

Abstract
In musculoskeletal tissues like bone, chemotherapy can impair progenitor cell differentiation and proliferation, resulting in decreased bone growth and mineralization throughout a patient's lifetime. In the current study, we investigated the effects of chemotherapeutics on adipose-derived stem cell (ASC) function to determine whether this cell source could be a candidate for repairing, or even preventing, chemotherapy-induced tissue damage. Dose-dependent proliferation rates of ASCs and normal human fibroblasts (NHFs) were quantified after treatment with cytarabine (CY), etoposide (ETO), methotrexate (MTX), and vincristine (VIN) using a fluorescence-based assay. The influence of MTX on the multipotency of ASCs and freshly isolated stromal vascular fraction (SVF) cells was also evaluated using lineage-specific stains and spectrophotometry. ASC and NHF proliferation were equally inhibited by exposure to CY and ETO; however, when treated with MTX and VIN, ASCs exhibited greater resistance. This was especially apparent for MTX-treated samples, with ASC proliferation showing no inhibition for clinically relevant MTX doses ranging from 0.1 to 50 M. Additional experiments revealed that the differentiation potential of ASCs was not affected by MTX treatment and that upregulation of dihydrofolate reductase possibly contributed to this response. Moreover, SVF cells, which include ASCs, exhibited similar resistance to MTX impairment, with respect to cellular proliferation, clonogenicity, and differentiation capability. Therefore, we have shown that the regenerative properties of ASCs resist the cytotoxicity of MTX, identifying these cells as a potential key for repairing musculoskeletal damage in patients undergoing chemotherapy.
Keywords: chemotherapy, mesenchymal stem cell, tissue regeneration, bone, cell transplantation
Introduction
The influence of chemotherapy on cellular function is of increasing interest to the stem cell field. While the effects of chemotherapeutics on bone marrow stem cells (BMSCs) and hematopoietic stem cells have been studied for a number of years (Gardner, et al., 1993; J. Li, et al., 2004), researchers have only recently begun investigating the effects of these drugs on adipose-derived stem cells (ASCs) (Liang, et al., 2011; Zimmerlin, et al., 2011). Advancements in drug formulations have resulted in increased patient survival, but unfortunately many of these chemotherapeutics also have severe side effects, such as long-term musculoskeletal damage (Schriock, et al., 1991). Reports suggest that these impairments result from the slowed proliferation, restricted differentiation, and apoptosis of progenitor cells, such as BMSCs (Georgiou, et al., 2012). Investigating the influence of chemotherapeutics on other stem cell types, like ASCs, will help identify whether these adverse side effects are universal or whether susceptibility is specific to BMSCs. Furthermore, the possibility of identifying alternative mesenchymal stem cell sources resistant to the cytotoxic effects of these drugs would provide a means to treat, or even prevent, tissue damage. The regenerative properties of ASCs make them an attractive cell population for these therapies. Moreover, they have been shown to withstand other toxic environments like oxidative stress, suggesting ASCs might also resist functional damage induced by chemotherapeutics (Ertas, et al., 2012).
Only limited investigations have been made into the area of mesenchymal stem cell response to chemotherapeutics (Liang, et al., 2011; Qi, et al., 2012). The effects of these treatments on ASC proliferation and differentiation potential have not previously been investigated thoroughly. Differences in mechanism of action suggest that, like BMSCs, ASCs will vary in their susceptibility to chemotherapeutic agents (J. Li, et al., 2004). Therefore, it is necessary to evaluate many commonly used drugs in order to understand the impact of each individual treatment on ASC regenerative properties. Comparing chemotherapeutic effects on ASCs versus a normal, non-stem, somatic cell type provides a simple way to identify resistance or susceptibility to a given drug since neither cell type is directly targeted.
The goal of this study was to evaluate the effects of methotrexate (MTX), vincristine (VIN), cytarabine (CY), and etoposide (ETO) on ASC regenerative properties. Chemotherapeutic agents were chosen based on their diverse mechanisms of action and their application towards a wide range of cancers. MTX functions by binding to and inhibiting dihydrofolate reductase (DHFR), an essential protein for DNA synthesis (Cronstein, 1997). Conversely, VIN prevents microtubule formation, CY inhibits polymerase function, and ETO targets topoisomerase II (George, et al., 1965; D. D. Ross, et al., 1990; W. Ross, et al., 1984). After treatment with individual chemotherapeutics in vitro, ASC growth was compared with untreated ASCs and treated/untreated normal human fibroblasts (NHFs). These comparisons enabled us to identify significant deviation from normal growth trends and evaluate the resistance of ASCs to chemotherapeutics relative to untargeted, non-stem somatic cells. We also assessed how multilineage differentiation potential was affected by drugs that did not inhibit ASC proliferation, and investigated the possible mechanism behind this immunity. These results provide important insight towards understanding how ASC regenerative properties are affected by commonly used chemotherapeutics and identify mechanisms of chemotherapeutic resistance that could be useful in designing regenerative therapies for cancer patients.
Materials and Methods
Cell culture
ASCs, originally isolated from the subcutaneous fat of healthy human, non-diabetic, non-smoking female donors, aged 18-60 years (N = 7), were purchased from Zen-Bio, Inc. (superlot #36; Research Triangle Park, NC). ASCs were grown in expansion medium consisting of DMEM/F-12 (HyClone), 10% FBS (Zen-Bio), 1% antibiotic/antimycotic (HyClone), 0.25 ng/mL transforming growth factor-β1, 5 ng/mL epidermal growth factor, and 1 ng/mL fibroblast growth factor (R&D Systems) (B. T. Estes, et al., 2008). ASCs were maintained in humidified incubators at 37°C, 5% CO2 and passaged at 80% confluence with 0.25% trypsin-EDTA (HyClone). Experiments used ASCs at passage 4-6 (P4-6).
NHFs derived from neonatal human foreskins (a gift from Dr. Jeffrey Morgan) were expanded in high glucose DMEM (DMEM-HG, HyClone), 10% FBS (HyClone), and 1% penicillin/streptomycin (HyClone) (Youssef, et al., 2011). Experiments used NHFs at P6-9.
Isolation and culture of stromal vascular fraction (SVF) cells
All procedures were approved by the internal review board (IRB) at Rhode Island Hospital. Human lipoaspirate was obtained from female donors, aged 20-56 (N = 3). SVF cells were isolated using established protocols (B. T. Estes, et al., 2010). Briefly, liposuction waste tissue was washed thoroughly with phosphate-buffered saline (PBS) and digested for one hour at 37°C in an equal volume of 0.1% type I collagenase (Worthington Biochemical Corporation). Samples were centrifuged at 300 g for 5 minutes, and the pellet was resuspended in stromal medium (DMEM/F-12, 10% FBS, 1% antibiotic/antimycotic) to neutralized the enzyme. Cells were washed twice more and incubated for 10 minutes at room temperature in an erythrocyte lysis buffer (155 mM NH4Cl, 10 mM K2CO3, and 0.1 mM EDTA) to remove red blood cells. The remaining cell population was centrifuged and resuspended in stromal medium, then filtered sequentially through 100 μM and 70 μM strainers before cell counting. All samples were stored in liquid nitrogen using freezing medium containing 80% FBS, 10% dimethyl sulfoxide, and 10% expansion medium. Prior to experimentation, cells were quickly thawed and plated in expansion medium at 2,000 cells/cm2. SVF cells were treated with chemotherapeutics at P0. Clonogenicity and differentiation were assessed at P1.
Chemotherapeutic agents
The following chemotherapeutics (and concentrations) were used in our experiments: MTX (0.1-50 μM, MP Biomedicals), VIN (0.01-1 μM, Cayman Chemicals, Ann Arbor, Michigan), CY (1-100 μM, Accela ChemBio, Shanghai, China), and ETO (0.1-5 μM, MP Biomedicals). The concentration range for each drug was chosen based on clinically relevant doses reported in the plasma of patients receiving chemotherapy (J. Li, et al., 2004). While treatment times ranging from 48-72 hours have been used in the past (Georgiou, et al., 2012; J. Li, et al., 2004; Qi, et al., 2012), a duration of 24 hours was used in the current study based on pilot work showing a dramatic response of ASCs and NHFs to the listed chemotherapeutics within that time frame.
