

Short abstract
This study shows that only a small fraction of yeast protein complexes are coregulated at the transcriptional level.
Abstract
Background
Multiprotein complexes play an essential role in many cellular processes. But our knowledge of the mechanism of their formation, regulation and lifetimes is very limited. We investigated transcriptional regulation of protein complexes in yeast using two approaches. First, known regulons, manually curated or identified by genome-wide screens, were mapped onto the components of multiprotein complexes. The complexes comprised manually curated ones and those characterized by high-throughput analyses. Second, putative regulatory sequence motifs were identified in the upstream regions of the genes involved in individual complexes and regulons were predicted on the basis of these motifs.
Results
Only a very small fraction of the analyzed complexes (5-6%) have subsets of their components mapping onto known regulons. Likewise, regulatory motifs are detected in only about 8-15% of the complexes, and in those, about half of the components are on average part of predicted regulons. In the manually curated complexes, the so-called 'permanent' assemblies have a larger fraction of their components belonging to putative regulons than 'transient' complexes. For the noisier set of complexes identified by high-throughput screens, valuable insights are obtained into the function and regulation of individual genes.
Conclusions
A small fraction of the known multiprotein complexes in yeast seems to have at least a subset of their components co-regulated on the transcriptional level. Preliminary analysis of the regulatory motifs for these components suggests that the corresponding genes are likely to be co-regulated either together or in smaller subgroups, indicating that transcriptionally regulated modules might exist within complexes.
Background
Multiprotein complexes such as the ribosome, spliceosome, cyclosome, proteasome and the nuclear pore complex have an essential role in cellular processes [1-3]. Until recently, information about the building blocks of specific complexes has been rather selective, and the mechanisms underlying the formation of these complexes, and their regulation, lifetimes and degradation remain largely unknown.
One can surmise that the formation of multiprotein complexes might be regulated at different levels, including transcriptional regulation, post-translational modification and degradation. In prokaryotes a significant proportion of the genes that are co-regulated at the transcriptional level code for proteins that interact physically. This proportion is even higher for gene groups whose co-regulation is conserved in different genomes [4]. In some multiprotein complexes in bacteria, the individual components were reported to be expressed 'as needed', in a time-dependent fashion related to their role in the complex [5].
In eukaryotes, mainly limited to yeast, gene-expression profiles have been shown to correlate with protein function and protein-protein interactions [6-8]. More particularly, genes corresponding to components of multiprotein complexes were found to exhibit correlated expression profiles, especially for complexes that form over a wide range of cellular conditions [8]. In contrast, the relationships between gene expression and genome-scale two-hybrid interaction data appear to be more tenuous [6,7,9].
Yeast is an ideal model system in which to investigate the relations between protein interactions and gene co-regulation. It is one of the few organisms in which many individual protein complexes have been characterized by biochemical and other methods, with results available in the Comprehensive Yeast Genome Database (CYGD) [10]. In addition, two independent studies recently characterized multiprotein complexes in yeast by a large-scale experimental approach involving tandem affinity purification and MS analysis (TAP [11]) and high-throughput MS protein complex identification (HMS, [12]). Each study identified several hundred complexes, containing on average about eight and eleven polypeptides, respectively. Many of these were shown to be associated with known cellular processes.
Yeast has also served as a model for the analysis of gene expression [13-15] and transcriptional regulation [16,17]. Information about the target genes of transcription factors has been compiled in specialized databases such as TRANSFAC [18,19], SCPD [19], YPD [20] and aMAZE [21,22]. Most recently, the genes bound by 106 yeast transcription factors were identified by a high-throughput approach [16], producing for the first time a global view of the transcriptional regulation network in this organism.
Here we investigate the transcriptional regulation of multiprotein complexes in yeast. In particular we aimed at finding out to what extent components of such complexes are co-regulated. We first determined the overlap between known sets of co-regulated genes in yeast and groups of genes coding for components of individual multiprotein complexes. A set of co-regulated genes is defined here as the group of target genes of the same transcription factor, and is denoted a 'regulon', in agreement with the classical concept of Maas [23]. Two categories of regulons are considered. The manually curated regulons stored in the databases, and the regulons defined by the gene-factor associations identified in the high-throughput analyses mentioned above [16]. The protein complexes examined are those manually curated in databases and the two datasets derived from the recent genome-scale analyses.
We then applied pattern-discovery algorithms [24,25] to the upstream sequences of genes coding for the proteins involved in each of the complexes in the three datasets under consideration. These algorithms are used to detect sequence patterns shared by some or all of these genes, which are likely to represent binding sites for transcription factors. These patterns take the form of short oligonucleotides (hexamers or pairs of trimers) that occur much more frequently in the upstream regions of these genes than in the corresponding regions across the entire yeast nuclear genome.
We have shown recently that these algorithms have an important advantage of returning predictions with a very small rate of false positives (over-represented patterns in groups of randomly selected genes) when stringent enough statistical criteria are used [26]. Alternative methods based on matrix descriptions [27-31] allow a more refined description of pattern degeneracy, in which a given sequence position need not be strictly conserved. But, unlike the approach used here, they have the inconvenience of nearly always returning a prediction, even for random sequences. This is particularly problematic when analyzing large groups of genes, of which a sizable proportion might not be regulated at the transcriptional level, or at least not by the same transcription factor, as might be the case for many of the protein complexes examined here.
Using the set of patterns detected for each complex, we proceeded to predict the components of the complex that are likely to be co-regulated. This is a difficult task, as the upstream regions of genes often contain multiple binding sites for the same factor or can be regulated by a combination of different factors that bind to distinct sites [32,33]. In addition, pattern-discovery algorithms generally return a number of strongly overlapping patterns for a given transcription factor, indicating the presence of a partial degeneracy [24,25]. Therefore, identifying sets of co-regulated genes usually involves assembling the patterns into longer motifs, and searching for upstream regions that score highly against these motifs, an approach that often yields ambiguous results.
Here we use an alternative approach in which a discriminant analysis is performed directly on the detected short patterns and their multiple occurrences [26], thereby avoiding the difficult task of pattern assembly. This analysis is done for all the complexes considered and the results are discussed in terms of our current knowledge of these complexes and their regulation.
Together, the approaches presented here provide valuable insights into the transcriptional regulation of multiprotein complexes in yeast and help in extracting information on function from genome-scale datasets for these complexes.
Results
Correspondence between multiprotein complexes and known regulons
The genes coding for the components of each protein complex in the different datasets are compared to those in the known regulons, with the aim of detecting complex-regulon pairs where the overlap between the components is more extensive than would be expected by chance.
The analyzed datasets of complexes comprised 243 annotated protein complexes from CYGD [10] and 725 complexes identified by the high-throughput studies [11,12]. The complexes from the latter two studies were taken as defined by their authors, without further grouping [34]. The regulons datasets comprised the 200 annotated and the 106 high-throughput regulons [16].
To determine whether the number of common components for a given complex-regulon pair is above chance level, or statistically significant, we compute the expectation value (E-value) of observing at least that number by chance, and retain only pairs with an E-value below a certain threshold (see Materials and methods).
Correspondence between regulons and annotated protein complexes
Table 1 lists the complex-regulon pairs whose overlap is above chance level (E-value ≤ 0.01), obtained when mapping the annotated complexes onto the annotated (Table 1a) and high-throughput (Table 1b) regulons, respectively. It is striking to see that the 243 annotated complexes and 306 known regulons form a total of only 57 pairs with a statistically significant overlap. Forty of those are with the annotated regulons, and the remaining ones (only 17 in total) are with the high-throughput regulons. Those pairs involve only about 8% of complexes (20 out of 243) and 14% of the regulons (44 out of 306). The overlap between known regulons and annotated complexes is thus on the whole quite limited.
Table 1.
