

Abstract
While cellular LC3B and SQSTM1 levels serve as key autophagy markers, their regulation by different signaling pathways requires better understanding. Here, we report the mechanisms by which the Raf/MEK/ERK pathway regulates cellular LC3B and SQSTM1 levels. In different cell types, δRaf-1:ER- or B-RafV600E-mediated MEK/ERK activation increased LC3B-I, LC3B-II, and SQSTM1/p62 levels, which was accompanied by increased BiP/GRP78 expression. Use of the autophagy inhibitors chloroquine and bafilomycin A1, or RNA interference of ATG7, suggested that these increases in LC3B and SQSTM1 levels were in part attributed to altered autophagic flux. However, intriguingly, these increases were also attributed to their increased expression. Upon Raf/MEK/ERK activation, mRNA levels of LC3B and SQSTM1 were also increased, and subsequent luciferase reporter analyses suggested that SQSTM1 upregulation was mediated at transcription level. Under this condition, transcription of BiP/GRP78 was also increased, which was necessary for Raf/MEK/ERK to regulate LC3B at the protein, but not mRNA, level. This suggests that BiP has a role in regulating autophagy machinery when Raf/MEK/ERK is activated. In conclusion, these results suggest that, under a Raf/MEK/ERK-activated condition, the steady-state cellular levels of LC3B and SQSTM1 can also be determined by their altered expression wherein BiP is utilized as an effector of the signaling.
Keywords: Autophagy, LC3, SQSTM1, BiP/GRP78, Raf, ERK1/2
Introduction
Autophagy is a constitutive lysosomal degradation pathway by which cells recycle cytoplasm and organelles to maintain cellular homeostasis. Under stress conditions, autophagy is upregulated to generate resources for the maintenance of essential cellular functions [1,2]. Autophagy can affect cellular differentiation, development, and homeostasis, and its altered activity is often associated with several diseases, including cancer, which are mostly attributed to aberrant signal transduction [3]. Therefore, it is important to identify the signaling pathways that can affect cellular capacity for autophagy.
The Raf/MEK/extracellular signal-regulated kinases 1 and 2 (ERK1/2) pathway is a three-layered kinase cascade, serving as a key signal transducer of receptor tyrosine kinases and the small GTPase, Ras [4]. The Ser/Thr kinase Raf (c-Raf-1, Raf-B or Raf-A) activates the dual-specificity kinases MEK1 and MEK2 which, in turn, sequentially phosphorylate Tyr and Thr in the activation loop of the ubiquitously expressed Ser/Thr kinases ERK1 and its homolog ERK2. Activated ERK1/2 mediates diverse biological processes by activating/inactivating a wide variety of proteins, and different magnitudes of its activity can lead to distinct biological outputs [4]. The Raf/MEK/ERK pathway has pivotal roles in regulating cell survival, cell cycle progression and differentiation [5,6], and dysregulated Raf/MEK/ERK signaling is a central signature of many cancers [7]. Currently our understanding of the role of Raf/MEK/ERK pathway in the context of autophagy is limited.
BiP (HSPA5/GRP78), a member of the heat shock protein 72 (HSP72) family [8], is a molecular chaperone that facilitates the folding and assembly of newly synthesized proteins in endoplasmic reticulum (ER) [9] and mediates the unfolded protein response by regulating the ER stress transducers, including PERK, IRE1, and ATF6 [10]. Upon onset of the ER-associated unfolded protein response, these molecules are liberated from BiP and become activated for cytoprotection. Therefore, BiP is characterized as a master regulator of ER stress. Recently, it has been discovered that autophagy is activated upon ER stress to facilitate cell survival [11,12], and that BiP has a critical role in this activation of autophagy [13]. Because ER stress is known to be triggered upon aberrant Raf/MEK/ERK activation [14,15], it may be possible that BiP plays a role for Raf/MEK/ERK in a context relevant to autophagy.
Microtubule-associated protein-1 light chain-3 (LC3), a mammalian homolog of yeast Atg8, and the LC3-binding protein, SQSTM1/p62, are the two key components in the formation of autophagosomes [16]. During autophagy, the cytoplasmic form of LC3 (LC3-I,18 kDa) is recruited to the autophagosome, where LC3-II (16 kDa) is generated by site-specific proteolysis and lipidation near to the C-terminus [17]. SQSTM1, on the other hand, binds LC3 and recruits proteins into autophagosomes for degradation [2]. Thus, increased LC3-II and decreased SQSTM1 levels indicate autophagic activity whereas SQSTM1 accumulation indicates defective autophagy. Nevertheless, it is possible that a change in LC3 and SQSTM1 levels indicates not only activity of the standard autophagy pathway but also other non-canonical mechanisms that can also affect their cellular levels.
In this study, we demonstrate that aberrant Raf/MEK/ERK activation is sufficient to increase cellular levels of LC3B-I and LC3B-II, and SQSTM1. Intriguingly, these increases in protein levels were also attributed to increased mRNA levels of LC3B and SQSTM1. Further, these changes were accompanied by BiP expression and our promoter luciferase reporter analysis indicated that Raf/MEK/ERK can upregulate transcription of BiP and SQSTM1. In addition, our evaluation of BiP function using RNA interference and gene overexpression suggests that BiP has a specific role in regulating cellular levels of LC3B protein, but not mRNA, under Raf/MEK/ERK-activated conditions. This study reveals previously unknown Raf/MEK/ERK-regulated signaling, which may affect cellular capacity for autophagy.
Materials and methods
Cell culture, generation of stable lines, and reagents
The human prostate cancer line, LNCaP (ATCC, CRL-1740), was maintained in phenol red-deficient RPMI 1640 (Invitrogen, 32404-014) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin and 100 μg of streptomycin per ml. The human embryonic kidney epithelial cell line, HEK293, was maintained in MEM (Invitrogen, 11095080) supplemented with 10% FBS, 100 U of penicillin and 100 μg of streptomycin per ml. The primary normal human fibroblasts BJ and E1A-immortalized IMR90 (IMR90E1A) were grown in DMEM supplemented with 10% FBS, 1% sodium pyruvate and 1% non-essential amino acids. The human melanoma lines, SK-MEL-28 (ATCC), SK-MEL-1 (ATCC), and RPMI-7951 (ATCC) were maintained in MEM (Invitrogen) supplemented with 10% FBS, 100 U of penicillin and 100 μg of streptomycin per ml. The human melanoma line, A375 (ATCC), was grown in DMEM (Invitrogen) supplemented with 10% FBS. The LNCaP line stably expressing ΔRaf-1:ER (LNCaP-Raf:ER) was previously described [18]. ΔRaf-1:ER was activated with 1 μM 4-hydroxytamoxifen (Sigma, H7904). Other chemicals used are the following: U0126 (Cell Signaling, 9903), MG-132 (Sigma, M7449), lactacystin (Sigma, L6785), bafilomycin A1 (Sigma, B1793), chloroquine (Sigma, C6628), and AZD6244 (Selleck Chemicals, 918505-84-7).
