

Abstract
The compromised antioxidant defense system in chronic lymphocytic leukemia (CLL) suggested a potential use for Reactive Oxygen Species (ROS) generating Arsenic Trioxide (ATO) and Ascorbic Acid. While both ATO and ascorbic acid mediated cytotoxicity in CLL B cells as single agents, the efficacy of ATO is enhanced by ascorbic acid. This effect is dependent on increased ROS accumulation, as pretreatment of B-CLL cells with a glutathione reducing buthionine sulfoximine or catalase inhibiting aminotriazole, enhanced ATO/ascorbic acid mediated cytotoxicity. Petreatment with reducing agents such as catalase, or thiol anti-oxidant, N-acetyl cysteine or GSH also abrogated ATO/ascorbic acid mediated cytotoxicity. Furthermore, Hu1D10 mediated cell death was enhanced with ATO and ascorbic acid, thus justifying potential combination of ATO/arsenic trioxide therapy with antibodies such as Hu1D10 that also cause accumulation of ROS.
Keywords: CLL, Arsenic trioxide, ascorbic acid
Introduction
B-cell chronic lymphocytic leukemia (CLL) is one of the most common adult leukemias and has a high mortality rate. In recent years monoclonal antibody therapies, like Rituximab, have shown some promise in combating this disease; however the search for a more effective therapy is still continuing. In 1984, Farber et al 1 showed that B-lymphocytes from CLL patients lost viability rapidly compared to T-cells when incubated with a H2O2 generating system. Several reports have suggested that CLL cells have a compromised antioxidant defense system evident from the low activities of the major antioxidant enzymes superoxide dismutase and catalase and the accumulation of degradation products like malonaldialdehyde and 8-oxo-deoxyguanosine 2-4. However there were no documented pre-clinical reports attempting to exploit this phenomenon with therapeutic agents relevant to clinical investigation.
Arsenic trioxide, a potent carcinogen and environmental toxin, has the ability to induce apoptosis in several tumors/leukemias 5-10. It is highly active in acute promyelocytic leukemia (APL). 11-16 In APL, its cytotoxic effect is thought to be mediated via its ability to generate reactive oxygen species (ROS) 17, possibly through the membrane bound NADPH oxidases 18; and also due to its binding affinity to thiol groups which results in a decreased glutathione pool in the cells 19,20. The cytotoxic effect of arsenic trioxide has been shown to increase with the addition of ascorbic acid which presumably aids in its redox cycling 21,22. Based upon the susceptibility of CLL cells to oxidative stress previously demonstrated, we sought to determine the effect of arsenic trioxide with or without ascorbic acid treatment in CLL1.
Materials and Methods
Cells
Blood was obtained from patients with B-cell CLL with informed consent under a protocol approved by the hospital internal review board. All patients examined in this series had immunophenotypically defined CLL as outlined by the modified 96 National Cancer Institute criteria (add reference Cheson BD et al). Enriched B-lymphocyte fractions were prepared by using MACS negative selection kit (Miltenyi Biotec, Auburn, CA) or by “Rosette-Sep” kit (Stem Cell Technologies, Vancouver, British Columbia, Canada) according to the manufacturer's instructions.
Culture conditions
Cells were incubated at 1 × 106 cells/ml with 1μM arsenic trioxide (Sigma, City/State), 1mM ascorbic acid (Sigma, St. Louis, MO) and 10μg/ml Hu1D10 (Protein Design Laboratories, Fremont, CA) with 10μg/ml goat anti-human IgG, Fcγ fragment specific (Jackson Immuonotechnology, West Grove, PA) as cross linker. CLL B cells, human B-lymphocyte cell lines Raji, 697, Ramos, Wac or MC60A were cultured at 37°C in an atmosphere of 5% CO2 in RPMI 1640 media (Gibco/Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco). All other additions and treatments are noted in figure legends. Z-VAD-fmk, buthionine sulfoximine, aminotrizole, catalase, N-acetyl cystein and H2O2 (all from Sigma) were used at indicated concentrations.
Analysis of cell viability and apoptosis
Flow cytometric analysis of cell viability was carried out by dual staining with Annexin V conjugated to FITC and Propidium iodide according to the manufacturer's instructions (BD Pharmingen, San Diego, CA). Cells excluding both FITC and PI were considered to be viable.
Measurement of ROS accumulation
ROS generated was measured by flow cytometry. After incubation 10μM dihydroethidine (Molecular Probes, City/State) was added to the incubation mixtures and the cells were further incubated for 60 minutes. The non-fluorescent dihydroethidine reacts with cellular superoxide to form ethidium which binds to nucleic acids to give a fluorescent product detectable by flow cytometry.
Statistics
The statistical analysis was performed in consultation with the Center for Biostatistics, the Ohio State University. SPSS software (version 9.0, SPSS, Chicago, IL) and SAS (version 9.2, SAS Institute, Inc., Cary, NC) softwares were used for statistic analysis. For the multiple factor experiments, ANOVA and mixed linear models were used to account for correlated responses within individual patient's cells.
Results
Arsenic trioxide and ascorbic acid mediated cytotoxicity in CLL Cells In Vitro
To systematically analyze the cytotoxic effects of arsenic trioxide (ATO) and ascorbic acid in B-CLL cells, we isolated purified CD19+ B cells from B-CLL patients. The demographics of the patient samples used are shown in Table 1. Cells from 10 patients were incubated with increasing concentrations of ATO for 24 and 72 hours cell viability was analyzed by flow cytometry using propidium iodide (PI) and Annexin V conjugated FITC reagents. Populations excluding both PI and Annexin V-FITC staining were considered viable. Incubation of CLL B cells with 0.5μM, 1μM and 2μM of ATO (concentrations previously reported to be physiologically achievable 24) resulted in dose and time dependent decreased viability (16% and 49% reduction in viability with 1μM and 2μM ATO respectively at 24 hours; p ≤ 0.001). The effect of ATO was time dependent as >80% reduction in viability was observed at 72 hour cultures (Fig. 1a).
Table 1. Description of Rai stage, IgVH mutational status and Zap70 status of patient samples.
| Patient # | Rai stage | Ig VH status (%) | Zap70 (%+ve) |
|---|---|---|---|
| 1 | 1 | 0 | 11.3 |
| 2 | 2 | 0.4 | 7.5 |
| 3 | 0 | N.D | N.D |
| 4 | 2 | 0.4 | 32.9 |
| 5 | 0 | 6.3 | 55.9 |
| 6 | 2 | 0.7 | 90.7 |
| 7 | 0 | N.D | N.D |
| 8 | 0 | 0 | 79.2 |
| 9 | 0 | 0 | N.D |
| 10 | 2 | N.D | 28.5 |
| 11 | 2 | 4.8 | 14.7 |
| 12 | 1 | 0 | 32.4 |
| 13 | 0 | 7 | 21 |
| 14 | 2 | 0.4 | 0.9 |
| 15 | 1 | 2.1 | N.D |
| 16 | 2 | 0 | 26.7 |
| 17 | 1 | 2.8 | 63.2 |
| 18 | 3 | 0 | 79.2 |
Figure 1. Arsenic trioxide or ascorbic acid mediated cytotoxicity in B cells from B-CLL patients- Dose and time kinetic analysis.
