

Short abstract
This study characterizes the KAP protein family - a newly identified sub-group of the P-loop NTPases, which have transmembrane helices inserted into the P-loop NTPase domain. Their unusual phyletic distribution suggests KAP proteins were transferred from bacteria to animals by horizontal gene transfer.
Abstract
Background
Recent sequence-structure studies on P-loop-fold NTPases have substantially advanced the existing understanding of their evolution and functional diversity. These studies provide a framework for characterization of novel lineages within this fold and prediction of their functional properties.
Results
Using sequence profile searches and homology-based structure prediction, we have identified a previously uncharacterized family of P-loop NTPases, which includes the neuronal membrane protein and receptor tyrosine kinase substrate Kidins220/ARMS, which is conserved in animals, the F-plasmid PifA protein involved in phage T7 exclusion, and several uncharacterized bacterial proteins. We refer to these (predicted) NTPases as the KAP family, after Kidins220/ARMS and PifA. The KAP family NTPases are sporadically distributed across a wide phylogenetic range in bacteria but among the eukaryotes are represented only in animals. Many of the prokaryotic KAP NTPases are encoded in plasmids and tend to undergo disruption to form pseudogenes. A unique feature of all eukaryotic and certain bacterial KAP NTPases is the presence of two or four transmembrane helices inserted into the P-loop NTPase domain. These transmembrane helices anchor KAP NTPases in the membrane such that the P-loop domain is located on the intracellular side. We show that the KAP family belongs to the same major division of the P-loop NTPase fold with the AAA+, ABC, RecA-like, VirD4-like, PilT-like, and AP/NACHT-like NTPase classes. In addition to the KAP family, we identified another small family of predicted bacterial NTPases, with two transmembrane helices inserted into the P-loop domain. This family is not specifically related to the KAP NTPases, suggesting independent acquisition of the transmembrane helices.
Conclusions
We predict that KAP family NTPases function principally in the NTP-dependent dynamics of protein complexes, especially those associated with the intracellular surface of cell membranes. Animal KAP NTPases, including Kidins220/ARMS, are likely to function as NTP-dependent regulators of the assembly of membrane-associated signaling complexes involved in neurite growth and development. One possible function of the prokaryotic KAP NTPases might be in the exclusion of selfish replicons, such as viruses, from the host cells. Phylogenetic analysis and phyletic patterns suggest that the common ancestor of the animals acquired a KAP NTPase via lateral transfer from bacteria. However, an earlier transfer into eukaryotes followed by multiple losses in several eukaryotic lineages cannot be ruled out.
Background
The P-loop NTPase domains constitute one of the largest apparently monophyletic groups of globular protein domains in the proteomes of most cellular organisms [1,2]. These domains are implicated in nearly all biochemical and mechanical processes in the cell, including translation, transcription, replication and repair, intracellular trafficking, membrane transport, and activation of various metabolites [1,3]. At the sequence level, most of the P-loop domains are characterized by two conserved motifs, termed the Walker A and B motifs [4]. Structurally, P-loop domains adopt a globular fold with at least 5 α/β units (the P-loop NTPase fold), with the strands typically forming a core parallel sheet [5,6]. The Walker A motif (typically, Gx4GK[T/S], where x is any residue) encompasses the first strand and helix, and is involved in binding the triphosphate moiety of the substrate NTP. The Walker B motif (typically, hhhhD, where h is a hydrophobic residue) encompasses the third universally conserved strand in the P-loop NTPase fold and coordinates a Mg2+ ion which directs an attack on the bond between the β and γ phosphates of the NTP [1,3,4].
A series of recent comparative studies on the sequences and structures of P-loop NTPases defined the probable major evolutionary events in the diversification of these domains [6-12]. In particular, these studies delineated two major divisions of P-loop NTPases, the KG (kinase-GTPase) division and the ASCE division (for additional strand, catalytic E). The KG division includes kinases and GTPases that share many structural similarities, such as the adjacent placement of the P-loop and Walker B strands [9,10]. The ASCE division is characterized by an additional strand in the core sheet, which is located between the P-loop strand and the Walker B strand (Figure 1) [10]. As opposed to kinases and GTPases, ATP hydrolysis by the ASCE proteins typically depends on a conserved catalytic (proton-abstracting) acidic residue (usually glutamate) that primes a water molecule for the nucleophilic attack on the γ-phosphate group of ATP ([10] and references therein). As a consequence, ASCE division proteins typically are more active NTPases than those of the KG division and do not require accessory factors, such as GTPase-activating and GDP-exchange proteins [9]. In addition, most of the ASCE division NTPases possess a conserved polar residue at the carboxy terminus of strand 4, which is inserted between the strands associated with the Walker A and B motifs [10]. The ASCE division includes AAA+, ABC, PilT, superfamily 1/2 (SF1/2) helicases, and RecA/F1/F0 classes of ATPases, and a large assemblage of NTPases related to the AP(apoptotic) and NACHT families [6-8,11,13,14].
