. After 3 days, RNA was extracted from homogenized tissue using Trizol reagent and RNA easy columns. After dissolving RNA in RNase-free water, the concentration and purity of isolated RNA was measured using TECAN plate reader Nanoquant (company info). At least 1.2 μg of RNA was reverse transcribed using iScriptReverse transcription Supermix for RT-qPCR on a BioRad thermocycler (company info). 50 ng/well cDNA was then amplified using SYBR green Supermix and fluorescence signal was measured on a BioRAD real time PCR machine. TGFβ1 forward: GCTGAACCAAGGAGACGGAA, reverse: AGAAGTTGGCATGGTAGCCC. NF-κβ forward: ATGTAGTTGCCACGCACAGA, reverse: GGGGACAGCGACACCTTTTA. TIMP1 forward: AGACACACCAGAGATACCATGA, reverse: GAGGACCTGATCCGTCCACA. FGF-1 forward: TCTGAAGAGTGGGCGTAGGA, reverse: GGCTATTTGGGGCCATCGTA. FGF-2: MMP-9: TTGAGTCCGGCAGACAATCC, reverse: CCTTATCCACGCGAATGACG MMP-2 forward: GAGTTGGCAGTGCAATACCT, reverse: GCCGTCCTTCTCAAAGTTGT. TNF-α: forward: ACGGCATGGATCTCAAAGAC, reverse: AGATAGCAAATCGGCTGACG. VEGF forward: ATGCGGATCAAACCTCACCA, reverse: CCGCTCTGAACAAGGCTCAC. GAPDH forward: TGAAGCAGGCATCTGAGGG, reverse: CGAAGGTGGAAGAGTGGGAG TIMP-2 forward: CTCGCTGGACGTTGGAGGAA, reverse: CACGCGCAAGAACCATCACT. Expression was calculated using the 2-Cq method and normalized to GAPDH expression (n=8).
MMP-9 Activity
After 72 hours culture in serum-free media, culture media from mAECs or RAW 264.7 cells (3×105 cells/mL, with/without peptides or TNF-α/MMP-9 inhibitors) was collected, concentrated, and analyzed by zymography, as described previously.[45] Concentrated protein samples were incubated with non-reducing buffer at a ratio of 1:1. 9.5 μg total protein per lane were loaded onto a 7.5% polyacrylamide gel containing 0.1% (w/v) gelatin (Sigma). Gels were washed in dH2O containing 3.3% (v/v) Triton X-100, followed by 12-48 hour incubation in reaction buffer at 37 °C. After incubation, gels were placed in fixative (30% methanol, 10% acetic acid, and 60% dH2O) for 1 hour before staining with 4 parts Coomassie brilliant blue R-250 (Sigma) and 1 part methanol for 12 hours. Gels were destained in 25% methanol for 1 hr. The gelatinolytic activity of pro-MMP-9 was determined by densitometry of the 97KDa white band on a blue background using ImageJ (n=4 per treatment). MMP-9 expression was then normalized to the expression from the no inhibitor, no peptide treated group.
Phagocytic Activity
RAW 264.7 cells (3×105 cells/mL) were cultured with Ac-SDKP, C16, or the combination of C16 and Ac-SDKP peptides (75 μg/mL) in the presence or absence of TNF-α inhibitor or MMP-9 inhibitor for 72 hours. Vybrant phagocytosis assays were used to evaluate the inflammatory activity, as described above in the in vivo section. (n=4 per treatment).
Tubulogenesis
mAECs (3×105 cells/mL) were cultured on growth factor reduced matrigel (200 μL, BD Biosciences) for 6 hours before imaging for tube formation using a Nikon Eclipse Ti microscope (n=4 per treatment).
ELISA
To verify TNF-α inhibition with antibodies, an ELISA was performed using Mouse TNF-α ELISA MAX™ Deluxe (Biolegend) according to supplier's protocol. Secreted TNF-α in RAW 264.7 cell culture supernatant was measured by reading absorbance at 450nm using a TECAN M1000 plate reader and quantified against a standard curve (n=4 per treatment).
Statistics
To determine if statistical significance existed between groups, one-way ANOVA was performed between groups followed by Tukey's range tests for comparisons between groups. For all experiments, p < 0.05 was considered statistically significant and results were presented as means ± standard error of the mean.
Results
Injectable Polymer Fabrication and Characterization
The co-polymer of 21% PEG-79% PCL (%: molar ratio) was synthesized by reacting ε-caprolactone with methoxyPEG (mPEG) using tin(II) ethyl hexanoate as a catalyst (Fig. 1A). The number average molecular weight (Mn) of resulting polymer was 3571 Da as verified by NMR (Fig. S1). The polymer structure was verified by 1H NMR spectra: 1H NMR (CDCl3) = d 4.06 (t, 3H,-OCH2), 3.65 (s, 4H, -OCH2), 2.31(t, 2H, -CH2), 1.66 (m, 2H,-CH2), 1.37 (m, 4H, -CH2) ppm. This polymer was dissolved in water and easily mixed with peptides at 25°C. When the temperature increased to 37°C, it underwent successful sol-gel transition and formed a stable gel within 15 seconds (Fig. 1B).
Cell Viability with Injectable Polymer Microgels
To determine the biocompatibility of injectable polymer microgels, human coronary artery endothelial cells (HCAECs) were cultured on pre-gelled polymer microgels for three days. As seen in Fig. S2, HCAECs maintained viability (green by calcein AM) with only few dead cells (red by ethidium homodimer). Of note, some larger areas of red fluorescence are from the polymer itself due to autofluorescence, and not necessarily indicative of dead cells.
Peptide Uptake by Macrophages and Endothelial Cells
Fluorescein isothiocyanate (FITC)-tagged C16 and Ac-SDKP peptides were used in mouse aortic endothelial cells (mAECs) and macrophages (RAW 264.7 cells) cultures to examine cellular uptake of peptides. Both cell types internalized Ac-SDKP and C16 peptides as seen in Fig. S3 where C16 was confined in punctuate, distinct areas in the cells, while Ac-SDKP was diffusely present throughout the cells.
In Vivo Peptide Release from Injectable Polymer Microgels
To evaluate the ability of these peptide loaded-microgels to regulate angiogenesis and inflammation in PAD, a mouse model of hind limb ischemia was used. The A/J mouse strain was chosen due to its prolonged time course recovery from hind limb ischemia to mimic the delayed recovery seen in human PAD.[46] When polymer solutions were injected into the muscle at the site of femoral artery ligation, they rapidly formed a stable gel (Fig. 1C). Polymer microgels, mixed with or without peptides, were delivered either by a single, bulk, 10 μL injection or ten individual 1 μl injections into the adductor muscle adjacent to the ligation sites. Even 14 days after injection, the 10 μL bulk microgel was still visible without any significant change in size and gelation status. To measure peptide release from these injectable microgels in vivo, FITC-tagged Ac-SDKP was mixed with polymer solutions before injection. After 7 days, the single 10 μL bulk injection of microgel retained over 10 times more of the peptides than multiple 1 μL injections, indicating faster peptide release from multiple 1 μL injections than the single 10 μL bulk injection (Fig. 1D-E).
Macrophage Recruitment with Peptide-loaded Injectable Polymer Microgels
To investigate inflammatory responses to our peptide-loaded microgels at fourteen days after ligation and injection of microgels, the adductor muscle tissue adjacent to the microgels was sectioned and stained with hematoxylin and eosin (H&E) (Fig. 2A-F) and mouse macrophage marker F4/80 (Fig. 2G-I). Without peptide treatment, bulk injection of microgel resulted in muscle hypertrophy as indicated by varying muscle fiber size, replacement with fibrous and adipose tissues, and inflammatory cell infiltration (Fig. 2A, G); however, multiple 1μl microgel injections decreased this inflammatory response (Fig. 2C). With multiple 1μl microgel injections, the incorporation of anti-inflammatory Ac-SDKP peptides (Fig. 2D) further decreased inflammatory infiltration in the muscle tissue, while treatment with pro-angiogenic C16 (Fig. 2E) augmented macrophage infiltration by two fold as compared to no peptide treatment (Fig. 2C). However, co-treatment with C16 and Ac-SDKP successfully regenerated muscle tissue as indicated by centralized nuclei in regenerating muscle fibers in which macrophage infiltration was reduced by 70% in bulk injection (Fig. 2B, H) compared to no peptide treatment (Fig. 2A). The regeneration effect augmented further when co-treated in multiple, low volume microgel injections (Fig. 2F, I).
Figure 2. Macrophage infiltration with injectable microgels in ischemic muscle.