Recombinant lentiviral constructs and plasmids
The N-terminal HA-tagged BiP expression system constructed in the lentiviral pHAGE vector was previously described [19]. Constitutively active MEK2 in pHAGE (pHAGE-MEK2CA) was previously described [18]. pEGFP-LC3B was previously described [20]. pGRP78-promoter luciferase reporters were previously described [21]. SQSTM1-promoter luciferase reporter was previously described [22].
Small hairpin RNA (shRNA) expression construct
Lentiviral shRNA expression systems were constructed using the pLL3.7 vector (ATCC, VRMC-39). pLL3.7-shBIP#1, pLL3.7-shBIP#2, and shBIP#3 target three different regions of human BIP mRNA, GTTGTGGCCACTAATGGAGA, ACCTTCGATGTGTCTCTTC, and GGAGCGCATTGATACTAGA. pLL3.7-shERK1 and pLL3.7-shERK2 were previously described [18]. The pLKO.1-shRNA vectors targeting ATG7 were purchased from Thermo Scientific (TRCN7586 and TRCN7587). Specific knockdown of target proteins was confirmed by Western blot analysis.
Viral infection
For lentivirus production, 293T cells were co-transfected with pHAGE, pLKO.1, or pLL3.7, and packaging vectors, as previously described [23,24]. Viral supernatants were collected after 48–72 h and mixed with polybrene (Sigma, 107689) at 4–8 μg/ml before use. Viral titer was determined by scoring cells expressing GFP.
Reverse transcriptase polymerase chain reaction (RT-PCR) and quantitative real time PCR (qPCR)
Primers used for RT-PCR are CGGAGTCAACGGATTTGGTCGTAT and AGCCTTCTCCATGGTGGTGAAGAC (GAPDH), CCCGCGCGATGCCCTCAGACCGGCC and CTGGCTCAGAAGCCGAAGGTTTCCT (LC3A-v1), ATACAAGGGAAGTGGCTATC and TTACACTGACAATTTCATCC (LC3B), CTGCCCAGACTACGACTTGTGT and TCAACTTCAATGCCCAGAGG (SQSTM1), AAGCTCTCCCTGGTGGCCGCGATGCTGCTG and CTACAACTCATCTTTTTCTGCTGTATCCTC (BiP), AGCAACTCTGGATGGGATTG and CACTGCAGAGGTGTTTCCAA (ATG5), and ACCCAGAAGAAGCTGAACGA and AGACAGAGGGCAGGATAGCA (ATG7). Primers used for qPCR are CAACATGAGCGAGTTGGTCAAGA and ACTCACCATGCTGTGCTGGTTC (LC3A-v1), AGCAGCATCCAACCAAAATC and CTGTGTCCGTTCACCAACAG (LC3B), ATCGGAGGATCCGAGTGT and TGGCTGTGAGCTGCTCTT (SQSTM1), and TGCTGGCCTAAATGTTATG and TGGTGAGAAGAGACACATC (BiP). qPCR was performed by reverse transcription of 0.25 μg total RNA and subsequent polymerase chain reaction using the Mx3005P™ instrument (Stratagene, 401449) and Brilliant SYBR® Green QPCR Core Reagent Kit (Stratagene, 600546) according to the manufacturer's protocol. Thermocycling conditions were 10 min at 95 °C as first denaturation step, followed by 40 cycles at 95 °C for 30 s, 55 °C for 60 s and 72 °C for 30 s. For normalization, expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was measured.
Calculation of the gene copy number was carried out using comparative threshold cycle, as previously described [25]. Briefly, the mean threshold cycle (mCT) was obtained from triplicate amplifications during the exponential phase of amplification. Then, mCT value of the reference gene was subtracted from mCT value of the target gene to obtain ΔCT. ΔΔCT values of each sample was calculated from corresponding CT values; where ΔΔCT= [mCT target (normal sample) – mCTreference (normal sample)] – [mCT target (test sample) – mCT reference (test sample)]. The calculated ΔΔCT was converted to ratio using the ratio formula (Ratio=2T–ΔΔC).
Luciferase reporter assay
Cells were transfected with different luciferase reporter constructs using Lipofectamine 2000 (Invitrogen, 11668). Transfected master cultures were then divided in triplicate into 24-well plates for further treatments, as described in the text. Cell lysates were analyzed using the Luciferase Assay System (Promega, E1910) according to the manufacturer's instructions. Reporter activity data were normalized for protein concentration.
Immunoblot analysis
Cells harvested at various times were lysed and analyzed using the BCA reagent to determine the protein concentration (Pierce, 23225), as we previously described [18]. 50–100 µg of protein was resolved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, 162-0177), and stained with Fast Green reagent (Thermo Fisher Scientific, BP123-10). Membrane filters were then blocked in 0.1 M Tris (pH 7.5)–0.9% NaCl–0.05% Tween 20 with 5% nonfat dry milk, and incubated with appropriate antibodies. Antibodies were diluted as follows: ATG7 (Cell Signaling, 8558), 1:2000; ERK1/2 (Cell Signaling, 4695), 1:2500; phospho-ERK1/2 (Thr202/Tyr204, Cell Signaling, 9102), 1:2500; phospho-p90RSK (Thr359/Ser363, Cell Signaling, 9344), 1:2500; GAPDH (Cell Signaling, 2118), 1:5000; BiP (Cell Signaling, 3183), 1:2000; phospho-eIF2α (Ser51, Cell Signaling, 3398), 1:2000; SQSTM1 (Santa Cruz, sc-25575), 1:2000; CHOP/GADD153 (Santa Cruz, sc-575), 1:1000; anti-HA (Santa Cruz, sc-7392), 1:1000; LC3B (MBL International, PM036), 1:2000. The Supersignal West Pico (Pierce, 34077) and Femto chemiluminescence kits (Pierce, 34095) were used for visualization of the signal. Immunoblots were scanned and analyzed using Image Lab (BioRad, 170-9690).
Statistical analysis
Experiments were repeated at least three times. Statistical significance was determined by Student's t test for unpaired samples. P<0.05 was considered significant.
Results
Raf/MEK/ERK can increase LC3B and SQSTM1 levels in different cell types, which is accompanied by BiP expression
We determined the effect of Raf/MEK/ERK activation on LC3B, a major LC3 in the autophagosome [26], and SQSTM1 in different cell types, including the human prostate cancer line LNCaP, immortalized human kidney epithelial cell line HEK293, primary cultured normal foreskin fibroblast BJ, and immortalized lung fibroblast IMR90E1A, which have been used as models to study the mechanisms underlying cellular responses to aberrant Raf/MEK/ERK activation [6,18,27]. To specifically control the pathway activity, we used the tamoxifen-regulated ΔRaf-1:ER, a CR3 catalytic domain of Raf-1 fused to hormone binding domain of the estrogen receptor [28]. When ΔRaf-1:ER was activated by 1 µM 4-hydroxytamoxifen in LNCaP and HEK293, these cells exhibited significantly increased LC3B and SQSTM1 protein levels within 48 h, along with increased ERK1/2 phosphorylation, which was accompanied by a significant increase in BiP levels (Fig. 1A). Consistent with LNCaP and HEK293, BJ cells also exhibited similar responses, albeit to a lesser extent (Fig. 1A). These changes were specific to Raf activation because 4-hydroxytamoxifen alone did not induce any similar effects (Fig. 1A). Moreover, consistent with µRaf-1:ER, ectopically expressed oncogenic B-RafV600E induced similar effects in IMR90E1A cells (Fig. 1B). These data demonstrate that aberrant Raf activation can affect cellular levels of LC3B, SQSTM1, and BiP in different cell types.