Figure 1.

Multiple alignment of the KAP family NTPases. The secondary structure predicted by the PHD program is displayed above the alignment, where E designates a β-strand and H designates α-helix. The helix and strand numbering is given for the secondary structural elements of the conserved P-loop fold. The 80% consensus coloring reflects the following amino acid classes: h (hydrophobic residues: ACFILMVWY), a (aromatic residues: FHWY), and l (aliphatic residues: VIL) are shaded yellow; b (big residues: LIYERFQKMW) are shaded gray; p (polar residues: CDEHKNQRST), - (acidic residues: DE), + (basic residues: HKR) and c (charged residues:HRKDE) are colored magenta; o (alcohol-group-containing residues: ST) are colored blue; s (small: GASCVDNPT) and u (tiny: GAS) residues are colored green. The protein identifiers in the alignment include the name of the protein/gene, species abbreviation and the GenBank gi separated by underscores. The groups discussed in the text are indicated to the right in the last block of the alignment. The asterisk next to the rat sequence indicates a Kidins paralog with a potentially inactive NTPase domain. Species abbreviations are as follows: Atu: Agrobacterium tumefaciens, Ana: Anabaena sp pcc 7120, Ce: Caenorhabditis elegans, Cpe: Clostridium perfringens, Cgl: Corynebacterium glutamicum, Ceff: Corynebacterium efficiens, Dr: Deinococcus radiodurans, Dm: Drosophila melanogaster, Ec: Escherichia coli, Plaf: F plasmid, Gsu: Geobacter sulfurreducens, Hs: Homo sapiens, Kpne: Klebsiella pneumoniae, Lme: Leuconostoc mesenteroides, Mcsp: Magnetococcus sp mc-1, Mde: Microbulbifer degradans, Npu: Nostoc punctiforme, Pput: Pseudomonas putida, Pfl: Pseudomonas fluorescens, Psy: Pseudomonas syringae, Rme: Ralstonia metallidurans, Rn: Rattus norvegicus, Step: Staphylococcus epidermidis, Ssp: Synechocystis sp, Tm: Thermotoga maritima, Vpar: Vibrio parahaemolyticus, Vvul: Vibrio vulnificus.
Recognition of these distinctive sequence and structural features allows classification of uncharacterized P-loop NTPase families into one of the principal divisions and facilitates predictions of their potential catalytic capacity. Systematic analysis of the P-loop NTPases further demonstrated that most of the conserved families of the ASCE division ATPases could be confidently placed within one of the six large classes mentioned above [11]. However, several families of ASCE NTPases remained outside this classification scheme. Here, we apply sequence and structural analysis to characterize one such previously unexplored family, which includes animal proteins participating in neural development and receptor tyrosine kinase signaling, and prokaryotic plasmid-encoded proteins that confer resistance to bacteriophages. We investigate the evolutionary implications of their unusual phyletic distribution and their unique structural feature, namely the insertion of multiple transmembrane helices into the P-loop NTPase fold. We also present predictions regarding their potential biochemical roles in eukaryotes and bacteria.
Results and discussion
Identification and classification of the KAP family of predicted ATPases
During our systematic analysis of the P-loop NTPase fold, we detected the mammalian neuronal membrane protein named kinase D-interacting substance of 220 kDa (Kidins220) or ankyrin repeat-rich membrane spanning protein (ARMS) [15,16] in various searches initiated with position-specific scoring matrices (PSSMs) for different ASCE division ATPases, such as the AAA+ class. The alignments produced in these searches indicated that the ARMS protein contained the characteristic sequence signatures of the Walker A and B motifs. However, examination of these alignments also showed that ARMS contained one or more long inserts (>100 amino acid residues) within the potential P-loop NTPase domain.