Proliferation assay
To examine the effects of chemotherapeutics on cell proliferation, ASCs and NHFs were quantified on days 6-10 after treatment with various chemotherapeutic agents. Cells were plated at 2,000 cells/cm2 in 96-well plates, a cell density chosen since it was high enough to detect fluorescence and low enough to enable the greatest amount of proliferation prior to reaching confluence. After adhering overnight, cells were treated with MTX, VIN, CY, or ETO for 24 hours. Following treatment, samples were rinsed with PBS, and fresh expansion media were added to ASC and NHF wells. Cells were cultured for an additional 10 days, quantifying cell counts on days 6-10 using a spectrophotometer-based assay developed and fully validated in our lab (Supplemental Fig. 1, 2). In brief, cell nuclei were stained with 5 μg/mL Hoechst 33342 dye (Invitrogen) for 30 minutes, rinsed three times with warm PBS, and assessed by spectrofluorometry using adsorption and emission wavelengths of 350 nm and 461 nm, respectively. Cell numbers were quantified based on sample fluorescence using a reference curve. To assist in comparing data across sample sets, values were presented as a percentage of control counts determined on day 10 for each individual chemotherapeutic.
Cell viability
To assess how 24 hour MTX treatment (2.5 μM) affects cell viability, ASCs and NHFs were monitored using a live/dead assay. Cells were stained on the day of MTX treatment removal (day 0) as well as the following ten days by incubating cultures in 2 μM calcein AM (AnaSpec Inc, Fremont, CA), 4.5 μM propidium iodide (EMD Millipore), and 1 μg/mL Hoechst for 30 minutes. Cellular viability was quantified by taking 16-24 images/well (N = 3 wells) at 10× and counting stained cells (n > 200).
Cell division
To determine if MTX inhibits cell division, Ki-67 immunostaining was performed on untreated and 2.5 μM MTX-treated ASCs and NHFs on days 0-10. Samples were fixed with 4% paraformaldehyde in PBS for 15 minutes and permeabilized with 0.25% Triton X (Thermo Fisher Scientific) for 15 minutes. Cells were blocked in 3% bovine serum albumin (Thermo Fisher Scientific) for 1 hour, stained with a Ki-67 antibody (1:200, EMD Millipore) for 30 minutes, and then stained with an Alexa Fluor 488-conjugated secondary antibody (Life Technologies) for 30 minutes. Samples were incubated in 5 μg/mL 4′,6-diamino-2-phenylindole (DAPI, Thermo Fisher Scientific) to visualize all cell nuclei. To determine Ki-67 expression, 16-24 images/well (N = 3 wells) were taken at 10× and stained cells (N > 200) were counted.
Senescence-associated β-galactosidase activity
To investigate how MTX treatment affects cell senescence, β-galactosidase staining in ASCs and NHFs was quantified after treatment with 2.5 μM MTX on days 0-10. Cells were fixed with 0.5% glutaraldehyde for 5 minutes and then washed with PBS. Plates were then incubated in X-gal solution (1 mg/ml X-gal (Genesee Scientific Corp, San Diego, CA), 5 mM K4Fe(CN)6·H2O, 5 mM K3Fe(CN)6, 1 mM MgCl2 in PBS, pH 6) overnight at 37qC. Cell nuclei were stained with DAPI as described above. The number of X-gal-positive cells versus total cells was quantified by brightfield and fluorescence imaging, respectively. To determine the percentage of senescent cells, 10 images/well (N = 3 wells) were taken at 10× and stained cells (N > 200) were counted.
Multilineage differentiation
Adipogenic and osteogenic differentiation
To examine whether MTX affected differentiation potential, ASCs were treated with MTX as described previously and expanded until cultures were 80% confluent. For freshly isolated SVF cells, samples were thawed and plated at 2,000 cells/cm2 in T-25 flasks. After adhering overnight, cells were treated with 2.5 μM MTX for 24 hours. Once treatment was removed, cells were cultured for an additional 6 days, until reaching 80% confluence. For differentiation assessment, ASCs and SVF cells were uplifted and seeded at 8,000 cells/well in 96-well plates with expansion medium. Upon reaching 100% confluence, ASCs were exposed to 200 μL of adipogenic, osteogenic, or control (stromal) medium (Guilak, et al., 2006; Zheng, et al., 2006). Adipogenic medium contained DMEM/F-12, 10% FBS, 1 μM dexamethasone, 10 μM insulin, 0.5 μM isobutyl-1-methylxanthine, 200 μM indomethacin (Sigma), and 1% antibiotic/antimycotic. Osteogenic medium contained DMEM-HG, 10% FBS, 10 nM dexamethasone, 10 mM β-glycerophosphate, 0.15 mM ascorbate-2-phosphate, 10 nM 1,25-(OH)2 vitamin D3 (Sigma), and 1% antibiotic/antimycotic. Media were changed every 2-3 days for two (adipogenesis) or three (osteogenesis) weeks before fixing cell monolayers with 3.7% paraformaldehyde (Thermo Fisher Scientific). Oil Red O (ORO, Sigma) staining was used to assess lipid production in adipogenic and control cultures. Alizarin Red S (ARS, Sigma) staining was used to assess calcified matrix deposition in osteogenic and control cultures. After imaging stained wells, ORO and ARS dyes were eluted, and optical densities were measured with spectrophotometry at 500 and 540 nm, respectively. Optical densities were normalized to cell number by counting nuclei stained with DAPI (Carpenter, et al., 2006). Assessment of SVF cell osteogenesis was only conducted for one donor, since cells from the other donors became highly contractile during the differentiation procedure, balling up and preventing comparative matrix deposition measurements.
Chondrogenic differentiation
Untreated and MTX-treated ASCs and SVF cells were placed in V-bottomed, 96-well plates at 50,000 cells/well and centrifuged at 400g for 5 minutes to form pellets. Expansion medium was replaced with 200 μL of chondrogenic or control media. Chondrogenic medium contained DMEM-HG, 10% FBS, 10 ng/ml TGF- 1, 0.15 mM ascorbate-2-phosphate, 100 nM dexamethasone, 1% ITS+ Premix (BD Biosciences), and 1% antibiotic/antimycotic (Gonzalez-Cruz, et al., 2012). Media were changed every 2-3 days, and after 3 weeks of culture, pellets were digested with papain (Sigma). Sulfated glycosaminoglycan (sGAG) amounts were quantified using previously established protocols (Awad, et al., 2003; Guilak, et al., 2006). Briefly, 200 μL of DMMB dye (Polysciences, Inc., pH 1.5) was added to 50 μL of pellet digests in 96-well plates and optical densities were measured at 525 nm. sGAG values were normalized to DNA amounts, determined using the PicoGreen assay (Invitrogen), measuring fluorescence at 480 nm excitation and 520 nm emission.
Colony forming unit-fibroblast assessment
The effect of MTX on the clonogenicity of heterogeneous SVF cell populations was examined using the colony forming unit-fibroblast (CFU) assay. Untreated and MTX-treated SVF cells were plated at 1,000 cells/100 mm dish (BD Biosciences) in stromal medium. Cells were cultured for 2 weeks, fixed with methanol, and stained with 0.5% Crystal Violet (Thermo Fisher Scientific). Colonies containing at least 50 cells were counted using a dissection microscope.
Western blotting
To investigate the mechanism of action behind ASC MTX resistance, dihydrofolate reductase (DHFR) protein expression was assessed by western blot. DHFR is the target of MTX, and upregulation during MTX treatment has been noted for other resistant cell types (Goldman and Matherly, 1985). Untreated and MTX-treated ASCs, NHFs, and SVF cells were lysed on ice for 30 minutes in RIPA buffer (25mM Tris•HCl pH 7.6, 150mM NaCl, 1% nonidet P-40 [NP-40], 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) supplemented with HALT protease and phosphatase inhibitors (Pierce). Protein concentrations were determined using a BCA Assay (Pierce). Equal amounts of protein were separated on SDS-PAGE gels (Bio-Rad), transferred onto PVDF membranes (Millipore) and confirmed with Ponceau Red stain. Membranes were blocked with Blotto (50 mM Tris (pH 7.5), 185 mM NaCl, 0.05% Tween 20, 3% (w/v) nonfat dry milk) and incubated with DHFR (1:500, Abcam) or β-tubulin (1:2000, Developmental Studies Hybridoma Bank) antibodies overnight. Membranes were washed for one hour in Blotto and then stained with peroxidase-conjugated secondary antibodies (Sigma, eBioscience Inc., San Diego, CA). Membranes were washed for 30 minutes in Blotto without milk and then visualized by enhanced chemiluminescence (EMD Millipore).