Fig. 1.
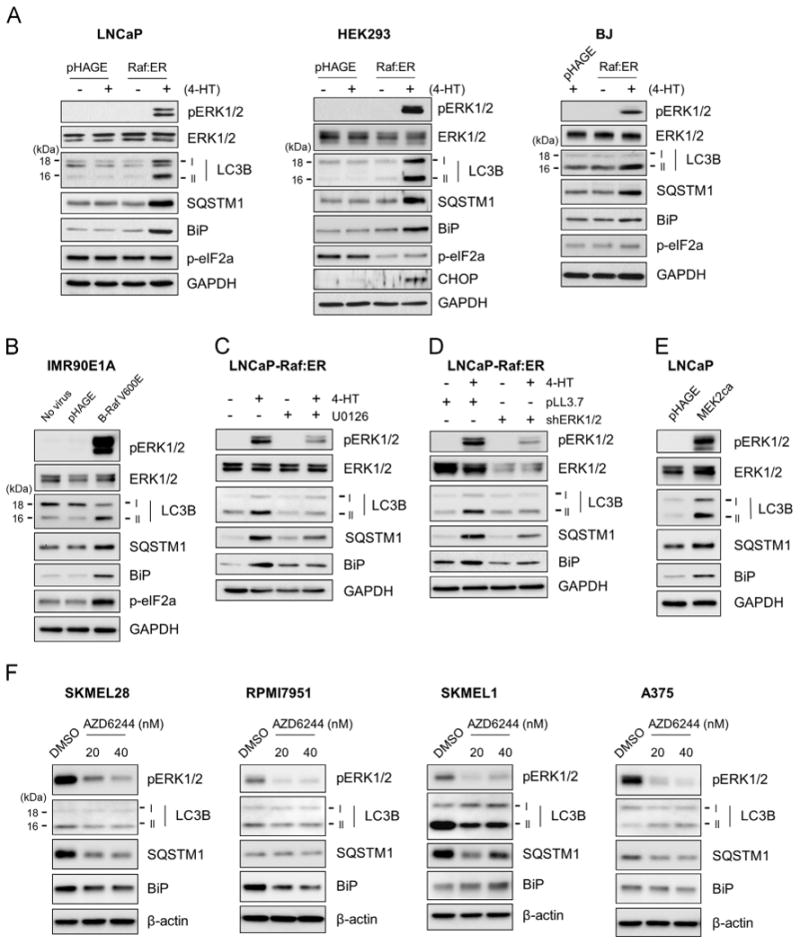
Raf/MEK/ERK activation increases the levels of LC3B, SQSTM1, and BiP in cells. Total cell lysates from each experiment were examined by Western blotting for expression of the indicated proteins. pERK1/2 indicates phosphorylated ERK1/2. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected to validate equal protein loading. (A) LNCaP, HEK293 and BJ cells infected with lentiviral pHAGE-Raf:ER were treated with 1 μM 4-hydroxytamoxifen (4-HT) for 2 days. (B) IMR90E1A cells were infected with lentiviral pHAGE-B-RafV600E for 2 days. (C) LNCaP-Raf:ER cells were treated with 1 μM 4-hydroxytamoxifen for 2 days in the presence of 10 μM U0126, a MEK1/2 inhibitor. (D) LNCaP-Raf:ER cells, co-infected with lentiviral pLL3.7 expressing shRNAs that target ERK1 and ERK2 (shERK1/2), were treated with 1 μM 4-hydroxytamoxifen for 2 days. (E) LNCaP cells infected with lentiviral pHAGE expressing a constitutively active MEK2 (MEK2CA) for 2 days. (F) B-RafV600E mutated human melanoma lines, SK-MEL28, RPMI7951, SK-MEL1, and A375, were treated with two different doses of AZD6244, a highly potent MEK1/2 inhibitor, for 2 days. Data shown are representative images of multiple independent experiments; A, LNCaP (N=5), HEK293 (N=3), BJ (N=2); B, N=2; C, N=2; D, N=2; E, N=2; F, N=2.
Although MEK/ERK is the major effector of Raf, it has been reported that Raf can mediate MEK/ERK-independent signaling, e.g., cell death responses [29]. Therefore, we determined whether MEK/ERK is necessary for Raf to regulate LC3B, SQSTM1, and BiP. When ΔRaf-1:ER was activated in LNCaP cells pretreated with the MEK1/2-specific inhibitor, U0126, Raf-induced upregulation of LC3B, SQSTM1 and BiP were significantly reduced (Fig. 1C). Consistent with this, shRNA-mediated knockdown of both ERK1 and ERK2 also effectively inhibited the effects of Raf activation (Fig. 1D). Conversely, ectopic expression of a constitutively active mutant of MEK2 (ΔN4/S222D/S226D) induced similar effects as Raf activation (Fig. 1E), indicating that MEK/ERK activation is necessary and sufficient for Raf to regulate LC3B, SQSTM1 and BiP.
To substantiate this observation, we also examined the effects of Raf/MEK/ERK inhibition on BiP, LC3 and SQSTM1 levels in B-RafV600E mutated human melanoma lines, SK-MEL28, SK-MEL1, A375, and RPMI7951. When these cells were treated with AZD6244, a highly potent MEK1/2-specific inhibitor, BiP levels were substantially downregulated in SK-MEL28 and RPMI7951 cells, although not significantly affected in A375 and SK-MEL1 cells (Fig. 1F). LC3B, especially LC3B-II, levels were also downregulated in SK-MEL1 cells while being mildly downregulated in SK-MEL28 and RPMI7951 cells (Fig. 1F). In A375 cells, LC3B-II levels were slightly upregulated while LC3B-I levels being downregulated. SQSTM1 levels were substantially downregulated in SK-MEL28, SK-MEL1, and A375 cells, although not in RPMI7951 cells (Fig. 1F). These data suggest that Raf/MEK/ERK activity can regulate BiP, LC3B and SQSTM1 levels in certain B-RafV600E-mutated melanoma cells, further supporting the ability of Raf/MEK/ERK to regulate BiP, LC3 and SQSTM1.
Raf/MEK/ERK mediates temporal regulation of BiP, LC3B and SQSTM1 in a manner dependent upon its signaling intensity
Because the Raf/ME/ERK pathway is known to mediate different, even opposing, contexts of signaling transduction depending upon the magnitude and duration of its activation [4,30], we examined the effects of different magnitude and duration of Raf/MEK/ERK activation using ΔRaf-1:ER and different tamoxifen doses. As determined in LNCaP cells (Fig. 2A), induction of LC3, SQSTM1, and BiP upregulation was commensurate with the intensity of ERK1/2 phosphorylation, which suggest that strong activity of the Raf/MEK/ERK pathway is required to induce these effects.
Fig. 2.