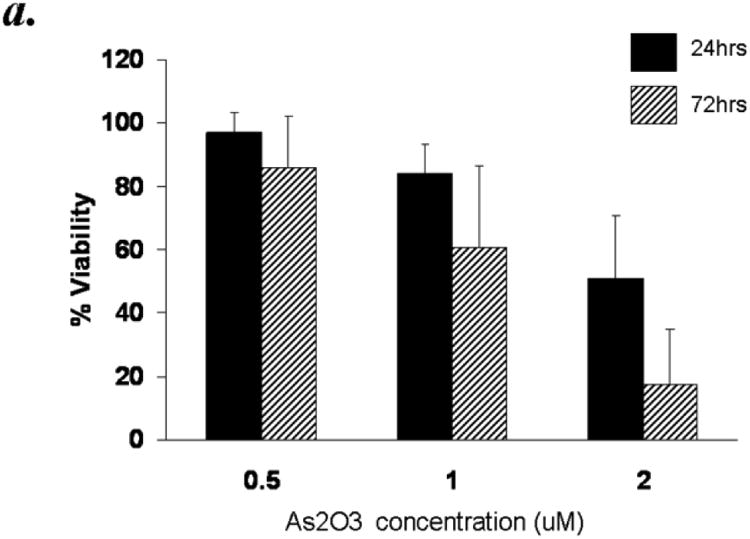
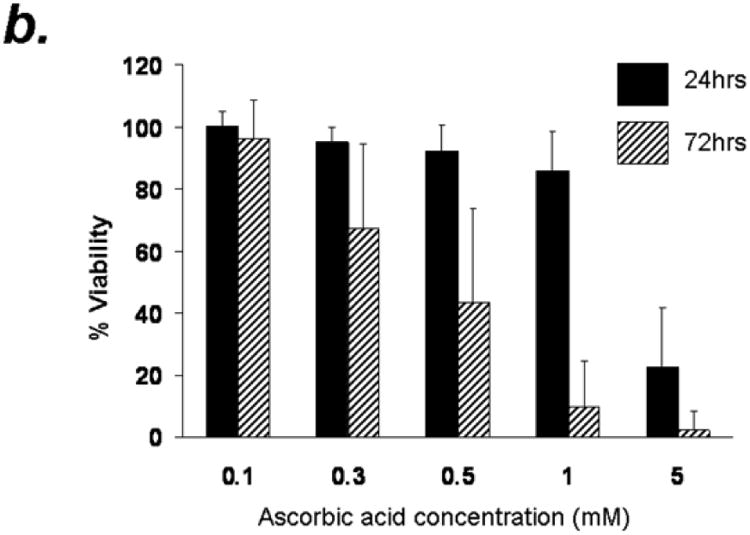
Purified B-lymphocytes (1 × 106cells/ml) from B-CLL patients were incubated in the presence of indicated concentrations of arsenic trioxide (Panel A) or ascorbic acid (Panel B) for indicated time periods. The cells were stained with Annexin-V-FITC and propidium iodide as described by us previously23. The cells were analyzed by flow cytometry and data collected under list mode. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. (n=10)
To determine the effect of ascorbic acid on B-CLL cells, purified CD19+ B cells from B-CLL patients were treated with increasing concentrations of ascorbic acid in the form of sodium ascorbic acid at final concentrations of 0.1, 0.3, 0.5, 1 and 5mM. Ascorbic acid treatment resulted in dose and time dependent decrease in viability (Fig. 1b). While 0.1, 0.3, 0.5 and 1mM ascorbic acid treatment resulted in minimal decrease in viable cells at 24 hours post treatment, 5mM induced significant reduction in viability (23±19% vs 85% viability in control groups). A more significant dose and time dependent reduction in viability was observed in these cells by 72hrs resulting in 2.4±6.3% viability. Interestingly a dramatic delayed decrease in viability was also observed with 1mM concentration of ascorbic acid (86± 12.6% at 24 hours to 10± 14.6% at 72 hours) (n=10; p ≤ 0.001) (Fig 1b).
Enhanced cytotoxicity by combined ATO and ascorbic acid treatment in primary CLL cells
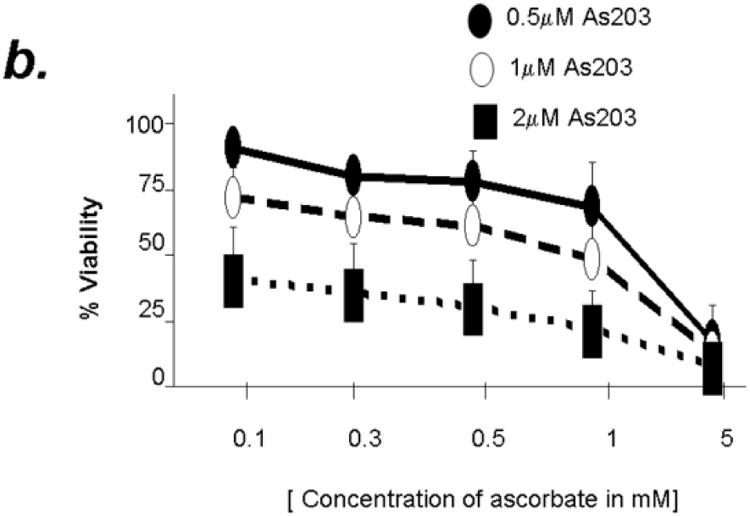
The cytotoxic effect of ATO has been shown to increase with ascorbic acid in multiple myeloma cells. In order to determine if combined treatment will result in enhanced cytotoxicity in primary CLL cells, we used combination of ATO (1uM) and ascorbic acid (1mM), at concentrations both of which were found to mediate minimal reduction in CLL B cell viability at 24 hours when tested independently (Fig 1). As shown in Fig 2a, combination of ATO and ascorbic acid resulted in enhanced cytotoxicity in a time dependent manner with viability decreasing by 40% at 24 hours over either agent alone (n=12, p=0.0009 for the interaction test of ATO and ascorbic acid). The combinatorial effect of ATO and ascorbic acid appears to be additive as evidenced by ∼20% decrease in viability with ATO or ascorbic acid independently by 24 hours and ∼44% decrease in viability in the presence of ATO and ascorbic acid together (Fig 2b). Detailed kinetics analysis revealed that the combined cytotoxic effects of ATO and ascorbic acid were dose dependent (Fig 2b). When ATO and ascorbic acid were incubated at the same ratio but at different concentrations, the viability of CLL B cells decreased with increased concentrations of ATO/ascorbic acid. Thus 96.2% ± 0.4 viability seen with 0.5μm ATO and 0.1mM ascorbic acid reduced to 44.4% ± 5.6 with 2μM ATO and 0.1mM ascorbic acid (Fig 2b).