Statistically significant associations between annotated complexes and known regulons
| (a) Associations between the annotated complexes and annotated regulons | |||||||
| Annotated complex | Annotated regulon | Components in complex | Genes in regulon | Common genes | E-value | Total overlap | |
| Permanent | 19-22S regulator | Rpn4 | 18 | 11 | 6 | 2E-11 | 6 |
| Cytochrome bc1 complex | Hap4 | 9 | 14 | 3 | 1E-04 | 2 | |
| Nucleosomal protein complex | Hir1 | 8 | 4 | 4 | 2E-10 | 7 | |
| Hir3 | 3 | 3 | 3E-07 | ||||
| Hta2 | 2 | 2 | 3E-04 | ||||
| Hta1 | 2 | 2 | 3E-04 | ||||
| Hir2 | 2 | 2 | 3E-04 | ||||
| Spt10 | 3 | 2 | 9E-04 | ||||
| Spt21 | 3 | 2 | 9E-04 | ||||
| RNA polymerase II | Abf1 | 13 | 37 | 3 | 1E-02 | 3 | |
| RNA polymerase III | Abf1 | 13 | 37 | 3 | 1E-02 | 3 | |
| Transient | 2 oxoglutarate dehydrogenase | Hap2 | 3 | 14 | 2 | 3E-03 | 2 |
| Hap3 | 15 | 2 | 3E-03 | ||||
| Alpha alpha trehalose phosphate synthase | Msn2 | 4 | 56 | 3 | 5E-04 | 3 | |
| Msn4 | 58 | 3 | 6E-04 | ||||
| Anthranilate synthase | Gcn4 | 2 | 40 | 2 | 8E-03 | 2 | |
| Fatty acid synthetase cytoplasmic | Reb1 | 2 | 19 | 2 | 2E-03 | 2 | |
| Ino2 | 19 | 2 | 2E-03 | ||||
| Ino4 | 19 | 2 | 2E-03 | ||||
| GAL80 complex | Mig1 | 3 | 26 | 2 | 1E-02 | 2 | |
| Glycine decarboxylase | Fau1 | 4 | 3 | 3 | 2E-08 | 3 | |
| Isocitrate dehydrogenase | Rtg3 | 2 | 6 | 2 | 2E-04 | 2 | |
| Rtg1 | 12 | 2 | 7E-04 | ||||
| Ribonucleoside diphosphate reductase | Dun1 | 4 | 3 | 3 | 2E-08 | 4 | |
| Rfx1 | 5 | 3 | 2E-07 | ||||
| Tup1 | 7 | 3 | 7E-07 | ||||
| Yku70 | 2 | 2 | 6E-05 | ||||
| Rad9 | 2 | 2 | 6E-05 | ||||
| Mbp1 | 6 | 2 | 9E-04 | ||||
| Others | Cdc28p complexes | Ndt80 | 10 | 11 | 5 | 3E-10 | 10 |
| Xbp1 | 5 | 3 | 6E-06 | ||||
| Swi6 | 10 | 3 | 7E-05 | ||||
| Mcm1 | 14 | 3 | 2E-04 | ||||
| Azf1 | 2 | 2 | 5E-04 | ||||
| Sit4 | 2 | 2 | 5E-04 | ||||
| Spt16 | 2 | 2 | 5E-04 | ||||
| Far1 | 2 | 2 | 5E-04 | ||||
| Cln3 | 2 | 2 | 5E-04 | ||||
| Bck2 | 2 | 2 | 5E-04 | ||||
| Glucan synthases | Swi4 | 5 | 8 | 2 | 3E-03 | 2 | |
| (b) Associations between annotated complexes and high-throughput regulons, identified by a genome-wide location analysis [16] | |||||||
| Annotated complex | High-throughput regulon | Components in complex | Genes in regulon | Common genes | E-value | Total overlap | |
| Permanent | Respiration chain complexes | ||||||
| Cytochrome bc1 complex | Hap4 | 9 | 69 | 7 | 5E-11 | 7 | |
| Cytochrome c oxidase | Hap4 | 8 | 69 | 8 | 2E-14 | 8 | |
| F0-F1 ATP synthase | Hap4 | 15 | 69 | 10 | 4E-15 | 10 | |
| Cytoplasmic ribosomes | |||||||
| Cytoplasmic ribosomal large subunit | Fhl1 | 81 | 194 | 67 | 4E-90 | 67 | |
| Rap1 | 209 | 46 | 3E-46 | ||||
| Yap5 | 107 | 10 | 1E-04 | ||||
| Pdr1 | 69 | 8 | 3E-04 | ||||
| Cytoplasmic ribosomal small subunit | Fhl1 | 57 | 194 | 53 | 2E-76 | 53 | |
| Rap1 | 209 | 30 | 4E-28 | ||||
| Yap5 | 107 | 8 | 5E-04 | ||||
| Nucleosomal-protein-complex | Hir2 | 8 | 21 | 6 | 2E-12 | 6 | |
| Hir1 | 30 | 6 | 2E-11 | ||||
| Transient | Fatty acid synthetase cytoplasmic | Ino2 | 2 | 11 | 2 | 3E-04 | 2 |
| Ino4 | 19 | 2 | 9E-04 | ||||
| Glycine decarboxylase | Bas1 | 4 | 44 | 3 | 1E-04 | 3 | |
| Ribonucleoside diphosphate reductase | Rfx1 | 4 | 32 | 3 | 5E-05 | 3 | |
| Others | Replication complexes | Mbp1 | 49 | 112 | 7 | 2E-03 | 7 |
Only the most statistically significant associations (E-value ≤ 0.01) between complexes and regulons are listed (see Additional data file 1 (Figure S2a-b) for a complete list). Each line lists the association detected between a multiprotein complex denoted by its CYGD name (column 2) and a regulon denoted by its common name (column 3). Column 4 lists the number of genes in the complex and column 5 lists the number of genes in the regulon. Column 6 lists the number of common genes between the regulon and complex, and column 7 lists the statistical significance criterion (E-value) for the detected overlap (see Materials and methods). The far right column lists the total number of genes in the complex that are common between it and all the regulons that map into it. Complexes have been subdivided into three categories, 'permanent', 'transient' or 'others', as indicated in column 1, and described in Materials and methods. When a smaller complex is completely included within a larger one and detected associations map into it, only the smaller complex is listed. For example, the larger assembly 'Cyclin-CDK complexes' is not listed because the detected association is with one of its components the 'Cdc28p complexes' only. When associations are detected with more than one complex of a larger assembly, as is the case for the small and large subunits of the cytoplasmic ribosomes, the name of the larger assembly is given first, with no details of the identified associations. But those are listed for each of the component complexes. Information on the annotated regulons in (a) was obtained from the TRANSFAC and aMAZE databases, from the list compiled by Young and colleagues [16,48] and from the recent literature.
Relating protein complexes to gene-expression data, Jansen et al. [7] found it useful to distinguish between two major categories of complexes. 'Permanent' complexes are defined as those that are detected under a wide range of different cellular conditions, whereas 'transient' ones are defined as complexes that form under a specific set of conditions. While keeping in mind that this division is probably oversimplified and could sometimes be misleading, we follow these authors in considering it a helpful working hypothesis. The list of complexes in each category was derived from Jansen et al. [7] with some editing. We classified complexes that did not clearly fit either of the first two categories, and some larger assemblies composed of several complexes, as 'other'.
Table 1 reveals that meaningful overlaps between complexes and known regulons occur for both permanent and non-permanent complexes. Associations with the annotated regulons involve fewer complexes of the permanent category than of non-permanent ones (Table 1a). In contrast, the associations with the high-throughput regulons involve more permanent complexes than transient ones (Table 1b), in better agreement with the reported stronger relations of permanent versus transient complexes with mRNA expression profiles [7].
Another interesting observation is that the set of complexes into which regulons map and the extent of overlap between complexes and regulons is also quite different for the annotated and high-throughput regulon datasets. Regulons from both datasets map into complexes such as cytochrome bc1, nucleosomal protein complex, ribonucleoside diphosphate reductase and fatty-acid synthetase. On the other hand, complexes such as the proteasome, the Cdc28p cyclins and RNA polymerase II are only involved in associations with annotated regulons (Table 1a), whereas the ribosomal subunits or cytochrome c oxidase complexes are only involved in associations with high-throughput regulons (Table 1b).
These and other differences are most likely to be due to the different composition of the regulon repertoires in the two datasets. The annotated dataset contains nearly twice as many regulons as the high-throughput one. But the regulons in the latter dataset are significantly larger, with on average six times more genes than in the annotated regulons (see Materials and methods). It is therefore not too surprising that for associations involving high-throughput regulons, the fraction of the components of a given complex covered by a regulon is in general higher than for annotated regulons. It should at the same time be cautioned that the high-throughput regulons probably contain a fair number of spurious members (false positives) [26].
Zoom-in on the overlaps between regulons and annotated complexes
We see that a complex is often associated with several regulons. This is due in part to the substantial overlap that often exists between the components of individual regulons. The most severe cases occur when different transcription factors are annotated as regulating the exact same set of genes, a situation that is often encountered for small regulons, and probably results from incomplete information or because some transcription factors act in combination or as complexes [35]. We see for example that seven regulons map into the nucleosomal protein complex, six map into the ribonucleoside diphosphate reductase complex, and as many as 10 regulons map into the modular Cdc28p cyclin complexes (Table 1a).
A given regulon also maps, in general, into more than one complex, often onto two, and occasionally onto three. These multiple associations form a patchy network, with several disconnected clusters, which link complexes to regulons. The network graphs built from the associations of the annotated complexes, with annotated and high-throughput regulons, respectively, are illustrated in Additional data file 1 (Figures S1 and S2).
Details of some of these clusters are illustrated in Figures 1 and 2, highlighting the common genes involved. The nucleosomal protein complex (Figure 1a) has seven out of its eight components in common with seven small regulons - Hta1/Hta2, Spt10/Spt21 and Hir1/Hir2/Hir3 - whose genes partially overlap one another. The ribonucleoside diphosphate reductase complex (Figure 1b) has all its four components in common with a total of six partially overlapping regulons. The picture is significantly more complicated for the cyclin-Cdc28p complexes (Figure 1c). As many as 10 regulons map into the 10 components of this complex: the Cln1 and Cln2 genes, which are regulated by as many as five different transcription factors, and two transcription-factor genes, Swi4 and Mcm1, also map into the glucan synthases and pre-replication complex, respectively.
Figure 1.