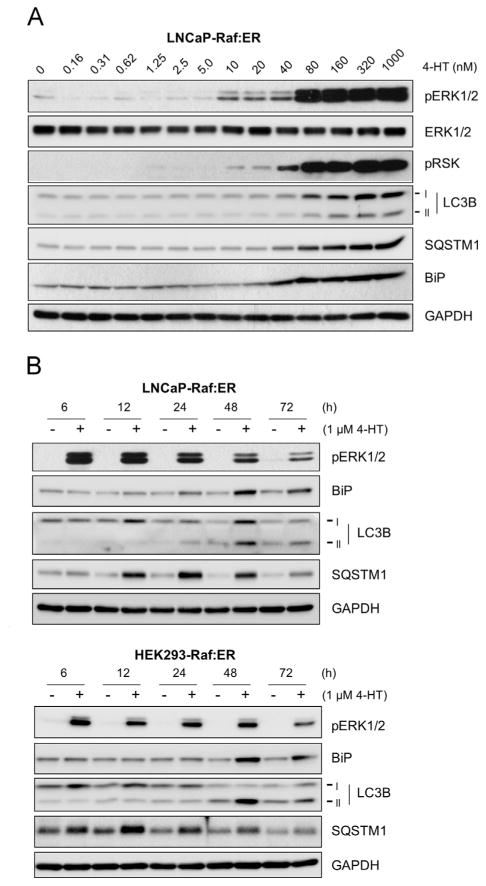
High magnitude Raf/MEK/ERK activity mediates temporal regulation of LC3B, SQSTM1, and BiP. (A) LNCaP-Raf: ER cells were treated with increasing doses of 4-hydroxy-tamoxifen for 2 days. (B) LNCaP-Raf:ER and HEK293-Raf:ER cells were treated with 1 μM 4-hydroxytamoxifen for indicated time periods. Total cell lysates harvested at different time courses were analyzed for expression of indicated proteins by Western blotting. Data shown are representative images of multiple independent experiments; A, N=2; B, N=3.
When a time-course study was conducted using LNCaP and HEK293 cells, the highest induction of BiP expression was detected around 48 h after Raf activation in both cell lines, which was concurrent with the emergence of LC3B-II (Fig. 2B). SQSTM1 increases occurred much earlier than expression of LC3B and BiP (Fig. 2B). After 24 h, the rates of SQSTM1 upregulation were significantly decreased whereas BiP and LC3B upregulation became more significant (Fig. 2B). These data indicate that Raf/MEK/ERK mediates a temporal regulation of BiP, LC3B, and SQSTM1, wherein regulation of SQSTM1 precedes other changes.
Raf-mediated increases in LC3B and SQSTM1 levels are augmented by autophagy inhibitors and ATG7 knockdown
The status of cellular autophagy can be more accurately determined by using the autophagy inhibitors or by modulating a key regulator of autophagy [31]. Chloroquine and bafilomycin A1 are the lysosomotropic agents which specifically block lysosomal degradation of autophagosomal contents [31]. When Raf was activated in cells in the presence of these chemicals at saturating doses, Raf-mediated increases in LC3B-II and SQSTM1 levels were strongly augmented (Fig. 3A; Supplemental Fig. S1). We also investigated the effects of knockdown of autophagy-related gene ATG7. ATG7 is an E1 like enzyme that conjugates LC3-I to phosphatidylethanolamine to produce LC3-II in collaboration with ATG3 (E2-like enzymes) [32]. Therefore, in the absence of ATG7, accumulation of LC3-I is expected. We depleted ATG7 in LNCaP cells, using two different lentiviral shRNA constructs that specifically target different mRNA regions of ATG7 (shATG7 #1 and #2). As expected, ATG7 knockdown inhibited Raf-induced conversion of LC3B-I to LC3B-II and increased protein levels of LC3B-I and SQSTM1 without significantly affecting ERK1/2 phosphorylation (Fig. 3B). These data suggested that Raf activation may be able to affect the autophagy flux.
Fig. 3.
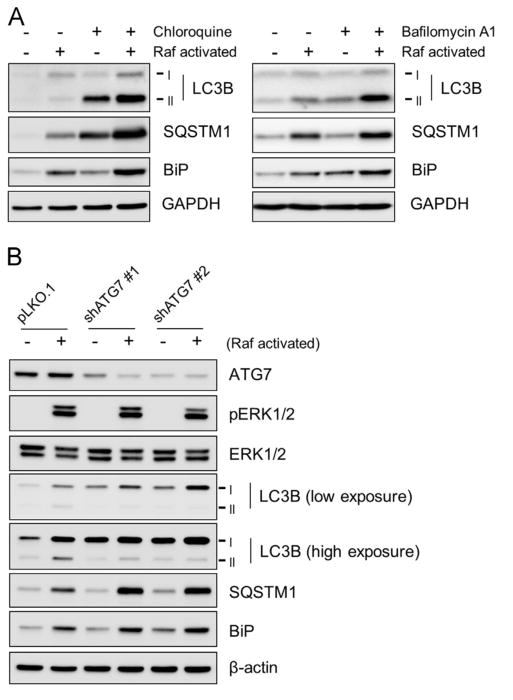
The effects of autophagy inhibitors and ATG7 knockdown on LC3B, SQSTM1, and BiP in Raf-activated cells. Total cell lysates were examined for expression of the indicated proteins by Western blotting. (A) LNCaP-Raf:ER cells were treated with 1 µM 4-hydroxytamoxifen for 1 day in the presence of 100 µM chloroquine (left panel) or 10 nM bafilomycin A1 (right panel). (B) LNCaP-Raf:ER cells, infected with lentiviral pLKO.1 expressing two different shRNAs that target ATG7 (shATG7 #1 and #2), were treated with 1 µM 4-hydroxytamoxifen for 2 days (Raf activated). Data shown are representative images of two independent experiments.
Raf/MEK/ERK can also increase LC3B and SQSTM1 levels partly by upregulating their expression
To further understand the nature of Raf/MEK/ERK-mediated regulation of LC3B and SQSTM1, we measured their mRNA levels in LNCaP cells using RT-PCR and quantitative PCR analyses, which were conduced using different primer sets. We found that Raf activation could significantly increase mRNA levels of LC3B and SQSTM1 (Fig. 4A; top panel, RT-PCR; bottom, qPCR), while U0126 blocked Raf-induced increases in LC3B and SQSTM1 mRNA levels (Fig. 4B; left, RT-PCR; right, qPCR), suggesting that Raf/MEK/ERK-induced increases in LC3B and SQSTM1 levels are also attributed to increased mRNA expression. Moreover, Raf activation mildly upregulated the activity of a luciferase reporter harboring 1.8 kb of human SQSTM1 promoter DNA (from –1781 to –7), which was consistently blocked by AZD6244 (Fig. 4C). This suggests that Raf/MEK/ERK can upregulate SQSTM1 transcription.
Fig. 4.