Figure 2. Combination of Arsenic trioxide and ascorbic acid enhance cytotoxicity in B cells from B-CLL patients.
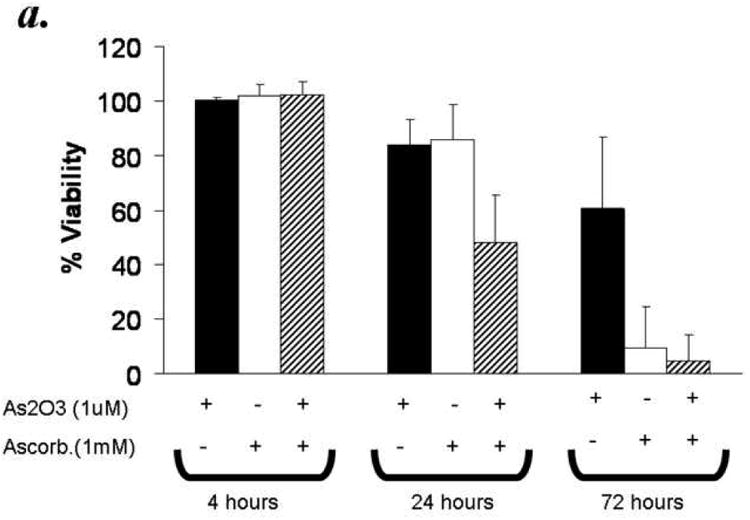
Panel a: Purified B-lymphocytes from CLL patients (1×106/ml media) were incubated with either arsenic trioxide [ATO] (1μM), ascorbic acid (1mM), or with both agents together for the indicated time periods. The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry and data collected under list mode. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. (n=12)
Panel b: Purified B-lymphocytes from CLL patients (1×106/ml media) were incubated with 0.5, 1 and 2μM arsenic trioxide [ATO] in conjunction with indicated concentrations of ascorbic acid for 24 hours. The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry and data collected under list mode. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. (n=12; p<0.001).
ATO and/or Ascorbic acid induced apoptosis in CLL Cells is dependent on activation of Caspase cascade
Apoptosis in B-CLL cells is mediated through various mechanisms involving activation of caspase dependent and/or independent pathways. In order to determine if the ATO and ascorbic acid induced cytotoxicity is medidated through caspase activation we tested the effect of pan caspase inhibitor z-VAD-fmk on ATO and/or ascorbic induced apoptosis. Concentrations of z-VAD-fmk that efficiently resuced the fludarbine induced apoptosis associated with prevention of cleavage of PAPR, a downsteam target of activated caspase 3, inhbited the ATO and/or ascorbic acid induced apoptosis (Fig 3a). Consistant with the activation of caspase cascade, ATO and ascorbic acid either alone or in combination induced PARP cleavage that is prevented by z-VAD-fmk (Fig 3b). These results indicate a critical role for caspase activaiton in ATO and ascorbic acid mediated cytotoxicity of primary CLL cells.
Figure 3. Arsenic trioxide and ascorbic acid induced cytotoxicity in CLL cells is dependent on activation of caspase.
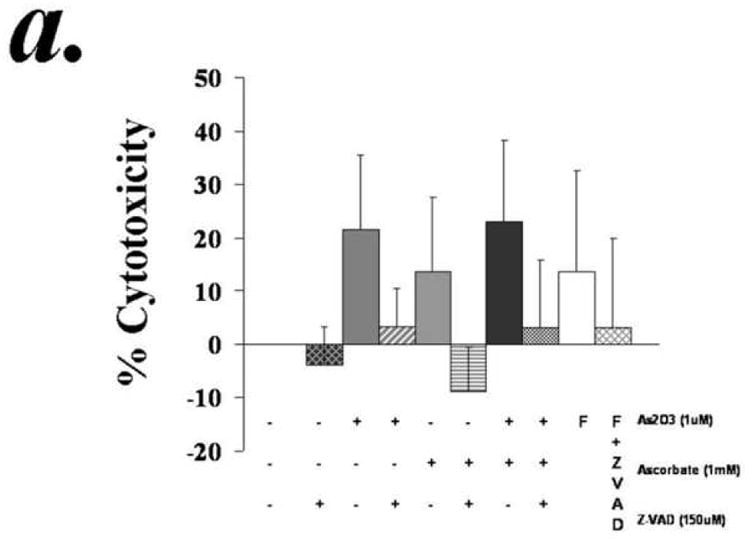

Purified B-lymphocytes from CLL patients (1×106/ml media) were incubated with 1μM arsenic and 1mM ascorbic acid either alone or in combination in the presence or absence of z-VAD-fmk (150uM) for 24 hours. The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry for apoptotic cells (panel a). The data shown represent % Annexin-V+/PI+ cells ± SD.(n=3). Panel B shows Westernblot analysis of the lysates from one of the above experiments was assessed for incleaved and cleaved PARP. Fludarabin is used as a control in these studies.
The cytotoxicity of ATO/ascorbic acid is dependent on the accumulation of Reactive Oxygen Species (ROS)
Arsenic trioxide exerts its cytotoxic effects in part by the accumulation of reactive oxygen species (ROS) 17,19,25,26; and ascorbic acid is also known to undergo autooxidation in the presence of transition metals. In both cases superoxide anion (O2-) and subsequently hydrogen peroxide (H2O2) is formed. For determining whether superoxide was formed, cells were treated with ATO/ascorbic acid for 2hours, washed in PBS and resuspended in PBS containing 10μM dihydroethidine and incubated for a further 60 minutes. The DHE+ cells were analyzed by flow cytometry. As shown in Fig. 4, the percentage of cells showing DHE fluorescence increased by 10 times when both ATO and ascorbic acid were added compared to ATO or ascorbic acid alone.
Figure 4. Accumulation of reactive oxygen species by arsenic trioxide and ascorbic acid.
