

Abstract
Background
Malaria, caused by Plasmodium sp. parasites, is a leading cause of global morbidity and mortality. Cerebral malaria, characterized by neurological symptoms, is a life-threatening complication of malaria affecting over 500,000 young children in Africa every year. Because of the prevalence and severity of cerebral malaria, a better understanding of the underlying molecular mechanisms of its pathology is desirable and could inform future development of therapeutics. This study sought to clarify the role of Toll-like receptors (TLRs) in promoting immunopathology associated with cerebral malaria, with a particular focus on the understudied TLR7.
Methods
Using the Plasmodium berghei ANKA mouse model of experimental cerebral malaria, C57BL/6 mice deficient in various TLRs were infected, and their resistance to cerebral malaria and immune activation through cytokine production were measured.
Results
Loss of TLR7 conferred partial protection against fatal experimental cerebral malaria. Additionally, loss of TLR signalling dysregulated the cytokine profile, resulting in a shift in the cytokine balance towards those with more anti-inflammatory properties.
Conclusion
This work identifies signalling through TLR7 as a source of pathology in experimental cerebral malaria.
Keywords: Cerebral malaria, Plasmodium berghei, Toll-like receptors, TLR7, Cytokines, Mouse
Background
Malaria, caused by protozoan parasites of the genus Plasmodium, is a major source of global morbidity and mortality, resulting in an estimated 154–289 million infections and 660,000 deaths in 2010 [1]. Approximately 12% of fatal infections in African children are caused by cerebral malaria, a severe neurological complication of Plasmodium falciparum infection characterized by coma (inability to localize a painful stimulus), presence of P. falciparum parasites in the blood, and exclusion of other causes of encephalopathy [2]. Without treatment, cerebral malaria is nearly universally lethal; with intervention, mortality is 15-20%. Furthermore, of children who survive cerebral malaria, approximately 15% exhibit neurological sequelae, from which a proportion of children experience permanent neurological impairment [3, 4]. Although this severe form of malaria represents only a modest proportion of cases, the overall high incidence of malaria results in an estimated 575,000 cases of cerebral malaria occurring annually in Africa [5]. The large number of cases, compounded by the severity of cerebral malaria, makes a better understanding of the underlying molecular mechanisms, and therapeutics based thereon, desirable.
The nature of the pathogenesis of cerebral malaria is controversial [6–12], but is thought to involve the excessive production of pro-inflammatory cytokines [13], the accumulation of leukocytes in the brain [14], and/or the sequestration of infected erythrocytes in the microvasculature of the brain [15, 16]. In the most widely used mouse model of cerebral malaria (infection of C57BL/6 mice with Plasmodium berghei strain ANKA, reviewed in [17]), it is clear that the activation of pro-inflammatory mechanisms results in cerebral immunopathology and symptoms. In particular, the balance of inflammatory Type I cytokines, such as interferon gamma (IFNG) and tumour necrosis factor (TNF), with Type II cytokines, e.g., interleukin 4 (IL4) and IL10, is thought to determine the lethality of cerebral malaria [18, 19]. In both humans and mouse models, high levels of TNF are correlated with cerebral malaria [20, 21]. Conversely, IL10 is thought to limit cerebral pathology in mice [22, 23], and IL10 polymorphisms are associated with cerebral malaria [24]. However, translation of these findings into anti-TNF trials in humans was unsuccessful [4, 25], highlighting the need to better understand the molecular drivers of cerebral pathology.
Toll-like receptors (TLRs) are a family of innate immune sensors that are prime candidates for initiating immune responses that promote cerebral malaria. Evidence for a TLR-dependent contribution to cerebral malaria pathology stems from the observation that mice lacking myeloid differentiation factor 88 (MYD88), a downstream adapter shared by most TLRs, are partially protected from developing cerebral malaria pathology during infection with P. berghei [26–29] (see [30] for dissenting evidence). There is a growing body of evidence supporting TLR recognition of a broad range of Plasmodium molecules [31–41]; however, controversy exists about individual receptor contributions to cerebral malaria pathology. For example, TLR2 is thought to recognize Plasmodium glycophosphatidylinositol [33, 39]; however, whereas studies have found that Tlr2-/- mice [26] and Tlr2-/-Tlr4-/- mice [29] escaped from initial cerebral malaria at a higher rate than wild-type mice, others have reported no differences between wild-type mice and mice lacking these sensors [28, 30, 42]. TLR9, an endosomal DNA sensor, is a more well-established sensor of both mouse and human malaria parasites [31, 32, 37, 40, 41]; however, similar to the controversy over the role of TLR2 in experimental cerebral malaria, some studies have found that TLR9 deficiency results in protection from disease [26, 28], whereas other studies have found no differences as compared with wild-type mice [30, 42]. In the studies that have reported a protective effect of TLR9 disruption, cerebral malaria was still more severe than that observed in MYD88-deficient mice, suggestive of signalling contributions from other MYD88-dependent pathways [28]. It is likely that, as with other parasitic infections, multiple TLRs contribute to the immune response [43–45], making it clear that quantitative measures of immune activation are needed to better understand the relative contributions of TLRs to immune activation during malaria infection.