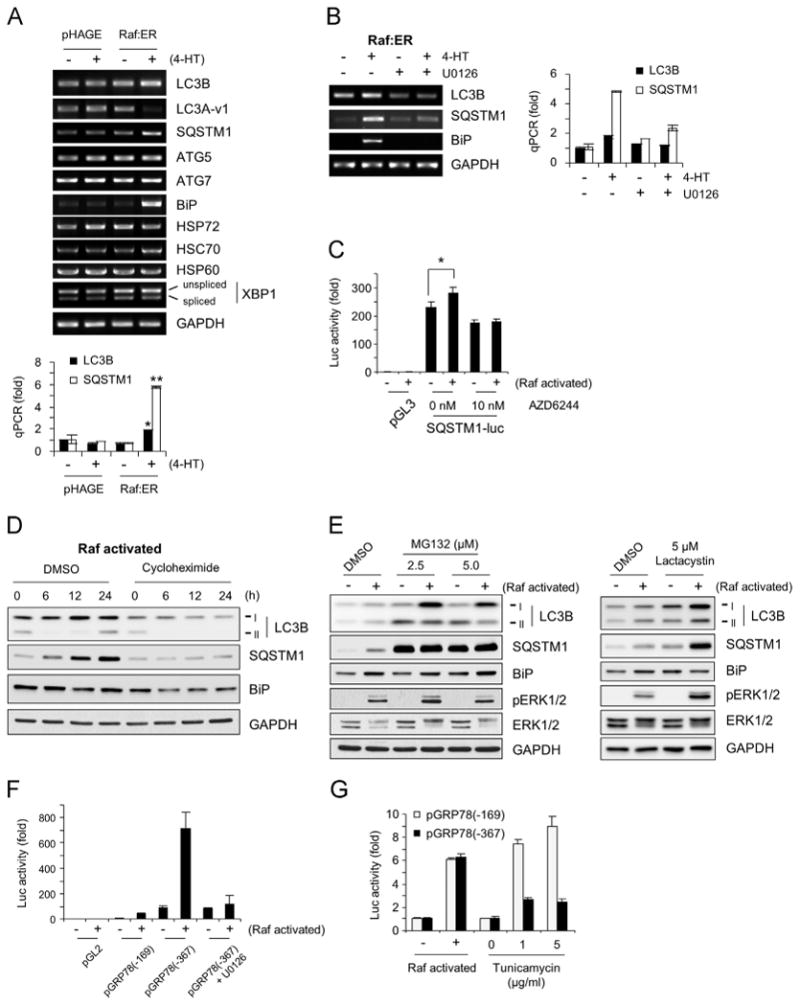
Raf/MEK/ERK upregulates expression of LC3B, SQSTM1, and BiP. (A) LNCaP cells infected with lentiviral pHAGE-Raf:ER or the control pHAGE were treated with 1 μM 4-hydroxytamoxifen for 2 days. Expression of mRNA of indicated targets was examined by RT-PCR and quantitative PCR. Data (mean±standard error) are from a representative experiment conducted in triplicates and are expressed as fold changes relative to the untreated cells harboring control plasmid. *P<0.05; **P<0.005 (Student's t test). (B) LNCaP-Raf:ER cells were treated with 1 μM 4-hydroxytamoxifen for 2 days in the presence or absence of 10 μM U0126. Expression of mRNA of indicated targets was examined by RT-PCR and quantitative PCR. (C) LNCaP-Raf:ER cells transfected with SQSTM1 promoter luciferase reporter were treated with 4-hydroxytamoxifen (Raf activated) for 1 day with or without the MEK1/2 inhibitor, AZD6244. Data (mean±standard error) are from a representative experiment conducted in triplicates and are expressed as fold changes relative to the untreated cells harboring control plasmid. *P<0.05 (Student's t test). (D) LNCaP-Raf:ER cells were treated with 1 μM 4-hydroxytamoxifen for indicated time periods in the presence of 50 μg/ml cycloheximide. (E) LNCaP-Raf:ER cells were treated with 4-hydroxytamoxifen for Raf activation for 1 day in the presence of different doses of proteasome inhibitors, MG132 and lactacystin. Total cell lysates were examined for expression of the indicated proteins by Western blotting. Equivalent volume of Dimethyl sulfoxide (DMSO) was used as the vehicle control. Data in (D) and (E) are representative images of two independent experiments. (F and G) LNCaP-Raf:ER cells transfected with BiP promoter luciferase reporters were treated with 4-hydroxytamoxifen for 1 day for Raf activation with or without tunicamycin. Data (mean±standard error) are from a representative experiment conducted in triplicates and are expressed as fold changes relative to the untreated cells harboring control plasmid (F) or as fold changes induced by Raf activation (G).
In support of the possibility that Raf/MEK/ERK can upregulate LC3B and SQSTM1 expression, Raf-induced upregulation of LC3B and SQSTM1 protein levels was significantly abolished in the presence of cycloheximide, an inhibitor of protein synthesis (Fig. 4D). Moreover, their upregulation by Raf was augmented in the presence of the proteasome inhibitors MG132 and lactacystin (Fig. 4E). Of note, contrary to the effect of autophagy inhibitors to increase LC3B-II levels (Fig. 3A), the proteasome inhibitors increased mainly LC3B-I levels (Fig. 4E), suggesting that the proteasome may also regulate the levels of unprocessed LC3B. These data demonstrate that Raf/MEK/ERK can regulate LC3B and SQSTM1 levels via non-canonical mechanisms other than autophagy.
While LC3B has been known as the major LC3 in the autophagosome, a recent study demonstrated that the other LC3 family member LC3A-vareint1 can also mediate autophagosome formation as efficiently as LC3B [26]. Contrary to LC3B regulation, Raf activation significantly decreased LC3A-vareint1 mRNA levels (Fig. 4A). However, Raf activation did not affect mRNA levels of the two key essential autophagy regulators, ATG5 and ATG7, whose transcriptional upregulation is required for prolonged autophagy [31] (Fig. 4A). These data suggest that Raf/MEK/ERK can selectively and differentially regulate the expression of certain autophagy machinery.
Raf/MEK/ERK activation upregulates BiP expression transcriptionally
Along with the changes in LC3B and SQSTM1 mRNA levels, Raf activation highly upregulated BiP mRNA levels (Fig. 4A), although this effect was blocked by U0126 (Fig. 4B). However, in contrast, Raf activation did not significantly affect mRNA levels of the two major HSP70 family molecular chaperones, HSP72 (heat-inducible) and HSC70 (constitutively expressed), or HSP60 (Fig. 4A), suggesting a specific requirement for BiP under Raf-activated conditions. Because BiP is known as a master regulator of ER stress response, we also examined whether Raf activation alters expression or splicing of the unfolded protein response-specific transcription factor, X-box binding protein 1 (XBP1), which indicates the onset of ER stress [10]. Raf activation did not upregulate XBP1 expression or splicing in LNCaP cells (Fig. 4A). Subsequently, we analyzed phosphorylation of eukaryotic translation initiator factor 2α (eIF2α) and expression of the pro-apoptotic transcription factor C/EBP-homologous protein (CHOP/GADD153), which also indicate ER stress [10]. In LNCaP, HEK293, and BJ cells, activation of ΔRaf-1:ER did not significantly increase eIF2α phosphorylation (Fig. 1A), although B-RafV600E induced eIF2α phosphorylation in IMR90E1A cells (Fig. 1B). Under these Raf-activated conditions, CHOP expression was induced only in HEK293 cells (Fig. 1A). These data therefore indicate that BiP expression is more closely correlated with the changes in LC3B and SQSTM1 than the changes in ER stress markers when Raf/MEK/ERK is activated.