To further investigate the structure and evolutionary connections of this protein, we performed PSI-BLAST searches (expectation value of 0.01 for inclusion of sequences into the PSSM, with the statistical correction for compositional bias) using as the query the sequence of the putative P-loop NTPase domain of ARMS (GenBank identifier gi: 14133247, residues 433-959). The first iteration of this search retrieved apparent orthologs of ARMS from other animals, such as Danio, Drosophila, Anopheles and Caenorhabditis, and a homolog from the cyanobacterium Anabaena. The subsequent iterations also detected, with significant E-values (e < 10-5) apparent divergent homologs from bacteria spanning a broad phyletic range (Figure 1). A possible pseudogene belonging to this family was also detected in the genomes of the archaea Methanococcus jannaschii and Methanosarcina (see below). The prokaryotic proteins detected in these searches included the PifA protein, which is encoded in the enterobacterial F plasmid and is required for exclusion of bacteriophage T7 [17,18]. All these proteins contain the typical Walker A and B signatures, suggesting that they are functional P-loop NTPases. In contrast to the animal ARMS orthologs, most of the bacterial proteins, except for those from Anabaena species, Geobacter sulfurreducens and Microbulbifer degradans, lacked the large inserts within the P-loop NTPase domain. Reciprocal PSI-BLAST searches initiated with these bacterial proteins as queries first retrieved a consistent set of proteins that included the animal ARMS orthologs before the retrieval of other ASCE NTPases, such as the AP/NACHT-NTPases, AAA+ and ABC classes. These observations suggested that ARMS homologs define a novel group of P-loop NTPases that is distinct from all the previously described classes of P-loop domains. Hereinafter, we refer to them as the KAP family of (predicted) NTPases (after Kidins220/ARMS and PifA). In addition, the above searches retrieved a vertebrate paralog of the ARMS protein (for example, Rattus norvegicus protein LOC308414), in which Walker A and B motifs are disrupted (Figure 1), indicating that, unlike other ARMS homologs, it might lack NTPase activity.
To further explore the functional features and evolutionary relationships of the KAP family, we constructed a multiple alignment of the KAP proteins and compared its sequence conservation pattern and predicted secondary structure with those of other P-loop NTPases (Figure 1). The Walker B motif in the KAP family sequences typically has the form hhhhD[D/G]hD (where h is any hydrophobic residue). The second aspartate (D) immediately after the Walker B aspartate (first aspartate) is present in most of the bacterial KAP domains but is replaced by a glycine or an alanine in the animal sequences (Figure 1). An acidic residue in this position is an ancestral feature of the ASCE division of ATPases, and the presence of an aspartate is specifically characteristic of the AP/NACHT-NTPases as opposed to the glutamate, which is most common in the SFI/II helicase and AAA+ ATPases [7,13,14,19,20]. Furthermore, the third aspartate located three positions downstream of the Walker B aspartate is a shared feature of the KAP and NACHT families [13]. In the KAP family proteins, one of these aspartates might function as the proton-abstracting negative charge in NTP hydrolysis. The KAP family proteins contain another conserved polar residue (typically, D) at the end of strand 4 (Figure 1). This feature is also characteristic of the ASCE NTPases and corresponds to the sensor I motif of the AAA+ domains and its counterparts in other proteins of the ASCE division [7,11,14]. These conserved features, together with the consistent detection of various ASCE NTPases in database searches with the profiles of KAP family PSSM, strongly suggest that this family belongs to the ASCE division.
The conserved core of the P-loop NTPase domain of the KAP family contains an α-helix amino-terminal of the Walker A strand and an α-helical extension with three to four predicted helical segments occurring carboxy-terminal of strand 5 (Figures 1, 2). Similar structural features are also seen in the AAA+ ATPases and the NACHT/AP-NTPases, suggesting that the KAP family might form a higher-order group with these classes of NTPase domains within the ASCE division [11,13]. However, the specific extended sequence signatures associated with the Walker B motif, strand 5 of the core P-loop NTPase domain, and the carboxy-terminal helical module (Figure 1) clearly distinguish KAP ATPases from all other ASCE NTPases. Although most proteins of the KAP family have a conserved lysine at the beginning of strand 5, this residue does not appear to be equivalent to the arginine finger, which is found in ring-forming ASCE NTPases, such as the AAA+ and VirD4-like ATPases [6,7,11,14]. This suggests that KAP ATPases do not have an arginine finger and are unlikely to function as oligomeric rings. However, the KAP family proteins contain a conserved arginine in the carboxy-terminal helical segment, which could potentially function similarly to the sensor-2 arginine of the AAA+ ATPases (Figure 1). Examination of the multiple alignment suggests that, in addition to the five conserved strands of the core P-loop domain, the KAP family NTPase domain contains an additional strand after the core strand 2 (Figure 1). By analogy with the RecA and VirD4/PilT classes, this additional extended segment might stack externally on the β-sheet alongside strand 2 (Figure 2) [6,8].
Figure 2.

Predicted topology of the KAP P-loop NTPases and comparison with other P-loop NTPases. The core conserved strands that are shared by all ASCE division NTPases are numbered 1-5, and X indicates additional strands that are observed only in certain NTPases.
