

Abstract
Background
Although pharmacological and psychological interventions are both effective for major depression, antidepressant drugs are frequently used as first‐line treatment in primary and secondary care settings. Milnacipran, a dual serotonin‐norepinephrine reuptake inhibitor (SNRI), is one of the antidepressant drugs that clinicians use for routine depression care.
Objectives
To assess the evidence for the efficacy, acceptability and tolerability of milnacipran in comparison with tricyclic antidepressants (TCAs), heterocyclics, SSRIs and other newer antidepressive agents in the acute‐phase treatment of major depression.
Search methods
The Cochrane Collaboration Depression, Anxiety & Neurosis review group Controlled Trials Register (CCDANCTR‐Studies and CCDANCTR‐References) were electronically searched in August 2008. References of relevant trials and other reviews were also checked. Trial databases of the drug‐approving agencies and ongoing clinical trial registers for all published and unpublished trials were hand‐searched in 2007. All relevant authors were contacted for supplemental data. No language restriction was applied.
Selection criteria
Randomised controlled trials comparing milnacipran with any other active antidepressive agents (including non‐conventional agents such as herbal products like hypericum) as monotherapy in the acute phase of major depression were selected.
Data collection and analysis
Two reviewers independently checked eligibility, assessed methodological quality and extracted data from the eligible trials using a standardised data extraction form. The number of participants who responded to treatment or those who achieved remission were calculated on an intention‐to‐treat basis. Random‐effects meta‐analyses were conducted, combining data from the included trials.
Main results
A total of 16 randomised controlled trials (n=2277) were included in the meta‐analysis.Despite the size of this sample, the pooled 95% confidence intervals were rather wide and there were no statistically significant differences in efficacy, acceptability and tolerability when comparing milnacipran with other antidepressive agents. However, compared with TCAs, patients taking milnacipran were associated with fewer dropouts due to adverse events (OR 0.55; 95%CI 0.35 to 0.85). There was also some weak evidence to suggest that patients taking milnacipran experienced fewer adverse events of sleepiness/ drowsiness, dry mouth or constipation compared with TCAs.
Authors' conclusions
Currently, there is inadequate evidence to conclude whether milnacipran is superior, inferior or the same as other antidepressive agents in terms of efficacy, acceptability and tolerability in the acute phase treatment of major depression. However, there is some evidence in favour of milnacipran over TCAs in terms of dropouts due to adverse events (acceptability) and the rates of experiencing adverse events (tolerability). Information about other clinically meaningful outcomes such as cost‐effectiveness and social functioning, including the ability to return to work, is lacking. Further study is needed to answer whether milnacipran would be the better choice of antidepressant for acute major depression.
Keywords: Humans; Antidepressive Agents; Antidepressive Agents/adverse effects; Antidepressive Agents/therapeutic use; Cyclopropanes; Cyclopropanes/adverse effects; Cyclopropanes/therapeutic use; Depressive Disorder, Major; Depressive Disorder, Major/drug therapy; Milnacipran; Randomized Controlled Trials as Topic; Selective Serotonin Reuptake Inhibitors; Selective Serotonin Reuptake Inhibitors/adverse effects; Selective Serotonin Reuptake Inhibitors/therapeutic use
Plain language summary
Milnacipran versus other antidepressive agents for depression
Major depression, also known as major depressive disorder or unipolar depression, is a common mental disorder characterised by a combination of symptoms that interfere with a person's ability to work, sleep, study, eat, and enjoy pleasurable activities. An episode of major depression may occur only once in a person's lifetime, but more often, it recurs throughout a person's life.
Antidepressant drugs are frequently used as first‐line treatment for major depression in primary and secondary care settings. Milnacipran, a dual serotonin‐norepinephrine reuptake inhibitor, is one of the antidepressant drugs that clinicians use for routine depression care in some countries. This systematic review investigated the efficacy, acceptability and tolerability of milnacipran compared to that of other antidepressive agents in the acute phase treatment of major depression. A total of 16 randomised controlled trials (2277 participants) were included in this review. When we brought together the results of approximately 2000 patients, we were unable to say whether milnacipran is better, worse or the same when compared to other antidepressive agents used in practice in terms of efficacy, acceptability and tolerability. However, there is some evidence that fewer people taking milnacipran stop taking the drug ('drop out') due to side effects and fewer people taking milnacipran experience side effects such as sleepiness, dry mouth or constipation than do people who take tricyclic antidepressants.