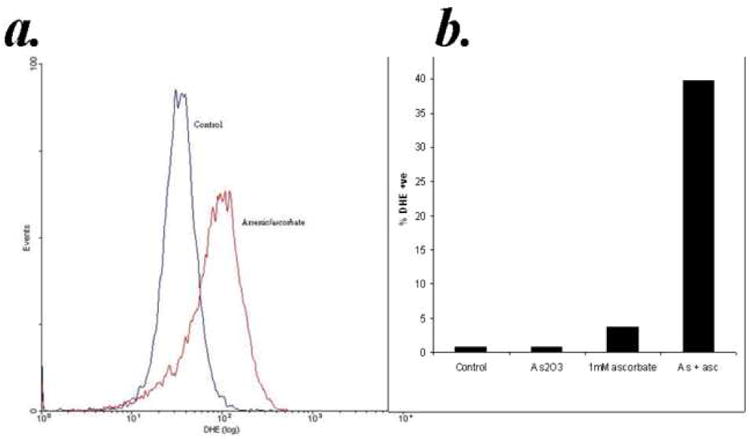
Purified B-lymphocytes from CLL patients (1×106/ml media) (1×106/ml) were treated with 1μM arsenic and 1mM ascorbic acid either alone or in combination. Subsequently DHE was added to 10μM final concentration and the cells were incubated for an additional 60 minutes. The cells were analyzed by flow cytometry and data collected under list mode. A representative histogram showing the vehicle control and arsenic trioxide [ATO]/ascorbic acid treated group is shown in the top panel. The bottom panel shows %DHE positive cells in media control, ATO (0.1uM), ascorbic acid (1mM) and ATO/ascorbic acid treated groups.
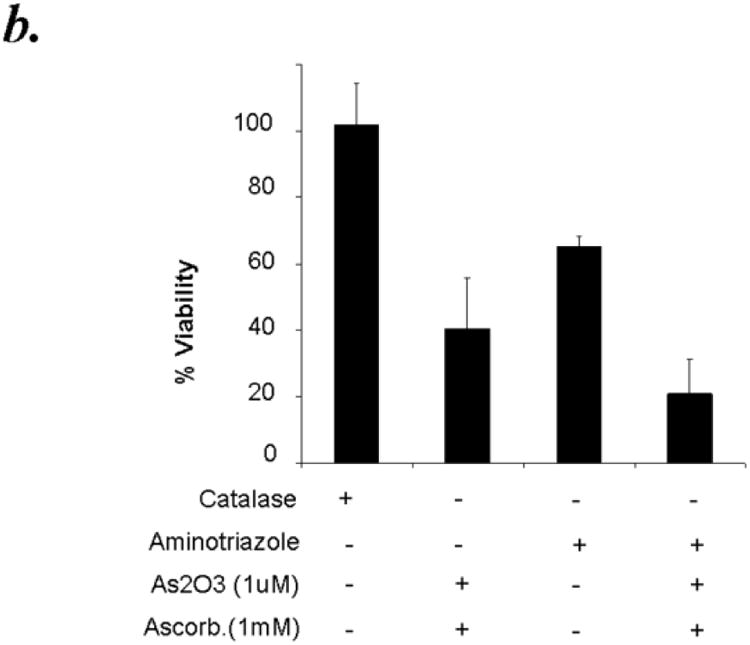
Hydrogen peroxide is the major ROS formed by ATO/ascorbic acid and it is also considerably more stable compared to the superoxide anion. Since catalase is the major enzyme involved in the detoxification of H2O2, we hypothesized that pretreatment of CLL patient derived B cells with catalase should protect them from the cytotoxic effects of ATO/ascorbic acid. Addition of 500 units of catalase per ml reaction mixture showed minimal effect on cell viability as detected by Annexin-V-/PI- populations. While ATO and ascorbic acid in combination resulted in 40.5% ± 15% viability, addition of purified catalase exhibited ∼45% reduction in the cytotoxic effect mediated by ATO/ascorbic acid resulting in 74.1% ± 13.4 in ATO/ascorbic acid + catalase treated cells p ≤ 0.001; n=6, p ≤ 0.009 for the interaction test of catalase and ATO/ascorbic acid)) [Fig. 5a]. The catalase mediated rescue in these studies is further confirmed to be dependent on the catalase enzyme activity, as heat inactivated catalase failed to exhibit any protective effect on ATO/ascorbic acid mediated cellular cytotoxicity (40.5% ± 15.3 viability in ATO/ascorbic acid treated cells compared to 26.7% ± 10.1 in ATO/ascorbic acid and heat inactivated catalase treated cells (n=6, p=0.143;), (Fig 5a). Further, inhibition of catalase using aminotriazole, a specific inhibitor of catalase, when its substrate H2O2 is present, provides further evidence of the protective effect of catalase and also shows the involvement of H2O2 as a result of ATO/ascorbic acid treatment. Thus, pre-treatment of CLL cells for 30 minutes with 100mM aminotriazole prior to adding ATO/ascorbic acid increased the cytotoxic effect of arsenic trioxide and ascorbic acid (Fig. 5b). Interestingly, aminotriazole alone also was cytotoxic towards CLL cells. (Fig. 5b).
Figure 5.
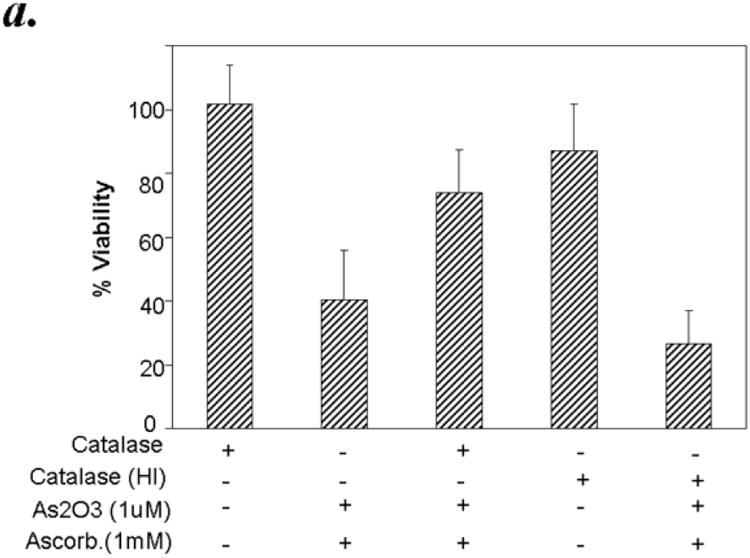
Panel a: Active but not heat inactivated catalase, protects against As2O3 and ascorbic acid mediated cytotoxicity in primary CLL B cells. Purified B-lymphocytes from CLL patients (1×106/ml media) were pretreated with 500 units of active catalase or heat inactivated catalase (HI) for 30 minutes prior to addition of arsenic trioxide [ATO] (1μM) and ascorbic acid (1mM). The cells were stained with Annexin-V-FITC and propidium iodide and the cells were analyzed by flow cytometry after 24 hours. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. (n=6; p<0.05).
Panel b: Catalase inhibitor aminotriazole enhances the cytotoxicity of As2O3 and ascorbic acid towards primary CLL B cells. Purified B-lymphocytes from CLL patients (1×106/ml media) were pretreated with 100mM aminotriazole for 30 minutes prior to addition of arsenic trioxide [ATO] (1μM) and ascorbic acid (1mM). The cells were stained with Annexin-V-FITC and propidium iodide and the cells were analyzed by flow cytometry after 24 hours. The data shown represent % Annexin-V-/PI-viable cells ± SD that are normalized to media control. (n=6; p<0.05).