Previous work from this lab showed that a deficiency in TLR7, which recognizes ribonucleic acid (RNA), leads to widespread immune dysfunction during Plasmodium chabaudi infection, although there was no impact on the clearance of parasites [41]. Similar results have been reported for TLR9 and the shared adapter molecule, MYD88; whereas loss of MYD88 or TLR9 diminished cytokine production during P. chabaudi infection, no changes in control of parasitaemia were seen [46]. Similarly, during P. berghei strain ANKA (referred to as ‘P. berghei’ hereafter) infection of the C57BL/6 mouse, in which Type I inflammation promotes a lethal infection with neurological symptoms consistent with cerebral malaria [18], a deficiency in either TLR9 or MYD88 partially protects against lethal disease [26, 28]. However, in contrast to findings with TLR9- and MYD88-deficient mice, in the only study to examine the role of TLR7-deficiency in cerebral malaria, TLR7 was found to be irrelevant for precipitation of fatal disease [26]. This study was performed using small cohorts of mice in an experimental design intended to screen several candidate strains, which allowed for the possibility that a role for TLR7 in cerebral malaria may have been overlooked. To more thoroughly interrogate the role of TLR7 signalling in experimental cerebral malaria in this present study, large sample groups and quantitative measures of immune activation were employed. The current findings demonstrate that the absence of TLR7 during experimental cerebral malaria shifts the balance of cytokines towards an anti-inflammatory state and confers protection from cerebral malaria lethality.
Methods
Ethics statement
All mouse work was conducted with the approval of the University of California, San Francisco (UCSF) Institutional Animal Care and Use Committee in strict accordance with the guidelines of the Office of Laboratory Animal Welfare.
Mice
The following mouse strains were used in this study: C57BL/6 (Jackson Laboratories or the National Cancer Institute); Tlr7−/− (Jackson Laboratories); Tlr9−/− (R Medzhitov, Yale University; with permission from S Akira, Osaka University, and T Taniguchi, University of Tokyo); Myd88−/− (J Cox, UCSF; with permission from S Akira). Tlr7-/-Tlr9-/-double knockout mice were bred in house. All strains were confirmed to have a C57BL/6 background of greater than 95% by microsatellite genotyping.
Parasites
All experiments were performed using P. berghei strain ANKA (MRA-311) parasites, which were obtained from the MR4 stock centre and maintained in C57BL/6 mice. Blood was harvested by cardiac puncture from an infected mouse on day 5 of infection, and 106 infected erythrocytes were introduced into a new mouse by intraperitoneal injection in 100 μl of Alsever’s solution. All infections were initiated at 14.00 hours. For cytokine analysis, blood was harvested by submandibular blood collection or cardiac puncture into K2EDTA for plasma isolation.
Infections
Infections were initiated as described above. Survival and signs of cerebral malaria were monitored daily, and twice daily during the peak of lethality (days 6 through 12). Animals that showed neurological symptoms, such as convulsions, ataxia or paralysis, or that died on or before day 12 post-infection, were considered to have cerebral malaria as previously described [26]. Parasitaemia (percentage of parasite-infected erythrocytes) was monitored daily by Giemsa-stained thin film blood smears. The significance of parasitaemia courses was assessed by the Mann–Whitney U test (α = 0.05). The significance of survival courses was assessed by comparing Kaplan-Meier curves using the log rank (Mantel-Cox) test (α = 0.05).
Cytokine detection
All plasma cytokines were measured by multiplexed cytometric bead immunoassay (Millipore) as per manufacturer instructions and detected on a MAGPIX (Luminex). Cytokine level significance was assessed using a Mann–Whitney U test (α = 0.05).
Results
Loss of TLR7 confers partial resistance to cerebral malaria lethality
Given the reduced levels of inflammatory cytokines observed in Tlr7-/- mice in response to several Plasmodium sp. in a previous work [41], it was hypothesized that TLR7 might play a role in the pathology of P. berghei cerebral malaria that was previously undetected. To test this, the survival of TLR7-deficient mice, as well as Tlr9-/-, Tlr7-/-Tlr9-/-, and Myd88-/- mice, infected with P. berghei parasites was monitored. As previously reported [26, 28], mice deficient in either TLR9 or MYD88 were partially protected from lethal infection with P. berghei when compared with wild-type mice, with Tlr9-/- mice less well protected than Myd88-/- mice (29% escape and 58% escape, respectively, with escape defined as survival past day 12 of infection; Figure 1A). These findings are similar to a previous report [28] in which the proportion of Myd88-/- mice that escaped from cerebral malaria was 1.5 times greater than the proportion of Tlr9-/- mice, suggesting the existence of additional MYD88-dependent sensors that promote cerebral malaria [26, 27, 29]. In support of the above hypothesis, Tlr7-/- mice were partially protected from lethality with approximately 24% escaping from cerebral malaria, as compared to 8% of wild-type mice, a difference that was detectable using large sample sizes (n = 71 for Tlr7-/- mice). Because TLR7 and TLR9 share the common signalling adapter MYD88, the possibility of a genetic interaction between these sensors was tested by generating mice lacking both sensors. As expected, Tlr7-/-Tlr9-/- mice demonstrated improved survival as compared to wild-type mice, with 24% escaping cerebral malaria. Notably, they were no more protected than mice with either deficiency alone and were also not as well protected as Myd88-/- mice. There was no significant difference in survival between females and males of any given genotype (log rank [Mantel-Cox] test; α = 0.05). Additionally, protected mice did not show symptoms of cerebral malaria, such as ataxia, hemi- or paraplegia, seizures or coma. This protection was not due to improved parasite restriction; all animals eventually succumbed to hyperparasitaemia after the initial escape from cerebral malaria, as observed in other immunodeficient mice [26, 47] (Figure 1B). The simplest interpretation of these data is that TLR7 and TLR9 synergistically signal to promote cerebral malaria, with both sensors being required for full elaboration of lethal pathology (and conversely, loss of both sensors not conferring more protection than loss of either sensor alone). In addition, it is likely that other MYD88-dependent, but TLR7- and TLR9-independent, mechanisms also promote pathology.
Figure 1.