Background
Description of the condition
Major depression, also known as major depressive disorder or unipolar depression, is a common metal disorder characterised by a combination of persistent symptoms (including depressed mood, loss of interest, loss of appetite, insomnia, fatigue, poor concentration, extreme guilt and suicide ideation) that interfere with a person's ability to work, study and enjoy pleasurable activities (APA 1994). Compared with other medical diagnoses, depression is very common. Lifetime prevalence estimates for major depression in the community range from 15 to 17% (APA 1994), 12‐month prevalence from 6 to 7% (Kessler 2003). The prevalence of major depression in the medical outpatient is 5 to 13% (Coyne 1994). Major depression is the third leading cause of burden among all diseases after lower respiratory infections and diarrhoeal diseases, accounting for 4.3% of human suffering in terms of illhealth; moreover, it is expected to show a rising trend during the coming 20 years (WHO 2004). This condition is associated with a marked personal, social and economic morbidity, loss of functioning and productivity, and creates significant demands on service providers in terms of workload (NICE 2007). In the USA, the economic burden of depression has been estimated at just over $83 billion in 2000, of which $26 billion were direct treatment costs, $5 billion were suicide‐related costs, and $52 billion were workplace costs (Greenberg 2003). It is also suspected that these figures are still underestimates of the true economic burden of the disease, which may in addition involve burden on family members and caregivers, the cost of lost productivity while at work, and cost associated with those who remain untreated (Greenberg 2005).
Description of the intervention
Although pharmacological and psychological interventions are both effective for major depression, in primary and secondary care settings antidepressant (AD) drugs remain the mainstay of treatment (APA 2000; Ellis 2004; NICE 2007) (see below for other references to the relevant evidence). Amongst ADs many different agents are available, including tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), serotonin‐noradrenaline reuptake inhibitors (SNRIs: venlafaxine, duloxetine, milnacipran), and other newer agents (mirtazapine, reboxetine, bupropion). In many western countries, during the last 20 years, ADs consumption has dramatically risen, mainly because of the increasing consumption of SSRIs and newer ADs, which have progressively become the most commonly prescribed ADs (Ciuna 2004; Guaiana 2005). SSRIs are generally better tolerated than TCAs (Barbui 2007), and there is evidence of similar efficacy (Anderson 2000a; Geddes 2000; Williams 2000). However, head‐to‐head comparisons provide contrasting findings. Amitriptyline, for example, may have the edge over SSRIs in terms of efficacy (Guaiana 2007), and individual SSRIs and SNRIs may differ in terms of efficacy and tolerability (Cipriani 2005; Smith 2002).
How the intervention might work
Milnacipran has been available as an antidepressant since 1997 in many countries including France and Japan (34 countries and regions as of 2006). Milnacipran appears to act exclusively at presynaptic sites to inhibit noradrenaline (norepinephrine) and serotonin uptake (Moret 1985), but unlike TCAs, has no significant effect on any neurotransmitter receptor (Briley 1996). Thus, compared with TCAs, milnacipran has shown a lower incidence of anticholinergic‐like side effects, less sedation due to histamine H1‐receptor binding and lower incidence of postural hypotension due to alpha‐1 adrenoceptor antagonism (Spencer 1998). The pharmacokinetic profile of the drug indicates that milnacipran has a high bioavailability, low plasma protein binding (13%) and is mostly eliminated in urine: 50% as the unchanged drug, 30% as a glucuronide (main metabolite) and the remaining 20% by oxidative transformation (Puozzo 1996). Milnacipran does not affect the activities of CYP‐2D6, 2C19, 1A2 and 3A4 isoforms, and its pharmacokinetics are not modified in poor metabolizers of CYP‐2D6 and CYP‐2C9 (Puozzo 1996; Sawada 2001; Puozzo 2005). Furthermore, studies in patients with liver dysfunction suggest that dose adjustment is not necessary or to be minor when milnacipran is administered to these patients (Puozzo 1996).