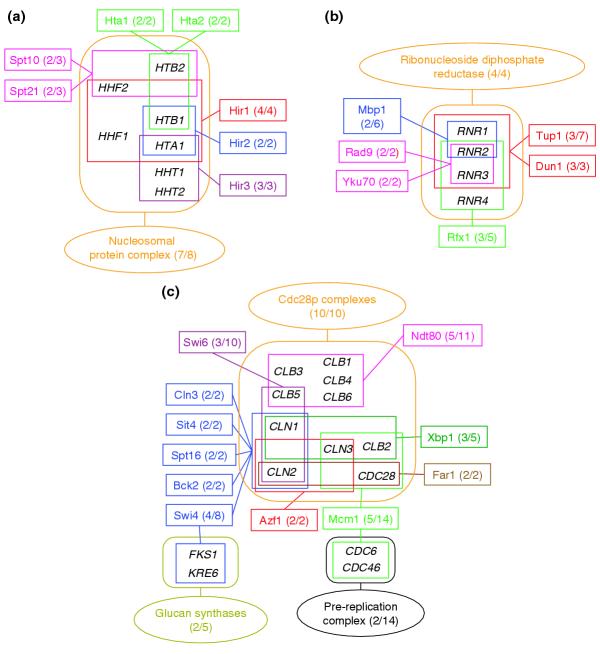
Detailed view of the main clusters in the network linking annotated protein complexes and regulons. The network (shown Additional data file 1 (Figure S1)) was built from the multiple links corresponding to associations with E-value ≥ 0.1, identified between the 243 CYGD yeast multiprotein complexes and the 200 annotated regulons (see text). Ellipsoid frames represent complexes, rectangular frame represent regulons, with individual complexes and regulons appearing in different colors in a given cluster. Individual complexes are identified by their name in the CYGD complexes catalog [10] and regulons are denoted by the name of the bound transcription factor. Genes involved in complexes or regulons are enclosed, respectively, in rounded frames or rectangles of the same color as the complex or regulon, and are displayed by their common name. The two digits given in parentheses indicate the number of genes involved in this cluster for the complex or regulon, and the total number of genes in the complex or regulon, respectively. (a) Cluster involving associations between three groups of regulons (Hta1-Hta2, Hir1-2-3, and Spt10-Spt21) and seven of the eight genes of the nucleosomal protein complex. (b) The ribonucleoside diphosphate reductase cluster, involving associations between all four genes of the corresponding complex and four groups of co-regulated genes belonging to six regulons. (c) Cluster involving associations between all the 10 components of the Cdc28p complexes, and seven distinct groups of genes belonging to 11 regulons. Five regulons - Cln3, Sit4, Spt16, Bck2, and Swi4 - map onto the exact same cyclin genes (CLN, CLN2). Two regulons, Swi4, and Mcm1, map also into the glucan synthases and pre-replication complex, respectively.
Figure 2.
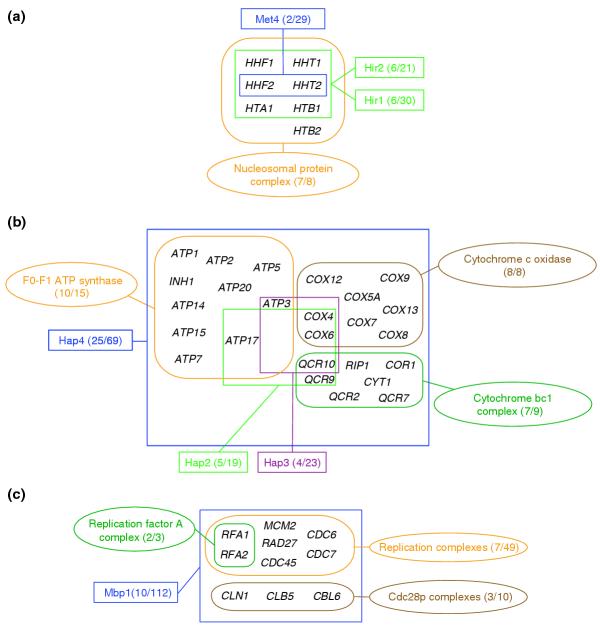
Detailed view of the main clusters in the network linking annotated protein complexes and high-throughput regulons. The network was built considering all the associations with E-value ≤ 0.1; regulons and complexes are denoted and depicted as described in the legend of Figure 1. (a) Cluster of associations involving seven of the eight components of the nucleosomal protein complex. Unlike in the equivalent cluster of Figure 1a, here only two distinct groups of, respectively, two and six genes belonging to three rather large regulons (respectively, Met4 and Hir1-2) map into this complex. Note that here Hir1-2 comprises a much larger group of genes than in the annotated regulons. (b) Cluster of the respiratory chain complexes. It comprises three complexes: the F0-F1-ATP-synthase complex, and the cytochrome bc1- and cytochrome c oxidase complexes. Twenty-five genes of the Hap4 regulon, and four and five genes of the Hap3 and Hap2 regulons, respectively, map into these complexes. As noted in the text, the Hap4 transcription factor is known as a respiratory-chain activator that does not bind DNA but fosters DNA binding by Hap2 and Hap3 [45]). The reasons for the more limited overlap between these latter two regulons and components of the respiration complexes are not clear. (c) An interesting cluster where the main node is the large Mbp1 regulon of 112 genes, of which 10 overlap with components of three complexes: the small replication factor A complex (3 genes), the replication complexes (49 genes) and the Cdc28p complexes (10 genes).
Correspondence between regulons and high-throughput protein complexes
The total number of statistically significant overlaps (E-value ≤ 0.01) is also very low (66 in total) when the known regulons are mapped onto TAP complexes and HMS complexes, even though the number of complexes considered is much larger (725).
The majority of the complex-regulon pairs with meaningful overlap (53) involve annotated regulons, whereas only 13 pairs involve high-throughput regulons. Matches with regulons from either dataset generally involve only a very small subset of the complex components, and there are twice as many matches with complexes from the HMS than from the TAP datasets, in line with the larger size of the former dataset (for a complete list of associations, see Additional data file 2 (Table S2)).
Owing to the appreciable overlap between the components of different complexes within and between the TAP and HMS datasets, the network of associations between these complexes and the regulons is much more intricate than for the annotated complexes. A network graph was built from the larger set of 125 complex-regulon pairs with meaningful overlaps (E-value ≤ 0.1) involving the annotated regulons (Figure 3). This network features seven separate dense clusters of connections (Figure 3a-g). Details of the regulon-complex overlaps in some of these clusters, highlighting the common genes involved, are depicted in Figure 4a-c. The remaining clusters are detailed in Additional data file 1 (Figure S3). In Figure 4h the set of remaining very small clusters, each involving mostly one or two connections, is grouped.
Figure 3.
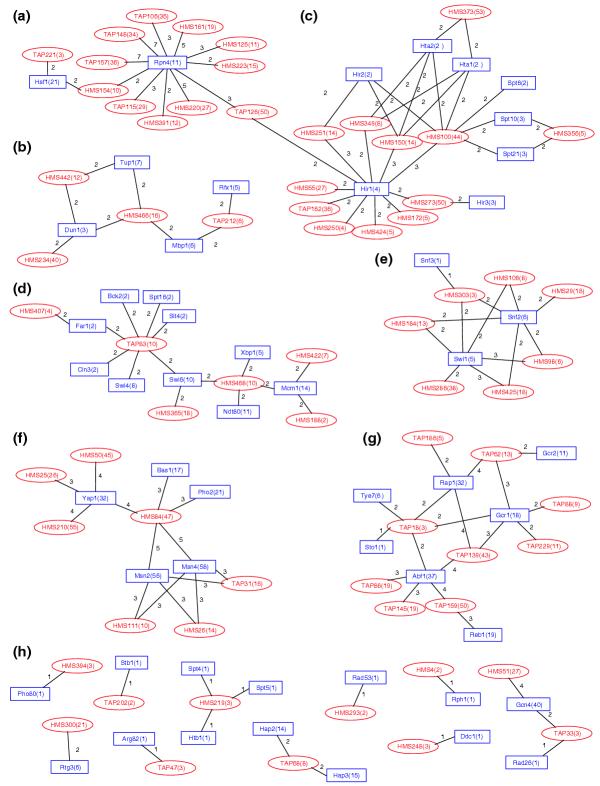
Network graph of the statistically significant links between the TAP and HMS complexes and annotated regulons. Each node represents a complex (red ellipse) or a regulon (blue rectangle). Individual complexes are identified by a number, prefixed by TAP [11] or HMS [12]. Regulons are denoted by the name of the bound transcription factor. The number of genes in each group (complex or regulon) is given in parentheses. The number of genes common to a given complex-regulon pair is indicated along the lines (arcs) joining the pair. (a-g) Seven dense clusters of connections. (h) The set of remaining very small clusters are grouped, each involving mostly one or two connections. Clusters (a-c) are detailed in Figure 4.
Figure 4.
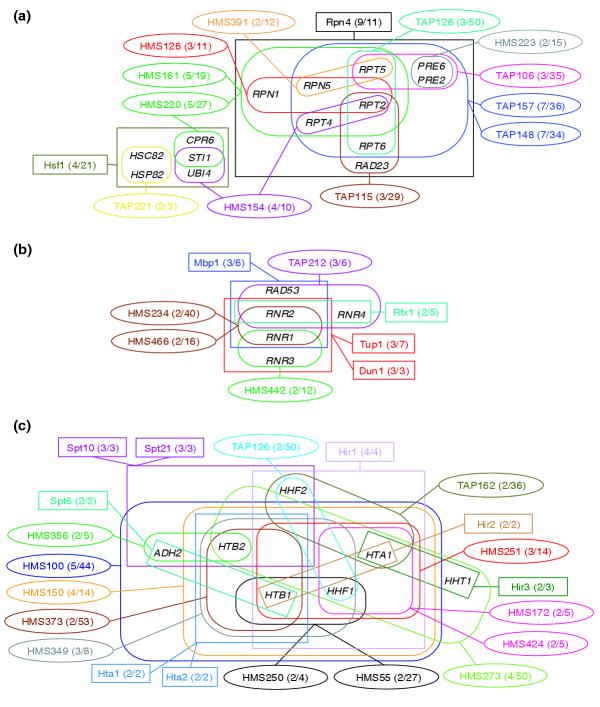
Details of three major clusters in the network linking the TAP and HMS complexes with annotated regulons (Figure 3). Individual complexes are identified by their alphanumerical code as in Figure 3 and depicted by ellipsoid frames, whereas regulons are denoted by the name of the bound transcription factor and depicted as rectangles. Individual complexes and regulons appear in different colors in a given cluster. Genes involved in complexes or regulons are enclosed by frames as in Figure 1. (a) Yeast proteasome cluster; (b) ribonucleoside diphosphate reductase cluster; (c) histone cluster.