Most of the NTPases of the KAP family have a variable α-helical insert amino-terminal to the Walker B motif. Remarkably, all animal KAP NTPases and three bacterial ones, those from Anabaena, G. sulfurreducens and Microbulbifer, contain two membrane-spanning helices inserted in this region (Figures 1, 2). The animal proteins additionally contain two more transmembrane helices inserted in the region between helix 1 (associated with the Walker A motif) and strand 2 of the core NTPase domain. Insertion of membrane-spanning helices into globular domains is extremely rare in proteins [21], and, to our knowledge, the KAP family is the first such instance among P-loop NTPase domains. In the NTPase domains that do not form ring structures, most residues involved in NTP-binding and hydrolysis are located at the carboxy termini of the strands forming the core parallel β-sheet (Figures 1, 2). This causes a polarity in the structure of the NTPase domain with respect to the location of catalytic surface, thus allowing it to accrete inserts in regions that are spatially disjointed from this catalytic surface. This might explain the ability of the KAP NTPase domain to retain its structural and functional integrity despite the insertion of transmembrane helices. Superposition of the multiple alignment of the KAP family onto known structures of the P-loop NTPase domains suggests that the membrane-spanning inserts project outward from the conserved intracellular globular core, probably from the surface opposite to the NTP-binding surface (Figure 2).
Prediction of functional features of the KAP NTPases
In mammals, Kidins220/ARMS localizes to the tips of neurites and is abundantly expressed in the neural tissues in regions that are enriched in receptors for ephrins and ligands of the neurotrophin family. Furthermore, Kidins220/ARMS physically interacts with TrkA and p75 neurotrophin receptors and is phosphorylated upon activation of the neutrophin and ephrin receptors [15,16]. Kidins220/ARMS also appears to be a physiological substrate for protein kinase D, suggesting that it might be a key target for multiple neuronal signaling cascades [15,16]. Kidins220/ARMS and all its animal orthologs contain 10 or more amino-terminal ankyrin repeats [22], while the Anabaena homolog with transmembrane segments contains approximately 40 TPR repeats amino-terminal to the P-loop NTPase domain [23]. Similarly, the membrane-associated KAP proteins from Microbulbifer and G. sulfurreducens contain a large amino-terminal segment with predicted coiled-coil structure. Phosphorylation of Kidins220/ARMS by various kinases suggests that this protein might function as a signaling nexus associated with the cell membrane. The α-superhelical structure domains present in animal (and some bacterial) KAP NTPases, such as ankyrin and TPR repeats, could provide extended surfaces to mediate interactions with various protein complexes. The likely function for the KAP NTPase domain is the regulation of assembly/disassembly of these complexes in an NTP-dependent manner. In particular, Kidins220/ARMS and the orthologous KAP NTPases in other animals might regulate the assembly of neurite-membrane-associated signaling complexes that are positioned downstream of different receptor tyrosine kinases in the respective signaling pathways. Consistent with this proposal, the high-throughput screens for protein-protein interactions in Drosophila recovered the PDZ-domain protein Dlg, which binds the carboxy-terminal tails of neural membrane proteins, as an interacting partner for the Kidins220/ARMS ortholog [24]. The vertebrate paralogs of Kidins220/ARMS with apparently inactive NTPase domains lack the ankyrin repeats and might function as dominant-negative regulators of active KAP NTPases.
The bacterial KAP proteins without the transmembrane regions contain a variable helical insert (Figure 1), which could function as a site for interactions with other proteins. The prokaryotic KAP family members have not been characterized biochemically, but potential leads to their functions are suggested by the available data on the PifA protein, which is encoded in enterobacterial F plasmids and is required for exclusion of bacteriophage T7 from plasmid-containing cells [17,18]. The exclusion process involves interactions between PifA and the products of T7 genes 1.2 and 10, which code for the major phage capsid proteins, and is accompanied by an increase in membrane permeability [17,25]. These observations imply that PifA might reorganize certain membrane-associated complexes in an ATP-dependent manner and thereby disrupt the T7 life cycle. While it is not clear whether the principal function of PifA is in bacteriophage exclusion, some other lines of circumstantial evidence support this possibility.
The sporadic distribution of the KAP family in prokaryotes and its presence on plasmids (and a filamentous phage in Vibrio) in various species (Figure 3) suggests that it was widely disseminated by these laterally mobile replicons. Protection of bacterial cells from phages could be one of the functions of KAP NTPases in prokaryotes, a role that is conducive to rapid horizontal spread, by analogy with the dissemination of antibiotic-resistance determinants. In at least six prokaryotes, including both occurrences in archaea, the genes for KAP NTPases were disrupted by frameshifts. Although some sequencing errors cannot be ruled out, it seems extremely unlikely that such errors occurred independently in homologous genes in several species. Furthermore, on several occasions, species or strains closely related to those that harbor a frameshift in the KAP gene have an intact counterpart, suggesting multiple recent pseudogene formation events in the KAP family. Inactivation of KAP NTPases might be driven by phages acquiring resistance to the KAP-mediated pathways, thereby rendering KAP genes superfluous. Coexpression of PifA with plasmids encoding genes 1.2 and 10 of T7 resulted in lethality in Escherichia coli [26]. Such deleterious effects of KAP NTPases under certain circumstances, such as expression of high levels of certain phage proteins, could be an alternative selective pressure for their inactivation.