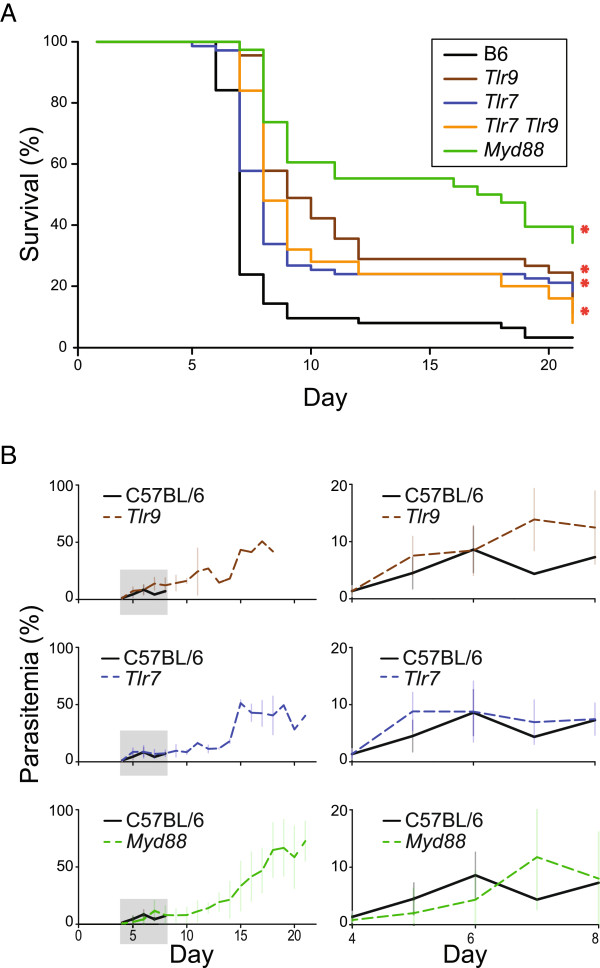
Survival of TLR-deficient mice during cerebral malaria. (A) C57BL/6 (B6), Tlr7 -/-, Tlr9 -/- , Tlr7 -/- Tlr9 -/- , and Myd88 -/- mice were challenged with 106 P. berghei erythrocytes and monitored daily for survival (n = 63 B6, n = 71 Tlr7 -/-, n = 45 Tlr9 −/−, n = 25 Tlr7 -/- Tlr9 -/- , n = 38 Myd88 −/−). *, p < 0.05 (log rank [Mantel-Cox] test). Data were pooled from six experiments. (B) Infected C57Bl/6, Tlr7 −/−, Tlr9 -/- , and Myd88 −/− mice were monitored for parasitaemia. Right, zoomed-in plot of region contained within gray box on left. (n = 25 B6, n = 25 Tlr7 -/-, n = 16 Tlr9 −/−; n = 21 Myd88 −/−). Data were pooled from three independent experiments.
In contrast to the current findings, Tlr7-/y mice were not found to be protected from cerebral malaria in the single study that has previously examined P. berghei infection in TLR7-deficient mice [26]. In the current study, the experimental hazard ratio between B6 and Tlr7-/- mice was determined to be 2.847, with a probability of 0.888 that a subject of either genotype would succumb to cerebral malaria by the end of the experiment. Based on these numbers, the sample size of five in each group used in the previous study would only provide a statistical power of 0.26 (α = 0.05). In order to detect the difference observed in this study, a minimum sample size of 33 mice is necessary [48]. Based on these calculations, it is possible that the contribution of TLR7 to P. berghei pathogenesis was overlooked in the previous work.
To better understand the inflammatory response as related to lethality of P. berghei infection in Tlr7-/-, Tlr9-/-, Tlr7-/-Tlr9-/-, and Myd88-/- mice, the levels of cytokines that have previously been associated with susceptibility to cerebral malaria [21, 49–52] were measured at three days post-infection, which is at the onset of parasite patency, and six days post-infection, which is approximately 12 to 24 hours before the onset of neurological symptoms in wild-type mice. A subset of both pro-inflammatory cytokines (IFNG, TNF, macrophage inflammatory protein 1 beta [MIP1B], and IL6) and anti-inflammatory cytokines (IL10 and IL4) was found to be dysregulated at various points in the absence of TLR7 and/or TLR9 signalling (Figure 2A and 2B).
Figure 2.