The first cluster (Figure 4a) corresponds chiefly to the overlap between the Rpn4 regulon and 12 rather large complexes (six TAP and six HMS complexes). Nine of the 11 genes of this regulon map onto these complexes. All the complexes contain components of the yeast proteasome, and some other functionally related proteins in variable proportions. Interestingly, six of the nine common genes correspond to proteins from the 19S regulatory subunit, encoding four of the six ATPases in the subunit (Rpt2, Rpt4, Rpt5, Rpt6). A further two genes, PRE6 and PRE2, code, respectively, for alpha and beta subunits of the catalytic domain [36], and another gene (RAD23) encodes a ubiquitin-like protein, which links DNA repair to the ubiquitin/proteasome pathway [37].
The second cluster (Figure 4b) involves four partially overlapping regulons of three genes each, totaling five genes. These genes map into three medium-sized complexes (6-16 genes) and one large complex of 40 genes, with no more than two to three genes mapping into the same complex. Here, too, the majority of the five genes correspond to a biologically active assembly - the ribonucleoside diphosphate reductase complex and associated kinase. The third cluster (Figure 4c) involves genes of the nucleosomal protein complex. A similar analysis can be made for the remaining four clusters (data not shown), and similar observations are made when analyzing the largest clusters in the network graph built from the 46 statistically significant overlapping pairs (E-value ≤ 0.1) involving the TAP and HMS complexes and high-throughput regulons (see Additional data files 1 and 2 (Figures S4, S5 and Table S2d, respectively)).
This detailed analysis shows that although the subset of the components of the multiprotein complexes that corresponds to known regulons is usually quite small, it tends to be composed of proteins with close physical interactions and/or clear functional relations. We also find that the bulk of the overlaps involve genes that map into both permanent complexes such as the proteasome or the nucleosomal-protein complex, as well as into non-permanent ones, such as the ribonucleoside diphosphate reductase and the cyclin-Cdc28p complexes. No clear trends can therefore be identified from these data on the regulation of any one category of complexes in particular.
Prediction of cis-acting regulatory elements in genes of multiprotein complexes
The very limited overlap between complexes and regulons detected above might be biologically meaningful, or might be due to the limited information that is currently available on the nature of protein complexes and regulatory networks in yeast. Given these uncertainties, it seemed of interest to complement the above analyses by an approach in which regulons are directly predicted from the components of protein complexes.
If the components of a given protein complex are co-regulated on the transcriptional level, one would expect to find common regulatory sequence elements, corresponding to transcription factor binding sites, in the upstream regions of the corresponding genes. The problem of identifying regulatory sites is notoriously difficult [33]. To tackle it we applied algorithms for the discovery of oligonucleotides (here, hexanucleotides) [24] and spaced pairs of trinucleotides [25], which occur more frequently in the upstream regions of the genes coding for the components of each complex than in the corresponding regions across the entire yeast nuclear genome. For this approach we considered only complexes with at least five components.
Highly significant patterns are detected for only a small subset of the complexes
Figure 5 plots the number and fraction of the protein complexes in each of the three datasets examined (the annotated, TAP and HMS complexes) for which regulatory-sequence patterns were identified by our prediction method using three different reliability thresholds. Plotted alongside are the corresponding results obtained here for sets of randomly selected genes (used as negative control) and results for known regulons (positive control) obtained in another study [26].
Figure 5.
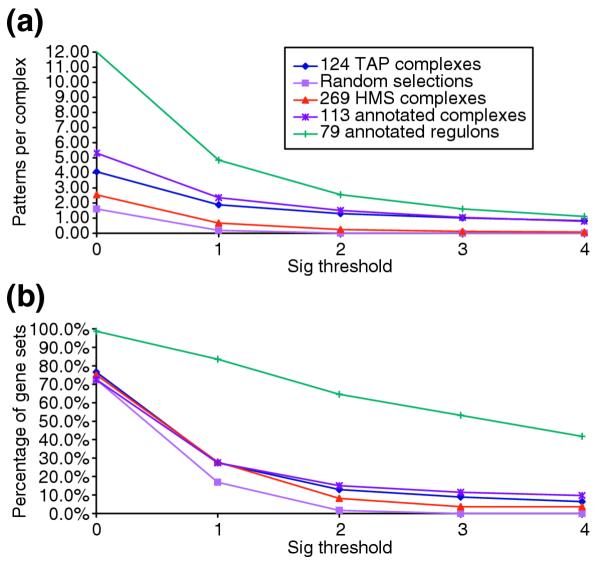
Prediction of regulatory motifs by analysis of upstream sequences. (a) The average number of patterns discovered per gene group and (b) the average fraction (%) of the genes groups in the dataset in which at least one pattern was discovered are plotted as a function of the statistical significance of the patterns (Sig) (see Materials and methods for detail). The five plots displayed in each panel represent results obtained for the different datasets analyzed. These comprise three datasets of multiprotein complexes: the curated complexes from the CYGD catalogue [10], and the complexes identified by the TAP [11] and HMS [12] genome-scale analyses (see Materials and methods). The number of gene groups in these sets is 113, 124 and 269, respectively. The remaining two plots represent results obtained for the 79 annotated regulons (used as positive control), and for groups of genes of the same size as the considered complex or regulon, randomly selected from the yeast genome (used as negative control, as described in the text). Only regulons or complexes containing at least of five genes/proteins were considered.
A first observation is that the fraction of complexes for which regulatory patterns are identified with some reliability is quite low. No more than 27-28% of the complexes from either of the three analyzed datasets have at least one pattern with statistical significance Sig ≥ 1 (corresponding to an E-value ≤ 0.1). At this threshold the fraction of complexes with identified patterns is nonetheless about 7-10% higher than for gene groups selected at random. With the more stringent significance threshold (Sig ≥ 2), the fraction of complexes with at least one pattern drops further, but less for the curated (15%) and TAP complexes (13%), than for the HMS complexes (8%).
We recently applied the same algorithms to the dataset of annotated regulons [26]. As the genes belonging to the same regulon are expected to be co-regulated and hence to exhibit common regulatory-sequence patterns, our algorithms should perform well on these genes. This was indeed the case. Patterns with Sig = 1 were identified in as many as 84% of the annotated regulons, as illustrated in Figure 5.
The fraction of the complexes in which regulatory patterns can be reliably detected is thus clearly much smaller, confirming that the components of complexes are on average much less consistently co-regulated than the genes that belong to known regulons.
Assigning components of protein complexes to putative regulons on the basis of predicted patterns
Having shown that highly reliable regulatory patterns can be detected in genes corresponding to at least a fraction of the complexes, we now proceeded to determine, for each complex, which of its components are likely to be co-regulated, and what fraction of the complex they represent. To this end, complexes with at least five component genes, featuring at least one significant pattern (Sig ≥ 1), are selected. A stepwise linear discriminant analysis [38] with a leave-one-out procedure is applied to assign a gene involved in a given complex, either to its original complex or to a control group of randomly selected genes, according to the number of occurrences of the discovered patterns in its upstream region. The assigned group (complex or control) is then compared to the group from which the gene was drawn to evaluate the coverage and positive predictive power (PPP) of the assignment. Coverage is defined as the fraction of the total number of genes in the complex that were reassigned to it by the discriminant procedure. PPP is defined as the fraction of total number of genes assigned to the complex that actually belong to it (see Materials and methods for details).
Figure 6 displays the coverage versus PPP values for a total of 140 individual complexes from the three datasets analyzed here (34 TAP, 75 HMS, and 31 annotated ones). The coverage obtained for these complexes has a mean value of 48%, and a standard deviation of about 25%. The mean PPP is 80%, with a standard deviation of about 10%, and only a single case with perfect assignment (PPP = 100%). There is very little difference between the results obtained for the annotated, TAP, and HMS complexes (see Additional data file 2 (Table S3) for details). It is noteworthy that significantly higher average values for the coverage and PPP (72% and 92% respectively) were obtained by applying the same procedure to the annotated regulons [26].
Figure 6.