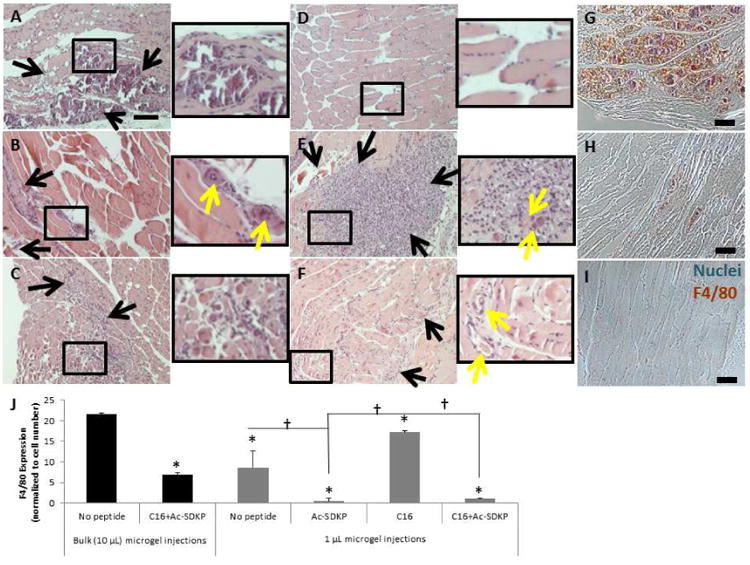
Sections of adductor muscle tissue adjacent to peptide-loaded injectable polymer microgels were stained with hematoxylin and eosin (H&E, A-F) or rat anti-mouse biotinylated F4/80 antibodies (a macrophage marker), as visualized by brown color in images (G-I). Nuclei were counterstained blue with hemalum. Scale bar =100 μm. Black rectangles indicate magnified area. Black arrows indicate inflammatory infiltration. Yellow arrows indicate blood vessels. Bulk 10 μL injection of microgels without peptide resulted in a high level in inflammatory infiltration (A, G), which was decreased with the co-treatment of C16 and Ac-SDKP peptides (B, H). Multiple 1 μL microgel injections (C) decreased the inflammatory response compared to the bulk injection (A). When delivered with multiple microgel injections, anti-inflammatory Ac-SDKP (D) minimized the inflammatory response, while C16 (E) increased the inflammatory response. The co-treatment (F, I) prevented inflammatory exacerbation while still forming blood vessels. (J) F4/80 staining was quantified by calculating the area of positively stained pixels divided by the total number of cells per image as measured by hematoxylin nuclear stain. Data are means ± SEM from four mice per condition. *p<0.05 vs. bulk injection with no peptide; †p<0.05 vs. groups connected by lines (Tukeys' Range Test).
Even with this co-treatment of the peptides in bulk injection (Fig 2B, H), the level of macrophage infiltration was still higher than the control PBS injection without microgel or peptides (Fig. S5F). When multiple 1 μL polymer microgel injections were used (Fig. 2I), the level of macrophage infiltration was successfully minimized to a similar level to the control PBS injection without polymer (Fig. S5J). These results suggest that multiple small volume injections of peptide-loaded microgels are more effective than bulk injection in attenuating the inflammatory response.
The recruitment of inflammatory cells also followed similar trends when the peptide-loaded implantable polymer scaffolds (Fig. S5A-D) were compared to the injectable polymer microgels (Fig. 2C-F), with C16 augmenting macrophage infiltration while Ac-SDKP diminished macrophage infiltration. The low level of macrophage infiltration observed under treatment of Ac-SDKP alone (Fig.2D) was preserved with the co-treatment of C16 and Ac-SDKP (Fig. 2F), confirming the therapeutic ability of these peptides for PAD. Therefore, multiple small volume microgel injections were used to deliver peptides in the following studies unless the injection method was specifically mentioned.
Perfusion Recovery with Peptide-loaded Microgels
Laser Doppler perfusion imaging (LDPI) and optical coherence tomography (OCT) were used to monitor blood perfusion to the ischemic hind limb over the course of 14 days. LDPI was conducted on the foot pads of the mouse hind limbs on 1, 3 7, and 14 days after femoral artery ligation. To quantify perfusion recovery in the ischemic hind limb, the perfusion in the right foot, in which the femoral artery and vein were ligated, was compared to perfusion in the left foot, which was left un-operated as an internal control. Without peptide or microgel treatment, perfusion in the ischemic right foot slightly increased over the course of 14 days, indicating minimal spontaneous recovery of function to the hind limb (Fig. 3 and Fig. S6). However, mice treated with microgels loaded with pro-angiogenic C16 had 29% higher perfusion compared to mice treated with microgels without peptides (Fig. 3). Anti-inflammatory Ac-SDKP loaded microgels did not increase perfusion compared to no peptide treatment (Fig. 3). Interestingly, the combination C16 and Ac-SDKP loaded microgels restored perfusion more effectively than any other treatment over the 14 day time course, suggesting a synergistic increase in tissue recovery with the dual peptide treatment (Fig. 3). LDPI also followed similar trends when the peptide-loaded implantable polymer scaffolds were compared to the injectable polymer microgels, with C16 increasing perfusion while Ac-SDKP decreased perfusion (Fig. S6). However, the combination of C16 and Ac-SDKP maintained the highest level of perfusion observed. The co-treatment of C16 and Ac-SDKP also preserved the minimal amount of macrophage infiltration (Fig. S5), confirming the therapeutic ability of these peptides.
Figure 3. Laser Dopper Perfusion Imaging (LDPI) of perfusion recovery from injection of peptide-loaded microgels.
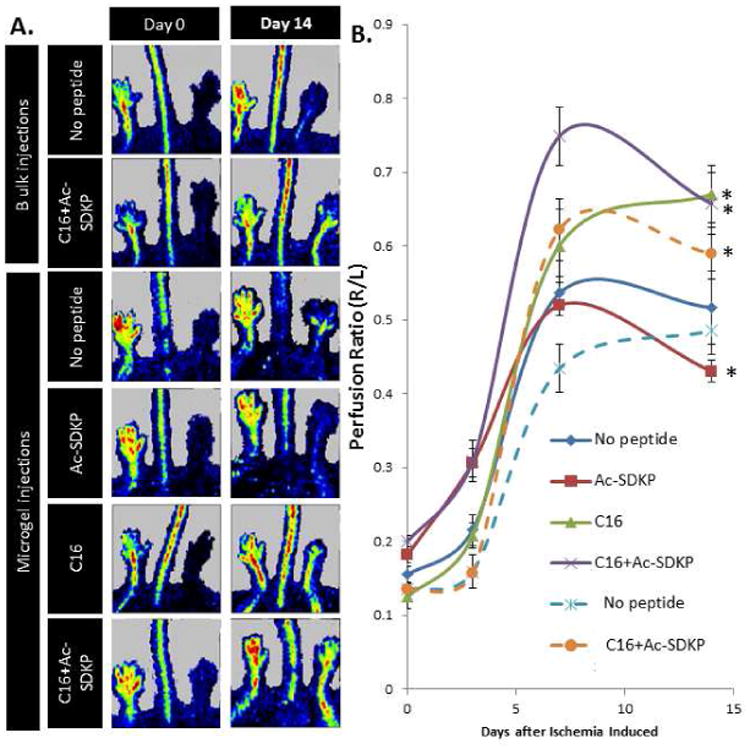
A) LDPI images of ischemic (right) and un-operated control (left) hind limbs at day 14 after femoral artery ligation and polymer injection with or without peptide loading. B) Perfusion was quantified as the ratio of right to left foot at each time point. Dashed lines represent a single bulk (10 μL) microgel injection, solid lines represent ten, individual 1 μL microgel injections. Data are means ± SEM from six mice per condition. *p<0.05 vs. no peptide treatment with microgels at day 14 (Tukeys' Range Test).
Doppler and speckle-variance OCT were also used to non-invasively image blood flow and vessel morphology in the hind limb, respectively. An increase in both the number and size of blood vessels in the ischemic calf muscle from 1 day to 13 days post-surgery with all treatment conditions was observed in cross-sectional Doppler OCT scans (Fig. S7). Quantification of Doppler OCT scans with implantable scaffolds revealed a similar trend to LDPI measurements: treatment with C16 alone or the combination of C16 and Ac-SDKP resulted in a greater than two fold increase in the total area of vessels over the time course compared to treatment with Ac-SDKP or no treatment (Fig. S7). No significant difference was observed between C16 alone and the co-treatment of C16 and Ac-SDKP, indicating the ability of this co-treatment to promote blood vessel formation. With injectable microgels, speckle-variance OCT volume scans of the calf muscle revealed Ac-SDKP or no peptide treatment formed few blood vessels with limited branching, while C16 or the combination of C16 and Ac-SDKP formed many vessels with a high degree of branching (Fig. 4A).
Figure 4. Angiogenesis and phagocytic activity in injectable microgels.