Why it is important to do this review
Given that the most recent available evidence refers to the SSRIs as an homogeneous group (Arroll 2005; Geddes 2000; Hansen 2005), it is still unclear how each newer antidepressive agent compares with other antidepressants in terms of effects and adverse events. A group of researchers therefore agreed to join forces under the rubric of the Multiple meta‐Analyses of New Generation Antidepressants (MANGA) Study to systematically review all available evidence for each specific newer antidepressant.
In terms of milnacipran, only limited evidence has been established regarding the efficacy, acceptability and tolerability in comparison with other antidepressive agents, to date. Some RCTs have reported that milnacipran has an antidepressant efficacy similar to other antidepressants, such as imipramine (Tignol 1998; Van Amerongen 2002; Lopez‐Ibor 2004), clomipramine (Leinonen 1997; Steen 1997), fluoxetine (Guelfi 1998), fluvoxamine (Clerc 2001) and paroxetine (Sechter 2004). In a systematic review (Puech 1997), milnacipran has shown superior antidepressant efficacy in comparison with SSRIs and the tolerability has been comparable to that of the SSRIs. However, this review was sponsored by a pharmaceutical company marketing milnacipran and was published more than a decade ago. Therefore, there is a good reason to conduct an up‐to‐date comprehensive systematic quantitative review using currently best‐available evidence on comparative efficacy and adverse effects of milnacipran against other antidepressive agents.
The primary objective of this systematic review is to assess the evidence for the efficacy, acceptability and tolerability of milnacipran in comparison with TCAs, heterocyclics, SSRIs and other newer antidepressive agents, including non‐conventional agents, in the acute‐phase treatment of major depression.
Objectives
To determine the efficacy of milnacipran in comparison with other antidepressive agents in alleviating the acute symptoms of depression.
To review acceptability of treatment with milnacipran in comparison with other antidepressive agents.
To investigate the adverse effects of milnacipran in comparison with other antidepressive agents.
Methods
Criteria for considering studies for this review
Types of studies
Only randomised controlled trials were included. Quasi‐randomised trials, such as those allocating by using alternate days of the week, were excluded. For trials which have a crossover design only results from the first randomisation period were considered.
Types of participants
Patients aged 18 or older, of both sexes with a primary diagnosis of major depression. Studies adopting any standardised criteria to define patients suffering from unipolar major depression were included. Studies from the 1990s onwards were likely to have used DSM‐IV (APA 1994) or ICD‐10 (WHO 1992) criteria. Ealier studies may have used ICD‐9 (WHO 1978), DSM‐III (APA 1980) / DSM‐ III‐R (APA 1987) or other diagnostic systems. ICD‐9 is not operationalised criteria, because it has only disease names and no diagnostic criteria, so studies using ICD‐9 were excluded. On the other hand, studies using Feighner Criteria or Research Diagnostic Criteria were included. We included the following depression subtypes: chronic, with catatonic features, with melancholic features, with atypical features, with postpartum onset, and with seasonal pattern. Studies in which less than 20% of the participants may be suffering from bipolar depression were included. A concurrent secondary diagnosis of another psychiatric disorder was not considered as exclusion criteria.
Major depression with psychotic features were excluded. A concurrent primary diagnosis of Axis I or II disorders was an exclusion criteria. Antidepressant trials in depressive patients with a serious concomitant medical illness were also excluded.
Types of interventions
Experimental intervention
Milnacipran (as monotherapy). No restrictions on dose, frequency, intensity and duration were applied.
Comparator intervention
Other active agents in the treatment of acute major depression, including:
TCAs (imipramine, clomipramine, amitriptyline)
Heterocyclic antidepressants (mianserin)
SSRIs ( fluvoxamine, fluoxetine, paroxetine, sertraline, citalopram, escitalopram)
Newer antidepressants (SNRIs such as venlafaxine and duloxetine, MAOIs or newer agents such as mirtazapine, bupropion, reboxetine
Non‐conventional antidepressive agents such as herbal products like hypericum (Linde 2008) and fish oil (Appleton 2006).
No restrictions on dose, frequency, intensity and duration were applied.
Other type of psychopharmacological agent such as anxiolytics, antic‐convulsants, anti‐psychotics or mood‐stabilizers were excluded. Trials in which milnacipran was used as an augmentation strategy were excluded. Placebo‐controlled trials were also excluded.