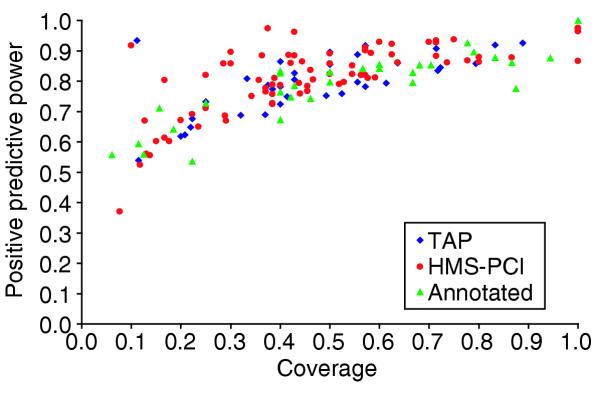
Assignments of genes to complexes by discriminant analysis. The assignment by discriminant analysis was made on the basis of the number of predicted regulatory-sequence patterns (see text). The assignment Coverage (x-axis) is the proportion of genes from the complex which were reassigned to it by the discriminant analysis (see Materials and methods for details). Positive predictive power (PPP) (y-axis) is the proportion of the genes assigned to the complex which originally belonged to it. Assignment results are for multiprotein complexes. Annotated complexes [10] are drawn as green triangles, TAP complexes [11] as blue diamonds and HMS complexes [12] as red circles.
Putative regulons in the annotated complexes
We determined whether the putative regulons identified by our procedures can provide useful information on the transcriptional regulation of protein complexes. As a first step, we discuss several aspects of the prediction results for patterns and putative regulons obtained for the annotated complexes, summarized in Table 2. This lists the results for all the complexes for which at least one statistically significant (Sig ≥ 1) regulatory pattern has been detected. A complete list of the predicted co-regulated components in each of the complexes considered is given in Additional data file 2 (Table S4).
Table 2.
Results of the pattern discovery and discriminant analysis for annotated complexes
| Annotated complex | ORFs | Coverage | Max sig | Discovered patterns | |
| Permanent | 26S proteasome | 36 | 0.83 | 15.84 | 24 |
| 19-22S regulator | 18 | 0.94 | 8.76 | 16 | |
| 20S proteasome | 15 | 0.87 | 5.76 | 13 | |
| Respiration chain complexes | |||||
| Cytochrome bc1 complex | 9 | 0.67 | 1.13 | 4 | |
| Cytochrome c oxidase | 8 | 0.88 | 1.51 | 2 | |
| F0-F1 ATP synthase | 15 | 0.67 | 4.01 | 4 | |
| Cytoplasmic ribosomes | 138 | 0.68 | 19.49 | 151 | |
| Cytoplasmic ribosomal large subunit | 81 | 0.70 | 11.67 | 77 | |
| Cytoplasmic ribosomal small subunit | 57 | 0.79 | 8.97 | 63 | |
| Cytoplasmic translation elongation | 9 | 0.78 | 2.35 | 10 | |
| Cytoplasmic translation initiation | 27 | 0.19 | 6.31 | 5 | |
| eIF3 | 7 | 0.43 | 1.24 | 2 | |
| Nucleosomal protein complex | 8 | 1.00 | 4.38 | 20 | |
| RNA polymerase III | 13 | 0.46 | 1.52 | 2 | |
| RNA polymerase II holoenzyme | 35 | 0.11 | 1.81 | 2 | |
| Transient | Microtubules | 32 | 0.16 | 1.45 | 5 |
| Pyruvate dehydrogenase | 5 | 0.60 | 1.81 | 1 | |
| Replication complexes | |||||
| Replication complex | 19 | 0.42 | 1.66 | 5 | |
| SAGA complex | 14 | 0.50 | 1 | 5 | |
| Spindle pole body | 32 | 0.13 | 2.93 | 8 | |
| SPB components | 16 | 0.25 | 3.38 | 8 | |
| Tim22p complex | 5 | 0.40 | 1.99 | 1 | |
| Others | Cdc28p complexes | 10 | 0.60 | 2 | 5 |
| H+transporting ATPase-vacuolar | 15 | 0.40 | 2.46 | 6 | |
| Nuclear splicing complexes-Spliceosome | 66 | 0.06 | 1.55 | 5 | |
| Other DNA repair complexes | 5 | 0.40 | 1.11 | 4 | |
| Replication complexes | 49 | 0.41 | 15.64 | 7 | |
| Replication fork complexes | 30 | 0.57 | 13.28 | 12 | |
| Respiration chain complexes | 37 | 0.59 | 8.49 | 11 | |
| rRNA processing complexes | 18 | 0.22 | 1.72 | 2 | |
| t-SNAREs | 10 | 0.40 | 1.45 | 3 |
Column 2 lists the names of the analyzed complexes. Columns 3 and 4 list, respectively, the number of components in the complex and the coverage of the assignment, defined as the fraction of the components of a given complex that are predicted to be part of a putative regulon. Max sig (column 5) is the highest significance index in patterns detected for this complex, and column 6 lists the number of discovered patterns for the complex. Complexes have been subdivided into three categories - 'permanent', 'transient' or 'others' - as indicated in column 1, and described in Materials and methods. Assemblies such as 'Respiration chain complexes', which contain several groups of complexes and hence do not represent a single physical entity, were classified here in the 'others' category. However, smaller complexes, which are part of this assembly and for which putative regulons were identified, were classified as 'permanent'.
Table 2 reveals a clear difference between results for the permanent and the non-permanent complexes. Most strikingly, the fraction of the components of a given complex covered by our putative regulons is noticeably higher for most permanent complexes (0.7-1.0) than for the non-permanent ones (0.06-0.6). The number of significant regulatory patterns and the significance value of the 'best' pattern are also generally higher in theses complexes. Among the complexes with the highest coverage by putative regulons and a large number of statistically significant patterns we find the proteasome, the large and small subunits of the cytoplasmic ribosome, three complexes of the respiratory chain, the translation elongation complex, as well as the nucleosomal protein and cyclin Cdc28p complexes. To illustrate the information provided by our approach, we will discuss in detail our findings for the nucleosomal protein complex and the replication fork complexes.
Nucleosomal protein complex
This complex has all of its eight components predicted to be part of a regulon, with a large number (20) of significant patterns. Details of the patterns discovered, of which the most statistically significant are spaced dyads, and their locations in the upstream regions of the corresponding genes are shown in the feature map (Figure 7). All but one of these dyads are mutually overlapping, and can be aligned to form the larger motif cGCGAan{5}caGAACg, where upper-case letters denote the most conserved residues, which seem to be the 'core' of the binding site, and the number in brackets is the length of the spacer in terms of the number of intervening nucleotides. The feature map shows that each upstream sequence contains at least two occurrences of this 'core', with some differences in the bases flanking this core. Although several regulons - Hta1/Hta2, Hir2/Hir3/Hir4 and Spt10/Spt21 - are known (and were found here) to map into this complex, covering a total of seven out of the eight components of the complex (Figures 1, 2), our findings represent the first instance where a regulatory motif is proposed for all the members of the nucleosomal complex.
Figure 7.
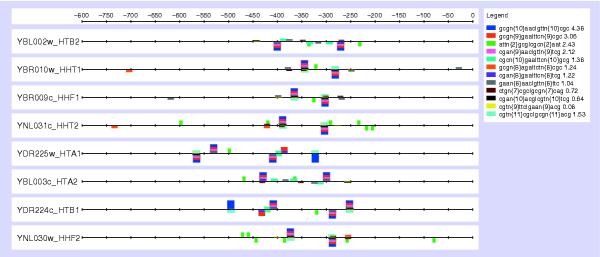
Feature map of the patterns (spaced dyads) discovered in the 800 bp upstream regions from the genes involved in the annotated nucleosomal protein complex. Each dyad is represented as a box of a given color, whose height is proportional to the significance level of the pattern, as indicated on the legend. The identified over-represented patterns are indicated in the inset on the upper right-hand side; n stands for any nucleotide residue and n{x}, with x from 2 to 11, indicates the spacer length in terms of the number of wild-card positions.
Replication fork complexes
The replication fork complex is an assembly of proteins involved in DNA replication (Table 2). It is encoded by a total of 30 genes, which can be subdivided into several smaller complexes such as the DNA polymerase δ, DNA polymerase ε, DNA α1 primase and replication factor C complexes. Analysis of the entire assembly detected 12 patterns with a maximum significance of 13.3, corresponding to an E-value of 2 × 10-13. The discriminant analysis carried out on the basis of these patterns allowed us to assign about half (17) of the 30 components of this assembly to putative regulons (Table 2).
Table 3 lists the probabilities for individual components to be assigned to the complex by the discriminant analysis. It reveals a striking observation: the predicted co-regulated genes correspond almost perfectly to seven out of the 14 individual complexes or entities that make up the assembly. The 17 genes that belong to the putative regulons include three of the four components of the DNA polymerase α1 primase complex, all the components of the DNA δ and ε complexes, the replication factor A and topoisomerase complexes, as well as the proliferating cell nuclear antigen (PCNA) and exonucleases. Furthermore, the majority of these genes were assigned to the replication fork assembly with high probability (0.8-0.99).
Table 3.