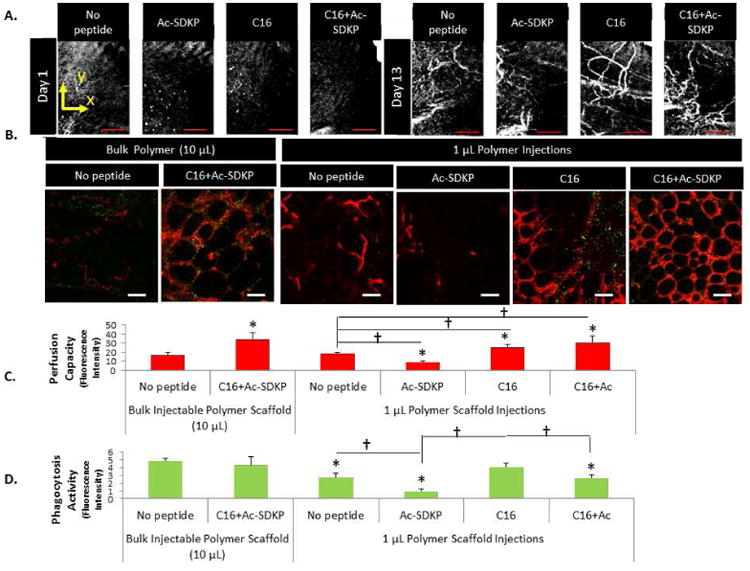
A) Maximum intensity projections of 3D speckle-variance OCT scans were taken of the mouse calf muscle at day 14 after femoral artery ligation and ten, 1 μL intramuscular injections of peptide-polymer microgels around the site of femoral artery ligation. Scale bar = 1mm. B) Blood vessel formation (red) and macrophages phagocytosing E.coli particles (yellow-green) were visualized in tissue with peptide-loaded injectable polymer microgels. Scale bar = 50 μm. C) Vessel perfusion capacity as measured by red fluorescence intensity of perfused microspheres extracted from microgels. D) Phagocytic activity (fluorescence intensity per image field). (C,D) Data are means ± SEM from four-six mice per condition. *p < 0.05 vs. no peptide treatment in same group (Bulk injection or 1μL injections of microgels); *p<0.05 vs. bulk injection with no peptide; †p<0.05 vs. groups connected by lines (Tukeys' Range Test).
Angiogenesis and Phagocytosis in Peptide-loaded Microgels
Fluorescence microangiography and a Vybrant phagocytosis assay were used to quantify angiogenesis and phagocytic activities, respectively in the tissue around microgels at the site of femoral artery ligations. We chose to measure phagocytosis because it is a crucial and potent indicator of inflammatory cell activation.[47] C16-loaded microgels enhanced angiogenesis and macrophage activities in the ischemic hind limb, while Ac-SDKP-loaded microgels reduced both responses in comparison to microgels without peptide loading (Fig. 4B-C). Slightly increased angiogenesis was observed upon PBS injections of C16 peptides compared to PBS alone (Fig. S8 A, C), while microgel-mediated C16 peptide delivery increased angiogenesis almost 1.5 fold versus microgels without peptides (Fig. 4B-C). Unfortunately, C16 delivered via microgels stimulated the inflammatory response as evidenced by higher phagocytic activity than no peptide controls. However, the incorporation of Ac-SDKP peptides abated this inflammatory response, lessening the phagocytic activity to levels comparable to PBS only. Co-delivery of both Ac-SDKP and C16 peptides increased perfusion capacity 1.7 fold versus microgels without peptides, while reducing phagocytosis to levels similar to no microgel controls, confirming our previous findings.[12]
Inflammatory activation, as quantified by a Vybrant phagocytosis assay, was highest with bulk injection of microgels, and was attenuated remarkably with multiple, low volume injections of microgels (Fig. 4C). With multiple small volume injections, phagocytosis decreased and perfusion capacity increased, indicating the improved therapeutic efficiency of these peptides when delivered via multiple small volume injections versus a single bulk injection.
In Vitro Evaluation of Angiogenesis and Inflammation by Gene Expression
Since the therapeutic efficiency of these peptides was demonstrated using injectable microgels in a mouse PAD model, we sought to elucidate a mechanism of uncoupling angiogenesis and inflammation. First, we analyzed expression of angiogenic and inflammatory genes (i.e. MMP-9, TIMP-1, and TNF-α) in mouse aortic endothelial cells (mAECs) and RAW 264.7 macrophages as an in vitro model of angiogenesis and inflammation, respectively (Fig. 5). Compared to no peptide treatment, MMP-9 expression in mAECs increased with C16 or co-treatment (both C16 and Ac-SDKP) over 2-fold, while its expression decreased with Ac-SDKP treatment over 50% (Fig. 5A), following trends similar to angiogenesis in vivo with peptide treatments. The expression of TIMP-1, an inhibitor of MMP-9, showed the opposite trend to MMP-9 expression in response to the peptide treatments in mAECs (Fig. 5B) as C16 and the combination of C16 and Ac-SDKP decreased TIMP-1 expression significantly. In RAW 264.7 macrophages, no significant differences were detected in MMP-9 or TIMP-1 expression, although there were some variations in TIMP-1 expression among the test groups (Fig. 5D,E). The overall expression level of TIMP-1 in RAW 264.7 was negligible when compared to that of mAECs and the overall expression level of MMP-9 in RAW 264.7, suggesting a minute role of TIMP-1 in MMP-9 activation in RAW 264.7 cells. These results illustrate the influence of MMP-9 on angiogenic processes within endothelial cells, while not having a significant effect on inflammatory cells or inflammatory processes in the peptide treatment conditions. However, expression of TNF-α in both mAECs and RAW 264.7 macrophages directly correlated with inflammatory activation as observed in vivo phagocytic activity and macrophage infiltration (Fig. 2 and Fig. 4D). C16 increased TNF-α expression, while Ac-SDKP and the combined peptide treatment decreased TNF-α expression in comparison to no peptide treatment (Fig. 5C,F), suggesting a regulatory role of TNF-α in the in vivo inflammatory response observed from the mouse PAD model.
Figure 5. In Vitro Evaluation of Angiogenesis and Inflammation by Gene Expression.
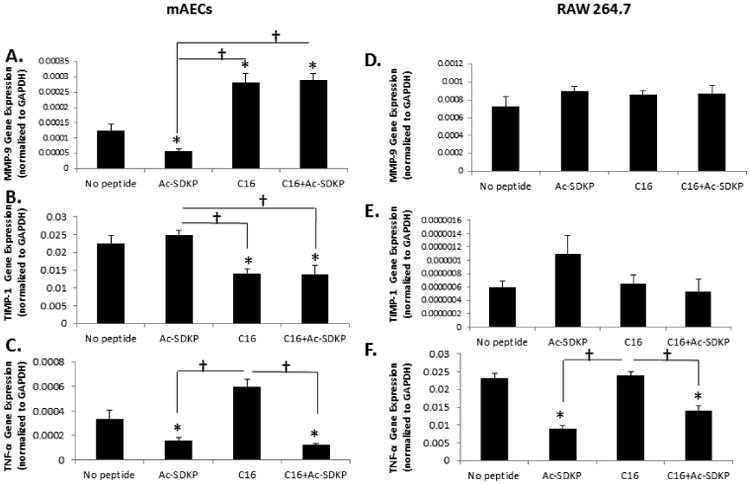
Mouse aortic endothelial cells (mAECs) or RAW 264.7 macrophages were cultured with 75 μg/mL of Ac-SDKP, C16, or the combination of C16 and Ac-SDKP peptides for 72 hours before isolating RNA and quantitative RT-PCR investigation. Gene expression of matrix metalloproteinase-9 (MMP-9) (A,D), tissue inhibitor-1 (TIMP-1), an inhibitor of MMP-9 (B, E), and tumor necrosis factor-α (TNF-α) (C, F) in mAECs (A, B,C) and RAW 264.7 macrophages (D, E, F). Expression was normalized to GAPDH. Data are means ± SEM from eight replicate experiments. (n=8) *p<0.05 vs. no peptide; † p<0.05 between groups connected by lines (Tukeys' Range Test).
TNF-α and MMP-9 Inhibition
To further investigate these mechanisms, inhibitors of TNF-α and MMP-9 were used in cell culture studies. To verify TNF-α inhibition, an ELISA assay was performed. The use of TNF-α antibodies as natural TNF-α inhibitors successfully abrogated cellular production of TNF-α in all the test conditions (Fig. 6A). Regardless of TNF-α inhibitor treatment, Ac-SDKP and the combination peptide–treated macrophages secreted the least amounts of TNF-α (Fig. 6A). To determine if TNF-α influenced MMP-9 activation in response to the peptide treatments, the amount of active MMP-9 produced from macrophages in each condition of peptide treatment was measured by zymography after culturing for 72 hours with or without treatment of TNF-α inhibitors. No significant differences were detected in the MMP-9 activities between the no inhibitor and TNF-α inhibitor-treated groups of macrophages in all the test conditions of peptide treatment (Fig. 6B-C). Regardless of TNF-α inhibition, significantly higher levels of MMP-9 activity were observed in macrophages treated with C16 and with the co-treatment of C16 and Ac-SDKP compared to macrophages without peptide treatment. These results indicate that TNF-α inhibition did not influence MMP-9 activity in the peptide treatment conditions. When phagocytic activities were measured using Vybrant phagocytosis assay, TNF-α inhibition minimized the macrophage phagocytic activities in all the test peptide treatment conditions (Fig. 7), confirming that TNF-α is a major regulator of inflammatory responses under peptide treatments (Fig 5. C, F).
Figure 6. Macrophage response to TNF-α inhibition.