Types of outcome measures
Efficacy, acceptability and tolerability during and at the end of acute‐phase treatment trials, defined as 6 to 12 weeks, was our outcome of interest. However, when data from trials longer than 12 weeks were available, we also included them.
Primary outcomes
Number of patients who responded to treatment, showing a reduction of at least 50% on the Hamilton Rating Scale of Depression (HAM‐D) (Hamilton 1960) or Montgomery‐Asberg Depression Scale (MADRS) (Montgomery 1979), or "much or very much improved" (score 1 or 2) on CGI‐Improvement (Guy 1970) out of the total number of randomised patients. HAM‐D has been the golden standard measure of depression severity for the clinical trials of antidepressants (Williams 2001).Therefore, we used the HAM‐D for judging response whenever possible, even when we needed to impute SDs or response rates according to the procedures described in the Methods below.
When studies reported response rates at various time points of the trial, we subdivided the treatment indices as follows, according to criteria decided a priori:
Early phase treatment: between 1 and 4 weeks (preference was given to the time point closest to 2 weeks);.
Acute phase treatment : between 6 and 12 weeks (preference was given to the study endpoint) ;
Follow‐up phase treatment: between 4 and 6 months (preference was given to the time point closest to 24 weeks)..
Secondary outcomes
Number of patients who achieved remission. The cutoff point for remission was set a priori (1) at 7 or less for the 17‐item HAM‐D and at 8 or less for all the other longer versions of HAM‐D, or (2) at 12 or less on the MADRS (Zimmerman 2004), or (3) "not ill or borderline mentally ill" (score 1 or 2) on CGI‐Severity (Guy 1970). We used the HAM‐D for judging remission whenever possible.
Severity of depression at the end of the trial as measured on continuous scale such as HAM‐D, MADRS, etc. We applied 'loose' ITT analyses, whereby all the patients with at least one post‐baseline measurement were represented by their last observations carried forward.
Social adjustment, social functioning including the Global Assessment of Function (GAF) (Luborsky 1962) scores.
Health‐related quality of life : We will limit ourselves to SF‐12/SF‐36 (Ware 1993), HoNOS (Wing 1994) and WHO‐QOL (WHOQOL Group 1998).
Costs to health care services
-
Acceptability measures
Number of patients who dropped out during the trial as a proportion of the total number of randomised patients ‐ due to any cause
Number of patients who dropped out during the trial as a proportion of the total number of randomised patients ‐ due to inefficacy
Number of patients who dropped out during the trial as a proportion of the total number of randomised patients ‐ due to adverse events
-
Tolerability measures:
Total number of patients experiencing at least some adverse events
Total number of patients experiencing the following specific adverse events was sought for:
sleepiness/drowsiness
insomnia
dry mouth
constipation
urination problems
hypotension
agitation/anxiety
suicide wishes/gestures/attempts
completed suicide
vomiting/nausea
diarrhoea
In order not to miss any relatively rare or unexpected yet important adverse events, in the data extraction phase, we collected all adverse event data reported in the literature and discussed ways to summarize them post hoc.
Search methods for identification of studies
Electronic searches
We searched using the Cochrane Collaboration Depression, Anxiety & Neurosis Controlled Trials Registers (CCDANCTR‐Studies and CCDAN‐References) (searched in December 2006; updated in August 2008). This register of randomised controlled trials is compiled by methodical searches of CENTRAL, AMED, CINAHL, EMBASE, LiLACS, MEDLINE, PSYCINFO, PSYNDEX supplemented with hand searching of both journals and conference proceedings.
CCDANCTR‐Studies were searched using the following search strategy: Diagnosis = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder" or "Affective Symptoms" and Intervention = Milnacipran
CCDANCTR‐References were searched using the following search strategy: Keyword = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder" or "Affective Symptoms" and Free‐Text = Milnacipran
Trial databases of the following drug‐approving agencies ‐ (the Food and Drug Administration (FDA) in the USA, the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK, the European Medicines Agency (EMEA) in the EU, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, the Therapeutic Goods Administration (TGA) in Australia and ongoing trial registers (clinicaltrials.gov in the USA, ISRCTN and National Research Register in the UK, Nederlands Trial Register in the Netherlands, EUDRACT in the EU, UMIN‐CTR in Japan and the Australian Clinical Trials Registry in Australia) were hand‐searched for published, unpublished and ongoing controlled trials
Searching other resources
Hand‐searching
Appropriate journals and conference proceedings relating to milnacipran treatment for depression have been hand‐searched and incorporated into the CCDANCTR databases up until August 2008.