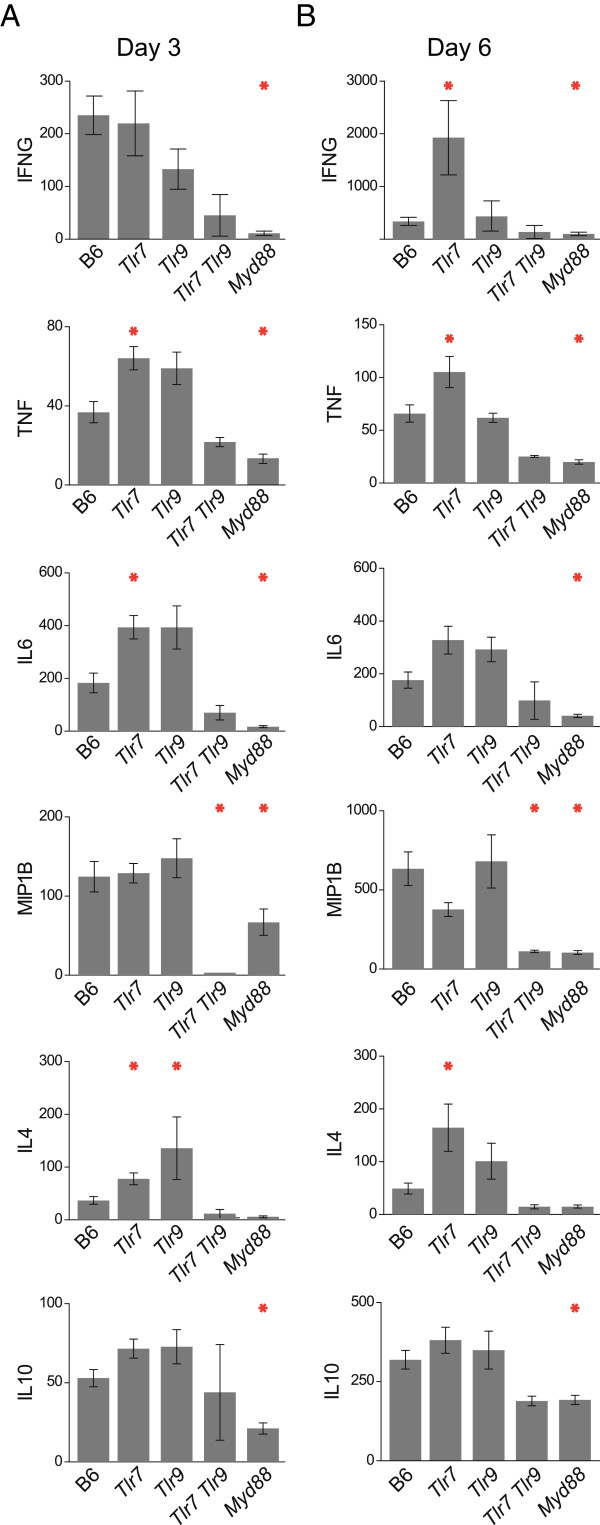
TLR-dependence of cytokine production during Plasmodium berghei infection. (A, B) Levels of IFNG, IL6, IL4, TNF, MIP1B, and IL10 were measured by cytometric bead array in plasma collected from mice of the indicated genotypes three days (A) or six days (B) after infection with 106 P. berghei-infected erythrocytes. All units are pg/mL. Means with SE are shown. *, p < 0.05 (Mann-Whitney) (n = 30 B6, n = 22 Tlr7 -/-, n = 11 Tlr9 −/−, n = 3 Tlr7 -/- Tlr9 -/-, n = 15 Myd88 −/−). Data were pooled from three independent experiments.
Specifically, at both days 3 and 6 post-infection, IFNG, IL6, TNF, MIP1B and IL10 were all significantly diminished in MYD88-deficient mice as compared to wild-type mice. MIP1B was also significantly diminished in Tlr7-/-Tlr9-/- mice on both days, as compared to wild-type mice. All of the other cytokines also followed the same trend of being reduced in Tlr7-/-Tlr9-/- as compared to wild-type mice, although they did not reach statistical significance. In contrast, none of these cytokines were significantly diminished in mice singly deficient in either TLR7 or TLR9. In fact, many cytokines were produced at higher levels in Tlr7-/- mice, including TNF, IFNG, and IL6. Tlr9-/- mice appeared to follow similar trends for these cytokines, but did not reach significance. These observations are similar to those from early P. chabaudi infection [41], wherein mice lacking TLR9 overproduce cytokines; this increase might be a result of decreased competition for endosomal trafficking between TLR7 and TLR9 [53]. Additionally, IL4 was increased in both Tlr7-/- and Tlr9-/- mice on day 3, as well as in Tlr7-/- mice on day 6. Importantly, unlike the synergistic interaction between TLR7 and TLR9 suggested by the survival data, these results are consistent with a model in which TLR7 and TLR9 signal redundantly through MYD88 to promote cytokine production during experimental cerebral malaria.
To assess the relative contributions of each cytokine to the pathogenesis of cerebral malaria, the average plasma levels of each cytokine in each genotype were examined for correlation with the percentage of that genotype that escaped from cerebral malaria. Consistent with the discrepancy between survival and cytokine production, none of the correlations reached a p value of less than 0.1, although r2 values for IFNG and MIP1B were above 0.5 (days 3 and 6, respectively; Figure 3A). These modest correlations led to the consideration of alternative explanations for the differential survival. Other studies have found that the ratio of certain Type I (i.e., pro-inflammatory) to Type II (anti-inflammatory) cytokines is more strongly associated with severe malaria disease than any single cytokine [54, 55]. Given the established role of pro-inflammatory cytokines in promoting experimental cerebral malaria and anti-inflammatory cytokines in suppressing lethal pathology [18, 19, 22, 47], it was hypothesized that the ratio of such cytokines to one another would be more strongly correlated with survival than the correlation observed for any individual cytokine. Because calculation of ratios results in propagation of error, a less stringent alpha (α = 0.1) was used to test for significant correlations between cytokine ratios and survival. Strikingly, three cytokine log ratios were significantly correlated on day 3 (p < 0.1) (Figure 3B), with correlation coefficients (r) of greater than 0.8 (Figure 3C). Additionally, these three ratios, IL4/IFNG, TNF/IFNG, and IL10/IFNG, were all significantly increased in Tlr7-/-, Tlr9-/-, Tlr7-/-Tlr9-/-, and Myd88-/- mice as compared to B6 on day 3 (with the exception of IL4/IFNG in Tlr7-/-Tlr9-/-, which did not reach significance; Figure 3D). Of these three ratios, two represent ratios of anti-inflammatory cytokines (IL4 and IL10) to the pro-inflammatory cytokine, IFNG, suggesting that the balance of these cytokines may be a driver of pathology. The most strongly correlated ratio on day 3, IL4/IFNG, also remained the most strongly correlated on day 6 (just prior to the onset of symptoms in unprotected mice). Notably, these findings might be analogous to those from a study that found the IL4/IFNG ratio to be associated with cerebral malaria in humans [54].To further assess whether these cytokine ratios would delineate susceptibility to cerebral malaria without regard to genetic makeup, mice of all genotypes were grouped by survival status. All three cytokine ratios were significantly elevated in mice that escaped cerebral malaria, as compared to those that succumbed (Figure 3E). These data suggest that TLR signalling in wild-type mice may promote the development of a pathological Type I immune response, whereas the cytokine response in mice deficient in TLR and MYD88 signalling is skewed toward a Type II response that protects against cerebral malaria. Furthermore, regardless of genotype, relative levels of cytokines, particularly the ratio of IL4 to IFNG, are more strongly associated with protection from cerebral malaria than levels of any single cytokine alone.