To understand the mechanisms underlying Raf/MEK/ERK-induced BiP expression, we investigated whether Raf activation could upregulate the activity of the luciferase reporters containing BiP promoter DNA. The BiP promoter reporters, pGRP78 (-367)-luc and pGRP78 (-169)-luc were previously used to identify the location of ER-stress response elements within the -169 region from the transcriptional initiation site [21]. We found that pGRP78(-367)-luc expressed higher basal and Raf-induced luciferase activity than pGRP78(-169)-luc when compared to the empty control vector, pGL2 (Fig. 4F). Further, U0126 blocked Raf-induction of pGRP78(-367)-luc activity, indicating that Raf/MEK/ERK can upregulate BiP at transcription level (Fig. 4F). Of note, despite their different activities, both reporters showed similar rates of fold induction in response to Raf activation, and this was contrasted to their responses to the ER stress inducer tunicamycin, which increased pGRP78(-169)-luc activity higher than pGRP(-367)-luc activity (Fig. 4G). These different responses suggest that Raf/MEK/ERK may regulate BiP transcription via a mechanism different from the one regulated by ER stress.
Of note, contrary to the augmenting effects of chloroquine, bafilomycin A1, and ATG7 knockdown on Raf-induced BiP upregulation (Fig. 3A and B), the proteasome inhibitors did not significantly affect BiP induction by Raf (Fig. 4E). Therefore, BiP levels under Raf-activated conditions may be determined by a balance between increased transcription and autophagic degradation.
BiP is necessary for Raf/MEK/ERK to regulate LC3B at protein levels, but not at mRNA levels
To determine the role of BiP in Raf-mediated regulation of LC3 and SQSTM1, we depleted BiP in LNCaP cells, using three different lentiviral shRNA constructs that specifically target different mRNA regions of BiP (shBiP #1, #2, and #3). All these constructs substantially abrogated both Raf-induced BiP expression and increases in LC3B-I and LC3B-II protein levels, indicating that BiP is required for Raf-mediated LC3B regulation (Fig. 5A for day 2 effects; Supplemental Fig. S2 for day 1 effects). Nevertheless, BiP depletion did not significantly affect Raf-mediated regulation of LC3B mRNA levels (Fig. 5B; left panel, RT-PCR; right, qPCR), indicating that BiP is specifically required for Raf to regulate LC3B at protein levels.
Fig. 5.
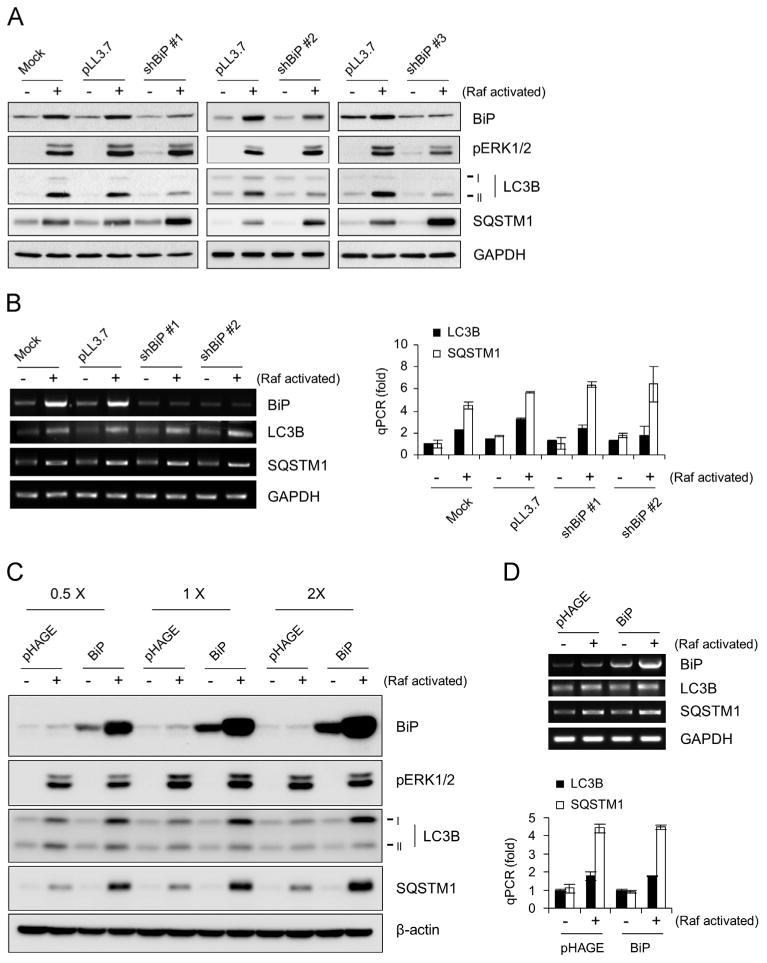
The effects of BiP knockdown or overexpression on LC3B and SQSTM1 in Raf-activated cells. (A and B) LNCaP-Raf:ER cells, infected with lentiviral pLL3.7 expressing three different shRNAs that target BiP (shBiP #1, #2, and #3), were treated with 1 µM 4-hydroxytamoxifen for 2 days (Raf activated). (A) Total cell lysates were examined by Western blot analysis for expression of the indicated proteins. Consistent results were also obtained at day 1 (Supplemental Fig. S1). (B) Expression of mRNAs of the indicated targets was analyzed by RT-PCR (left panel) and quantitative PCR (right panel). (C and D) LNCaP-Raf:ER cells, infected with lentiviral pHAGE expressing BiP at three different titers (0.5 ×, 1 ×, and 2 ×), were treated with 1 µM 4-hydroxytamoxifen for 2 days (Raf activated). (C) Total cell lysates were examined by Western blot analysis for expression of the indicated proteins. (D) Expression of mRNAs of the indicated targets was analyzed by RT-PCR (top panel) and quantitative PCR (bottom panel). Data (mean±standard error) are from a representative experiment performed in triplicate. Data in (A) and (C) are representative images of two independent experiments.
Conversely, overexpression of BiP (evaluated at three different expression levels) markedly increased the level of Raf-induced upregulation of LC3B-I, but not LC3B-II in LNCaP cells (Fig. 5C), without significantly affecting Raf-regulated LC3B mRNA expression (Fig. 5D; top panel, RT-PCR; bottom, qPCR) or autophagic flux under Raf-activated condition, as determined by using chloroquine (Supplemental Fig. S3). Of note, BiP overexpression by itself was not sufficient to increase LC3B-I levels, indicating that BiP may have a role specific to Raf/MEK/ERK activation. Importantly, BiP knockdown or overexpression did not affect ERK1/2 phosphorylation (Fig. 5A and C), indicating that the BiP effects were not due to altered Raf/MEK/ERK activity in cells but were mediated at a downstream level of the pathway signaling. These data suggests that BiP comprises a downstream pathway for Raf/MEK/ERK to regulate LC3B protein, although Raf/MEK/ERK regulates LC3 mRNA levels independently of BiP (depicted in Fig. 6).
Fig. 6.