Figure 3.

Phylogenetic tree and domain architectures of KAP NTPases. Proteins are denoted by their gene names and species abbreviations. Plasmid-borne genes are denoted by red asterisks, and phage genes are denoted by a red +; the eukaryotic branches are colored green. Species abbreviations are as in Figure 1. Filled yellow circles indicate nodes with bootstrap support of greater than 75% in the full maximum-likelihood analysis. The bootstrap values obtained through different methods (Full maximum likelihood, Rell bootstrap with Protml/Rell BP, Puzzle bootstrap/Puzzle-B, Neighbor Joining, Minimum evolution) are specifically shown for the clade that includes animal and bacterial proteins. In the schematics of protein and gene structure, conserved operons are shown as boxed arrow, and transmembrane regions inserted into the KAP domain are shown in blue. DRC0009-C and PifA-C refer to carboxy-terminal globular regions shared by the DRC0009-C and PifA subfamily KAP ATPases. Note that CPE1287 and Lmes0002 do not have the PifA-C domain.
In prokaryotic genomes, genes coding for functionally interacting proteins often co-occur in conserved operons or form gene fusions to give rise to a single gene. Consequently, evolutionarily conserved juxtaposition of functionally uncharacterized genes with genes whose functions are known has the potential to throw light on the functions of the former [27-29]. In the case of KAP NTPases, a conserved gene neighborhood was detected in E. coli (strain cft073), Deinococcus radiodurans plasmid CP1, and Agrobacterium tumefaciens plasmid AT, in which the gene for the KAP NTPase is located next to genes encoding a TIM barrel DNase of the TatD family [30] and an ATP pyrophosphohydrolase of the PP-loop fold [31]. Although the exact functional implications of this linkage are unclear, it seems likely that these enzymes cooperate with the KAP NTPases in the inhibition of phage reproduction; the DNase, in particular, is a candidate for a role in degradation of phage DNA.
Evolution of the KAP NTPase family
Phylogenetic trees of the conserved NTPase domain of the KAP family were constructed using the maximum likelihood, neighbor-joining, and minimum evolution methods (see Materials and methods for details). The trees constructed with each of these methods had similar topologies and suggested existence of several subfamilies within the KAP family. One of these, the ARMS subfamily, includes all animal KAP proteins and three bacterial members, those from M. degradans, G. sulfurreducens and Anabaena (Figure 3). In this case, phylogenetic analysis strongly supported monophyly of this group, which was independently suggested by their shared derived character, the insertion of transmembrane helices into the P-loop domain. A second subfamily consists of proteins from phylogenetically diverse bacteria, such as E. coli (strain cft073), D. radiodurans plasmid CP1, A. tumefaciens plasmid AT, Ralstonia and Magnetococcus, and is also supported by an apparent shared derived character, a carboxy-terminal globular domain that is unique to this subfamily. This bacterial subfamily groups with the ARMS subfamily, to the exclusion of homologs from all other prokaryotes (Figure 3). The third major subfamily includes the F-plasmid-borne PifA and its homologs from plasmids and chromosomes of Klebsiella, Pseudomonas, Corynebacterium, Nostoc, Thermotoga, Clostridium and Leuconostoc. The validity of this family is supported by the presence of a unique carboxy-terminal domain that shows no obvious relationships with any previously conserved globular domains.
Thus, on more than one occasion, the phylogenetic tree of the KAP family brings together phylogenetically distant bacteria (for example, Deinococcus, Agrobacterium and E. coli) in well-supported clades, strongly suggesting a major role of plasmid-mediated horizontal transfer in the evolution of this family (Figure 3). The most striking feature of the tree is the nesting of the animal ARMS homologs within a clade containing bacterial members. Among the currently available members of the KAP family, the greatest diversity is seen in bacteria, and almost all subfamilies contain multiple plasmid-borne members. It seems likely that the original KAP NTPase evolved on a bacterial plasmid and had a role in the modification of the bacterial membrane that results in exclusion of bacteriophages from the plasmid-carrying bacteria. Subsequently, the KAP NTPase in one of the bacterial lineages acquired the pair of transmembrane helices inserted into the P-loop domain, which made it an integral membrane protein. The apparent preponderance of horizontal gene transfer in the evolution of the KAP family and the phylogenetic affinities of the animal KAP NTPases suggest that the gene for a membrane-spanning KAP NTPase was laterally transferred to eukaryotes before the divergence of the major animal lineages, probably from a bacterial plasmid or chromosome. As no eukaryotes other than animals are currently known to have a KAP NTPase, it seems likely that this gene transfer occurred relatively late in evolution - that is, after the separation of the lineage leading to the animals from other crown-group eukaryotes. However, given the sparse sampling of large eukaryotic genomes from different crown-group lineages, the possibility remains that the transfer occurred earlier, but KAP genes have been lost in the currently sampled taxa.