Details of the discriminant analysis results for the replication fork complexes
| Complex | Gene | ORF | P(da) |
| Components predicted to be co-regulated | |||
| DNA polymerase αI primase complex | POL1 | YNL102W | 0.95155315 |
| PRI2 | YKL045W | 0.92757655 | |
| POL12 | YBL035C | 0.8740073 | |
| PRI1 | YIR008C | 0.14922267 | |
| DNA polymerase δIII | POL32 | YJR043C | 0.88528264 |
| HYS2 | YJR006W | 0.79712089 | |
| CDC2 | YDL102W | 0.79079457 | |
| DNA polymerase εII | POL2 | YNL262W | 0.85845235 |
| DPB2 | YPR175W | 0.7575824 | |
| DPB3 | YBR278W | 0.64557167 | |
| Exonucleases | RAD27 | YKL113C | 0.99432872 |
| PCNA | POL30 | YBR088C | 0.69668482 |
| Replication factor A complex | RFA1 | YAR007C | 0.98673268 |
| RFA2 | YNL312W | 0.87875323 | |
| RFA3 | YJL173C | 0.73210048 | |
| Topoisomerases | TOP1 | YOL006C | 0.82395424 |
| TOP2 | YNL088W | 0.73032433 | |
| Components excluded from the predicted regulon group | |||
| DNA helicases | ECM32 | YER176W | 0.23499023 |
| DNA2 | YHR164C | 0.05406037 | |
| DNA ligases | CDC9 | YDL164C | 0.03708159 |
| DNA polymerase βIV | POL4 | YCR014C | 0.03919368 |
| DNA polymerase γ | MIP1 | YOR330C | 0.03182497 |
| DNA polymerase ζ | REV7 | YIL139C | 0.05724429 |
| REV3 | YPL167C | 0.04090946 | |
| Replication factor C complex | RFC4 | YOL094C | 0.50870756 |
| RFC5 | YBR087W | 0.33589936 | |
| RFC3 | YNL290W | 0.26089619 | |
| RFC2 | YJR068W | 0.10584985 | |
| RFC1 | YOR217W | 0.05246123 | |
| RNase H1 | RNH1 | YMR234W | 0.02989109 |
The CYGD names of the subcomplexes or independent genes of the replication fork complexes are listed in column 1. The second and third columns list, respectively, the common gene name and ORF identifier. The posterior probability P(da), computed by the discriminant analysis, with which the listed gene/ORF was assigned to the putative regulon is listed in column 4. All the components predicted to be co-regulated (probability > 0.5 for most of the genes) are listed in the top part of the table, while those excluded from the predicted regulon group are listed in the lower part of the table. The two genes whose assignment is different from those of other related components of the subcomplex are marked in bold.
Interestingly, Jansen et al. [7] reported a poor correlation with expression data for the replication complex, a large complex that partially overlaps the replication fork assembly discussed above. They showed, however, that a much better correlation with expression profiles could be obtained for subgroups of components within the complex. Two such subgroups were the DNA polymerase δ and DNA polymerase ε complexes. These are also part of the co-regulated subgroup that we identify in the replication fork assembly, except that our observations suggest that their transcriptional regulation is coupled to that of 11 other genes (the remaining genes with probabilities > 0.5 in Table 3) also involved in DNA replication, and that the transcriptional regulation of all 17 genes is controlled by the same factor, or set of factors.
Our findings therefore agree with the conclusions drawn from relationships between complexes and mRNA expression profiles, namely that the so-called permanent complexes display a more marked trend to be co-regulated en bloc than non-permanent ones, but that non-permanent complexes often contain subgroups of co-regulated components [7]. Our findings furthermore suggest that subsets of the components of a given multiprotein assembly, whose genes display distinct expression patterns, can be under common transcriptional control. This can happen when the same transcription factor requires the presence of different associated factors when binding to each group of co-regulated components, thereby producing different expression profiles for the individual gene groups (see, for example [39]).
Putative regulons in the high-throughput complexes
Interpretation of the predicted regulons for the TAP and HMS complexes is more complicated. Owing to the appreciable overlap between the complexes, significant overlap is also observed between the predicted sets of regulatory patterns and between the corresponding groups of putative co-regulated components in different complexes. These overlaps create a dense network of connections between the complexes. The bulk of the network comprises two large separate clusters of interconnected complexes and a few very small clusters. The complete list of predicted co-regulated components for each complex, and the network graph can be found in Additional data file 2 (Table S4) and Additional data file 1 (Figure S6).
One of the largest clusters comprises a group of nine complexes (four HMS and five TAP) that involve the proteasome genes. The subsets of putative co-regulated genes in each complex and the pattern of overlap between them are illustrated in Figure 8. Remarkably, the vast majority of the genes predicted as being co-regulated in all the nine complexes are annotated as belonging to the proteasome, whereas the majority of remaining genes, not classified with putative regulons, are not annotated as proteasome genes. For most complexes, the fraction of genes assigned to regulons is furthermore quite high (65% on average). The most statistically significant regulatory patterns shared by the genes in the corresponding complexes (Table 4) display a substantial degree of overlap and can be assembled into a larger pattern (TTTGCCACC/GGTGGCAAA). This pattern corresponds to the binding site of the Rpn4 transcription factor, known to be involved in the regulation of proteasome genes [40]. It is present in the upstream regions of nearly all the proteasome genes, with the exception of RPN10, RPN13, PRE5, PUP1, RPN8 and NAS6. But the latter gene group exhibits patterns differing by only one nucleotide from the Rpn4-binding sequence, and might hence still be regulated by this factor or a closely related one. Thus, quite strikingly, the overlap between the complexes and putative regulons is much more extensive here than in the proteasome-related cluster shown in Figures 3 and 4a, which links TAP and HMS complexes to annotated regulons.
Figure 8.
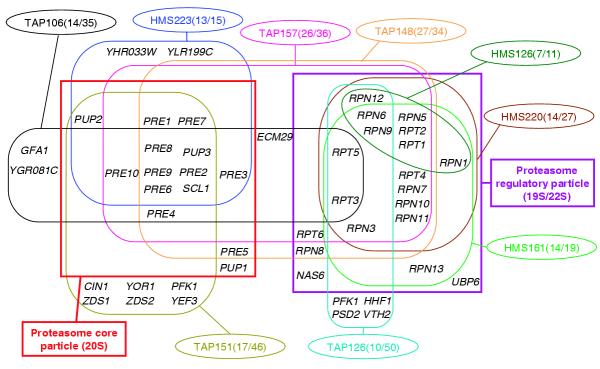
Details of overlaps between predicted co-regulated genes from proteasome-related complexes. Complexes are displayed as ellipses, with individual complexes identified by the prefix TAP or HMS followed by a number (see Figure 3 legend for details). The numbers in parentheses are the number of genes in the predicted co-regulated set for the complex and the total number of genes in the complex, respectively; the ratio represents the proportion of the components of the complex that is predicted to be co-regulated (see text and Figure 6). The set of predicted co-regulated genes in each complex is enclosed in a rounded frame of the same color as the ellipse of the corresponding complex. Two large rectangular frames enclose the subset of co-regulated genes that code for the components of the proteasome core particle (left) and the proteasome regulatory particle (right).
Table 4.
Regulatory patterns discovered in the protein complexes of the proteasome system
| Pattern | TAP106 | TAP126 | TAP148 | TAP151 | TAP157 | HMS126 | HMS161 | HMS220 | HMS223 |
| ....GCCACC.. | 4.23 | 1.54 | 13.99 | 3.41 | 15.74 | 1.21 | 6.35 | 4.67 | 7.62 |
| ...TGCCAC... | 3.08 | 11.72 | 1.14 | 12.26 | 0.56 | 4.3 | 3.34 | 6.25 | |
| ..TTGNCAC... | 10.61 | 9.56 | 1.11 | 2.47 | 2.56 | 3.43 | |||
| ..TTGNNACC.. | 0.27 | 8.68 | 9.86 | 2.96 | 2.44 | 1.68 | |||
| ...TGCNACC.. | 2.38 | 9.14 | 2.25 | 9.74 | 2.36 | 1.63 | 4.33 | ||
| ..TTGCCA.... | 6.3 | 5.46 | 0.21 | 2.62 | 1.45 | 1.55 | |||
| .TTTGCC..... | 5.62 | 4.82 | 0.22 | 2.84 | 3.1 | 0.71 | |||
| .TTTNNNACC.. | 4.27 | 5.97 | 1.16 | 1.94 | 0.33 | ||||
| .....CCACCG. | 2.03 | 2.68 | 0.51 | 0.46 | 1.49 | ||||
| .TTTNCCA.... | 2.13 | 1.84 | |||||||
| ....GCCNCCG. | 0.78 | 1.26 | |||||||
| .TTTNNCAC... | 1.6 | 0.96 | |||||||
| ATTNGCC..... | 1.13 | 0.41 | |||||||
| ......CACCGG | 0.86 | 0.05 | |||||||
| GGTNNNNAAA | 0.36 | 0.94 | |||||||
| .GTGNNNAAA | 2.77 | 0.82 | 0.22 | ||||||
| .GGGTAA | 1.44 | 0.95 | 1.27 | ||||||
| CGGGTA. | 0.68 | 0.05 | 0.61 | ||||||
| AGGGCA | 1,56 | 0.36 | |||||||
| AATNNNNNNNNNNACC | 0.75 |
Column 1 lists the predicted regulatory-sequence pattern. The patterns are aligned to indicate how individual patterns could be assembled. Distinct groups of patterns are separated by horizontal spaces. The complexes in which the pattern has been identified are listed in the top row. These complexes are denoted as in Figure 3 and the text. The listed values are the statistical significance (Sig), computed as the logarithm of the E-value. Patterns with Sig ≤ 0.5 are not listed. Those with Sig ≥ 2 or higher are highly significant. Rows corresponding to highly significant patterns identified in at least six complexes are in bold.