Figure 3.
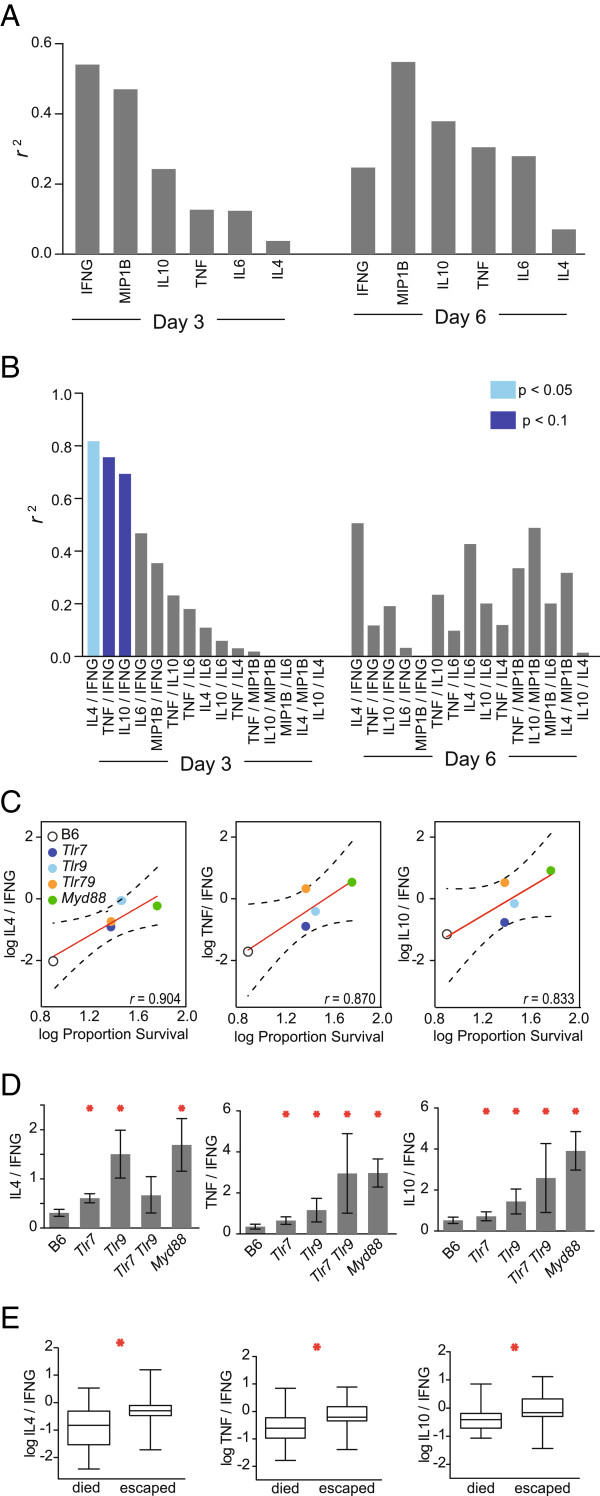
Association of cytokine ratios with outcomes of experimental cerebral malaria. (A) For each genotype (B6, Tlr7 -/-, Tlr9 -/-, Tlr7 -/- Tlr9 -/- , and Myd88 -/-) a Pearson’s product moment correlation was used to determine the relationship between the average log10 (plasma cytokine concentration + 1) and the log10 fraction of each genotype that escaped cerebral malaria (survived until day 12). r 2 values were calculated from a linear regression of the above. (B) As above, except using ratios of cytokines. A less stringent statistical threshold (α = 0.1) was tested to account for the propagation of error when calculating ratios. Light blue, p < 0.05; dark blue, p < 0.1 (Pearson’s correlation). (C) Significant log10 plasma cytokine ratios from (B) as a function of average survival for each genotype. The linear regression across genotypes is shown with 95% confidence intervals. (D) Significant cytokine ratios from (B) by genotype. Errors represent SE and all statistical comparisons are to B6 mice. *, p < 0.05 (Mann–Whitney). (E) Box plots of significantly correlated, log10 cytokine ratios for all mice, grouped by escape from cerebral malaria (survival past day 12). *, p < 0.05 (Mann–Whitney).
Discussion
In this highly powered study, it has been shown that signalling through TLR7 contributes to lethality from cerebral malaria. The loss of both TLR7 and TLR9 signalling leads to cytokine dysregulation during the course of P. berghei ANKA infection and protects against symptoms of cerebral malaria. Interestingly, the absence of TLR7 or TLR9 results in overproduction of multiple cytokines, but loss of both TLR7 and TLR9 results in reduced cytokine production (Figure 2). TLR7 and TLR9 have previously been shown to compete via the shared endosomal trafficking molecule, UNC93b1 [53]. It is possible that in mice singly deficient for either TLR7 or TLR9, enhanced signalling by the intact sensor leads to increased cytokine production in response to P. berghei, whereas loss of both TLR7 and TLR9 leads to decreased cytokine production. This phenomenon leads to an apparent discrepancy between cytokine production and survival data that can be explained through the consideration of ratios of cytokines to one another, rather than analysis of any single cytokine in isolation.