Statistical analysis
Three-way ANOVA (cell type, concentration, day) was used to assess proliferation data. Two-way ANOVA was used to assess differentiation data of ASCs (treatment, differentiation condition), SVF CFU and proliferation data (donor, treatment), as well as SVF osteogenesis (treatment, differentiation condition). Three-way ANOVA was used to assess SVF adipogenesis and chondrogenesis (donor, treatment, differentiation condition), as well as viability, cell division, and senescence (cell type, treatment, day). Following each ANOVA, the significance level of individual comparisons was determined using a Tukey post-hoc test.
Results
Cell growth after chemotherapeutic treatment
To compare how ASC and NHF proliferation is influenced by chemotherapeutics, cells were treated with individual drugs and counted on days 6-10. ASC proliferation was not inhibited by MTX treatment, whereas NHF proliferation was. No significant reductions in ASC counts were determined for any time point or concentration of MTX (Fig. 1A). Interestingly, cell counts of ASCs after 2.5 or 50 μM MTX treatments were 30-40% higher than non-treated controls on day 10 and days 8-10, respectively (p < 0.05), although results after 50 μM MTX treatment were variable. Conversely, NHF cell counts were reduced from control values at all time points by 80-95% after 2.5 or 50 μM MTX (p < 0.001). ASCs and NHFs treated with 0.1 μM MTX were not significantly different from their respective control values. These effects did not change when ASCs were treated and cultured in NHF medium or when NHFs were treated and cultured in ASC medium (Supplemental Fig. 2).
Figure 1.

ASC (red lines) and NHF (blue lines) proliferation were differentially inhibited by chemotherapeutic agents. ASCs and NHFs were quantified 6-10 days after a 24 hour treatment with different concentrations of (A) methotrexate, (B) vincristine, (C) cytarabine, (D) or etoposide. Cell counts are presented as the percent of ASC or NHF untreated control counts on day 10. ASC growth was not inhibited by any concentration of methotrexate, while NHFs were impaired after 2.5 and 50 μM treatments. Similarly, ASC proliferation was more resistant to vincristine exposure than NHFs. However, both cell types were equally impaired by cytarabine and etoposide. Error bars show standard error.
Similar to MTX, ASC proliferation was less inhibited by VIN (p < 0.001, Fig. 1B). Treatment with 0.01 μM reduced ASC counts from controls by 55-77% (p < 0.001), and higher doses of 0.125 or 1 μM inhibited ASC growth by 75-79% (p < 0.001). Conversely, all concentrations of VIN reduced NHF counts from controls by 79-90% (p < 0.001). Treatment with CY caused similar inhibition of ASC and NHF growth (Fig. 1C). ASC and NHF counts were significantly reduced from control counts by 55-95% after treatment with concentrations ranging from 1 to 100 μM (p < 0.001). As with CY, ETO treatment resulted in similar growth inhibition of ASCs and NHFs (Fig. 1D). Counts were significantly reduced from control values following treatment with ETO concentrations ranging from 0.1-5 μM (p < 0.05). Specifically, 0.1 μM ETO lowered cell counts of ASCs and NHFs by a maximum of only 16% (p < 0.05), while 1.25 and 5 μM ETO reduced counts by 70-75% (p < 0.001).
MTX effects on cell expansion properties
To understand the effects of 2.5 μM MTX on ASC and NHF expansion properties, viability, cell division, and senescence were examined on days 0-10 after treatment. Results from the live/dead assay revealed that MTX had disparate effects on ASCs versus NHFs (Fig 2A). Statistical analysis indicated no significant differences existed between untreated and MTX-treated ASCs at any time point (p = 0.682). Conversely, the viability of MTX-treated NHFs was significantly reduced from untreated NHFs by 10-30% on days 2-10, consistently decreasing over time and reaching viabilities as low as 62% on day 10 (p < 0.001). MTX-treated NHF viability was also 9-37% lower than MTX-treated and untreated ASC viability on days 2-10 (p < 0.001). Results also showed that untreated NHF viability was 9-12% lower than untreated ASCs (p < 0.001), although this viability percentage of ∼90% was relatively constant for days 4-10.
Figure 2.

Methotrexate effects on cell viability, cell division, and senescence revealed clear differences between ASCs and NHFs. (A) Live/dead staining indicated that ASC viability remained at 100% following 2.5 μM MTX treatment while NHF viability steadily dropped over time, Representative images of live (green) and dead (red) cells at day 10 indicate differences in ASC and NHF samples following MTX treatment (insets show controls). B) Ki-67 immunostaining confirmed that MTX had no effect on ASC division. Conversely, cell division in NHFs was significantly reduced compared to controls. Representative images of Ki-67 (green) and DAPI (blue) staining on day 10 suggest prevalent Ki-67 expression in ASCs, with minimal expression in NHFs. C) X-gal staining determined that MTX did not affect ASC senescence. However, MTX-treated NHF samples had significantly more senescent cells than untreated controls, with nearly all cells senescing by day 8. Images of senescent (blue) cells on day 10 indicate a greater percentage of senescence in NHFs than ASCs. (Scale bar indicates 200 μm, *p < 0.001 between untreated and MTX-treated NHFs).
Immunostaining of Ki-67 indicated that MTX treatment reduced cell division in NHF samples but had no effect in ASCs (Fig. 2B). Untreated and MTX-treated ASCs showed no statistically significant differences at any time point (p = 0.457). Conversely, Ki-67 expression in MTX-treated NHFs was significantly reduced compared to untreated NHFs by 25-99% on days 2-10, reaching percent expressions as low as 0.25% on day 10 (p < 0.001), which suggests no active cell division at all. Furthermore, MTX-treated NHF Ki-67 expression was 30-99% lower than untreated and MTX-treated ASC populations on days 2-10 (p < 0.001). Comparisons between untreated ASCs and NHFs indicated that NHF Ki-67 expression was 5-65% lower than ASC expression on days 2-10 (p < 0.001), which is to be expected since NHFs are larger in size than ASCs and reach confluence sooner in control conditions.
Quantifying β-galactosidase expression of ASCs and NHFs indicated that MTX did not influence ASC senescence but did increase NHF senescence. Untreated and MTX-treated ASCs showed no statistical differences in senescence at any time point (Fig. 2C). For NHFs, senescence of MTX-treated cells increased 5-22-fold over untreated NHFs on days 2-10, reaching 95% positive staining for β-galactosidase by day 8 (p < 0.001). Moreover, senescence of MTX-treated NHFs was 9-55-fold greater than untreated and MTX-treated ASCs (p < 0.001). No differences between untreated ASCs or NHFs were observed, although senescence of both cell types increased at later time points as cultures became confluent.
MTX effects on ASC differentiation potential
To determine whether ASC differentiation potential is resistant to MTX, commercially available, enriched populations of ASCs were treated with 2.5 μM MTX and then expanded before replating for differentiation. Results from the adipogenic, osteogenic, and chondrogenic assays indicated that the multipotency of ASCs was not inhibited by 2.5 μM MTX treatment. Histological stains of lipid and calcified matrix revealed comparable amounts of metabolite production between untreated and MTX-treated ASCs (Fig. 3A). Both untreated and MTX-treated adipogenic populations produced significantly more lipid than their respective non-induced controls (p < 0.001). While MTX treatment resulted in a slight increase in lipid production for adipogenic (11%) and non-induced (29%) samples over non-MTX-treated samples (Fig. 3B, p < 0.05), the average increases in lipid production over non-induced controls were comparable for untreated and MTX-treated populations (0.5-fold and 0.65-fold increase, respectively). Analysis of osteogenesis and chondrogenesis showed that increases in metabolite production over non-induced controls were comparable for untreated and MTX-treated ASCs in osteogenic (22.5-23.5-fold increase, p < 0.001) and chondrogenic (1.4-1.8- fold increase, p < 0.05) culture environments. Calcified matrix and sGAG production amounts were also similar for untreated and MTX-treated samples (Fig. 3C, D).
Figure 3.