Role of GSH on the cytotoxicity of arsenic/ascorbic acid mediated cytotoxicity
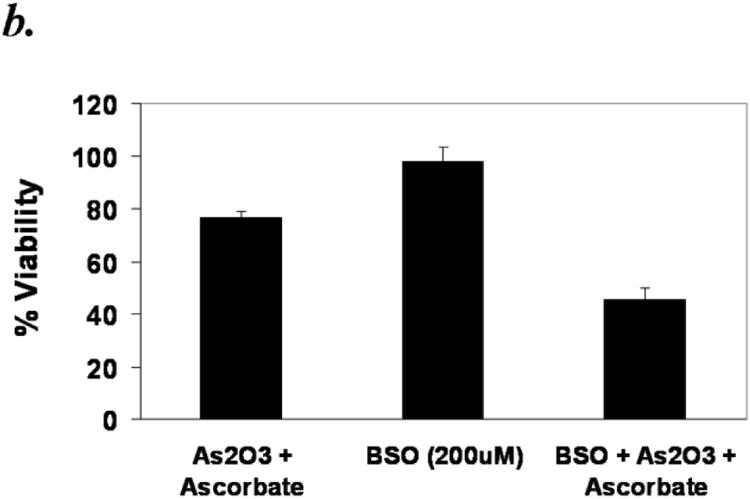
Reduced glutathione (GSH) is the major antioxidant present in cells. Its concentration can reach up to millimolar levels in healthy cells and it plays a major role in the detoxification of xenobiotics. Arsenic or ascorbic acid is known to bind thiol groups and reduce glutathione levels in the cells 27,28. We hypothesized that GSH and also N-acetyl cysteine (NAC), a known thiol antioxidant, would have a protective effect against ATO/ascorbic acid induced cytotoxicity either by scavenging ROS or by directly binding ATO. The protective effect of NAC can also be due to its ability to increase the levels of cellular thiols. Treating primary CLL cells with 1mM NAC or GSH effectively abrogated the ATO/ascorbic acid mediated cytotoxicity, by increasing the viability from 41 ± 13.7% (in ATO/ascorbic acid treated cells) to 78 ± 9.4 % with 1mM NAC and 80 ± 8.5% with 1mM GSH (Fig 6a). To determine if depletion of thiol levels would enhance the cytotoxicity of ATO/ascorbic acid, cells were grown in the presence of 200μM buthionine sulfoximine, which inhibits a key enzyme in glutathione biosynthesis and thus reducing the cellular free thiol pool. As shown in Fig 6b, depletion of GSH by buthionine sulfoximine treatment decreased the viability due to ATO/ascorbic acid by 40% (p ≤ 0.05). Buthionine sulfoximine by itself failed to show any significant toxicity compared to the ATO/ascorbic acid control.
Figure 6.
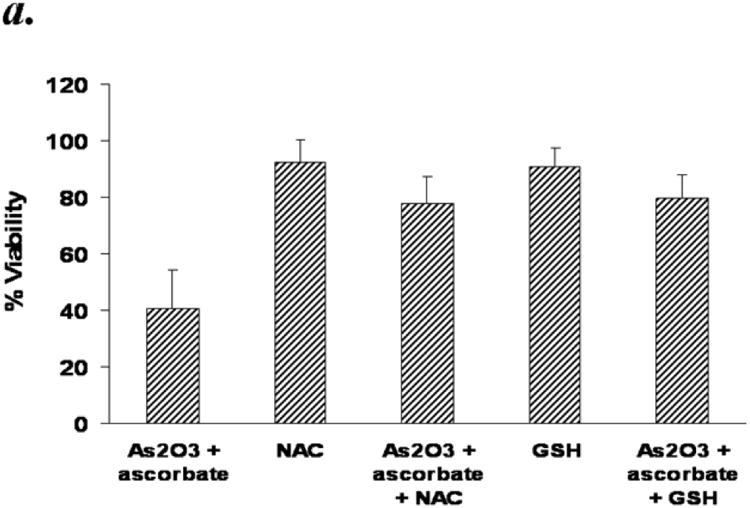
Panel a: N-acetyl cysteine (NAC) or Glutathione (GSH) mediated abrogation arsenic trioxide/ascorbic acid induced cytotoxicity in primary CLL B cells. Purified B-lymphocytes from CLL patients (1×106/ml media) were pretreated with Glutathione [GSH (1mM)] or N-acetyl cysteine (NAC) for 30 minutes prior to addition of ATO (1μM) and ascorbic acid (1mM). The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry after 24 hours. The data shown represent % Annexin-V-/PI- viable cells + SD that are normalized to media control. (n=6; p<0.05).
Panel b: Depletion of GSH by buthionine sulfoximine (BSO) enhances the As2O3 and ascorbic acid mediated cytotoxicity. Purified CLL B cells (1×106/ml) were grown in the presence of 200μM BSO for 24 hours to deplete GSH levels. The cells were analyzed by flow-cytometry 24 hours after addition of ATO/ascorbic acid as described above.