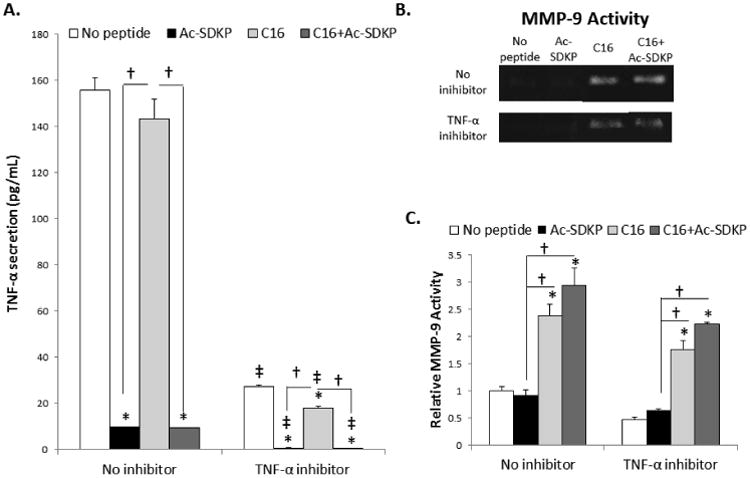
RAW 264.7 macrophages were cultured with 75 μg/mL of Ac-SDKP, C16, or the combination of C16 and Ac-SDKP peptides for 72 hours with TNF-α antibodies as soluble TNF-α inhibitors (5 μg/mL). A) TNF-α activity in cell culture supernatants was measured by ELISA. B) MMP-9 activity was measured by zymography. C) MMP-9 activity was quantified by densiometry using Image J and normalized to the average activity of no peptide/no inhibitor treatment condition. Data are means ± SEM from four replicate experiments. *p<0.05 vs. no peptide in same condition (no inhibition or TNF-α inhibition); † p<0.05 between groups connected by lines; ‡p<0.05 vs. no inhibition with same peptide treatment (Tukeys' Range Test).
Figure 7. Phagocytic Activity of RAW 264.7 macrophages with TNF-α inhibition.
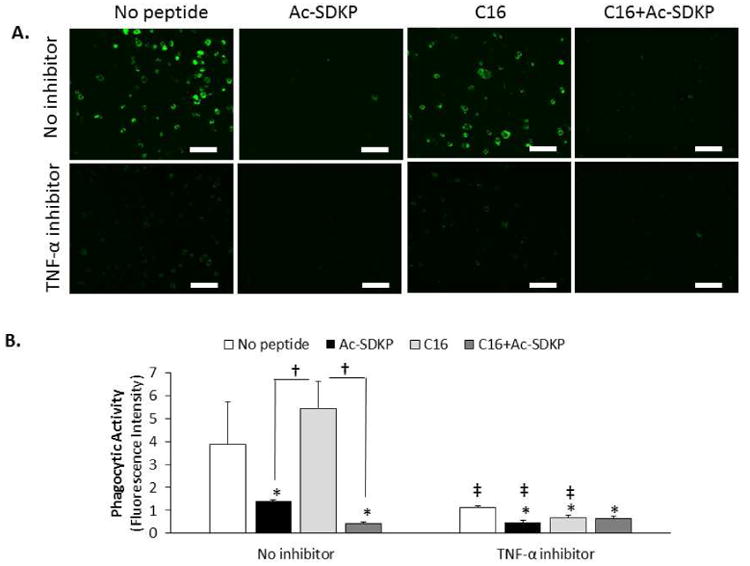
A) RAW 264.7 macrophages were cultured with peptides (75 μg/mL) and TNF-α antibodies (5 μg/mL) as an inhibitor of TNF-α for 72 hours. Images representative of phagocytic activity of macrophages after incubating with green fluorescent E. coli particles (n=4). Scale bar = 100 μm. B) Phagocytic activity was quantified by the green fluorescence intensity. Data are means ± SEM. *p < 0.05 vs. no peptide treatment in same condition (no inhibition or TNF-α inhibition); † p<0.05 between groups connected by lines; ‡p<0.05 vs. same peptide treatment without inhibitor (n=4; Tukeys' Range Test).
MMP-9 activity was also investigated using a molecular inhibitor of MMP-9.[43] In endothelial cells, MMP-9 inhibition was verified by the attenuation of MMP-9 activity as measured by zymography (Fig. 8A-B). While TNF-α inhibition did not significantly influence MMP-9 activity or tubulogenesis in endothelial cells, these activities were significantly reduced when C16 and MMP-9 inhibitor were co-treated (Fig. 8A-C). Without TNF-α inhibitors, MMP activation and expression, as well as tubulogenesis upon peptide treatments followed similar trends to angiogenesis in vivo. Particularly, C16 or the co-treatment of C16 and Ac-SDKP augmented MMP-9 activity and tubulogenesis, while these effects were diminished with Ac-SDKP treatment (Fig. 8). MMP-9 inhibition reduced these activities significantly to similar levels of Ac-SDKP treatment. These results indicate that angiogenesis was regulated by MMP-9 independently of TNF-α, thereby serving a major mechanism for uncoupling angiogenesis and inflammation under the co-treatment of C16 and Ac-SDKP.
Figure 8. mAECs response to TNF-α and MMP-9 inhibition.
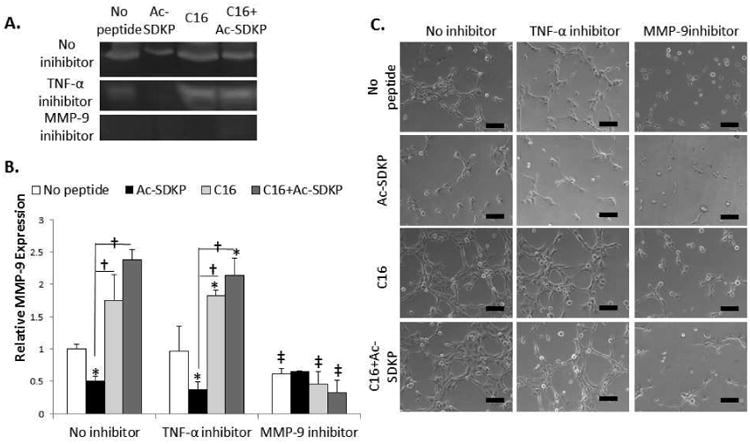
A-B) Mouse aortic endothelial cells (mAECs) were cultured with 75 μg/mL of Ac-SDKP, C16, or the combination of C16 and Ac-SDKP peptides for 72 hours with MMP-9 inhibitors (5 μM) or TNF-α antibodies as soluble TNF-α inhibitors (5 μg/mL). A) MMP-9 activity was measured by zymography. B) MMP-9 activity was quantified by densiometry using Image J and normalized to the average activity of no peptide/no inhibitor treatment. C) mAECs were cultured on growth factor reduced matrigel for 6 hours before imaging tube formation. Images are representative of 4 replicate experiments. Scale bar = 100 μm. Data are means ± SEM. *p<0.05 vs. no peptide in same condition (no inhibition or TNF-α inhibition); †p<0.05 between groups connected by lines; ‡p<0.05 vs. no inhibition with same peptide treatment (Tukeys' Range Test).
Discussion
We first demonstrated the new therapeutic effect of combined C16 and Ac-SDKP on PAD when delivered via an injectable polymer scaffold system to mouse hind-limb ischemia. This new therapeutic approach promotes angiogenesis while reducing inflammation in a mechanistically uncoupled manner, providing a new idea to the field of PAD therapy with high translational potential.
Although several surgical and non-surgical treatments are available for patients with PAD, there is an unmet need to restore blood flow to ischemic tissues while avoiding detrimental inflammation and other side effects in a minimally-invasive format for the 50% of patients with PAD that are ineligible for surgical interventions. While other studies have used VEGF, FGF, PDGF, GM-CSF, MCP-1, or bFGF in animal models of PAD,[41, 48-54] only bFGF has been used thus far in clinical trials.[55-57] The first of these trials showed no adverse effects in the short term study;[55] however, the second trial by Cooper et al. in 2001 found no positive effects with bFGF treatment and reported the negative side effect of severe proteinuria (excess proteins excreted in urine).[56] For these reasons the study was terminated prematurely. The third clinical trial did report increased peak walking time without increased incidence of death or cardiac events in patients treated with bFGF, but also noted the high incidence of proteinuria.[57]
Biomaterial systems have also been used to control the delivery of bFGF via gelatin microspheres in a phase 1 clinical trial.[58] While this trial demonstrated promising results of improved perfusion and transcutaneous oxygen pressure, no placebo controls were used in this study to verify these findings. Other randomized clinical trials of bFGF administration in PAD patients have not demonstrated improvement versus placebo controls.[2] In addition, the synthesis of the gelatin microspheres required the use of glutaraldehyde - a highly cytotoxic crosslinking agent.[59, 60] The injectable microgel system used in our study avoids toxic agents and instead utilizes biocompatible polymer systems consisting of PEG and PCL which can be tuned for controlled peptide release without the need for chemical crosslinking. Our study also reduces the cost of treatment by utilizing economical peptides in lieu of costly proteins such as bFGF.