Additionally, other MYD88-dependent signalling cannot be explained by TLR7 and TLR9 (Figure 1), indicating contributions from other MYD88-dependent sensors. Based on other studies, TLR2 is likely to account for the bulk of the additional MYD88-dependent contribution [26, 29, 33, 39]. Previous studies using the P. berghei mouse model of cerebral malaria have shown that multiple pro-inflammatory cytokines, chemokines and leukocyte populations drive the observed rapid lethality [47, 56–58], whereas anti-inflammatory cytokines confer protection from lethality [22, 58, 59]. Consistent with these reports, mice lacking both TLR7 and TLR9, or lacking MYD88, exhibit cytokine profiles skewed toward anti-inflammatory cytokine production and are protected from lethal cerebral malaria. Furthermore, the strong association of cytokine ratios with survival in TLR- and MYD88- deficient mice suggests that protection against pathology may be conferred by the ratio of anti-inflammatory to pro-inflammatory cytokines produced during infection.
Interestingly, one study reported that mice overexpressing TLR7 were also partially protected from cerebral malaria [60]. Although further experiments are needed to reconcile these findings, it is possible that the increased baseline levels of IL10 found in these mice may protect them from subsequent immunopathology. Consistent with this possibility, recombinant IL10 treatment can suppress P. berghei lethality [22]. Notably, similar to the protection seen in TLR7-deficient mice, TLR7 overexpression did not have any effect on parasite load [60], indicating that this protection is a consequence of increasing host tolerance to cerebral malaria without affecting host resistance [61].
The mechanistic study of cerebral malaria in humans is difficult, as this disease may be considerably more heterogeneous than is currently appreciated [62, 63]. Given the observation of neurological sequelae following treatment and convalescence in a subset of patients that no longer harbour parasites [64], it is reasonable to expect that some proportion of cases may occur as a result of immunopathology. It is further likely that different sensors contribute to the overall pathology of cerebral malaria differentially in ethnically and genetically diverse individuals, making the identification of all potentially pathogenic molecules desirable. This work identifies TLR7 as yet another molecule involved in the pathological response to Plasmodium parasites, and supports the notion that immune responses to Plasmodium must be finely tuned to effect parasite clearance while minimizing immunopathology.
Conclusions
Loss of TLR7 signalling confers partial protection against fatal experimental cerebral malaria, while having no effect on parasite restriction. TLR7 signalling promotes lethal pathology in a manner that is synergistic with TLR9 signalling. The protection conferred by the loss of TLR7 is correlated with a shift towards an anti-inflammatory cytokine profile during P. berghei infection.
Acknowledgements
We thank Bryan Greenhouse for data analysis advice, and Eunice Chen, Chuck Chan, Angela Lee, Moena Nishikawa, Yuvadee Srijongsirikul, Kim D’Costa, and Ali Esmaeili for technical assistance. This work was supported by NIAID T32 AI 060537 (MFF), the Dean’s Office Medical Student Research Program at the University of California San Francisco (AB), and NIAID R00AI085035 (CCK). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- RNA
Ribonucleic acid
- TLRs
Toll-like receptors
- IFNG
Interferon gamma
- TNF
Tumor necrosis factor
- IL
Interleukin
- MYD88
Myeloid differentiation factor 88
- MIP1B
Macrophage inflammatory protein 1 beta.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AB and CCK conceived and designed experiments; AB, MFF, BWH, and CCK performed the experiments; AB, BWH and CCK analysed data; AB and CCK wrote the manuscript; all authors read and approved the final manuscript.
Contributor Information
Alyssa Baccarella, Email: baccarellaa@medsfgh.ucsf.edu.
Brian W Huang, Email: brianwhuang@gmail.com.
Mary F Fontana, Email: fontanam@medsfgh.ucsf.edu.
Charles C Kim, Email: charlie.kim@ucsf.edu.
References
- 1.WHO . World Malaria Report 2012. Geneva: World Health Organization; 2012. [Google Scholar]
- 2.Murphy SC, Breman JG. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg. 2001;64(1–2 Suppl):57–67. doi: 10.4269/ajtmh.2001.64.57. [DOI] [PubMed] [Google Scholar]
- 3.Mung’ala-Odera V, Snow RW, Newton CRJC. The Burden of the neurocognitive impairment associated with Plasmodium falciparum malaria in sub-Saharan Africa. Am J Trop Med Hyg. 2004;71(2 Suppl):64–70. [PubMed] [Google Scholar]
- 4.Van Hensbroek MB, Palmer A, Jaffar S, Schneider G, Kwiatkowski D. Residual neurologic sequelae after childhood cerebral malaria. J Pediatr. 1997;131:125–129. doi: 10.1016/S0022-3476(97)70135-5. [DOI] [PubMed] [Google Scholar]
- 5.Idro R, Marsh K, John CC, Newton CR. Cerebral malaria; mechanisms of brain injury and strategies for improved neuro-cognitive outcome. Pediatr Res. 2010;68:267–274. doi: 10.1203/PDR.0b013e3181eee738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White NJ, Turner GDH, Medana IM, Dondorp AM, Day NPJ. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26:11–15. doi: 10.1016/j.pt.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson MM, Gros P, Olivier M, Fortin A, Serghides L. Cerebral malaria: human versus mouse studies. Trends Parasitol. 2010;26:274–275. doi: 10.1016/j.pt.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Milner DA. Rethinking cerebral malaria pathology. Curr Opin Infect Dis. 2010;23:456–463. doi: 10.1097/QCO.0b013e32833c3dbe. [DOI] [PubMed] [Google Scholar]