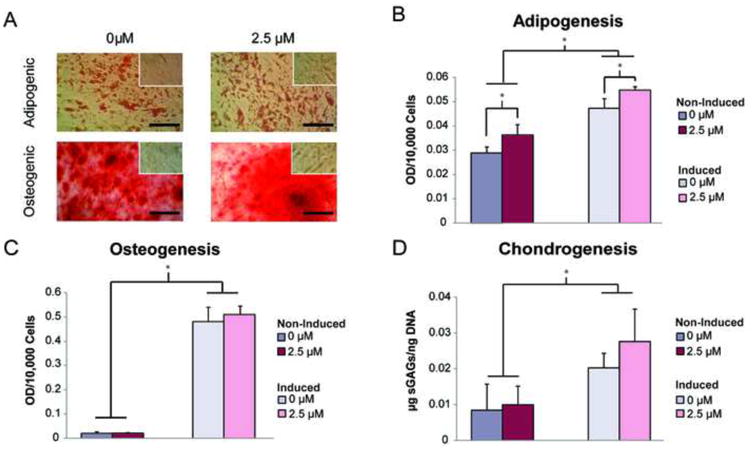
Treatment with 2.5 μM MTX had no negative effect on ASC multilineage differentiation potential. (A) Oil Red O and Alizarin Red S stains confirmed comparable lipid and calcified matrix production for induced and non-induced (insets), untreated and methotrexate-treated ASCs. Optical densities of eluted, metabolite-specific stains were quantified to determine differences in (B) adipogenic, (C) osteogenic, and (D) chondrogenic differentiation potential. While MTX caused statistically significant increases in lipid production for induced and non-induced samples over untreated wells, these differences were minor. No differences were observed between untreated and methotrexate-treated osteogenic or chondrogenic differentiation potentials. Error bars show standard deviation. (Scale bar indicates 50 μm, *p < 0.05)
MTX effects on SVF properties
While properties of expanded ASCs were shown to be largely unaffected by MTX, it was unknown whether freshly isolated cells would also resist inhibitory effects of the drug. Therefore, we investigated the influence of MTX on freshly isolated, heterogeneous SVF populations obtained from 3 different donors. After expanding and counting SVF cells treated with 2.5 μM MTX, results showed that that the drug actually increased proliferation of all donor populations, with MTX-treated samples having 17-50% more cells than untreated samples (Fig. 4A, p < 0.05). Moreover, colony counts for all MTX-treated donor populations were 50-100% higher than untreated populations (Fig. 4B, p < 0.001). Despite these other changes, MTX treatment did not affect the differentiation potential of SVF cells. Metabolite production for all lineages was significantly higher in induced samples than non-induced controls for both untreated and MTX-treated groups. Histological images illustrated that similar amounts of lipids and calcified matrix were produced by untreated and MTX-treated samples (Fig. 5A). These findings were supported by quantitative data showing that lipid, calcified matrix, and sGAG levels were comparable within both the induced and non-induced control sample conditions (Fig. 5B-D, p < 0.05). As mentioned in the methods, osteogenesis was only included for donor 1, since SVF cells from donors 2 and 3 could not maintain a monolayer morphology throughout the 3-week induction period, preventing accurate comparisons across donors.
Figure 4.

MTX treatment increased the proliferation and clonogenicity characteristics of SVF cell populations. (A) After monolayer expansion of untreated and MTX-treated SVF populations, cell counts of MTX-treated populations were higher than untreated populations. (B) Likewise, colony counts of treated SVF populations were significantly higher than untreated populations when assessed via a CFU assay. Error bars show standard deviation. (*p < 0.05)
Figure 5.
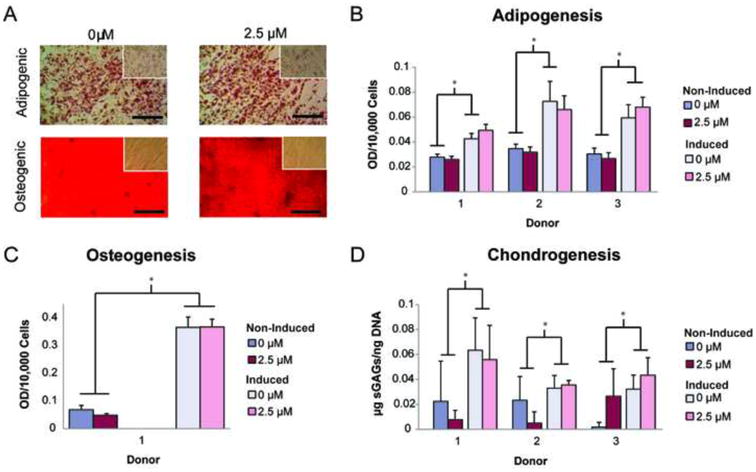
Treatment with 2.5 μM MTX had no negative effect on SVF cell multilineage differentiation potential. (A) Oil Red O and Alizarin Red S staining confirmed comparable lipid and calcified matrix production for induced and non-induced (insets), untreated and methotrexate-treated SVF cells (representative images for one donor). Optical densities of eluted, metabolite-specific stains were quantified to determine differences in (B) adipogenic, (C) osteogenic, and (D) chondrogenic differentiation potential of three, unique, SVF cell populations. No differences were observed between untreated and methotrexate-treated adipogenic, osteogenic, or chondrogenic differentiation potentials. Osteogenic assessment was only conducted on donor 1, as donors 2 and 3 became overly contractile and could not maintain a monolayer throughout three weeks of matrix deposition. Error bars show standard deviation. (Scale bar indicates 50 μm, *p < 0.05)
DHFR protein expression after MTX treatment
Previous studies suggested that DHFR protein upregulation may be an MTX-specific mechanism of resistance in multiple cell types. Therefore, we conducted western blot experiments to explore the role of DHFR in the MTX resistance exhibited by freshly isolated (SVF cells) and monolayer expanded ASCs. Protein expression was compared between untreated and 2.5 μM MTX-treated SVF cells, ASCs and NHFs. DHFR expression was upregulated differentially between SVF cells, ASCs, and NHFs (Fig. 6A). Quantification of protein levels via densitometry determined that average DHFR upregulation in SVF cells and ASCs following MTX treatment was at least 11-fold greater than that observed in NHFs (Fig. 6B). Furthermore, SVF upregulation was 2-fold greater than ASC upregulation, on average.
Figure 6.
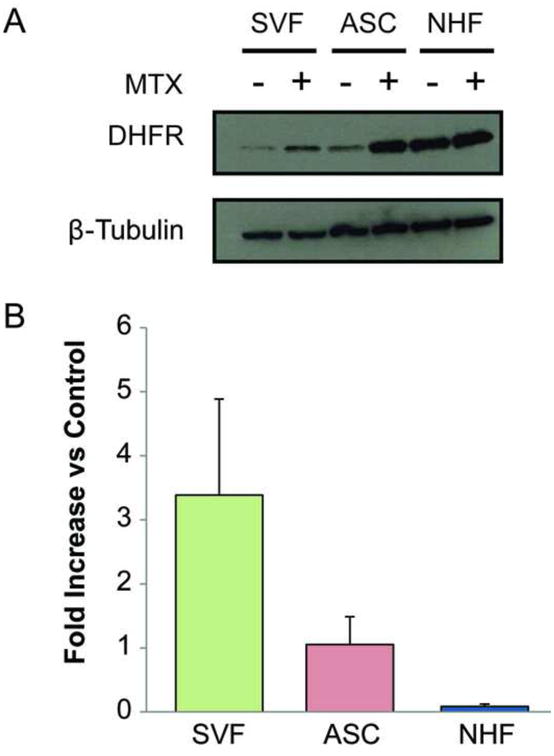
DHFR upregulation in SVF cells and ASCs was greater than NHFs after MTX treatment. (A) Western blot was used to compare protein expression of DHFR in untreated and 2.5 μM MTX-treated SVF cells, ASCs, and NHFs. (B) Quantification of band intensity revealed that SVF cells and ASCs upregulate DHFR 11-fold more than NHFs in the presence of MTX. Error bars show standard deviation.