Interestingly, the TAP126 and TAP151 complexes, which are part of the proteasome cluster in Figure 8, stand out as the two complexes for which the predicted regulons are less consistent with the functional annotations. We see, however, that these regulons include at most a third of the components of the complexes, and that the PPPs of regulon assignment for these complexes are significantly below average (62% and 69%, respectively), suggesting that the corresponding predictions might be less reliable. On the other hand, two genes of unknown function (YHR033W and YLR199C) are predicted to be co-regulated with other proteasome genes in the HMS233 complex, for which the coverage and PPP values are high (87% and 88%, respectively). The possibility that these orphan genes might be co-regulated with other proteasome components is therefore a reasonable hypothesis that can be tested experimentally.
These various observations indicate that combining pattern discovery and discriminant analysis to predict regulons should be a promising avenue for the interpretation of high-throughput datasets on protein complexes, in terms of biological function and transcriptional regulation.
Discussion
We used two approaches to investigate transcriptional regulation of multiprotein complexes in yeast. First, sets of genes representing known targets of yeast transcription factors, the regulons, were mapped onto both manually curated protein complexes and those identified by genome-wide pull-down experiments. Second, a predictive approach was applied to the same set of complexes. In this approach, string-based techniques for the discovery of regulatory patterns were combined with a discriminant analysis in order to assign genes to regulons on the basis of the predicted patterns.
The straightforward mapping approach revealed that only very few of the complexes had a good fraction of their components belonging to known regulons, whereas for the majority of the complexes only a very partial overlap with regulons was detected. Following previous work [7], we subdivided the complexes into two categories: permanent complexes that exist under a wide range of cellular conditions; and transient complexes that form under particular conditions only. Overall, we found little difference in the overlap with regulons between complexes of the two categories, except maybe when matching the annotated complexes against the high-throughput regulons dataset. In the latter case, a more extensive overlap with regulons was observed for the permanent complexes than for the transient ones (Table 1b). But this observation should be considered with caution, taking into account the sizable level of noise in the corresponding regulon dataset.
Considering that the above results mainly reflect the limited information currently available on transcription factor binding sites in yeast, we applied our predictive approach. The pattern-discovery procedures identified statistically significant regulatory patterns in only a small fraction of the complexes (8-15%). Putative regulons were identified in this small fraction on the basis of the patterns discovered, in agreement with our analysis of the overlaps between complexes and known regulons. The identified regulons included on average nearly half (47%) of the components in the annotated complexes, with, nonetheless, a large spread in component coverage, ranging between 6% and 100% of the proteins/genes of a complex.
More interestingly, in annotated complexes in which regulatory patterns were predicted, the fraction of the components belonging to putative regulons, the number of regulatory patterns, and the statistical significance value for the 'best' pattern in each complex were generally substantially higher for the permanent complexes than for complexes from other categories. This result is in good agreement with previous reports on the better correlation between the mRNA expression profiles of genes coding for the components of permanent complexes than for their transient counterparts [7]. It suggests, furthermore, that complexes such as the proteasome, the respiratory-chain complexes, the cytoplasmic ribosome or the nucleosomal protein complex, which are present under a range of different cellular conditions, are actively regulated at the transcriptional level.
Detailed analysis of the predicted regulons in some of the non-permanent complexes, such as the replication fork complexes (Table 3), also suggests that even in cases in which only a fraction of the components belong to regulons, these components are likely to be co-regulated, either together or in smaller subgroups, thereby revealing the existence of transcriptionally regulated modules within complexes. Interestingly, in the case of the replication fork complex, the detected modules contain groups of genes reported to display distinct mRNA expression patterns [7], an indication that gene groups with different expression patterns might nevertheless be under common transcriptional control. These findings are clearly preliminary and should be confirmed by a systematic comparison with mRNA expression data for genes involved in all the complexes for which putative regulons were identified. In particular, this comparison should consider separately the mRNA expression profiles measured under different experimental conditions rather than combine them as was done previously [7], as we might expect the transcriptional regulation of certain complexes to vary with these conditions.
It is useful at this point to discuss the reliability of our prediction procedure. Recent benchmarks performed on the annotated regulons in Saccharomyces cerevisiae [26], showed that our combined approach produces highly specific regulon assignments, with good coverage. On average, 91% of the genes assigned to a regulon were actually part of it, indicating that our approach produces a very low rate of false positives, and hence that it would rarely assign to a regulon genes that do not belong to it. The coverage, or the fraction of the genes in a regulon that could be reassigned to it, was lower (73% on average), suggesting that the regulons detected by our approach might not include all their actual members, a much less serious shortcoming than making false-positive predictions.
More important still, the same benchmarks showed that our approach maintained on average a similar level of coverage and reached PPP values as high as 84% when applied to gene groups in which known regulons were mixed with an equal number of random genes, a situation more closely resembling that of the multiprotein complexes analyzed here. We therefore believe that our predictive methods yield quite reliable regulon predictions, particularly for larger complexes in which only about 50% or more of the genes may be co-regulated.
Our methods do, however, have obvious limitations. One is that they would miss altogether sites located outside the 800 base-pair (bp) limit of the analyzed upstream sequences. Another potential shortcoming is their limited capacity to detect regulatory sites that have a high degree of sequence variability, something that matrix-based methods could do better [27-31]. It could therefore be possible that we tend to find fewer putative regulons in the so-called transient complexes than in their permanent counterparts because the regulatory mechanisms of these complexes are more specific, involving a larger variety of transcription factors or a more diverse pattern of interactions.
On the other hand, a clear advantage of our pattern-discovery procedure is its low rate of false-positive predictions. As a result, the most reliable patterns discovered here should be a good starting point for deriving the full regulatory motif. This requires that the discovered patterns be extended and their sequence degeneracy characterized [41,42], a difficult task that is often considered an obligatory step in identifying regulons. Here, this difficulty was circumvented by our cross-validated discriminant analysis, which we applied directly to the small sequence motifs. Combining the two approaches in the future should lead to improvements.
Lastly, we show that useful information could also be obtained by building the network graph representing the multiple links between protein complexes and predicted regulons, and analyzing the common genes participating in these links (Additional data file 1 (Figure S6)). This was illustrated for one of the large clusters of this graph, representing the proteasome-related complexes (Figure 7), but was also observed for other regulon-complex relationships discovered in this study (data not shown). Such networks are complementary to the recently described regulatory network of gene-transcription factor interactions [16,43] and should provide insights into the functional relationships between complexes.
The various observations made here are consistent with the results of a related study [44], in which pattern-discovery methods were applied to sets of yeast genes and those sets with common patterns in their upstream regions were scored against interacting proteins from the TAP and HMS protein complexes or closely linked proteins in the metabolic network.
Materials and methods
Data on multiprotein complexes
Three different datasets on protein complexes from the yeast S. cerevisiae were analyzed. One comprises a set of 243 protein complexes manually curated from the literature and retrieved from the complexes catalog in CYGD [10,45]. Those are referred to as annotated complexes. This set includes complexes that are known to form a single physical entity under some experimental conditions, as well as larger assemblies composed of several complexes whose formation is thought to be interdependent. Components of complexes encoded by mitochondrial genes were not considered, as the analysis of their regulatory motifs requires a different background model than for the nuclear genes. The complete list of annotated complexes used in this study is given in Additional data file 2 (Table S1).
The other two datasets comprise a total of 725 protein complexes identified respectively in the tandem affinity purification and MS analysis (TAP, 232 complexes) [11], and in the high-throughput MS protein complex identification (HMS, 493 complexes) [12]. These analyses probed only a subset of the proteins comprising many of the phosphatases, kinases and proteins involved in DNA repair. This subset (which corresponds to the so-called bait proteins [11,12]) represents about 25% of all yeast proteins in the TAP study, and about 10% in the HMS study. The composition of individual complexes from these studies was downloaded from the websites indicated in the original papers [46,47].
Data on co-regulated genes
Information on co-regulated genes in S. cerevisiae was obtained from two types of data on gene-transcription factor associations. One corresponds to 1,406 gene-factor associations manually curated from the literature, which were obtained from the TRANSFAC [18] and aMAZE [21,22] databases, from the list compiled by Young and colleagues, which excludes gene-factor associations deduced from sequence analysis [16,48], and from additional literature searches.
The manually curated gene-factor associations were grouped into 200 regulons, with on average seven genes per regulon, and are referred to as annotated regulons in this study. A regulon is defined here as the set of genes that bind the same transcription factor, and is denoted by the name of the transcription factor polypeptide. We considered individual polypeptides as distinct transcription factors, ignoring the fact that the actual active species might in some cases be a complex of several polypeptides (for example Hap2, Hap3, Hap4 [49]).
The second set of gene-factor associations is data obtained by Lee et al. [16] using a high-throughput ChIP approach under specific cellular and culture conditions; it was downloaded from the authors' website [50]. Here we consider only the associations with the highest statistical significance (P-value ≤ 10-3), as defined by Lee et al. [16]. Those number 4,433, and correspond to 106 distinct regulons, with 41.8 genes on average. Only 185 of these gene-factor associations are the same as those of the manually curated dataset.
Correspondence between protein complexes and regulons
To evaluate the correspondence between protein complexes and regulons, their gene compositions are compared and a statistical significance criterion (E-value) is computed using the hypergeometric formula implemented in the software Compare-Classes. The complexes and the regulons are considered as independent samples of genes from the complete yeast genome (n = 6,450). For a complex containing a genes, and a regulon b genes, the probability of finding exactly c common genes between them is
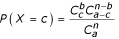 ,
,
where 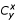 is the binomial coefficient. The probability of observing at least c genes in common by chance is given by
is the binomial coefficient. The probability of observing at least c genes in common by chance is given by
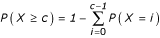 .