- 9.Langhorne J, Buffet P, Galinski M, Good M, Harty J, Leroy D, Mota MM, Pasini E, Renia L, Riley E, Stins M, Duffy P. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10:23. doi: 10.1186/1475-2875-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, Turner G, Langhorne J, participants of the Hinxton Retreat meeting on Animal Models for Research on Severe Malaria The role of animal models for research on severe malaria. PLoS Pathog. 2012;8:e1002401. doi: 10.1371/journal.ppat.1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rénia L, Grüner AC, Snounou G. Cerebral malaria: in praise of epistemes. Trends Parasitol. 2010;26:275–277. doi: 10.1016/j.pt.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Riley EM, Couper KN, Helmby H, Hafalla JCR, de Souza JB, Langhorne J, Jarra WB, Zavala F. Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010;26:277–278. doi: 10.1016/j.pt.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 13.John CC, Panoskaltsis-Mortari A, Opoka RO, Park GS, Orchard PJ, Jurek AM, Idro R, Byarugaba J, Boivin MJ. Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am J Trop Med Hyg. 2008;78:198–205. [PMC free article] [PubMed] [Google Scholar]
- 14.Patnaik JK, Das BS, Mishra SK, Mohanty S, Satpathy SK, Mohanty D. Vascular clogging, mononuclear cell margination, and enhanced vascular permeability in the pathogenesis of human cerebral malaria. Am J Trop Med Hyg. 1994;51:642–647. [PubMed] [Google Scholar]
- 15.MacPherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119:385–401. [PMC free article] [PubMed] [Google Scholar]
- 16.Haldar K, Murphy SC, Milner DA, Taylor TE. Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol Mech Dis. 2007;2:217–249. doi: 10.1146/annurev.pathol.2.010506.091913. [DOI] [PubMed] [Google Scholar]
- 17.De Souza JB, Hafalla JCR, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology. 2010;137:755–772. doi: 10.1017/S0031182009991715. [DOI] [PubMed] [Google Scholar]
- 18.de Kossodo S, Grau GE. Profiles of cytokine production in relation with susceptibility to cerebral malaria. J Immunol. 1993;151:4811–4820. [PubMed] [Google Scholar]
- 19.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/S1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 20.Prakash D, Fesel C, Jain R, Cazenave P-A, Mishra GC, Pied S. Clusters of cytokines determine malaria severity in Plasmodium falciparum–infected patients from endemic areas of Central India. J Infect Dis. 2006;194:198–207. doi: 10.1086/504720. [DOI] [PubMed] [Google Scholar]
- 21.Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60:1623–1635. doi: 10.1007/s00018-003-2347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology. 1997;91:536–540. doi: 10.1046/j.1365-2567.1997.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckwalanga M, Marussig M, Tavares MD, Bouanga JC, Hulier E, Pavlovitch JH, Minoprio P, Portnoï D, Rénia L, Mazier D. Murine AIDS protects mice against experimental cerebral malaria: down-regulation by interleukin 10 of a T-helper type 1 CD4+ cell-mediated pathology. Proc Natl Acad Sci U S A. 1994;91:8097–8101. doi: 10.1073/pnas.91.17.8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apinjoh TO, Anchang-Kimbi JK, Njua-Yafi C, Mugri RN, Ngwai AN, Rockett KA, Mbunwe E, Besingi RN, Clark TG, Kwiatkowski DP, Achidi EA, Malaria GEN Consortium Association of cytokine and Toll-like receptor gene polymorphisms with severe malaria in three regions of Cameroon. PLoS One. 2013;8:e81071. doi: 10.1371/journal.pone.0081071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, Memming H, Frenkel J, Enwere G, Bennett S, Kwiatkowski D, Greenwood B. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis. 1996;174:1091–1097. doi: 10.1093/infdis/174.5.1091. [DOI] [PubMed] [Google Scholar]
- 26.Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, Kawai T, Takeuchi O, Hisaeda H, Horii T, Akira S. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol. 2007;19:67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 27.Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataíde MA, de Oliveira RB, Golenbock DT, Gazzinelli RT. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci. 2011;108:3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffith JW, O’Connor C, Bernard K, Town T, Goldstein DR, Bucala R. Toll-like receptor modulation of murine cerebral malaria is dependent on the genetic background of the host. J Infect Dis. 2007;196:1553–1564. doi: 10.1086/522865. [DOI] [PubMed] [Google Scholar]
- 29.Kordes M, Matuschewski K, Hafalla JCR. Caspase-1 activation of interleukin-1β (IL-1β) and IL-18 is dispensable for induction of experimental cerebral malaria. Infect Immun. 2011;79:3633–3641. doi: 10.1128/IAI.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Togbe D, Schofield L, Grau GE, Schnyder B, Boissay V, Charron S, Rose S, Beutler B, Quesniaux VFJ, Ryffel B. Murine cerebral malaria development is independent of Toll-Like receptor signaling. Am J Pathol. 2007;170:1640–1648. doi: 10.2353/ajpath.2007.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol. 2004;172:4926–4933. doi: 10.4049/jimmunol.172.8.4926. [DOI] [PubMed] [Google Scholar]
- 32.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. 2005;201:19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280:8606–8616. doi: 10.1074/jbc.M413541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 35.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couper KN, Barnes T, Hafalla JCR, Combes V, Ryffel B, Secher T, Grau GE, Riley EM, de Souza JB. Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog. 2010;6:e1000744. doi: 10.1371/journal.ppat.1000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, Gowda NM, Kumar S, Gowda DC. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol. 2010;184:4338–4348. doi: 10.4049/jimmunol.0903824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gowda NM, Wu X, Gowda DC. The nucleosome (histone-DNA complex) is the TLR9-specific immunostimulatory component of Plasmodium falciparum that activates DCs. PLoS One. 2011;6:e20398. doi: 10.1371/journal.pone.0020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, Krishnegowda G, Li G, Gowda DC. Proinflammatory responses by glycosylphosphatidylinositols (GPIs) of Plasmodium falciparum are mainly mediated through the recognition of TLR2/TLR1. Exp Parasitol. 2011;128:205–211. doi: 10.1016/j.exppara.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol Baltim Md 1950. 2012;188:5073–5085. doi: 10.4049/jimmunol.1102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baccarella A, Fontana MF, Chen EC, Kim CC. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun. 2013;81:4431–4442. doi: 10.1128/IAI.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lepenies B, Cramer JP, Burchard GD, Wagner H, Kirschning CJ, Jacobs T. Induction of experimental cerebral malaria is independent of TLR2/4/9. Med Microbiol Immunol (Berl) 2008;197:39–44. doi: 10.1007/s00430-007-0057-y. [DOI] [PubMed] [Google Scholar]