Discussion
The results of this study indicate that ASCs are resistant to high-dose MTX treatment, whereas clinically relevant concentrations of VIN, CY, and ETO inhibit cellular growth. The effects of chemotherapy on mesenchymal stem cells, such as ASCs, is an important area of investigation since these cells play a critical role in tissue maintenance and tissue regeneration (Liang, et al., 2011). However, differences in mechanism of action suggest that not all chemotherapeutics will uniformly affect ASC function. Therefore, examining the influence of individual agents on ASC regenerative properties is necessary to determine drug-specific resistance or sensitivity for these cells. The goal of this study was to assess the response of ASCs versus an untargeted, somatic, non-stem cell population (NHFs) following in vitro exposure to common chemotherapeutics. We sought to identify resistance or susceptibility of ASCs to the tested drugs and improve upon our current understanding of chemotherapy effects. Furthermore, we aimed to investigate a potential mechanism behind any drug resistance to elucidate the phenomena observed in our results. Initial experiments used monolayer-expanded ASCs, which are more homogeneous than freshly isolated cells, to examine the effects of chemotherapeutics on regenerative properties. To investigate whether these effects were conserved for a more complex cell population, subsequent experiments used heterogeneous, SVF cells to examine the proliferation and differentiation capabilities of drug-treated samples.
To determine the effects of MTX, VIN, CY, and ETO on ASC and NHF proliferation, cells were counted on days 6-10 following treatment with specified drug concentrations. Most interestingly, we observed that ASC growth was not inhibited by MTX at any concentration (0.1-50 μM). Conversely, NHF growth was inhibited after treatment with as low as 2.5 μM, which is within the clinically relevant range (Kearney, et al., 1979; J. Li, et al., 2004). While the current study showed no dose dependent impairment for ASCs exposed to MTX, Qi et al. observed decreases in ASC proliferation when treating with 550 μM MTX for 48 hours, suggesting that longer exposure at much higher drug concentrations can negatively affect ASC growth (Qi, et al., 2012). The other chemotherapeutics investigated in this study, VIN, CY, and ETO, all inhibited ASC proliferation, although variability existed among drug type and concentrations. ASCs and NHFs responded comparably to CY and ETO, suggesting similar susceptibility to these drugs, which prevent DNA synthesis via inactivation of polymerase and inhibition of topoisomerase II, respectively (D. D. Ross, et al., 1990; W. Ross, et al., 1984). However, cellular response to VIN was not as uniform. While most drug concentrations resulted in greatly decreased proliferation, these decreases were less for ASCs than NHFs. Therefore, ASCs may be better equipped to rectify inhibition of microtubule formation, the mechanism of action for VIN (George, et al., 1965). This is supported by a study by Liang et al. that found ASCs could recover after exposure to 0.1 μM VIN (Liang, et al., 2011). Discrepancies between these findings and our own, which showed no recovery after exposure to 0.125 μM VIN, could be due to the slightly higher VIN-treatment concentration or other differences in the medium composition, such as serum fraction. It remains to be examined whether the superior resistance of ASCs over NHFs is conserved at even lower concentrations of VIN. However, those results may not be of great translational interest since ASCs and NHF growth was inhibited at clinically relevant VIN concentrations (0.1 μM) (J. Li, et al., 2004). The variability among ASC response to MTX, VIN, CY, and ETO suggests that ASCs are not impervious to all chemotherapeutics. In particular, high concentrations of VIN, CY, and ETO reduced cell counts by 70-95%. The clinically relevant range of doses used in this study illustrate the impact chemotherapeutics can have on the function of ASCs and other somatic cells such as NHFs. ASC senescence during chemotherapy could impair their ability to maintain tissue integrity, leading to degradation akin to the effects of chemotherapy on BMSCs and osteoblasts in bone. However, additional experiments are necessary to determine whether our in vitro results are indicative of the in vivo response. Cumulative effects of chemotherapeutics and the influence of the surrounding microenvironments during long-term in vivo treatment may result in different findings, as has been shown previously to occur with BMSCs (Georgiou, et al., 2012).
While we have discussed the impact of clinically relevant chemotherapeutic concentrations on ASC and NHF proliferation, it is important to note that these concentrations are based off of plasma levels and not necessarily the concentrations within adipose tissue. Reports on methotrexate and vincristine indicate that drug concentrations are 3-10-fold lower in adipose tissue than plasma (Anderson, et al., 1970; Behan, et al., 2010). These data suggest that the concentrations used in this study could be higher than what some cells in adipose tissue experience during treatment. However, since it has been suggested that ASCs reside within the adipose vasculature (Tang, et al., 2008), they may be exposed to concentrations closer to plasma levels. Regardless, the range of doses used in our study, which includes levels above and below physiologically relevant plasma concentrations, encompasses the most likely exposures ASCs would see in adipose tissue. Of particular relevance to our findings involving methotrexate, we showed that even at high concentrations relative to physiological dosing, ASCs exhibited a remarkable resistance to the negative effects of the drug.
To further understand the impact of MTX on ASC and NHF expansion, cells were treated with 2.5 μM MTX and assessed for viability, cell division (Ki-67), and senescence (X-gal) on the day that treatment was removed (day 0) as well as the subsequent 10 days. Results confirmed that MTX had no effect on ASC viability, cell division, or senescence. Conversely, MTX-treated NHF viability and cell division were significantly reduced from untreated levels, and the fraction of senescent cells was significantly increased. These data confirm that ASC expansion properties are resistant to 2.5 μM MTX in vitro, and reveal why MTX-treated ASC counts were not lower than untreated ASC counts in our proliferation assay. The cell death and growth arrest observed for NHFs is consistent with previous studies and indicates why low NHF counts existed after MTX treatment. Published findings suggest that exposure to MTX can cause either cell death or senescence, although this response is dependent on MTX treatment concentration and time. For example, Hattangadi, et al. determined that MTX caused 40% of MCF-7 breast tumor cells to apoptose and a greater fraction of cells to undergo growth arrest (Hattangadi, et al., 2004). While untreated ASC and NHF division was reduced and senescence was increased over time, this can be attributed to the cells reaching confluence (Fagman, et al., 2003; Severino, et al., 2000).
In addition to proliferation and growth characteristics, we investigated the effects of MTX on cellular differentiation potential. We compared the adipogenic, osteogenic, and chondrogenic differentiation outcomes of untreated and MTX-treated, homogeneous ASC populations. Analysis of normalized, metabolite levels revealed MTX caused no change in osteogenic or chondrogenic differentiation, while slightly increasing adipogenic response. These results suggest that ASC differentiation potential is not impaired by 24 hour exposure to 2.5 μM MTX treatment in vitro. Combined with our proliferation findings, ASCs appear to be a promising stem cell source that is less sensitive to the negative effects of this commonly used chemotherapeutic agent compared to other somatic cell types. This is important since previously published studies have shown that BMSCs are susceptible to MTX. In vitro treatment of BMSCs with 0.1 μM MTX for 48 hours resulted in increased adipogenesis but had no effect on osteogenesis. When replicated in vivo, treatment still increased adipogenesis but showed significant inhibition of osteogenesis (Georgiou, et al., 2012). The authors proposed that MTX-impaired BMSC function contributes to bone disorders arising after MTX treatment (Georgiou, et al., 2012). To determine superior MTX resistance of ASCs over BMSCs, experiments directly comparing these cell types must be conducted.
Given the abilities of ASCs to maintain their differentiation potential after short-term treatment with concentrations 25-fold higher than clinically relevant doses (Kearney, et al., 1979) they could be an attractive source for regenerative therapies for patients undergoing MTX treatment. Acute lymphoblastic leukemia, the most common form of childhood cancer, is one disease primarily treated with this drug. Currently, young patients undergoing chemotherapy face the risk of never recovering MTX-induced, bone mineral density loss, leaving them at higher risk for osteoporosis and bone fractures throughout their lifetime (Mandel, et al., 2004). However, it is possible that treatment with a resistant stem cell population, like ASCs, could ameliorate these defects by regenerating tissue damage after MTX exposure or even preventing bone loss altogether if introduced prophylactically. To verify that ASCs are a promising cell source for these regenerative therapies, additional investigations into the extent of ASC MTX-resistance is necessary. Studies should examine the effects of longer treatments and whether ASC resistance to MTX extends to the in vivo environment.