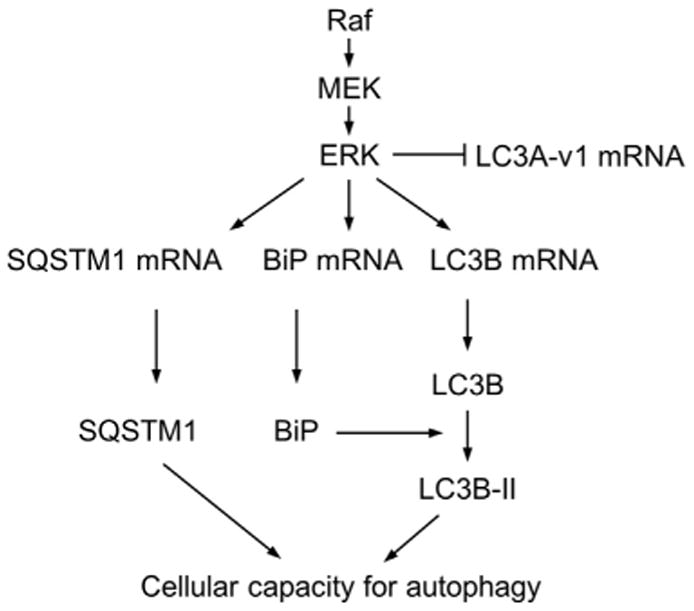
A model for Raf/EMK/ERK-mediated regulation of LC3 and SQSTM1. Raf/MEK/ERK activation is sufficient to induce alterations in LC3B and SQSTM1 levels, which is similar to the typical patterns indicating autophagy. Nonetheless, our results suggest that the net steady-state levels of LC3B and SQSTM1 under a Raf/MEK/ERK-activated condition are determined by not only the standard autophagic pathway but also by a regulation at mRNA levels and by an involvement of BiP. This signaling may be important in determining cellular capacity for autophagy under a Raf/MEK/ERK-activated condition.
Interestingly, both BiP knockdown and overexpression conditions further increased SQSTM1 protein levels in Raf-activated cells but did not affect its basal levels in the control cells (Fig. 5A and C). In contrast, Raf-mediated upregulation of SQSTM1 mRNA levels was not affected under any of these conditions (Fig. 5B and D). Therefore, either the absence of BiP or surplus of BiP may result in increased accumulation of SQSTM1 in Raf-activated cells. It is possible that these increases in SQSTM1 protein levels are due to limited LC3B supply under the BiP-depleted condition and due to inefficient processing of LC3B-I to LC3B-II under BiP-overexpressed condition.
Lastly, sustained Raf/MEK/ERK activation can paradoxically induce growth inhibition in different cell types, which is currently recognized as an anti-oncogenic mechanism in the face of aberrant Raf/MEK/ERK signal [33,34]. We previously reported that Raf/MEK/ERK can induce similar responses in LNCaP cells [18,19,27]. Therefore, we evaluated the effect of ATG7 knockdown or BiP knockdown on Raf-induced growth inhibition. In LNCaP cells, depletion of ATG7 or BiP neither significantly affected Raf-induced cell cycle arrest in G0/G1 phase nor altered apoptotic index (Supplemental Figs. S4 and S5), suggesting that regulation of BiP and autophagy machinery is not indispensable for Raf/MEK/ERK to mediate growth inhibitory signaling.
Discussion
Our present study demonstrate that aberrant Raf/MEK/ERK activation is sufficient to induce signals that trigger an alteration in LC3B and SQSTM1 levels in a similar pattern indicating autophagy. Indeed, the effects of autophagy inhibitors and ATG7 knockdown suggest that Raf/MEK/ERK may be able to alter autophagic activity in cells. However, other data in this report suggest that Raf/MEK/ERK-induced changes in these autophagy markers are also attributed to other mechanisms. Of particular interest, Raf/MEK/ERK activation led to upregulation of LC3B and SQSTM1 at mRNA levels, and the increases in SQSTM1 mRNA levels were due to increased transcription. These results suggest that, under a Raf/MEK/ERK-activated condition, the net steady-state levels of LC3B and SQSTM1 can be determined by multiple mechanisms including a non-canonical regulation of the autophagic machinery at an expression level (Fig. 6). In support of the complexity in SQSTM1 regulation, it has recently been reported that not only autophagic degradation but also increased transcription can affect its net steady-state levels under a prolonged starvation condition [35]. A cautious interpretation of changes in LC3B and SQSTM1 levels is therefore required when studying the effect of the Raf/MEK/ERK pathway on autophagy.
Intriguingly, Raf/MEK/ERK activation also led to upregulation of the ER chaperone, BiP, along with LC3B and SQSTM1, and this addresses a mechanism by which Raf/MEK/ERK regulates LC3B processing. BiP appears to have a specific role for LC3B regulation in response to Raf/MEK/ERK activation because BiP expression was necessary for Raf-induced LC3B processing. LC3B is expressed as pro-LC3B, which is cleaved into LC3B-I by ATG4. LC3B-I is conjugated with phosphatidylethanolamine for its conversion into LC3B-II, a step mediated by ATG5 and ATG7. LC3B-II is then utilized for the formation of autophagosome [2]. ER is the reservoir of Ca2+ and its release can activate calpain which inactivates ATG5 via proteolysis and, subsequently, inhibits LC3B processing [36]. Since BiP is the ER chaperone, these events may potentially be related. Indeed, under Raf/MEK/ERK-activated conditions, BiP overexpression was associated with LC3B-I accumulation while BiP knockdown was associated with depletion of both intermediates of LC3B. Therefore, BiP may affect LC3B processing via ER activity although it may also regulate additional mechanisms, e.g., LC3B translation and/or protein stability. Intriguingly, BiP did not affect Raf-regulated LC3B mRNA levels in this study, although it was reported that ER stress can upregulate LC3B transcription via the PERK/ATF4 cascade, a key effector of BiP in mediating ER stress [37]. Moreover, Raf-induced LC3B processing was not strongly correlated with any obvious changes of ER stress markers other than BiP. Therefore, ER stress may not be necessary for aberrant Raf/MEK/ERK activity to induce LC3B processing, and the primary reason for Raf/MEK/ERK to induce BiP may be mainly to regulate LC3B. In support of this, our promoter reporter analysis suggests that Raf/MEK/ERK upregulates BiP transcription via a responsive element(s) different from those regulated by ER stress. Taken all, BiP function for Raf/MEK/ERK-mediated LC3B regulation is not entirely consistent with its role for ER stress-induced autophagy. Indeed, recent studies have demonstrated that BiP has additional functions in different subcellular locations other than ER [38,39]. This possibility needs to be explored in our future study.