Hu1D10 induced cytotoxicity is enhanced by ATO/ascorbic acid in CLL B cells
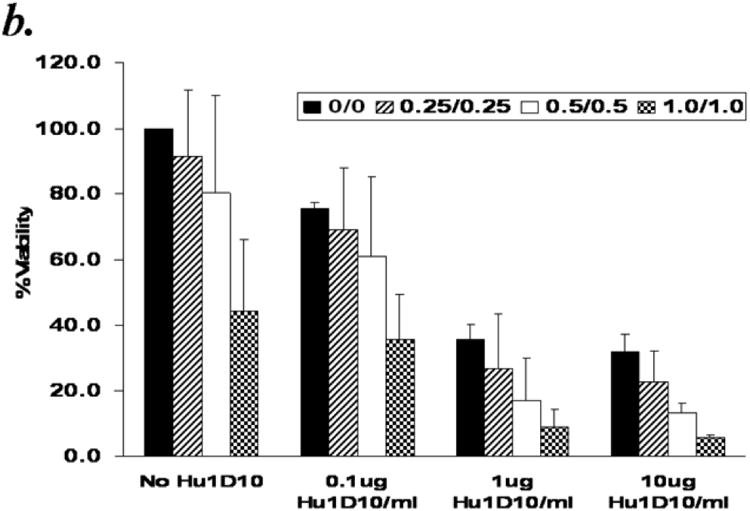
Hu1D10 (apolizumab), a humanized HLA-DR beta-chain-specific antibody directed to the 1D10 antigen, has been shown to be cytotoxic towards primary B-cell chronic lymphocytic leukemia (CLL) cells and is currently in clinical trials for this disease. We previously reported that in vitro Hu1D10 treatment of CLL cells resulted in accumulation of reactive oxygen species (ROS)23. We hypothesized that CLL cells would be susceptible to damage by ROS generating agents, and that ROS generating agents would enhance the Hu1D10 mediated cytotoxicity. To test this hypothesis we investigated the effect of ATO/ascorbic acid on Hu1D10 induced cytotoxicity in B-CLL cells. Primary CD19+ B cells isolated from CLL patients were treated with Hu1D10 ± ATO/ascorbic acid for 48 hours (Fig 7a.) While Hu1D10 or ATO/ascorbic acid resulted in comparable levels of cytotoxicity, the combination of HuID10 and ATO/ascorbic acid resulted in increased cytotoxicity, compared to individual agents [% viability in Hu1D10=61.9±19.5%; ATO/ascorbic acid= 64.8±21.5%; ATO/ascorbic acid +Hu1D10=38±25.7%). (n=1p < 0.0001) for the interaction test of Hu1D10 and ATO/ascorbic acid). This is further confirmed by detailed dose kinetic analysis of ATO/ascorbic acid dependent enhancement in Hu1D10 cytotoxicity (Fig 7b). Hu1D10 induced dose dependent cytotoxicity at 0.1, 1 and 10μg/ml concentrations. The Hu1D10 induced cytotoxicity at each of these concentrations was enhanced by increasing concentrations (0.25, 0.5 and 1mM) ATO/ascorbic acid (Fig. 7b). Thus, the viability decreased with increasing concentrations of ATO/ascorbic acid and Hu1D10; at the highest concentrations of ATO(1μm)/ascorbic acid (1mM), the viability decreased from 44.4% ± 5.6 in the absence of Hu1D10 to 5.7% ± 0.7 in the presence of 10μg/ml of Hu1D10. Similar dose dependent enhancement by ATO/ascorbic acid was also observed at 0.1 and 1μg/ml of Hu1D10 (44.4± 5.6 in the absence of Hu1D10 reduced to 35.7 ± 9.5% and 5.7±0.7% at 0.1μg/ml and 1ug./ml of Hu1D10 respectively. (Fig 7b).
Figure 7.
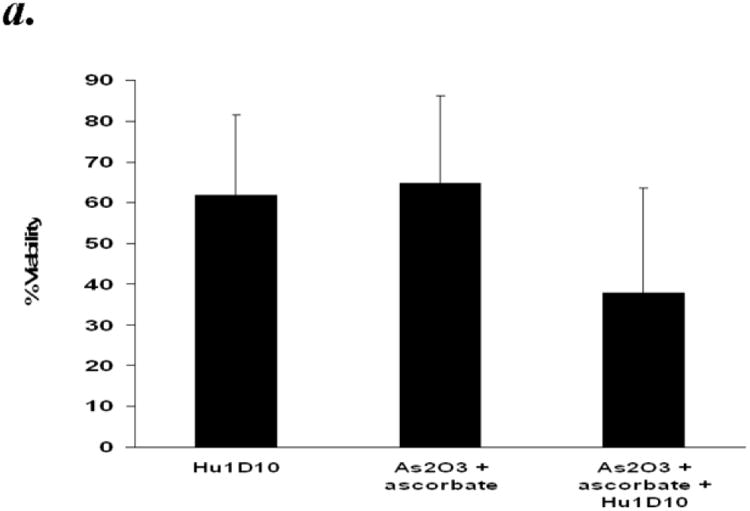
Panel a: Arsenic trioxide and ascorbic acid enhance Hu1D10 mediated cytotoxicity of primary CLL B cells. Purified B-lymphocytes from CLL patients (1×106/ml media) were treated with Hu1D10 (10ug/ml), arsenic trioxide [ATO](1μM)/ascorbic acid (1mM) or Hu1D10 and ATO/ascorbic acid. The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry as described above. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. (n=11).
Panel b: Arsenic trioxide and ascorbic acid enhance the cytotoxicity of Hu1D10 in a dose dependent manner. Purified B-lymphocytes from CLL patients (1×106/ml media) were treated with indicated concentrations of Hu1D10, arsenic trioxide [ATO] and ascorbic acid. The cells were stained with Annexin-V-FITC and propidium iodide and analyzed by flow cytometry as described above. The data shown represent % Annexin-V-/PI- viable cells ± SD that are normalized to media control. Varying arsenic trioxide/ascorbic acid and Hu1D10 concentrations show that even if Hu1D10 concentration is lowered 10 fold, the cytotoxicity in conjunction with ATO/ascorbic acid is significantly enhanced.
Discussion
In the present work we have shown that the susceptibility of CLL B-lymphocytes to ROS can be exploited with arsenic trioxide, a therapeutic agent currently approved for clinical use in acute promyelocytic leukemia. Furthermore, we have demonstrated that CLL is similar to multiple myeloma where the cytotoxic effect of arsenic trioxide is greatly enhanced by the addition of ascorbic acid. Diverse forms of ROS (O2-, OH, H2O2, 1O2, etc.) can be formed due to ATO/ascorbic acid. We have demonstrated that O2- is formed when these agents are used in combination. Most ROS have a very short biological half-life but among these H2O2 is comparatively long-lived and has the potential to do the most damage. Catalase, the major enzyme which scavenges H2O2, is a major component of the cellular antioxidant defense system. The susceptibility of CLL cells to H2O2 has been shown previously 1 and also that these cells have a compromised catalase activity 2,3. The addition of exogenous catalase helped CLL cells to survive in vitro 4. The importance of catalase is further borne out by our observation that aminotriazole, which inhibits catalase in the presence of H2O2 by itself is also cytotoxic to CLL cells. It is clear that endogenous H2O2 is formed at least when CLL cells are cultured in vitro and it contributes to and may be even accelerates their eventual mortality.
The reduced form of γ-glutamyl cysteinyl glycine (glutathione, GSH) is the major antioxidant in the cell and was able to protect against ATO/ascorbic acid cytotoxicity. The levels of GSH in CLL cells are comparable to healthy cells and sometimes even greater 2,29-31. This could be in part due to the cell's attempt to compensate for its compromised antioxidant enzyme activity. ATO or ascorbic acid is known to deplete GSH levels in the cells and thus increase its susceptibility to various toxicants. Indeed, depletion of GSH levels by BSO, which inhibits γ-glutamyl cysteinyl synthetase, a key enzyme in GSH biosynthesis, increased the cytotoxicity mediated by ATO/ascorbic acid. N-acetyl cysteine, a thiol antioxidant in wide use also effectively reduced the cytotoxicity due to ATO/ascorbic acid. Therefore ATO and ascorbic acid is able to serve a dual purpose, on one hand they lower the cellular thiol pool and thus make them more susceptible to ROS, and on the other hand they produce ROS which can exert their cytotoxicity without interference from GSH. Karasavvas and collaborators have demonstrated that a reduced form of ascorbic acid protects against ATO induced toxicity36. While the present study shows the opposite effect, it should be pointed out that we have used the oxidized form of ascorbic acid. The differences of both forms of Vitamin C toward the amplification or blockage of ATO redox cycle remains to be evaluated as this may justify the use of oxidized instead of reduced form.