.
To correct for multi-testing (a given complex is compared to several hundred regulons), P is converted to an E-value (expected value), E-value = R * P(X ≥ c), where R is the total number of regulons.
Pattern discovery in upstream sequences
Upstream sequences are analyzed using the regulatory sequence analysis tools (RSAT) [51,52]. For each complex, the corresponding upstream sequences are retrieved over, at most, 800 bp from the start codon, clipping the sequence when necessary to avoid including upstream open reading frames (ORFs). Large redundant fragments, which may result from very recent duplications or from the presence of two genes transcribed in opposite directions, are discarded using Mkvtree and Vmatch [53].
Two pattern-discovery algorithms are applied to each sequence set to detect oligonucleotides [24], and dyads (spaced pairs of short oligonucleotides) [25], respectively, which occur more frequently (are over-represented) in these regions in comparison to upstream regions of all the genes from the S. cerevisiae nuclear genome. The degree of over-representation of a given pattern is determined by a statistical significance criterion Sig = -log(E-value). The larger the co-regulated gene group, the more reliable the prediction becomes. From previous experience we chose to analyze only gene groups with at least five members.
Discriminant analysis
Linear discriminant analysis [38] is used to classify the genes involved in a given set (protein complex or regulon) according to the type and number of statistically significant oligonucleotide and dyad sequence patterns that occur in their upstream regions.
A detailed description of the approach is given elsewhere [26]. In summary, two gene groups are defined. Group 1 comprises the g genes of a given protein complex, in which p over-represented patterns are identified. Group 2 is a control group of 3g genes, selected at random from the yeast nuclear genome. Upstream regions of the genes in both groups are analyzed to count the number of occurrences for all the identified p patterns, taken as variables. A linear discriminant function is then built, which optimally separates the genes from groups 1 and 2 into their respective groups in the p-dimensional space of the considered variables. To avoid over-fitting, the most discriminant variables are identified in a stepwise approach.
To assign individual genes from a given complex to either group, a leave-one-out procedure is carried out, whereby the genes are removed from the complex one at a time, a discriminant function is built each time with a different gene removed and used to assign the removed gene to groups 1 or 2.
To minimize fluctuation in the results, the entire procedure is repeated 100 times for each complex, using different random selections of genes for group 2, and the probability that a given gene is part of the complex is computed as the mean of the posterior probabilities evaluated in all the trials. Genes with mean posterior probability > 0.5 are assigned to the group of putative co-regulated genes, or regulons. The discriminant analysis was performed with the R package [54].
To assess the performance of the discriminant analysis procedure the group (complex or control) to which a given gene has been assigned is compared to the group from which it was originally drawn and two quantities are evaluated. One is the coverage, defined as TP/(TP + FN), where TP is the number of genes assigned to a given complex that were originally part of it, and FN is the number of genes that are part of the original complex, but which were (incorrectly) assigned to the control group. The other is the positive predictive power (PPP), defined as TP/(TP + FP), with FP being the number of genes that were (incorrectly) assigned to the complex.
Additional data files
The following additional data files are available with the complete version of this article, online: a PDF file (Additional data file 1) containing supplementary Figures S1 to S6 and their legends, and a Word file (Additional data file 2) containing supplementary Tables S1 to S4 and their legends.
Supplementary Material
A PDF file containing supplementary Figures S1 to S6 and their legends
A Word file containing supplementary Tables S1 to S4 and their legends
Acknowledgments
Acknowledgements
We thank Michael Holger and Edgar Wingender for access to TRANSFAC, and Jean Richelle and Raphael Leplae for help with the computer systems. N.S. is grateful to Didier Croes for Java lessons and support. This work was funded by the European Communities, grant QLRI-199-01333 to the CYGD project, by the Action de Recherches Concertées de la Communauté Française de Belgique, and by the Government of the Brussels Region.
References
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Shin TH, Galova M, Obermaier B, Nasmyth K. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science. 1996;274:1201–1204. doi: 10.1126/science.274.5290.1201. [DOI] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J Cell Biol. 2000;148:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen M, Snel B, Lathe W, 3rd, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalir S, McClure J, Pabbaraju K, Southward C, Ronen M, Leibler S, Surette M, Alon U. Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria. Science. 2001;292:2080–2083. doi: 10.1126/science.1058758. [DOI] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann SA, Babu MM. Conservation of gene co-regulation in prokaryotes and eukaryotes. Trends Biotechnol. 2002;20:407–410. doi: 10.1016/S0167-7799(02)02032-2. [DOI] [PubMed] [Google Scholar]
- Gerstein M, Jansen R. The current excitement in bioinformatics - analysis of whole-genome expression data: how does it relate to protein structure and function? Curr Opin Struct Biol. 2000;10:574–584. doi: 10.1016/S0959-440X(00)00134-2. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/S0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang MQ. SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics. 1999;15:607–611. doi: 10.1093/bioinformatics/15.7.607. [DOI] [PubMed] [Google Scholar]
- Costanzo MC, Crawford ME, Hirschman JE, Kranz JE, Olsen P, Robertson LS, Skrzypek MS, Braun BR, Hopkins KL, Kondu P, et al. YPD, PombePD and WormPD: model organism volumes of the BioKnowledge library, an integrated resource for protein information. Nucleic Acids Res. 2001;29:75–79. doi: 10.1093/nar/29.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J, Naim A, Mancuso R, Eldridge M, Wernisch L, Gilbert D, Wodak SJ. Representing and analysing molecular and cellular function using the computer. Biol Chem. 2000;381:921–935. doi: 10.1515/BC.2000.113. [DOI] [PubMed] [Google Scholar]
- van Helden J, Naim A, Lemer C, Mancuso R, Eldridge M, Wodak S. From molecular activities and processes to biological function. Brief Bioinform. 2001;2:81–93. doi: 10.1093/bib/2.1.81. [DOI] [PubMed] [Google Scholar]
- Maas WK. Studies on the mechanism of repression of arginine biosynthesis in Escherichia coli. II. Dominance of repressibility in diploids. J Mol Biol. 1964;8:365–370. doi: 10.1016/s0022-2836(64)80200-x. [DOI] [PubMed] [Google Scholar]
- van Helden J, Andre B, Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol. 1998;281:827–842. doi: 10.1006/jmbi.1998.1947. [DOI] [PubMed] [Google Scholar]
- van Helden J, Rios AF, Collado-Vides J. Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res. 2000;28:1808–1818. doi: 10.1093/nar/28.8.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis N, Wodak SJ, Cohen GN, Van Helden J. Combining pattern discovery and discriminant analysis to predict gene co-regulation. Bioinformatics. 2004 doi: 10.1093/bioinformatics/bth252. DOI: 10.1093/bioinformatics/bth252. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Liu JS, Lawrence CE. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci. 1995;4:1618–1632. doi: 10.1002/pro.5560040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Thijs G, Lescot M, Marchal K, Rombauts S, De Moor B, Rouze P, Moreau Y. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001;17:1113–1122. doi: 10.1093/bioinformatics/17.12.1113. [DOI] [PubMed] [Google Scholar]
- Jones EW, Pringle JR, Broach JR. The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression. New York: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Bucher P. Regulatory elements and expression profiles. Curr Opin Struct Biol. 1999;9:400–407. doi: 10.1016/S0959-440X(99)80054-2. [DOI] [PubMed] [Google Scholar]
- Krause R, von Mering C, Bork P. A comprehensive set of protein complexes in yeast: mining large scale protein-protein interaction screens. Bioinformatics. 2003;19:1901–1908. doi: 10.1093/bioinformatics/btg344. [DOI] [PubMed] [Google Scholar]
- Manke T, Bringas R, Vingron M. Correlating protein-DNA and protein-protein interaction networks. J Mol Biol. 2003;333:75–85. doi: 10.1016/j.jmb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature. 1998;391:715–718. doi: 10.1038/35661. [DOI] [PubMed] [Google Scholar]
- Huberty CJ. Applied Discriminant Analysis. New York: Wiley; 1994. [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett . 1999;450:27–34. doi: 10.1016/S0014-5793(99)00467-6. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Ettwiller LM, Rung J, Birney E. Discovering novel cis-regulatory motifs using functional networks. Genome Res. 2003;13:883–895. doi: 10.1101/gr.866403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive Yeast Genome Database: yeast cellular substructures, protein complexes http://mips.gsf.de/proj/yeast/catalogues/complexes
- MDS Proteomics: Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry http://www.mdsproteomics.com/yeast [DOI] [PubMed]
- YEAST protein complex database http://yeast.cellzome.com
- Previous evidence http://staffa.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=17&f=evidence
- Gancedo JM. Yeast carbon catabolite repression. Microbiol Mol Biol Rev. 1998;62:334–361. doi: 10.1128/mmbr.62.2.334-361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Transcriptional Regulatory Networks http://web.wi.mit.edu/young/regulator_network/
- van Helden J. Regulatory Sequence Analysis Tools. Nucleic Acids Res. 2003;31:3593–3596. doi: 10.1093/nar/gkg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulatory Sequence Analysis Tools (RSAT) http://rsat.ulb.ac.be/rsat/
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The R Project for Statistical Computing http://www.r-project.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A PDF file containing supplementary Figures S1 to S6 and their legends
A Word file containing supplementary Tables S1 to S4 and their legends