Evidence of independent insertion of transmembrane helices in other P-loop NTPase domains
In search of other possible instances of insertion of transmembrane segments into P-loop NTPase domains we analyzed all uncharacterized NTPase domains detected in our searches using the TMHMM program for transmembrane helix prediction. As a result, we identified another small family of predicted NTPases containing transmembrane helices inserted into the P-loop domain. This family is present in several bacteria and includes the yobI gene of Bacillus subtilis and its orthologs from Clostridium perfringens, Bacteroides thetaiotaomicron and Streptococcus mutans (Figure 4). All these proteins contain a pair of predicted transmembrane helices inserted after the second conserved strand-helix unit of the NTPase core. The location of this insert thus differs from that seen in the ARMS subfamily of the KAP family, where the transmembrane helices are inserted immediately after the Walker A associated strand-helix unit (Figures 1, 4). The P-loop domain of these proteins shows the hallmarks of the ASCE division but no specific affinity with the KAP family, suggesting an independent origin of the inserts. In addition, these proteins contain a large conserved carboxy-terminal extension that is predicted to adopt an α-superhelical structure. The presence of these predicted NTPases in a taxonomically disjointed set of bacteria suggest a horizontal mode of dissemination similar to that discussed above for the KAP family.
Figure 4.
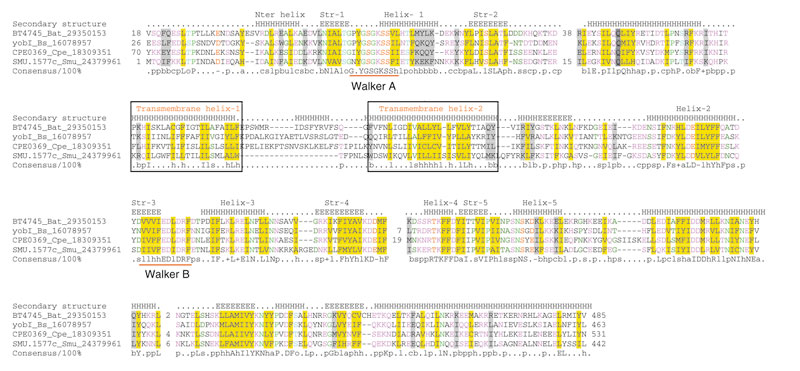
Multiple alignment of the YobI family NTPases. The coloring scheme and labeling conventions are as in Figure 1. Species abbreviations are as follows: Bs: Bacillus subtilis, Bat: Bacteroides thetaiotaomicron, Cpe: Clostridium perfringens, Smu: Streptococcus mutans.
Conclusions
We describe here a previously unnoticed family of P-loop NTPases that displays unusual structural features and phyletic patterns. The P-loop NTPase domain of this family, designated the KAP family, belongs to the ASCE division of P-loop NTPases and might be distantly related to the AAA+ and AP/NACHT NTPases [10,11,13]. All eukaryotic and several bacterial members of the KAP family contain two or four transmembrane segments inserted into the P-loop NTPase domain and, accordingly, are predicted to be integral membrane proteins, with the P-loop domain attached to the intracellular side of the membrane. In addition, we identified another small family of predicted bacterial NTPases, which do not seem to be specifically related to the KAP family, but also contain two transmembrane helices inserted into the P-loop domain. Insertion of transmembrane helices into globular domains is generally rare and, to our knowledge, has not been described in P-loop NTPases so far. It is well known, however, that the P-loop domain tolerates extremely long inserts of hydrophilic domains, such as the coiled-coil domains in the SMC family ATPases involved in chromatin dynamics and repair [32,33]. Furthermore, many P-loop NTPases are involved in membrane transport and secretion. In particular, these are the principal functions of the ABC-class ATPases, and some of these, such as the CFTR protein in animals, contain multiple transmembrane helices, which, however, are located outside the P-loop domain [34]. The discovery of two families of predicted P-loop NTPases with transmembrane helices inserted into the P-loop domain itself unifies these two structural themes and further expands our notion of the enormous structural and functional plasticity of this widespread domain.