CLL cells by virtue of their compromised antioxidant defense system would be more susceptible than healthy cells. However it is likely that the indiscriminate nature of ROS will make healthy cells potential target for these agents albeit to a lesser extent. Monoclonal antibody therapy is attractive because of its specificity and is increasingly used in various cancers 32. In CLL the anti CD20 antibody, rituximab, has shown remarkable success 33,34. Another monoclonal antibody, the humanized 1D10 (Hu1D10; apolizumab) is among the antibodies that are currently under evaluation in phase II clinical trials in CLL patients. The 1D10 antibody is directed against a polymorphic epitope on the beta-chain of human leucocyte antigen (HLA) class II 35. Our laboratory had previously demonstrated the efficacy of Hu1D10 against CLL 23, and there were indications that ROS might be involved in this cytotoxicity. However N-acetyl cysteine failed to abrogate the cytotoxicity of Hu1D10. We have shown that in both cell lines expressing the 1D10 antigen and also in primary CLL cells Hu1D10 by itself shows considerable cytotoxicity which is enhanced by the addition of ATO/ascorbic acid, indicating a promise for combining a targeted therapy such as Hu1D10 with a non-targeted therapeutic approach such as ATO and ascorbic acid or another ROS generating agent. The cytotoxic pathways followed by these agents are recently beginning to be unraveled and it appears that there are many diverse pathways including, caspase dependant or independent, apoptotic or necrotic pathways, which ultimately lead to the demise of the cancer cells. B-CLL cells are dependent on cell to cell communication events (follicular dendritic cells or other stromal cells) to fully “activate” their anti-apoptotic machinery. The effect of ATO/Ascorbic acid on B-CLL cells in the context of stromal cells remains to be tested. Recently, the ATO has been shown to induce apoptosis preferentially in B-CLL cells of patients with unfavorable prognostic factors including del17p13. 37 Given the defects in specific cell death pathway in CLL contributing to resistance to spontaneous and treatment induced apoptosis, combinatorial therapeutic approaches aimed at both targeted and a non-targeted cell death induction strategies are very promising.
Acknowledgments
N.M and J.C.B. contributed equally to this work as co-senior authors. This work was supported by National Cancer Institute P01 CA95426, The Leukemia and Lymphoma Society of America and The D. Warren Brown Foundation (JCB and NM).
Footnotes
Authors' Contributions: SB contributed to the design of the study and acquisition of data, and analysis and drafted the first draft of the article. XBZ and APM contributed to the acquisition of data. XM and DJ performed the statistical analysis. MV contributed to patient demographic analysis, NM and JCB contributed to the conception and design of the study, analysis and interpretation of data and revising the manuscript critically for important intellectual content and final submission of the manuscript.
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Farber CM, Liebes LF, Kanganis DN, Silber R. Human B lymphocytes show greater susceptibility to H2O2 toxicity than T lymphocytes. J Immunol. 1984;132:2543–2546. [PubMed] [Google Scholar]
- 2.Oltra AM, Carbonell F, Tormos C, Iradi A, Saez GT. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med. 2001;30:1286–1292. doi: 10.1016/s0891-5849(01)00521-4. [DOI] [PubMed] [Google Scholar]
- 3.Farber CM, Kanganis DN, Liebes LF, Silber R. Antioxidant enzymes in lymphocytes from normal subjects and patients with chronic lymphocytic leukaemia: increased glutathione peroxidase activity in CLL B lymphocytes. Br J Haematol. 1989;72:32–35. doi: 10.1111/j.1365-2141.1989.tb07647.x. [DOI] [PubMed] [Google Scholar]
- 4.Moran EC, Kamiguti AS, Cawley JC, Pettitt AR. Cytoprotective antioxidant activity of serum albumin and autocrine catalase in chronic lymphocytic leukaemia. Br J Haematol. 2002;116:316–328. [PubMed] [Google Scholar]
- 5.Ivanov VN, Hei TK. Arsenite sensitizes human melanomas to apoptosis via tumor necrosis factor alpha-mediated pathway. J Biol Chem. 2004;279:22747–22758. doi: 10.1074/jbc.M314131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu J, Okumura H, Ohtake S, Nakamura S, Nakao S. The molecular mechanism of arsenic trioxide-induced apoptosis and oncosis in leukemia/lymphoma cell lines. Acta Haematol. 2003;110:1–10. doi: 10.1159/000072407. [DOI] [PubMed] [Google Scholar]
- 7.Akay C, Gazitt Y. Arsenic trioxide selectively induces early and extensive apoptosis via the APO2/caspase-8 pathway engaging the mitochondrial pathway in myeloma cells with mutant p53. Cell Cycle. 2003;2:358–368. [PubMed] [Google Scholar]
- 8.Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q, Chen Z, Tong JH, Dai W, Chen GQ. Arsenic trioxide-induced mitotic arrest and apoptosis in acute promyelocytic leukemia cells. Leukemia. 2003;17:1333–1337. doi: 10.1038/sj.leu.2402983. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Okumura H, Ohtake S, Nakamura S, Nakao S. Arsenic trioxide induces apoptosis in leukemia/lymphoma cell lines via the CD95/CD95L system. Oncol Rep. 2003;10:705–709. [PubMed] [Google Scholar]
- 10.Lehmann S, Bengtzen S, Paul A, Christensson B, Paul C. Effects of arsenic trioxide (As2O3) on leukemic cells from patients with non-M3 acute myelogenous leukemia: studies of cytotoxicity, apoptosis and the pattern of resistance. Eur J Haematol. 2001;66:357–364. doi: 10.1034/j.1600-0609.2001.066006357.x. [DOI] [PubMed] [Google Scholar]
- 11.Lazo G, Kantarjian H, Estey E, Thomas D, O'Brien S, Cortes J. Use of arsenic trioxide (As2O3) in the treatment of patients with acute promyelocytic leukemia: the M. D. Anderson experience. Cancer. 2003;97:2218–2224. doi: 10.1002/cncr.11314. [DOI] [PubMed] [Google Scholar]