One explanation for the limited success of the use of single growth factors may be that PAD treatments are complicated by the interplay between angiogenesis and inflammation in this pathogenesis.[61-67] Careful regulation of inflammation and angiogenesis is needed to treat PAD as some level of inflammation is needed for the initiation of angiogenesis to promote collateral vessel formation and restore blood flow to ischemic tissues. In this study we used small peptides – pro-angiogenic C16 and anti-inflammatory Ac-SDKP - which take into consideration for controlling both angiogenic and inflammatory responses in PAD. The incorporation of this dual peptide treatment into an injectable polymer microgel proved to promote recovery of ischemic hind limbs in a mouse model of PAD while minimizing inflammatory responses.
The current study used LDPI and OCT imaging techniques to image blood flow and perfusion recovery in the model of PAD. These methods are advantageous over traditional imaging methods such as MRI and CT as they can be performed non-invasively in vivo and do not require a contrast agent.[36, 68] LDPI is non-invasive and semi-quantitative, with its ease of use making LDPI the gold standard for measuring recovery from hind limb ischemia. OCT is more sensitive than LDPI, with a higher resolution; however it is also depth limited. While LDPI could only accurately measure surface perfusion (within 200 μm) in the footpad, OCT can be used to image blood vessels up to 2 mm in depth of the ischemic calf muscle. According to results from LDPI, OCT, fluorescent microangiography, histological approaches, and Vybrant phagocytosis assays, the perfusion recovery and inflammatory activation with peptide-loaded implantable scaffolds were similar to trends seen with multiple 1 μL injections of peptide-loaded microgels. Specifically, C16 and C16 in combination with Ac-SDKP restored perfusion in the hind limb to the highest levels of all treatments tested (Fig. S6, S7, S8). Ac-SDKP decreased perfusion compared to no peptide treatment (Fig. S6, S7, S8). Phagocytic activity and macrophage infiltration also increased with C16 compared to no peptide treatment whereas Ac-SDKP decreased these inflammatory responses (Fig. S5, S8). The combination of C16 and Ac-SDKP maintained the low levels of phagocytic activity and macrophage infiltration observed with Ac-SDKP treatment alone.
Injectable polymer microgels provide an effective method for delivering functional peptides to the site of ischemia. Microgels were fabricated from biocompatible, biodegradable, combinatorial polymers PEG and PCL (Fig. 1A). [19, 20] The sol-gel transition of the injectable polymer microgels used in this study allows for easy mixing of peptides into the polymer solution at room temperature. When injected to a target site, the peptides are stably encapsulated in a gel at the body temperature, keeping them in close proximity to the site of arterial blockage. The rapid sol-gel transition observed with the 21%PEG-79% PCL polymer minimizes the flow of injectable microgel and a loss of peptides. As compared to peptides delivered in PBS solution, injectable polymer microgels resulted in greater effects on angiogenesis and inflammation, indicating the need of our injectable polymer microgels to retain peptides at the site of ischemia for improved therapeutic efficiency. However, peptide delivery via a single bulk injection of polymer microgel did not alter angiogenesis or inflammation as significantly as multiple, small volume injections of microgels (ten, 1 μL injections). Small volume microgel injections may have released peptides more rapidly due to the increased surface area to volume ratio, as evident by IVIS imaging of peptide release (Fig. 1D-E). The PBS injection may release peptides too quickly to have pro-longed effects on angiogenesis or inflammation. Multiple small volume injections of polymer microgel provide an ideal time course for peptide release in this model.
Peptides were used for this study in lieu of growth factors due to their lower cost. Without loading in microgels, peptides injected in PBS did not significantly alter any of the measured outcomes, indicating the need for microgels to sustain release of peptides to the surrounding tissue. When incorporated into polymer microgels, anti-inflammatory Ac-SDKP decreased phagocytic activity and macrophage infiltration, successfully minimizing the host inflammatory response to the polymer microgels and avoiding potential aggravation of inflammatory activated endothelium in occluded blood vessels. However, Ac-SDKP treatment alone slightly decreased angiogenesis or perfusion in the hind limb, suggesting the treatment of Ac-SDKP alone is not suitable for restoring function to ischemic limbs affected by PAD. Pro-angiogenic C16 loaded microgels increased angiogenesis and perfusion to the ischemic hind limb; however they also resulted in increased inflammation with increased phagocytic activity and macrophage infiltration compared to no peptide treatment. This high level of inflammatory response is concerning when considering translating these therapies to human patients with inflamed arteries. Therefore, a pro-angiogenic treatment without inflammatory exacerbation was sought. Microgels loaded with C16+Ac-SDKP resulted in increased blood perfusion to the ischemic hind limb, as evaluated by LDPI, OCT, and fluorescent microangiography, as well as limited inflammatory response as evaluated by Vybrant phagocytosis, histology, and F4/80 staining. The treatment with pro-angiogenic C16 in combination with anti-inflammatory Ac-SDKP provided optimal collateral angiogenesis without detrimental inflammation, suggesting an ideal treatment for PAD by regulating angiogenesis and inflammation independently. Quantification of perfusion capacity and phagocytic activity directly correlated with results obtained from peptide-loaded scaffolds in a subcutaneous model, which confirms our previous work.[12] The site-specific delivery of these peptides prevents unintended vascularization or inflammatory reduction in other tissues - such as retinal neovascularization or reduction of alveolar macrophage activity.[51] At the site of ischemia, however, blood flow was increased, fibrosis and detrimental inflammation (as measured by phagocytic activity and macrophage infiltration) were minimized, and tissue necrosis was prevented by our minimally-invasive, site-specific delivery of the therapeutic peptides.
A mechanism of uncoupling angiogenesis and inflammation by co-treatment of C16 and Ac-SDKP was investigated in vitro. The overall expression level of MMP-9 in Raw 264.7 was higher than that of mAECs (Fig, 5A, D), and the overall expression level of TIMP-1 in Raw 264.7 was negligible compared to that of mAECs (Fig. 5 B,E), indicating that macrophages might be a major regulator of MMP-9 activation in vivo. However, the numbers of F4/80 positive cells (Fig. 2G-J) and angiogenic endothelial cells forming vessels (Fig. 3&4) at the injection sites of the mouse PAD model varied significantly among the peptide conditions. Considering these facts, it is suggested that MMP-9 activation regulated by its gene expression and TIMP-1 together with the number of cells producing MMP-9 played a cooperative role in modulating angiogenic response at the ligation sites in vivo. When TNF-α inhibitors were co-treated with peptides, MMP-9 activation did not change in both macrophages and endothelial cells with intact tubulogenesis, but the phagocytic activity of macrophages significantly decreased. On the other hand, MMP-9 inhibition significantly reduced tubulogenic activity of endothelial cells. Taken together, these results suggest that the inflammatory effects of peptide treatments were mediated by TNF-α secretion, while the angiogenic effects were mediated by MMP-9 activity. We also demonstrated that the regulation of inflammation through TNF-α was independent of MMP-9 mediated angiogenesis.
These findings are consistent with a recent study by Camargo et al. which proved independent modulation of TNF-α without affecting NF- κβ, a transcription factor for MMP-9.[69] Many other factors are known to regulate MMP-9 besides TNF-α, including the inflammatory cytokines IL-1β and IL-1α.[26, 70, 71] In fact, in a pivotal study by Bond et al, IL-1β was proven to be a more potent promoter of MMP-9 than TNF-α.[71] Without the synergistic effects of PDGF or bFGF, TNF-α did not significantly stimulate MMP-9 activity, indicating the need for combined cytokines and growth factors to stimulate maximal MMP-9 secretion.[71] However, IL-1α alone did stimulate low levels of MMP-9 activity, and even higher levels when combined with PDGF or bFGF. TNF-α and IL-1α activate Nf-κβ, whereas bFGF and PDGF activate the ERK-1/ERK-2 MAPK pathway resulting in activation of AP-1, another transcription factor of MMP-9.[71, 72] As explained by Bond et al., their results indicate that both AP-1 and NF-κβ are required for MMP-9 activation.[71] The binding regions of these promoters are proximal to each other, allowing for interaction. Therefore multiple signal transduction pathways are needed for MMP-9 expression, with either TNF-α or IL-β required for Nf-κβ activation. In our study, inflammation was modulated independently of angiogenesis. MMP-9 expression was maintained during inhibition of TNF-α, possibly due to alternate mechanisms of NF-κβ activation by IL-1 and/or AP-1 stimulation by PDGF or bFGF. The independent control of angiogenesis and inflammation should contribute to clinical translation of our approach as an optimal PAD treatment.
Future work will investigate the influence NF-κβ, AP-1, IL-1, PDGF, and bFGF on our pro-angiogenic and anti-inflammatory peptide effects. Our study design did not determine whether changes in the dose and duration or repeated administration enhanced the therapeutic benefit of Ac-SDKP and C16. Further optimization of the dose and time course of delivery of these peptides may greatly improve the therapeutic effect and translational potential. Although this treatment alone may not to be sufficient to recover limb function in patients with CLI, in combination with minimally-invasive bypass grafting, stenting, or other procedures/therapies this peptide-polymer delivery system may be useful in a clinical setting.