While enriched, homogeneous ASC populations were resistant to the effects of MTX, it was unknown whether cells comprising freshly isolated, heterogeneous SVF populations would also withstand MTX cytotoxicity. SVF cells from lipoaspirate are heterogeneous, consisting of ASCs, adipocytes, smooth muscle cells, endothelial cells, and fibroblasts (Yoshimura, et al., 2006). ASC purification approaches use a variety of strategies, the most effective of which is often serial passaging. Rapid enrichment of freshly isolated ASCs is problematic since there are no universally accepted surface markers for mesenchymal stem cells (Lv, et al., 2014). If cells are to be isolated from a patient and re-implanted with no extensive, monolayer expansion steps, then SVF cells are an important population to examine. Moreover, a secondary aim of this study was to determine whether the regenerative properties of SVF samples could be enhanced by MTX exposure based on the hypothesis that resident ASCs would proliferate while non-stem cells would not, effectively enriching the sample for our cell type of interest. Freshly isolated cells were treated with MTX and then analyzed for their clonogenicity and differentiation potential. Quantifying the number of colonies formed by untreated and treated SVF cells revealed that MTX increased colony counts by 50-100%. Since ASC quantity and CFUs correlate, our results indicate that MTX treatment effectively enriched ASCs within the heterogeneous populations. This enrichment should equate to a more robust differentiation response following chemical induction. However, analysis of adipogenic, osteogenic, and chondrogenic differentiation potential for untreated and MTX-treated SVF samples showed that lineage-specific metabolite production did not increase as a consequence of treatment. We hypothesize that ASC proportions in these heterogeneous cell populations, while higher, was not increased enough to see an effect on the metabolite production of the overall SVF samples. Interestingly, these findings suggest that the various cell types in SVF combine to exhibit a similar, resistant response to MTX as the more homogeneous, monolayer-expanded ASCs.
Experimental results also showed that ASC resistance to MTX is not passage-dependent or donor-dependent. Groups have reported that monolayer expansion can influence many characteristics of ASCs including surface marker expression, proliferation, and differentiation potential (Mitchell, et al., 2006; Schellenberg, et al., 2011; Zhao, et al., 2012). However, increased CFU counts and uninhibited differentiation potential of MTX-treated, freshly isolated SVF cells suggests that monolayer expansion does not cause MTX resistance of ASCs. This response was consistent for cells from multiple, human donors. While differentiation potential has been determined to be donor dependent (Guilak, et al., 2006), ASC MTX resistance is hypothesized to be an inherent function of this specific cell type, not influenced by differences in donor physiology.
Data showed that in response to drug treatment ASCs and SVF cells upregulated DHFR protein, the target of MTX and hypothesized mechanism behind ASC resistance (Cronstein, 1997). DHFR is involved in DNA biosynthesis and is essential for proliferation. During treatment, MTX binds available DHFR, inhibiting its function and leading to cell senescence or apoptosis. Therefore, DHFR upregulation due to an increase in gene expression infers cellular resistance, as this provides excess, unbound DHFR for continuation of necessary bioprocesses (Goldman and Matherly, 1985). Quantification of protein amounts for SVF cells, ASCs, and NHFs revealed that the increase in DHFR expression from baseline levels is at least 11-fold greater in ASCs and SVF cells than MTX-sensitive NHF populations. These results suggest that DHFR upregulation in the presence of MTX likely contributes to ASC resistance, though studies examining DHFR gene expression should be conducted to further confirm this.
Western blot experiments also revealed that SVF cell DHFR upregulation was 2-fold greater, on average, than upregulation of purified ASCs, despite SVF populations containing a greater fraction of terminally differentiated cells than ASC populations. Two reasons for the disparate responses of SVF and ASC DHFR upregulation have been hypothesized. First, ASCs are exposed to culture medium supplemented with growth factors, which have been suggested to improve chondrogenesis, while potentially reducing ASC “stemness” (R. T. Estes, et al., 2006). Loss of “stemness” may prevent ASCs from upregulating DHFR as much as the ASCs present in SVF populations. An additional contributing factor could be that freshly isolated SVF populations are more prolific than expanded ASCs. Studies have shown that non-passaged SVF cells express surface markers associated with proliferation that are lost during expansion (Suga, et al., 2009). As such, while both expanded and freshly isolated ASCs are able to upregulate DHFR and resist adverse MTX effects, the metabolic and prolific properties of freshly isolated ASCs could enable them to upregulate DHFR more than passaged cells.
It is possible that DHFR upregulation is not exclusively responsible for ASC resistance but may contribute in conjunction with complementary mechanisms. Other studies have reported that MTX resistance could be a result of low reduced folate carrier expression, overexpression of breast cancer resistance protein, or upregulation of multidrug resistance-associated protein 3 (Cowman and Foote, 1990; Guo, et al., 1999; Wall and Rubnitz, 2003). Superior DNA repair abilities of ASCs may also contribute to resistance, since accumulation of single- and double-strand breaks causes apoptosis during chemotherapy (J. C. Li and Kaminskas, 1984). Furthermore, differences in growth rates could contribute to the superior chemotherapeutic resistance of ASCs over NHFs. Chemotherapeutic agents are designed specifically to target rapidly dividing cells, while slowly dividing cells are not as susceptible to damage. Since ASCs have a longer doubling time than NHFs, 36 versus 24 hours, respectively, it is possible that this difference renders ASCs more resistant to these treatments than NHFs. Further study is warranted, since inducing targeted, MTX resistance via chemical treatment would be a more straight-forward, therapeutic approach than cellular transplantation.
A possible consequence of ASC upregulation of DHFR is increased proliferation rates. Results showed that ASC growth after MTX treatment determined that ≥2.5 μM MTX increased cell numbers 30-40% from control levels. This trend was conserved for SVF populations, as well, with cell counts of 2.5 μM MTX-treated populations being consistently higher than counts of untreated populations. DHFR overexpression has been implicated previously in increased proliferation, with cells in zebra fish embryos proliferating faster when DHFR is overexpressed (Sun, et al., 2011). While DHFR expression in NHFs was also elevated in the presence of MTX, these cells were still impaired by drug treatment. This is not unexpected since DHFR has previously been noted to increase protein expression in MTX-treated osteosarcoma cells, while mRNA expression decreased (Yoon, et al., 2010). Therefore, it is possible that DHFR protein upregulation was an initial response of the cells to the treatment but could not be sustained. NHF cell senescence after treatment suggests that expression is eventually lost, due to the reliance of cell proliferation on DHFR. Examining DHFR levels at a later time point could confirm this hypothesis.
Conclusion
The effects of chemotherapeutic agents on ASC proliferation and differentiation potential are imperative to understanding the musculoskeletal consequences of high-dose treatments and potentially identifying preventative treatments for patients receiving chemotherapy. This study revealed differential resistance and susceptibility of ASCs to commonly used chemotherapeutics. We have identified multiple drugs that impair ASC proliferation, revealing concerns for patients with respect to mesenchymal stem cell function. However, we also determined that ASC regenerative properties were unaffected by MTX, a chemotherapeutic known to cause long-term musculoskeletal injury. This study's findings support further investigation into the applicability of ASCs for regenerative therapies targeting patients undergoing MTX treatment.
Supplementary Material
Supplemental Figure 1. A novel, plate-based cell counting method was validated for reproducibility and accuracy in comparison to hemocytometer and software-based approaches. A) A standard curve was determined for MG-63 cells (cell line used in preliminary studies) correlating fluorescent units with cell counts. Known cell numbers were plated in 96-well plates at values ranging from 5,000-50,000 cells. Following overnight adherence, cells were stained with Hoechst and fluorescence was quantified using spectrophotometry. Values were plotted to obtain an equation correlating fluorescence values with cell counts. B) Our plate-based counting technique was compared with established methods to confirm consistency. Known cell amounts were plated in 96-well plates and allowed to proliferate. After 24 hours, cells were counted using plate-based (fluorescence of Hoechst-stained nuclei), hemocytometer-based (manual counting), and software-based (imaging and quantifying DAPI-stained nuclei) approaches. Statistical analysis confirmed that no significant differences existed between plate and hemocytometer-based approaches. Conversely, the software-based technique yielded counts significantly lower than plate and hemocytometer-based methods, due to difficulties identifying overlapping nuclei as individual cells. These data confirm that our plate-based approach is comparable to the commonly used hemocytometer-based counting technique. Furthermore, the plate-based approach is potentially more accurate than the software-based approach, a less conventional, but validated method. (*p < 0.001 between software-based and plate/hemocytometer-based counts)
Supplemental Figure 2. To assist with cell counting, a simple fluorescence-based assay was developed in our lab. Standard curves for (A) ASCs and (B) NHFs were generated by plating known numbers of each cell type (ranging from 0-50,000 cells) in 96-well plates. The following day, nuclei were stained with Hoechst dye, and spectrofluorometry was used to quantify relative fluorescence units. Data were plotted to obtain an equation correlating relative fluorescence units with cell counts. This assay was extensively validated for each cell type and served as a reliable method for quantifying proliferation. While relative fluorescence units could also be used to assess proliferation, converting to cell numbers provided for more intuitive descriptions.