- 43.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol Baltim Md 1950. 2006;177:3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 44.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6:895–906. doi: 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- 45.Petzke MM, Brooks A, Krupna MA, Mordue D, Schwartz I. Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J Immunol Baltim Md 1950. 2009;183:5279–5292. doi: 10.4049/jimmunol.0901390. [DOI] [PubMed] [Google Scholar]
- 46.Franklin BS, Rodrigues SO, Antonelli LR, Oliveira RV, Goncalves AM, Sales-Junior PA, Valente EP, Alvarez-Leite JI, Ropert C, Golenbock DT, Gazzinelli RT. MyD88-dependent activation of dendritic cells and CD4(+) T lymphocytes mediates symptoms, but is not required for the immunological control of parasites during rodent malaria. Microbes Infect. 2007;9:881–890. doi: 10.1016/j.micinf.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 47.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc Natl Acad Sci U S A. 1989;86:5572–5574. doi: 10.1073/pnas.86.14.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schoenfeld D. The asymptotic properties of nonparametric tests for comparing survival distributions. Biometrika. 1981;68:316–319. doi: 10.1093/biomet/68.1.316. [DOI] [Google Scholar]
- 49.Rudin W, Favre N, Bordmann G, Ryffel B. Interferon-γ is essential for the development of cerebral malaria. Eur J Immunol. 1997;27:810–815. doi: 10.1002/eji.1830270403. [DOI] [PubMed] [Google Scholar]
- 50.Angulo I, Fresno M. Cytokines in the pathogenesis of and protection against malaria. Clin Diagn Lab Immunol. 2002;9:1145–1152. doi: 10.1128/CDLI.9.6.1145-1152.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jain V, Armah HB, Tongren JE, Ned RM, Wilson NO, Crawford S, Joel PK, Singh MP, Nagpal AC, Dash AP, Udhayakumar V, Singh N, Stiles JK. Plasma IP-10, apoptotic and angiogenic factors associated with fatal cerebral malaria in India. Malar J. 2008;7:83. doi: 10.1186/1475-2875-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, Kone A, Harley R, Plowe CV, Doumbo OK, Sztein MB. Serum levels of the proinflammatory cytokines interleukin-1 Beta (IL-1β), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun. 2004;72:5630–5637. doi: 10.1128/IAI.72.10.5630-5637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukui R, Saitoh S, Matsumoto F, Kozuka-Hata H, Oyama M, Tabeta K, Beutler B, Miyake K. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–1350. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tangteerawatana P, Pichyangkul S, Hayano M, Kalambaheti T, Looareesuwan S, Troye-Blomberg M, Khusmith S. Relative levels of IL4 and IFN-γ in complicated malaria: Association with IL4 polymorphism and peripheral parasitemia. Acta Trop. 2007;101:258–265. doi: 10.1016/j.actatropica.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 55.May J, Lell B, Luty AJ, Meyer CG, Kremsner PG. Plasma interleukin-10:Tumor necrosis factor (TNF)-alpha ratio is associated with TNF promoter variants and predicts malarial complications. J Infect Dis. 2000;182:1570–1573. doi: 10.1086/315857. [DOI] [PubMed] [Google Scholar]
- 56.Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, Hooks JJ, Detrick B. Beta interferon suppresses the development of experimental cerebral malaria. Infect Immun. 2011;79:1750–1758. doi: 10.1128/IAI.00810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yañez DM, Batchelder J, van der Heyde HC, Manning DD, Weidanz WP. Gamma delta T-cell function in pathogenesis of cerebral malaria in mice infected with Plasmodium berghei ANKA. Infect Immun. 1999;67:446–448. doi: 10.1128/iai.67.1.446-448.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol Baltim Md 1950. 1996;157:1620–1624. [PubMed] [Google Scholar]
- 59.Tan RS-P, Kara AU, Feng C, Asano Y, Sinniah R. Differential interleukin-10 expression in interferon regulatory factor-1 deficient mice during Plasmodium berghei blood-stage infection. Parasite Immunol. 2000;22:425–435. doi: 10.1046/j.1365-3024.2000.00312.x. [DOI] [PubMed] [Google Scholar]
- 60.Waisberg M, Tarasenko T, Vickers BK, Scott BL, Willcocks LC, Molina-Cruz A, Pierce MA, Huang C, Torres-Velez FJ, Smith KGC, Barillas-Mury C, Miller LH, Pierce SK, Bolland S. Genetic susceptibility to systemic lupus erythematosus protects against cerebral malaria in mice. Proc Natl Acad Sci U S A. 2011;108:1122–1127. doi: 10.1073/pnas.1017996108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakaria AK, Mahajan SK, Desai RR, Shah KB. Delayed cerebellar ataxia: A rare self limiting complication of Plasmodium falciparum malaria. Adv Biomed Res. 2013;2:27. doi: 10.4103/2277-9175.107997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brewster DR, Kwiatkowski D, White NJ. Neurological sequelae of cerebral malaria in children. Lancet. 1990;336:1039–1043. doi: 10.1016/0140-6736(90)92498-7. [DOI] [PubMed] [Google Scholar]
- 64.Senanayake N, de Silva HJ. Delayed cerebellar ataxia complicating falciparum malaria: a clinical study of 74 patients. J Neurol. 1994;241:456–459. doi: 10.1007/BF00900965. [DOI] [PubMed] [Google Scholar]