Conclusion
We developed a clinically translatable microgel system that employs a unique peptide combination to achieve the dual therapeutic effects, promoting angiogenesis and minimizing inflammatory responses in an uncoupled fashion. The results suggest a promising option for potential treatment of PAD in a minimally-invasive way due to the gel injectability at the microscale. By regulating pathways involved in anti-inflammatory and pro-angiogenic effects independently, the dual peptide treatment will significantly improve safe recovery of ischemic tissues in patients with PAD without invasive surgery, thereby addressing the issues noticed from the current clinical trials.
Supplementary Material
Figure S1. Characterization of polymer microgel system. NMR spectra of injectable microgel co-polymer. Number average molecular weight (Mn) was determined to be 3571 Da as calculated by the molar ratio between PEG and PCL.
Figure S2. Biocompatibility of injectable polymer microgels. Human coronary artery endothelial cells (HCAECs) were cultured on top of pre-gelled injectable polymer microgels. Live cells were stained by calcein AM (green, top row) and dead cells were stained by ethidium homodimer (red, middle row). The injectable polymer microgels exhibit some red autofluorescence (left column) that is not indicative of dead cells. Images are representative of n=4 biological replicates. Scale bar = 50 μm.
Figure S3. Peptide Uptake by Macrophages and Endothelial Cells. RAW 264.7 macrophages or mouse aortic endothelial cells (mAECs) were prelabeled with DilC12 (red, top row) and cultured with FITC-labeled Ac-SDKP or C16 (green, middle row) for 72 hours. Images are representative of n=4 biological replicates. Scale bar = 20 μm.
Figure S4. Implantable Scaffold Fabrication and Characterization. A) Synthesis schematic of combinatorial polymers for implantable scaffold fabrication. B) SEM of porous structure of implantable scaffolds after salt leaching. Scale bar = 200 μm.
Figure S5. Macrophage infiltrations into implantable scaffolds in ischemic muscle. Sections of adductor muscle tissue adjacent to the implantation site of peptide-loaded scaffold or the injection site of peptide-loaded PBS were stained with hematoxylin and eosin (A-D) or rat anti-mouse biotinylated F4/80 antibodies (a macrophage marker), as visualized by brown color in images (E-J). Nuclei were counterstained blue with hemalum. The conditions include with scaffold (A-D, F-J), PBS without scaffold (E), no peptide (A, E, F), Ac-SDKP (B, G), C16 (C, I), C16+Ac (D, J), and LPS (H). F4/80 staining was quantified by calculating the area of positively stained pixels divided by the total number of cells per image as measured by hematoxylin nuclear stain (K). n=4 mice per condition. Scale bar = 100 μm.
Figure S6. Laser Dopper Perfusion Imaging (LDPI) of perfusion recovery in implantation of peptide-loaded scaffolds. A) LDPI images of ischemic (right) and control (left) hind limbs after femoral artery ligation and scaffold implantation with or without peptide loading. As controls, peptides were intramuscularly injected in PBS without scaffolds. B) Perfusion was quantified as the ratio of right to left foot at each time point. Dashed lines represent peptides in PBS injections without scaffolds; solid lines represent peptide-loaded scaffold treatments. n=6 mice per condition. *p<0.05 vs. no peptide treatment with scaffolds at day 14.
Figure S7. Optical Coherence Tomography (OCT) imaging of blood vessel formation with implantation of peptide-loaded scaffolds. A) Doppler OCT B-scans were taken of the calf muscle of mice after femoral artery ligation and peptide-loaded scaffold implantation. Blood vessels appear red or blue (depending on direction of blood flow) overlaid on tissue morphology (gray). B-scans show the surface of the skin at the top of the image and increase in depth into the calf muscle until signal can no longer be accurately detected at the bottom of the image. Scans are 1.8 mm in depth and 4 mm in width. B) Quantification of vessel formation was performed by dividing the total area of vessels in each image by the total imaged area. All groups are with implanted scaffolds. n=6 mice per condition. *p<0.05 vs. no peptide on day 13 after femoral artery ligation and scaffold implantation.
Figure S8. Angiogenesis and phagocytosis assay in peptide-loaded implanted scaffolds. Blood vessel formation (red) and macrophages phagocytosing E.coli particles (yellow-green) were visualized in tissue after injection of peptides or PBS vehicle (A) without or (B) with implanted polymer scaffolds. Scale bar=50 μm. C) Vessel perfusion capacity as measured by red fluorescence intensity of perfused microspheres extracted from scaffolds. D) Phagocytic activity (fluorescence intensity per image field). (C, D) *p < 0.05 vs. no peptide treatment in same group (without or with scaffold).
Acknowledgments
This study was supported by NIH HL091465, NSF DMR 1006558, and AHA Predoctoral Fellowship 12PRE12070196. The authors would also like to acknowledge the use of resources at the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE), a facility renovated under NSF ARI-R2 DMR-0963361. Confocal images were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). 1H-NMR was conducted in the Small Molecule NMR Facility Core, and the Translational Pathology Shared Resources Core assisted in the preparation and staining of histological specimens. The authors would also like to acknowledge Dr. Ambra Pozzi for providing mouse aortic endothelial cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Angela L. Zachman, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Xintong Wang, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA.
Jason M. Tucker-Schwartz, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Sean T. Fitzpatrick, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Sue H. Lee, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Scott A. Guelcher, Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA
Melissa C. Skala, Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacilli A, Faggioli G, Stella A, Pasquinelli G. An Update on Therapeutic Angiogenesis for Peripheral Vascular Disease. Ann Vasc Surg. 2010;24:258–68. doi: 10.1016/j.avsg.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Gardner AW, Parker DE, Montgomery PS, Sosnowska D, Casanegra AI, Ungvari Z, et al. Gender and racial differences in endothelial oxidative stress and inflammation in patients with symptomatic peripheral artery disease. J Vasc Surg. doi: 10.1016/j.jvs.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Signorelli SS, Fiore V, Malaponte G. Inflammation and peripheral arterial disease: the value of circulating biomarkers (Review) Int J Mol Med. 2014;33:777–83. doi: 10.3892/ijmm.2014.1657. [DOI] [PubMed] [Google Scholar]
- 5.Llorente-Cortes V, de Gonzalo-Calvo D, Orbe J, Paramo JA, Badimon L. Signature of subclinical femoral artery atherosclerosis in peripheral blood mononuclear cells. Eur J Clin Invest. 2014;44:539–48. doi: 10.1111/eci.12267. [DOI] [PubMed] [Google Scholar]
- 6.Alexandru N, Popov D, Georgescu A. Platelet dysfunction in vascular pathologies and how can it be treated. Thromb Res. 2011 doi: 10.1016/j.thromres.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 7.Bennett PC, Silverman S, Gill P, Lip GYH. Peripheral Artery Disease and Angiogenesis: A Link Between Angiogenesis and Atherothrombosis. In: Slevin M, editor. Therapeutic Angiogenesis for Vascular Diseases. Springer; Netherlands: 2011. pp. 339–59. [Google Scholar]
- 8.Grisar JC, Haddad F, Gomari FA, Wu JC. Endothelial progenitor cells in cardiovascular disease and chronic inflammation: from biomarker to therapeutic agent. Biomark Med. 2011;5:731–44. doi: 10.2217/bmm.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleiner M, Kummer M, Mirlacher M, Sauter G, Cathomas G, Krapf R, et al. Arterial neovascularization and inflammation in vulnerable patients - Early and late signs of symptomatic atherosclerosis. Circulation. 2004;110:2843–50. doi: 10.1161/01.CIR.0000146787.16297.E8. [DOI] [PubMed] [Google Scholar]
- 10.Bailey LO, Washburn NR, Simon CG, Chan ES, Wang FW. Quantification of inflammatory cellular responses using real-time polymerase chain reaction. J Biomed Mater Res A. 2004;69A:305–13. doi: 10.1002/jbm.a.20134. [DOI] [PubMed] [Google Scholar]
- 11.Hu WJ, Eaton JW, Tang LP. Molecular basis of biomaterial-mediated foreign body reactions. Blood. 2001;98:1231–8. doi: 10.1182/blood.v98.4.1231. [DOI] [PubMed] [Google Scholar]