Supplemental Figure 3. To confirm that adipose-derived stem cell and normal human fibroblast media were not contributing to the effects of methotrexate, cells were treated with the drug in swapped media. (A) ASCs cultured and treated in NHF medium were resistant to methotrexate treatment. NHFs cultured and treated in ASC medium were inhibited by 2.5 and 50 μM methotrexate. (B) Swapped media results were consistent with findings for cells treated in matched media conditions (from Fig. 1A).
Highlights.
Long-term effects of chemotherapeutics can include musculoskeletal dysfunction.
A screen of common drugs showed disparate effects on ASCs and fibroblasts.
One drug, methotrexate, did not impair ASC growth characteristics or multipotency.
Upregulation of dihydrofolate reductase may enable ASC methotrexate resistance.
ASCs thus pose a possible means to ameliorate long-term tissue damage.
Acknowledgments
The authors thank Aaron Chiou for his contributions to early proliferation studies, as well as Hetal Desai, Manisha Kanthilal, and Nicholas Labriola for their SVF cell isolation efforts. We also thank Dr. Kimberly Mowry and Samantha Jeschonek for their generous assistance with western blot experiments. Furthermore, we would like to thank Robert Gutiérrez for his work on validating the plate-based proliferation assay. This work was supported by awards from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR063642), National Institute of General Medical Sciences (P20GM104937), and National Science Foundation (CAREER Award, CBET1253189). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
Abbreviations
- ASC
adipose-derived stem cell
- BMSC
bone marrow-derived stem cell
- MTX
methotrexate
- CY
cytarabine
- ETO
etoposide
- VIN
vincristine
- SVF
stromal vascular fraction
- PBS
phosphate buffered saline
- ORO
Oil Red O
- ARS
Alizarin Red S
- sGAG
sulfated glycosaminoglycan
- DMMB
dimethylmethylene blue
- CFU
colony forming unit
- DHFR
dihydrofolate reductase
Footnotes
Author Contributions: OSB and EMD designed the study, analyzed data, and wrote the manuscript. OSB conducted all experimental work, including cell culture, proliferation tests, differentiation assays, and CFU analysis. VCF assisted with cell culture, proliferation tests, western blots, as well as conducting and interpreting the chondrogenic differentiation assay.
Disclosure of Interest: The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson LL, Collins GJ, Ojima Y, Sullivan RD. A study of the distribution of methotrexate in human tissues and tumors. Cancer Research. 1970;30:1344–8. [PubMed] [Google Scholar]
- Awad HA, Halvorsen YD, Gimble JM, Guilak F. Effects of transforming growth factor beta1 and dexamethasone on the growth and chondrogenic differentiation of adipose-derived stromal cells. Tissue Engineering. 2003;9:1301–12. doi: 10.1089/10763270360728215. [DOI] [PubMed] [Google Scholar]
- Behan JW, Avramis VI, Yun JP, Louie SG, Mittelman SD. Diet-induced obesity alters vincristine pharmacokinetics in blood and tissues of mice. Pharmacol Res. 2010;61:385–90. doi: 10.1016/j.phrs.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman AF, Foote SJ. Chemotherapy and Drug-Resistance in Malaria. International Journal for Parasitology. 1990;20:503–513. doi: 10.1016/0020-7519(90)90198-v. [DOI] [PubMed] [Google Scholar]
- Cronstein BN. The mechanism of action of methotrexate. Rheumatic Disease Clinics of North America. 1997;23:739–&. doi: 10.1016/s0889-857x(05)70358-6. [DOI] [PubMed] [Google Scholar]
- Ertas G, Ural E, Ural D, Aksoy A, Kozdag G, Gacar G, Karaoz E. Comparative analysis of apoptotic resistance of mesenchymal stem cells isolated from human bone marrow and adipose tissue. Scientific WorldJournal. 2012;2012:105698. doi: 10.1100/2012/105698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes BT, Diekman BO, Gimble JM, Guilak F. Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nature Protocols. 2010;5:1294–1311. doi: 10.1038/nprot.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes BT, Diekman BO, Guilak F. Monolayer cell expansion conditions affect the chondrogenic potential of adipose-derived stem cells. Biotechnol Bioeng. 2008;99:986–95. doi: 10.1002/bit.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes RT, Wu AW, Storms RW, Guilak F. Extended passaging, but not aldehyde dehydrogenase activity, increases the chondrogenic potential of human adipose-derived adult stem cells. J Cell Physiol. 2006;209:987–995. doi: 10.1002/jcp.20808. [DOI] [PubMed] [Google Scholar]
- Fagman H, Larsson F, Arvidsson Y, Meuller J, Nordling M, Martinsson T, Helmbrecht K, Brabant G, Nilsson M. Nuclear accumulation of full-length and truncated adenomatous polyposis coli protein in tumor cells depends on proliferation. Oncogene. 2003;22:6013–22. doi: 10.1038/sj.onc.1206731. [DOI] [PubMed] [Google Scholar]
- Gardner RV, Lerner C, Astle CM, Harrison DE. Assessing Permanent Damage to Primitive Hematopoietic Stem-Cells after Chemotherapy Using the Competitive Repopulation Assay. Cancer Chemotherapy and Pharmacology. 1993;32:450–454. doi: 10.1007/BF00685889. [DOI] [PubMed] [Google Scholar]
- George P, Journey LJ, Goldstein MN. Effect of vincristine on the fine structure of HeLa cells during mitosis. J Natl Cancer Inst. 1965;35:355–75. [PubMed] [Google Scholar]
- Georgiou KR, Scherer MA, Fan CM, Cool JC, King TJ, Foster BK, Xian CJ. Methotrexate chemotherapy reduces osteogenesis but increases adipogenic potential in the bone marrow. J Cell Physiol. 2012;227:909–18. doi: 10.1002/jcp.22807. [DOI] [PubMed] [Google Scholar]
- Goldman ID, Matherly LH. The Cellular Pharmacology of Methotrexate. Pharmacology & Therapeutics. 1985;28:77–102. doi: 10.1016/0163-7258(85)90083-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cruz RD, Fonseca VC, Darling EM. Cellular mechanical properties reflect the differentiation potential of adipose-derived mesenchymal stem cells. Proc Natl Acad Sci U S A. 2012;109:E1523–9. doi: 10.1073/pnas.1120349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilak F, Lott KE, Awad HA, Cao Q, Hicok KC, Fermor B, Gimble JM. Clonal analysis of the differentiation potential of human adipose-derived adult stem cells. J Cell Physiol. 2006;206:229–37. doi: 10.1002/jcp.20463. [DOI] [PubMed] [Google Scholar]
- Guo W, Healey JH, Meyers PA, Ladanyi M, Huvos AG, Bertino JR, Gorlick R. Mechanisms of methotrexate resistance in osteosarcoma. Clin Cancer Res. 1999;5:621–7. [PubMed] [Google Scholar]
- Hattangadi DK, DeMasters GA, Walker TD, Jones KR, Di X, Newsham IF, Gewirtz DA. Influence of p53 and caspase 3 activity on cell death and senescence in response to methotrexate in the breast tumor cell. Biochem Pharmacol. 2004;68:1699–708. doi: 10.1016/j.bcp.2004.06.033. [DOI] [PubMed] [Google Scholar]
- Kearney PJ, Light PA, Preece A, Mott MG. Unpredictable serum levels after oral methotrexate in children with acute lymphoblastic leukaemia. Cancer Chemother Pharmacol. 1979;3:117–20. doi: 10.1007/BF00254982. [DOI] [PubMed] [Google Scholar]
- Li J, Law HK, Lau YL, Chan GC. Differential damage and recovery of human mesenchymal stem cells after exposure to chemotherapeutic agents. Br J Haematol. 2004;127:326–34. doi: 10.1111/j.1365-2141.2004.05200.x. [DOI] [PubMed] [Google Scholar]
- Li JC, Kaminskas E. Accumulation of DNA strand breaks and methotrexate cytotoxicity. Proc Natl Acad Sci U S A. 1984;81:5694–8. doi: 10.1073/pnas.81.18.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Xia H, Li J, Zhao RC. Human adipose tissue derived mesenchymal stem cells are resistant to several chemotherapeutic agents. Cytotechnology. 2011;63:523–30. doi: 10.1007/s10616-011-9374-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv FJ, Tuan RS, Cheung KM, Leung VY. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408–19. doi: 10.1002/stem.1681. [DOI] [PubMed] [Google Scholar]