Our time-course data that SQSTM1 upregulation preceded the increases in LC3B and BiP may suggest a potential scenario in which these changes are important in the context of Raf/MEK/ERK signaling. A recent study has reported that, in addition to autophagosome formation, SQSTM1 facilitates nutrient sensing by serving as an integral scaffold of the mTORC1 complex required for activation of S6K1 and 4EBP1 [40]. The requirement of SQSTM1 for K-Ras tumorigenesis has also been demonstrated in a mouse lung tumor model [41]. Given these, it is conceivable that increased SQSTM1 expression may be due to cellular recognition of aberrant Raf/MEK/ERK activity as a mitogenic signal while dysregulation of this process may cause a metabolic stress, which induces BiP expression. Given that BiP confers stress tolerance in different cell types, including LNCaP cells [42], and that autophagy generates resources for cell survival under stress conditions [1,2], it is conceivable that cells may attempt to resist stress by increasing autophagy partly via BiP upregulation under aberrant Raf/MEK/ERK activation conditions. Of note, it has been reported that ERK1/2-mediated autophagy induction is a mechanism underlying the development of cisplatin resistance of ovarian cancer [43]. Our findings may have potential significance in this context, providing an insight into the possible mechanisms by which ERK1/2 regulates autophagy. Future study is thus required to determine whether ERK1/2 can mediate similar effects on BiP, LC3 and SQSTM1 in cisplatin-resistant tumors.
It has been reported that oncogenic Ras requires MEK/ERK activation to induce autophagy [44,45]. In this context, our results may provide an insight into the mechanism by which the Ras/Raf pathway regulates autophagy effectors. It appears that although Ras and Raf can commonly increase LC3B processing and expression, these two signal transducers affect autophagy effectors differently. For example, contrary to the Raf effects observed in this study, Ras activation decreased SQSTM1 protein levels [44], while increasing ATG7 mRNA levels in human diploid fibroblast cells [45]. Indeed, although aberrant Ras or Raf activation triggers largely similar physiological effects, many of these effects are mediated via different mechanisms because Ras can activate additional pathways other than Raf/MEK/ERK, including the PI3-kinase/AKT and RalGDS pathways [46]. It is therefore conceivable that Raf/MEK/ERK alone may not be able to mediate balanced autophagy while Ras does so by activating additional pathways. In support of this notion, additional alterations of different signaling pathways are often detected in B-RafV600E transformed tumors [47,48].
Our current study identifies BiP as a novel inducible effector of Raf/MEK/ERK. Considering that oncogenic Raf, such as B-RafV600E, is often detected in different tumor types [7], i.e., melanoma (≥ 70%), papillary thyroid cancer (≥ 50%), and colon cancer (≥ 10%), it is warranted to investigate BiP expression in these cancers in association with any autophagic alteration. In summary, our study demonstrates that (i) aberrant Raf/MEK/ERK activation is sufficient to increase not only LC3B processing and SQSTM1 accumulation but also their expression and (ii) Raf/MEK/ ERK induces BiP expression to regulate LC3B at protein levels. This study reveals novel Raf/MEK/ERK signaling, which may operate in a context relevant to autophagy.
Supplementary Material
Acknowledgments
We thank Amy Hudson (Medical College of Wisconsin) and Richard Mulligan (Harvard Univ.) for pHAGE, Clark Distelhorst (Case Western Reserve University) for pGRP78-luc, and James Brody (University of California, Irvine) for SQSTM1-luc. This work was supported by the National Cancer Institute (R01CA138441), American Cancer Society (RSGM-10-189-01-TBE), and FAMRI Young Investigator Award (062438) to J.P
Abbreviations
- 4-HT
4-hydroxytamoxifen
- ATG
autophagy-related gene
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HSP
Heat shock protein
- LC3
microtubule-associated protei-1 light chain-3
- SQSTM1
sequestosome 1
Footnotes
Disclosure statement: The authors declare no conflict of interest.
Appendix A. Supporting information: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.yexcr.2014.08.001.
References
- 1.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–119. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 5.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2009;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 7.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 8.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 9.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 11.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460–1471. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, Kaufman RJ, Bastian BC, Soengas MS. Antioncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- 15.Kaul A, Overmeyer JH, Maltese WA. Activated Ras induces cytoplasmic vacuolation and non-apoptotic death in glioblastoma cells via novel effector pathways. Cell Signal. 2007;19:1034–1043. doi: 10.1016/j.cellsig.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, Rubinsztein DC, Thumm M, Tooze SA. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 18.Hong SK, Yoon S, Moelling C, Arthan D, Park JI. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem. 2009;284:33006–33018. doi: 10.1074/jbc.M109.012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI, Mortalin A. /HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013;33:4051–4067. doi: 10.1128/MCB.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He H, McColl K, Distelhorst CW. Involvement of c-Fos in signaling grp78 induction following ER calcium release. Oncogene. 2000;19:5936–5943. doi: 10.1038/sj.onc.1203994. [DOI] [PubMed] [Google Scholar]
- 22.Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22:2322–2333. doi: 10.1038/sj.onc.1206325. [DOI] [PubMed] [Google Scholar]
- 23.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci USA. 2006;103:16406–16411. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott MX, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Bai H, Inoue J, Kawano T, Inazawa J. A transcriptional variant of the LC3A gene is involved in autophagy and frequently inactivated in human cancers. Oncogene. 2012;31:4397–4408. doi: 10.1038/onc.2011.613. [DOI] [PubMed] [Google Scholar]
- 27.Hong SK, Kim JH, Lin MF, Park JI. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res. 2011;317:2671–2682. doi: 10.1016/j.yexcr.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human Raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci USA. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abdalla EC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, Bast RC, Jr, Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen X, Chen X, Chen YG, Chen Y, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, Amelio MD, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farre JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesus L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, Gonzalez-Estevez C, Gonzalez-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hebert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hohfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Hoyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidore C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jaattela M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jiang X, Jimenez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhasz G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovacs AL, Kraft C, Krainc D, Kramer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Kruger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, Laszlo L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Low P, Lu B, Lu B, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KE, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Marino G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, Mclnerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Melendez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Muller S, Muller S, Munger K, Munz C, Murphy LO, Murphy ME, Musaro A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nurnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poggeler S, Poirot M, Poletti A, Pous C, Pozuelo-Rubio M, Praetorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Regnier-Vigouroux A, Reichert AS, Reiners JJ, Jr, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodriguez de Cordoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB, 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sanchez-Alcazar JA, Sanchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schatzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schuller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Stralfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takacs-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tanaka K, Tang D, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tonges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doom W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wang Z, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ, 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 33.Park JI. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front Biol. 2014;9:95–103. doi: 10.1007/s11515-014-1299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDuff FK, Turner SD. Jailbreak: oncogene-induced senescence and its evasion. Cell Signal. 2011;23:6–13. doi: 10.1016/j.cellsig.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTMl/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M, Ma X, Ma D, Yuan J. Control of basal autophagy by calpain 1 mediated cleavage of ATG5. Autophagy. 2010;6:61–66. doi: 10.4161/auto.6.1.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–2306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–15075. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37:230–236. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Li N, Zoubeidi A, Beraldi E, Gleave ME. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene. 2013;32:1933–1942. doi: 10.1038/onc.2012.212. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:17163–17173. doi: 10.1074/jbc.M114.558288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic ras-induced expression of noxa and beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeanblanc M, Ragu S, Gey C, Contrepois K, Courbeyrette R, Thuret JY, Mann C. Parallel pathways in RAF-induced senescence and conditions for its reversion. Oncogene. 2012;31:3072–3085. doi: 10.1038/onc.2011.481. [DOI] [PubMed] [Google Scholar]
- 47.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–344. doi: 10.1038/onc.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.