- 12.Zachman AL, Crowder SW, Ortiz O, Zienkiewicz K, Bronikowski CM, Yu SS, et al. Pro-angiogenic and anti-inflammatory regulation by functional peptides loaded in polymeric implants for soft tissue regeneration. Tissue Eng Part A. 2012 doi: 10.1089/ten.tea.2012.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuratomi Y, Nomizu M, Tanaka K, Ponce ML, Komiyama S, Kleinman HK, et al. Laminin gamma 1 chain peptide, C-16 (KAFDITYVRLKF), promotes migration, MMP-9 secretion, and pulmonary metastasis of B16-F10 mouse melanoma cells. Br J Cancer. 2002;86:1169–73. doi: 10.1038/sj.bjc.6600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponce ML, Nomizu M, Delgado MC, Kuratomi Y, Hoffman MP, Powell S, et al. Identification of endothelial cell binding sites on the laminin gamma 1 chain. Circ Res. 1999;84:688–94. doi: 10.1161/01.res.84.6.688. [DOI] [PubMed] [Google Scholar]
- 15.Malinda KM, Nomizu M, Chung M, Delgado M, Kuratomi Y, Yamada Y, et al. Identification of laminin alpha1 and beta1 chain peptides active for endothelial cell adhesion, tube formation, and aortic sprouting. FASEB J. 1999;13:53–62. [PubMed] [Google Scholar]
- 16.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, et al. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–36. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tziampazis E, Kohn J, Moghe PV. PEG-variant biomaterials as selectively adhesive protein templates: model surfaces for controlled cell adhesion and migration. Biomaterials. 2000;21:511–20. doi: 10.1016/s0142-9612(99)00212-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu E, Treiser MD, Patel H, Sung HJ, Roskov KE, Kohn J, et al. High-content profiling of cell responsiveness to graded substrates based on combinyatorially variant polymers. Comb Chem High Throughput Screen. 2009;12:646–55. doi: 10.2174/138620709788923737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu SS, Koblin RL, Zachman AL, Perrien DS, Hofmeister LH, Giorgio TD, et al. Physiologically Relevant Oxidative Degradation of Oligo(proline) Cross-Linked Polymeric Scaffolds. Biomacromolecules. 2011;12:4357–66. doi: 10.1021/bm201328k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta MK, Walthall JM, Venkataraman R, Crowder SW, Jung DK, Yu SS, et al. Combinatorial Polymer Electrospun Matrices Promote Physiologically-Relevant Cardiomyogenic Stem Cell Differentiation. PLoS One. 2011;6:e28935. doi: 10.1371/journal.pone.0028935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutowska A, Jeong B, Jasionowski M. Injectable gels for tissue engineering. Anat Rec. 2001;263:342–9. doi: 10.1002/ar.1115. [DOI] [PubMed] [Google Scholar]
- 22.Minh KN, Lee DS. Injectable Biodegradable Hydrogels. Macromol Biosci. 2010;10:563–79. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- 23.Chen LF, Greene WC. Shaping the nuclear action of NF-kappa B. Nat Rev Mol Cell Bio. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh S, Karin M. Missing pieces in the NF-kappa B puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Liang HP, Castrillon DH, DePinho RA, Olson EN, Liu ZP. FoxO4 regulates tumor necrosis factor alpha-directed smooth muscle cell migration by activating matrix metalloproteinase 9 gene transcription. Mol Cell Biol. 2007;27:2676–86. doi: 10.1128/MCB.01748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tseng HC, Lee IT, Lin CC, Chi PL, Cheng SE, Shih RH, et al. IL-1 beta Promotes Corneal Epithelial Cell Migration by Increasing MMP-9 Expression through NF-kappa B- and AP-1-Dependent Pathways. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. New Engl J Med. 1996;334:1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 28.Sherry B, Cerami A. Cachectin Tumor Necrosis Factor Exerts Endocrine, Paracrine, and Autocrine Control of Inflammatory Responses. J Cell Biol. 1988;107:1269–77. doi: 10.1083/jcb.107.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YCE, Mathison J, et al. Identity of Tumor Necrosis Factor and the Macrophage-Secreted Factor Cachectin. Nature. 1985;316:552–4. doi: 10.1038/316552a0. [DOI] [PubMed] [Google Scholar]
- 30.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: The next generation. Nat Rev Drug Discov. 2003;2:736–46. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 31.Maiuri MC, Tajana G, Iuvone T, De Stefano D, Mele G, Ribecco MT, et al. Nuclear factor-kappa B regulates inflammatory cell apoptosis and phagocytosis in rat carrageenin-sponge implant model. Am J Pathol. 2004;165:115–26. doi: 10.1016/s0002-9440(10)63280-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowder SW, Gupta MK, Hofmeister LH, Zachman AL, Sung HJ. Modular polymer design to regulate phenotype and oxidative response of human coronary artery cells for potential stent coating applications. Acta Biomaterialia. 2012;8:559–69. doi: 10.1016/j.actbio.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gimenez S, Ponsart S, Coudane J, Vert M. Synthesis, properties and in vitro degradation of carboxyl-bearing PCL. J Bioact Compat Pol. 2001;16:32–46. [Google Scholar]
- 34.Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Protoc. 2009;4:1737–48. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- 35.Mercanzini A, Reddy ST, Velluto D, Colin P, Maillard A, Bensadoun JC, et al. Controlled release nanoparticle-embedded coatings reduce the tissue reaction to neuroprostheses. J Control Release. 2010;145:196–202. doi: 10.1016/j.jconrel.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 36.Wardell K, Jakobsson A, Nilsson GE. Laser Doppler perfusion imaging by dynamic light scattering. IEEE Trans Biomed Eng. 1993;40:309–16. doi: 10.1109/10.222322. [DOI] [PubMed] [Google Scholar]
- 37.Poole KM, Tucker-Schwartz JM, Sit WW, Walsh AJ, Duvall CL, Skala MC. Quantitative optical imaging of vascular response in vivo in a model of peripheral arterial disease. Am J Physiol Heart Circ Physiol. 2013;305:H1168–80. doi: 10.1152/ajpheart.00362.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues - Potential role in capillary branching. Circ Res. 2004;94:262–8. doi: 10.1161/01.RES.0000111527.42357.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sung HJ, Johnson CE, Lessner SM, Magid R, Drury DN, Galis ZS. Matrix metalloproteinase 9 facilitates collagen remodeling and angiogenesis for vascular constructs. Tissue Eng. 2005;11:267–76. doi: 10.1089/ten.2005.11.267. [DOI] [PubMed] [Google Scholar]
- 40.Foukas LC, Katsoulas HL, Paraskevopoulou N, Metheniti A, Lambropoulou M, Marmaras VJ. Phagocytosis of Escherichia coli by insect hemocytes requires both activation of the Ras/mitogen-activated protein kinase signal transduction pathway for attachment and beta3 integrin for internalization. J Biol Chem. 1998;273:14813–8. doi: 10.1074/jbc.273.24.14813. [DOI] [PubMed] [Google Scholar]
- 41.Biscetti F, Pecorini G, Straface G, Arena V, Stigliano E, Rutella S, et al. Cilostazol promotes angiogenesis after peripheral ischemia through a VEGF-dependent mechanism. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.03.103. [DOI] [PubMed] [Google Scholar]
- 42.Dace DS, Khan AA, Kelly J, Apte RS. Interleukin-10 promotes pathological angiogenesis by regulating macrophage response to hypoxia during development. PLoS One. 2008;3:e3381. doi: 10.1371/journal.pone.0003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raghu H, Sodadasu PK, Malla RR, Gondi CS, Estes N, Rao JS. Localization of uPAR and MMP-9 in lipid rafts is critical for migration, invasion and angiogenesis in human breast cancer cells. Bmc Cancer. 2010;10 doi: 10.1186/1471-2407-10-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F, Li X, Wang LK, Wang LW, Han XQ, Zhang H, et al. Inhibitions of NF-kappaB and TNF-alpha Result in Differential Effects in Rats with Acute on Chronic Liver Failure Induced by D-Gal and LPS. Inflammation. 2014 doi: 10.1007/s10753-013-9805-x. [DOI] [PubMed] [Google Scholar]
- 45.Zachman AL, Page JM, Prabhakar G, Guelcher SA, Sung HJ. Elucidation of adhesion-dependent spontaneous apoptosis in macrophages using phase separated PEG/polyurethane films. Acta Biomater. 2013;9:4964–75. doi: 10.1016/j.actbio.2012.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chalothorn D, Faber JE. Strain-dependent variation in collateral circulatory function in mouse hindlimb. Physiol Genomics. 2010;42:469–79. doi: 10.1152/physiolgenomics.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta-Mol Basis Dis. 2003;1639:141–51. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Voskuil M, van Royen N, Hoefer IE, Seidler R, Guth BD, Bode C, et al. Modulation of collateral artery growth in a porcine hindlimb ligation model using MCP-1. American Journal of Physiology - Heart and Circulatory Physiology. 2003;284:H1422–H8. doi: 10.1152/ajpheart.00506.2002. [DOI] [PubMed] [Google Scholar]
- 49.Seidler RW, Allgauer S, Ailinger S, Sterner A, Dev N, Rabussay D, et al. In vivo human MCP-1 transfection in porcine arteries by intravascular electroporation. Pharm Res. 2005;22:1685–91. doi: 10.1007/s11095-005-6334-9. [DOI] [PubMed] [Google Scholar]
- 50.Sun H, Wang X, Hu X, Yu W, You C, Hu H, et al. Promotion of angiogenesis by sustained release of rhGM-CSF from heparinized collagen/chitosan scaffolds. J Biomed Mater Res B Appl Biomater. 2012;100:788–98. doi: 10.1002/jbm.b.32512. [DOI] [PubMed] [Google Scholar]