- Mandel K, Atkinson S, Barr RD, Pencharz P. Skeletal morbidity in childhood acute lymphoblastic leukemia. Journal of Clinical Oncology. 2004;22:1215–1221. doi: 10.1200/JCO.2004.04.199. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, McIntosh K, Zvonic S, Garrett S, Floyd ZE, Kloster A, Di Halvorsen Y, Storms RW, Goh B, Kilroy G, Wu X, Gimble JM. Immunophenotype of human adipose-derived cells: temporal changes in stromal-associated and stem cell-associated markers. Stem Cells. 2006;24:376–85. doi: 10.1634/stemcells.2005-0234. [DOI] [PubMed] [Google Scholar]
- Qi Z, Zhang Y, Liu L, Guo X, Qin J, Cui G. Mesenchymal stem cells derived from different origins have unique sensitivities to different chemotherapeutic agents. Cell Biol Int. 2012 doi: 10.1042/CBI20110637. [DOI] [PubMed] [Google Scholar]
- Qi Z, Zhang YM, Liu L, Guo X, Qin J, Cui GH. Mesenchymal stem cells derived from different origins have unique sensitivities to different chemotherapeutic agents. Cell Biology International. 2012;36:857–862. doi: 10.1042/CBI20110637. [DOI] [PubMed] [Google Scholar]
- Ross DD, Chen SR, Cuddy DP. Effects of 1-beta-D-arabinofuranosylcytosine on DNA replication intermediates monitored by pH-step alkaline elution. Cancer Res. 1990;50:2658–66. [PubMed] [Google Scholar]
- Ross W, Rowe T, Glisson B, Yalowich J, Liu L. Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 1984;44:5857–60. [PubMed] [Google Scholar]
- Schellenberg A, Lin Q, Schuler H, Koch CM, Joussen S, Denecke B, Walenda G, Pallua N, Suschek CV, Zenke M, Wagner W. Replicative senescence of mesenchymal stem cells causes DNA-methylation changes which correlate with repressive histone marks. Aging-Us. 2011;3:873–888. doi: 10.18632/aging.100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriock EA, Schell MJ, Carter M, Hustu O, Ochs JJ. Abnormal growth patterns and adult short stature in 115 long-term survivors of childhood leukemia. Journal of Clinical Oncology. 1991;9:400–5. doi: 10.1200/JCO.1991.9.3.400. [DOI] [PubMed] [Google Scholar]
- Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Experimental Cell Research. 2000;257:162–171. doi: 10.1006/excr.2000.4875. [DOI] [PubMed] [Google Scholar]
- Suga H, Matsumoto D, Eto H, Inoue K, Aoi N, Kato H, Araki J, Yoshimura K. Functional implications of CD34 expression in human adipose-derived stem/progenitor cells. Stem Cells Dev. 2009;18:1201–10. doi: 10.1089/scd.2009.0003. [DOI] [PubMed] [Google Scholar]
- Sun SN, Gui YH, Jiang Q, Song HY. Dihydrofolate reductase is required for the development of heart and outflow tract in zebrafish. Acta Biochimica Et Biophysica Sinica. 2011;43:957–969. doi: 10.1093/abbs/gmr098. [DOI] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–6. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall AM, Rubnitz JE. Pharmacogenomic effects on therapy for acute lymphoblastic leukemia in children. Pharmacogenomics Journal. 2003;3:128–135. doi: 10.1038/sj.tpj.6500174. [DOI] [PubMed] [Google Scholar]
- Yoon SA, Choi JR, Kim JO, Shin JY, Zhang X, Kang JH. Influence of reduced folate carrier and dihydrofolate reductase genes on methotrexate-induced cytotoxicity. Cancer Res Treat. 2010;42:163–71. doi: 10.4143/crt.2010.42.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura K, Shigeura T, Matsumoto D, Sato T, Takaki Y, Aiba-Kojima E, Sato K, Inoue K, Nagase T, Koshima I, Gonda K. Characterization of freshly isolated and cultured cells derived from the fatty and fluid portions of liposuction aspirates. Journal of Cellular Physiology. 2006;208:64–76. doi: 10.1002/jcp.20636. [DOI] [PubMed] [Google Scholar]
- Youssef J, Nurse AK, Freund LB, Morgan JR. Quantification of the forces driving self-assembly of three-dimensional microtissues. Proc Natl Acad Sci U S A. 2011;108:6993–8. doi: 10.1073/pnas.1102559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Waldman SD, Flynn LE. The effect of serial passaging on the proliferation and differentiation of bovine adipose-derived stem cells. Cells Tissues Organs. 2012;195:414–27. doi: 10.1159/000329254. [DOI] [PubMed] [Google Scholar]
- Zheng B, Cao B, Li G, Huard J. Mouse adipose-derived stem cells undergo multilineage differentiation in vitro but primarily osteogenic and chondrogenic differentiation in vivo. Tissue Engineering. 2006;12:1891–901. doi: 10.1089/ten.2006.12.1891. [DOI] [PubMed] [Google Scholar]
- Zimmerlin L, Donnenberg AD, Rubin JP, Basse P, Landreneau RJ, Donnenberg VS. Regenerative therapy and cancer: in vitro and in vivo studies of the interaction between adipose-derived stem cells and breast cancer cells from clinical isolates. Tissue Eng Part A. 2011;17:93–106. doi: 10.1089/ten.tea.2010.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. A novel, plate-based cell counting method was validated for reproducibility and accuracy in comparison to hemocytometer and software-based approaches. A) A standard curve was determined for MG-63 cells (cell line used in preliminary studies) correlating fluorescent units with cell counts. Known cell numbers were plated in 96-well plates at values ranging from 5,000-50,000 cells. Following overnight adherence, cells were stained with Hoechst and fluorescence was quantified using spectrophotometry. Values were plotted to obtain an equation correlating fluorescence values with cell counts. B) Our plate-based counting technique was compared with established methods to confirm consistency. Known cell amounts were plated in 96-well plates and allowed to proliferate. After 24 hours, cells were counted using plate-based (fluorescence of Hoechst-stained nuclei), hemocytometer-based (manual counting), and software-based (imaging and quantifying DAPI-stained nuclei) approaches. Statistical analysis confirmed that no significant differences existed between plate and hemocytometer-based approaches. Conversely, the software-based technique yielded counts significantly lower than plate and hemocytometer-based methods, due to difficulties identifying overlapping nuclei as individual cells. These data confirm that our plate-based approach is comparable to the commonly used hemocytometer-based counting technique. Furthermore, the plate-based approach is potentially more accurate than the software-based approach, a less conventional, but validated method. (*p < 0.001 between software-based and plate/hemocytometer-based counts)
Supplemental Figure 2. To assist with cell counting, a simple fluorescence-based assay was developed in our lab. Standard curves for (A) ASCs and (B) NHFs were generated by plating known numbers of each cell type (ranging from 0-50,000 cells) in 96-well plates. The following day, nuclei were stained with Hoechst dye, and spectrofluorometry was used to quantify relative fluorescence units. Data were plotted to obtain an equation correlating relative fluorescence units with cell counts. This assay was extensively validated for each cell type and served as a reliable method for quantifying proliferation. While relative fluorescence units could also be used to assess proliferation, converting to cell numbers provided for more intuitive descriptions.
Supplemental Figure 3. To confirm that adipose-derived stem cell and normal human fibroblast media were not contributing to the effects of methotrexate, cells were treated with the drug in swapped media. (A) ASCs cultured and treated in NHF medium were resistant to methotrexate treatment. NHFs cultured and treated in ASC medium were inhibited by 2.5 and 50 μM methotrexate. (B) Swapped media results were consistent with findings for cells treated in matched media conditions (from Fig. 1A).