- 12.Tsimberidou AM, Estey E, Whitman GJ, Dryden MJ, Ratnam S, Pierce S, Faderl S, Giles F, Kantarjian HM, Garcia-Manero G. Extramedullary relapse in a patient with acute promyelocytic leukemia: successful treatment with arsenic trioxide, all-trans retinoic acid and gemtuzumab ozogamicin therapies. Leuk Res. 2004;28:991–994. doi: 10.1016/j.leukres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Douer D, Hu W, Giralt S, Lill M, DiPersio J. Arsenic trioxide (trisenox) therapy for acute promyelocytic leukemia in the setting of hematopoietic stem cell transplantation. Oncologist. 2003;8:132–140. doi: 10.1634/theoncologist.8-2-132. [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, Chen Z, Lallemand-Breitenbach V, de The H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer. 2002;2:705–713. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 15.Mayorga J, Richardson-Hardin C, Dicke KA. Arsenic trioxide as effective therapy for relapsed acute promyelocytic leukemia. Clin J Oncol Nurs. 2002;6:341–346. doi: 10.1188/02.CJON.341-346. [DOI] [PubMed] [Google Scholar]
- 16.Bachleitner-Hofmann T, Kees M, Gisslinger H. Arsenic trioxide: acute promyelocytic leukemia and beyond. Leuk Lymphoma. 2002;43:1535–1540. doi: 10.1080/1042819021000002857. [DOI] [PubMed] [Google Scholar]
- 17.Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- 18.Chou WC, Jie C, Kenedy AA, Jones RJ, Trush MA, Dang CV. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc Natl Acad Sci U S A. 2004;101:4578–4583. doi: 10.1073/pnas.0306687101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- 20.Hirano S, Kobayashi Y, Cui X, Kanno S, Hayakawa T, Shraim A. The accumulation and toxicity of methylated arsenicals in endothelial cells: important roles of thiol compounds. Toxicol Appl Pharmacol. 2004;198:458–467. doi: 10.1016/j.taap.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 21.Bachleitner-Hofmann T, Gisslinger B, Grumbeck E, Gisslinger H. Arsenic trioxide and ascorbic acid: synergy with potential implications for the treatment of acute myeloid leukaemia? Br J Haematol. 2001;112:783–786. doi: 10.1046/j.1365-2141.2001.02608.x. [DOI] [PubMed] [Google Scholar]
- 22.Grad JM, Bahlis NJ, Reis I, Oshiro MM, Dalton WS, Boise LH. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood. 2001;98:805–813. doi: 10.1182/blood.v98.3.805. [DOI] [PubMed] [Google Scholar]
- 23.Mone AP, Huang P, Pelicano H, Cheney CM, Green JM, Tso JY, Johnson AJ, Jefferson S, Lin TS, Byrd JC. Hu1D10 induces apoptosis concurrent with activation of the AKT survival pathway in human chronic lymphocytic leukemia cells. Blood. 2004;103:1846–1854. doi: 10.1182/blood-2003-08-2836. [DOI] [PubMed] [Google Scholar]
- 24.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 25.Nakagawa Y, Akao Y, Morikawa H, Hirata I, Katsu K, Naoe T, Ohishi N, Yagi K. Arsenic trioxide-induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sci. 2002;70:2253–2269. doi: 10.1016/s0024-3205(01)01545-4. [DOI] [PubMed] [Google Scholar]
- 26.Woo SH, Park IC, Park MJ, Lee HC, Lee SJ, Chun YJ, Lee SH, Hong SI, Rhee CH. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int J Oncol. 2002;21:57–63. [PubMed] [Google Scholar]
- 27.Davison K, Cote S, Mader S, Miller WH. Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia. 2003;17:931–940. doi: 10.1038/sj.leu.2402876. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu M, Hochadel JF, Fulmer BA, Waalkes MP. Effect of glutathione depletion and metallothionein gene expression on arsenic-induced cytotoxicity and c-myc expression in vitro. Toxicol Sci. 1998;45:204–211. doi: 10.1006/toxs.1998.2539. [DOI] [PubMed] [Google Scholar]
- 29.Silber R, Farber CM, Papadopoulos E, Nevrla D, Liebes L, Bruck M, Brown R, Canellakis ZN. Glutathione depletion in chronic lymphocytic leukemia B lymphocytes. Blood. 1992;80:2038–2043. [PubMed] [Google Scholar]
- 30.Bakan N, Taysi S, Yilmaz O, Bakan E, Kuskay S, Uzun N, Gundogdu M. Glutathione peroxidase, glutathione reductase, Cu-Zn superoxide dismutase activities, glutathione, nitric oxide, and malondialdehyde concentrations in serum of patients with chronic lymphocytic leukemia. Clin Chim Acta. 2003;338:143–149. doi: 10.1016/j.cccn.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Ferraris AM, Rolfo M, Mangerini R, Gaetani GF. Increased glutathione in chronic lymphocytic leukemia lymphocytes. Am J Hematol. 1994;47:237–238. doi: 10.1002/ajh.2830470318. [DOI] [PubMed] [Google Scholar]
- 32.Mavromatis B, Cheson BD. Monoclonal antibody therapy of chronic lymphocytic leukemia. J Clin Oncol. 2003;21:1874–1881. doi: 10.1200/JCO.2003.09.113. [DOI] [PubMed] [Google Scholar]
- 33.O'Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J, Lerner S, Keating MJ. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol. 2001;19:2165–2170. doi: 10.1200/JCO.2001.19.8.2165. [DOI] [PubMed] [Google Scholar]
- 34.Byrd JC, Murphy T, Howard RS, Lucas MS, Goodrich A, Park K, Pearson M, Waselenko JK, Ling G, Grever MR, Grillo-Lopez AJ, Rosenberg J, Kunkel L, Flinn IW. Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. J Clin Oncol. 2001;19:2153–2164. doi: 10.1200/JCO.2001.19.8.2153. [DOI] [PubMed] [Google Scholar]
- 35.Stockmeyer B, Schiller M, Repp R, Lorenz HM, Kalden JR, Gramatzki M, Valerius T. Enhanced killing of B lymphoma cells by granulocyte colony-stimulating factor-primed effector cells and Hu1D10-a humanized human leucocyte antigen DR antibody. Br J Haematol. 2002;118:959–967. doi: 10.1046/j.1365-2141.2002.03722.x. [DOI] [PubMed] [Google Scholar]
- 36.Karasavvas N, Cárcamo JM, Stratis G, Golde DW. Vitamin C protects HL60 and U266 cells from arsenic toxicity. Blood. 2005 May 15;105(10):4004–12. doi: 10.1182/blood-2003-03-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merkel O, Heyder C, Asslaber D, Hamacher F, Tinhofer I, Holler C, Stöcher M, Prokesch A, Papak C, Scheideler M, Trajanoski Z, Greil R. Arsenic trioxide induces apoptosis preferentially in B-CLL cells of patients with unfavourable prognostic factors including del17p13. J Mol Med. 2008 May;86(5):541–52. doi: 10.1007/s00109-008-0314-6. [DOI] [PubMed] [Google Scholar]