- 51.Vajanto I, Rissanen TT, Rutanen J, Hiltunen MO, Tuomisto TT, Arve K, et al. Evaluation of angiogenesis and side effects in ischemic rabbit hindlimbs after intramuscular injection of adenoviral vectors encoding VEGF and LacZ. J Gene Med. 2002;4:371–80. doi: 10.1002/jgm.287. [DOI] [PubMed] [Google Scholar]
- 52.Grochot-Przeczek A, Dulak J, Jozkowicz A. Therapeutic angiogenesis for revascularization in peripheral artery disease. Gene. 2013;525:220–8. doi: 10.1016/j.gene.2013.03.097. [DOI] [PubMed] [Google Scholar]
- 53.Das S, Singh G, Baker AB. Overcoming disease-induced growth factor resistance in therapeutic angiogenesis using recombinant co-receptors delivered by a liposomal system. Biomaterials. 2014;35:196–205. doi: 10.1016/j.biomaterials.2013.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Wei Y, Liu K, Yuan C, Tang Y, Quan Q, et al. Synergistic effects of FGF-2 and PDGF-BB on angiogenesis and muscle regeneration in rabbit hindlimb ischemia model. Microvasc Res. 2010;80:10–7. doi: 10.1016/j.mvr.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 55.Lazarous DF, Unger EF, Epstein SE, Stine A, Arevalo JL, Chew EY, et al. Basic fibroblast growth factor in patients with intermittent claudication: results of a phase I trial. J Am Coll Cardiol. 2000;36:1239–44. doi: 10.1016/s0735-1097(00)00882-2. [DOI] [PubMed] [Google Scholar]
- 56.Cooper LT, Jr, Hiatt WR, Creager MA, Regensteiner JG, Casscells W, Isner JM, et al. Proteinuria in a placebo-controlled study of basic fibroblast growth factor for intermittent claudication. Vasc Med. 2001;6:235–9. doi: 10.1177/1358836x0100600406. [DOI] [PubMed] [Google Scholar]
- 57.Lederman RJ, Mendelsohn FO, Anderson RD, Saucedo JF, Tenaglia AN, Hermiller JB, et al. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. Lancet. 2002;359:2053–8. doi: 10.1016/s0140-6736(02)08937-7. [DOI] [PubMed] [Google Scholar]
- 58.Marui A, Tabata Y, Kojima S, Yamamoto M, Tambara K, Nishina T, et al. Novel approach to therapeutic angiogenesis for patients with critical limb ischemia by sustained release of basic fibroblast growth factor using biodegradable gelatin hydrogel - An initial report of the phase I-IIa study. Circulation Journal. 2007;71:1181–6. doi: 10.1253/circj.71.1181. [DOI] [PubMed] [Google Scholar]
- 59.Speer DP, Chvapil M, Eskelson CD, Ulreich J. Biological Effects of Residual Glutaraldehyde in Glutaraldehyde-Tanned Collagen Biomaterials. J Biomed Mater Res. 1980;14:753–64. doi: 10.1002/jbm.820140607. [DOI] [PubMed] [Google Scholar]
- 60.Huanglee LLH, Cheung DT, Nimni ME. Biochemical-Changes and Cytotoxicity Associated with the Degradation of Polymeric Glutaraldehyde Derived Cross-Links. J Biomed Mater Res. 1990;24:1185–201. doi: 10.1002/jbm.820240905. [DOI] [PubMed] [Google Scholar]
- 61.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 62.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 63.Libby P, Ridker PM. Inflammation and Atherothrombosis: From Population Biology and Bench Research to Clinical Practice. Journal of the American College of Cardiology. 2006;48:A33–A46. [Google Scholar]
- 64.Fan W, Li C, Qin X, Wang S, Da H, Cheng K, et al. Adipose stromal cell and sarpogrelate orchestrate the recovery of inflammation-induced angiogenesis in aged hindlimb ischemic mice. Aging Cell. 2013;12:32–41. doi: 10.1111/acel.12014. [DOI] [PubMed] [Google Scholar]
- 65.Hamburg NM, Leeper NJ. Therapeutic Potential of Modulating MicroRNA in Peripheral Artery Disease. Curr Vasc Pharmacol. 2013 doi: 10.2174/15701611113119990014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jaipersad AS, Lip GYH, Silverman S, Shantsila E. The Role of Monocytes in Angiogenesis and Atherosclerosis. Journal of the American College of Cardiology. 63:1–11. doi: 10.1016/j.jacc.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 67.Imhof BA, Aurrand-Lions M. Angiogenesis and inflammation face off. Nat Med. 2006;12:171–2. doi: 10.1038/nm0206-171. [DOI] [PubMed] [Google Scholar]
- 68.Drexler W. Ultrahigh-resolution optical coherence tomography. J Biomed Opt. 2004;9:47–74. doi: 10.1117/1.1629679. [DOI] [PubMed] [Google Scholar]
- 69.Camargo LD, Babelova A, Mieth A, Weigert A, Mooz J, Rajalingam K, et al. Endo-PDI is required for TNF alpha-induced angiogenesis. Free Radical Bio Med. 2013;65:1398–407. doi: 10.1016/j.freeradbiomed.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 70.Siwik DA, Chang DLF, Colucci WS. Interleukin-1 beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–65. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 71.Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. Febs Letters. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- 72.Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, et al. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappa B/p300 binding in human tracheal smooth muscle cells. Am J Physiol-Lung C. 2007;292:L799–L812. doi: 10.1152/ajplung.00311.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of polymer microgel system. NMR spectra of injectable microgel co-polymer. Number average molecular weight (Mn) was determined to be 3571 Da as calculated by the molar ratio between PEG and PCL.
Figure S2. Biocompatibility of injectable polymer microgels. Human coronary artery endothelial cells (HCAECs) were cultured on top of pre-gelled injectable polymer microgels. Live cells were stained by calcein AM (green, top row) and dead cells were stained by ethidium homodimer (red, middle row). The injectable polymer microgels exhibit some red autofluorescence (left column) that is not indicative of dead cells. Images are representative of n=4 biological replicates. Scale bar = 50 μm.
Figure S3. Peptide Uptake by Macrophages and Endothelial Cells. RAW 264.7 macrophages or mouse aortic endothelial cells (mAECs) were prelabeled with DilC12 (red, top row) and cultured with FITC-labeled Ac-SDKP or C16 (green, middle row) for 72 hours. Images are representative of n=4 biological replicates. Scale bar = 20 μm.
Figure S4. Implantable Scaffold Fabrication and Characterization. A) Synthesis schematic of combinatorial polymers for implantable scaffold fabrication. B) SEM of porous structure of implantable scaffolds after salt leaching. Scale bar = 200 μm.
Figure S5. Macrophage infiltrations into implantable scaffolds in ischemic muscle. Sections of adductor muscle tissue adjacent to the implantation site of peptide-loaded scaffold or the injection site of peptide-loaded PBS were stained with hematoxylin and eosin (A-D) or rat anti-mouse biotinylated F4/80 antibodies (a macrophage marker), as visualized by brown color in images (E-J). Nuclei were counterstained blue with hemalum. The conditions include with scaffold (A-D, F-J), PBS without scaffold (E), no peptide (A, E, F), Ac-SDKP (B, G), C16 (C, I), C16+Ac (D, J), and LPS (H). F4/80 staining was quantified by calculating the area of positively stained pixels divided by the total number of cells per image as measured by hematoxylin nuclear stain (K). n=4 mice per condition. Scale bar = 100 μm.
Figure S6. Laser Dopper Perfusion Imaging (LDPI) of perfusion recovery in implantation of peptide-loaded scaffolds. A) LDPI images of ischemic (right) and control (left) hind limbs after femoral artery ligation and scaffold implantation with or without peptide loading. As controls, peptides were intramuscularly injected in PBS without scaffolds. B) Perfusion was quantified as the ratio of right to left foot at each time point. Dashed lines represent peptides in PBS injections without scaffolds; solid lines represent peptide-loaded scaffold treatments. n=6 mice per condition. *p<0.05 vs. no peptide treatment with scaffolds at day 14.
Figure S7. Optical Coherence Tomography (OCT) imaging of blood vessel formation with implantation of peptide-loaded scaffolds. A) Doppler OCT B-scans were taken of the calf muscle of mice after femoral artery ligation and peptide-loaded scaffold implantation. Blood vessels appear red or blue (depending on direction of blood flow) overlaid on tissue morphology (gray). B-scans show the surface of the skin at the top of the image and increase in depth into the calf muscle until signal can no longer be accurately detected at the bottom of the image. Scans are 1.8 mm in depth and 4 mm in width. B) Quantification of vessel formation was performed by dividing the total area of vessels in each image by the total imaged area. All groups are with implanted scaffolds. n=6 mice per condition. *p<0.05 vs. no peptide on day 13 after femoral artery ligation and scaffold implantation.
Figure S8. Angiogenesis and phagocytosis assay in peptide-loaded implanted scaffolds. Blood vessel formation (red) and macrophages phagocytosing E.coli particles (yellow-green) were visualized in tissue after injection of peptides or PBS vehicle (A) without or (B) with implanted polymer scaffolds. Scale bar=50 μm. C) Vessel perfusion capacity as measured by red fluorescence intensity of perfused microspheres extracted from scaffolds. D) Phagocytic activity (fluorescence intensity per image field). (C, D) *p < 0.05 vs. no peptide treatment in same group (without or with scaffold).