Among eukaryotes, the KAP family is so far represented only in animals and is typified by the neuronal membrane protein Kidins220/ARMS and its paralog, which seems to have a catalytically inactive NTPase domain. In prokaryotes, KAP NTPases are often encoded by plasmids and might function in exclusion of bacteriophages from the plasmid-bearing bacterial cells. We predict that both eukaryotic and bacterial KAP NTPases regulate NTP-dependent assembly or disassembly of membrane-associated protein complexes. Phyletic pattern and phylogenetic analysis suggest that lateral transfer from bacteria to the animal lineage (or an earlier ancestral form) before the diversification of the latter gave rise to the ancestor of the eukaryotic KAP NTPases. However, given the evidence of rampant gene loss in diverse eukaryotes [35,36], it is conceivable that the KAP NTPases were acquired early in eukaryotic evolution and subsequently lost in several non-animal lineages. Regardless of the exact origin scenario, these NTPases provide a remarkable example of recruitment of a protein originally acquired from bacteria for animal-specific functions, such as receptor tyrosine kinase-mediated signaling in neural growth and development.
Materials and methods
The non-redundant (NR) database of protein sequences (National Center for Biotechnology Information, NIH, Bethesda) was searched using BLASTP [37]. Iterative database searches were conducted using PSI-BLAST with either a single sequence or an alignment used as the query, with the PSSM inclusion expectation (E) value threshold of 0.01 (unless specified otherwise); the searches were iterated until convergence [37]. For all searches with compositionally biased proteins, the statistical correction for this bias was used [38,39]. Multiple alignments were constructed using the T_Coffee or PCMA programs, followed by manual correction based on the PSI-BLAST results [40,41]. All large-scale sequence analysis procedures were carried out using the SEALS package [42]. Transmembrane regions were predicted in individual proteins using the TMPRED [43], TMHMM2.0 [44] and TOPRED1.0 [45] programs with default parameters. For TOPRED1.0, the organism parameter was set to 'prokaryote' or 'eukaryote' depending on the source of the protein.
Protein-structure manipulations were performed using the Swiss-PDB viewer program [46] and the ribbon diagrams were constructed using the MOLSCRIPT program [47]. Protein secondary structure was predicted using a multiple alignment as the input for the PHD program [48]. Similarity-based clustering of proteins was carried out using the BLASTCLUST program [49].
Phylogenetic analysis was carried out using the maximum-likelihood, neighbor-joining, and minimum evolution (least squares) methods. Maximum-likelihood distance matrices were constructed with the TreePuzzle 5 program using 1,000 replicates generated from the input alignment and used as the input for construction of neighbor-joining trees with the Weighbor program [50,51]. Weighbor uses a weighted neighbor-joining tree construction procedure that has been shown to correct effectively for long-branch effects [51]. The minimal evolution trees were constructed using the FITCH program of the Phylip package, [52] followed by local rearrangement using the Protml program of the Molphy package [53] to produce the maximum likelihood (ML) tree. The statistical significance of the internal nodes of the ML tree was assessed using the relative estimate of logarithmic likelihood bootstrap (Protml RELL-BP), with 10,000 replicates [53]. A full ML tree was constructed using the Proml program of the Phylip package [52]. This tree was used as the input tree to generate further full ML trees using the PhyML program with 100 bootstrap replicates generated from the input alignment [54]. The consensus of these trees was derived using the Consense program of the Phylip package to obtain the bootstrapped ML tree [52]. A gamma distribution with one invariant and eight variable sites with different rates was used in the ML analysis. Gene neighborhoods were determined by searching the NCBI PTT tables with a custom-written script. These tables can be accessed from the genomes division of the Entrez retrieval system.
References
- Saraste M, Sibbald PR, Wittinghofer A. The P-loop - a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-F. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L. Protein fold recognition using sequence profiles and its application in structural genomics. Adv Protein Chem. 2000;54:245–275. doi: 10.1016/s0065-3233(00)54008-x. [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A. Nucleoside triphosphate-binding proteins: different scaffolds to achieve phosphoryl transfer. Q Rev Biophys. 1999;32:1–56. doi: 10.1017/S0033583599003480. [DOI] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner-White EJ, Coggins JR, Anton IA. Evidence for an ancestral core structure in nucleotide-binding proteins with the type A motif. J Mol Biol. 1991;221:751–754. doi: 10.1016/0022-2836(91)80170-Y. [DOI] [PubMed] [Google Scholar]
- Lupas AN, Martin J. AAA proteins. Curr Opin Struct Biol. 2002;12:746–753. doi: 10.1016/S0959-440X(02)00388-3. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Leipe DD, Aravind L, Grishin NV, Koonin EV. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000;10:5–16. [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Aravind L. The NACHT family - a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci. 2000;25:223–224. doi: 10.1016/S0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure - diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- Iglesias T, Cabrera-Poch N, Mitchell MP, Naven TJ, Rozengurt E, Schiavo G. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J Biol Chem. 2000;275:40048–40056. doi: 10.1074/jbc.M005261200. [DOI] [PubMed] [Google Scholar]
- Kong H, Boulter J, Weber JL, Lai C, Chao MV. An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci. 2001;21:176–185. doi: 10.1523/JNEUROSCI.21-01-00176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CK, Kemp P, Molineux IJ. Genes 1.2 and 10 of bacteriophages T3 and T7 determine the permeability lesions observed in infected cells of Escherichia coli expressing the F plasmid gene pifA. J Bacteriol. 1991;173:6507–6514. doi: 10.1128/jb.173.20.6507-6514.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cram HK, Cram D, Skurray R. F plasmid pif region: Tn1725 mutagenesis and polypeptide analysis. Gene. 1984;32:251–254. doi: 10.1016/0378-1119(84)90053-2. [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. A novel superfamily of nucleoside triphosphate-binding motif containing proteins which are probably involved in duplex unwinding in DNA and RNA replication and recombination. FEBS Lett. 1988;235:16–24. doi: 10.1016/0014-5793(88)81226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins. 1993;17:363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Boguski MS, Goebl M, Hieter P. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell. 1990;60:307–317. doi: 10.1016/0092-8674(90)90745-z. [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Blumberg DD, Mabie CT, Malamy MH. T7 protein synthesis in F-factor-containing cells: evidence for an episomally induced impairment of translation and relation to an alteration in membrane permeability. J Virol. 1975;17:94–105. doi: 10.1128/jvi.17.1.94-105.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CK, Molineux IJ. Expression of gene 1.2 and gene 10 of bacteriophage T7 is lethal to F plasmid-containing Escherichia coli. J Bacteriol. 1991;173:1536–1543. doi: 10.1128/jb.173.4.1536-1543.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. doi: 10.1016/S0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- Huynen M, Snel B, Lathe W, 3rd, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Kondrashov AS, Koonin EV. Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome Res. 2001;11:356–372. doi: 10.1101/gr.GR-1619R. [DOI] [PubMed] [Google Scholar]
- Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, Berks BC, Palmer T. TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family proteins in sec-independent protein export. J Biol Chem. 2000;275:16717–16722. doi: 10.1074/jbc.M000800200. [DOI] [PubMed] [Google Scholar]
- Aravind L, Anantharaman V, Koonin EV. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implications for protein evolution in the RNA. Proteins. 2002;48:1–14. doi: 10.1002/prot.10064. [DOI] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SH, Krien MJ, O'Connell MJ. Structural maintenance of chromosomes (SMC) proteins, a family of conserved ATPases. Genome Biol. 2002;3:reviews3003.1–3003.5. doi: 10.1186/gb-2002-3-2-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland IB, Blight MA. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J Mol Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci USA. 2000;97:11319–11324. doi: 10.1073/pnas.200346997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortschak RD, Samuel G, Saint R, Miller DJ. EST analysis of the cnidarian Acropora millepora reveals extensive gene loss and rapid sequence divergence in the model invertebrates. Curr Biol. 2003;13:2190–2195. doi: 10.1016/j.cub.2003.11.030. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994;18:269–285. doi: 10.1016/0097-8485(94)85023-2. [DOI] [PubMed] [Google Scholar]
- Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Pei J, Sadreyev R, Grishin NV. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics. 2003;19:427–428. doi: 10.1093/bioinformatics/btg008. [DOI] [PubMed] [Google Scholar]
- Walker DR, Koonin EV. SEALS: a system for easy analysis of lots of sequences. Proc Int Conf Intell Syst Mol Biol. 1997;5:333–339. [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. TMbase - a database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;347:166. [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Claros MG, von Heijne G. TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci. 1994;10:685–686. doi: 10.1093/bioinformatics/10.6.685. [DOI] [PubMed] [Google Scholar]
- Peitsch MC. ProMod and Swiss-Model: internet-based tools for automated comparative protein modelling. Biochem Soc Trans. 1996;24:274–279. doi: 10.1042/bst0240274. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. doi: 10.1107/S0021889891004399. [DOI] [Google Scholar]
- Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- BLASTCLUST ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.txt
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Bruno WJ, Socci ND, Halpern AL. Weighted neighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol Biol Evol. 2000;17:189–197. doi: 10.1093/oxfordjournals.molbev.a026231. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Adachi J, Hasegawa M. MOLPHY: Programs for Molecular Phylogenetics. Tokyo: Institute of Statistical Mathematics; 1992. [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]