Personal communications
Pharmaceutical companies and experts in this field were asked if they knew of any study which meets the inclusion criteria of this review (contacted in May 2007).
Reference lists
Reference lists of the included studies, previous systematic reviews and major textbooks of affective disorder written in English were checked for published reports and citations of unpublished research. The reference of all included studies were checked via Science Citation Index for articles which had cited the included study.
Data collection and analysis
Selection of studies
Studies relating to milnacipran generated by the electronic search of the CCDANCTR‐Studies were scanned by one review authors (HMG).Full texts were retrieved of all those studies which met the following rough inclusion criteria:
Randomized trial
Comparing milnacipran against any other antidepressive agents
Patients with depression, regardless of the diagnostic criteria used.
Studies relating to milnacipran generated by the search strategies of the CCDANCTR‐References and the other complementary searches were checked by the CCDAN Trial Search Coordinator (HMG), who is an author of this review, and another independent review author (AN and NW) to see if they met the inclusion criteria, firstly based on the title and abstracts. All the studies rated as possible candidates by either of the two reviewers (AN and NW) were added to the preliminary list and their full texts were retrieved. All the full text articles in this preliminary list were then assessed by two review authors (AN and NW) independently to see if they met strict inclusion criteria. If the raters disagreed the final rating was made by consensus with the involvement (if necessary) of another member of the review group. Non‐congruence in selection of trials were reported as percentage disagreement. Considerable care was taken to exclude duplicate publications.
Data extraction and management
One review author (AN) first extracted data concerning participant characteristics (age, sex, depression diagnosis, comorbidity, depression severity, antidepressant treatment history for the index episode, study setting), intervention details (intended dosage range, mean daily dosage actually prescribed, co‐intervention if any, milnacipran as investigational drug or as comparator drug, sponsorship) and outcome measures of interest from the included studies. We planned at protocol stage to compare results with those in relevant completed reviews of individual antidepressants in the Cochrane Library and feed back any discrepancies to their authors: in the event, there were insufficient existing reviews to make this possible.
Assessment of risk of bias in included studies
We used the Cochrane risk‐of‐bias tool as recommended in RevMan 5.0.0 (Higgins 2008a; Higgins 2008b). This instrument consists of six items. Two of the items assess the strength of the randomisation process in preventing selection bias in the assignment of participants to interventions: adequacy of sequence generation and allocation concealment. The third item (blinding) assesses the influence of performance bias on the study results. The fourth item assesses the likelihood of incomplete outcome data, which raise the possibility of bias in effect estimates. The fifth item assesses selective reporting, the tendency to preferentially report statistically significant outcomes. It requires a comparison of published data with trial protocols, when such are available. The final item refers to other sources of bias that are relevant in certain circumstances, for example, in relation to trial design (methodological issues such as those related to crossover designs and early trial termination) or setting.
Two independent review authors (AN and NW) assessed risk of bias in each trial independently, in accordance with the Cochrane Handbook (Higgins 2008a). Where inadequate details of allocation concealment and other characteristics of trials were provided, the authors were contacted in order to obtain further information. If the raters disagreed the final rating was made by consensus with the involvement (if necessary) of another member of the review group.
Measures of treatment effect
Data were checked and entered into RevMan 5 software by two review authors (AN and NW) (double data entry). For dichotomous, or event‐like data, odds ratios (OR) were calculated with 95% confidence intervals. For continuous data, weighted mean differences (WMD) or standardized mean differences (SMD) (where different measurement scales are used) were calculated with 95% confidence intervals.
Unit of analysis issues
We planned at protocol stage to compare results from the initial randomisation phase of a crossover trial or a trial involving three (or more)‐armed trial with a placebo arm. However, none of the included studies required implementation of these plans.
Dealing with missing data
Responders and remitters to treatment were calculated on an intention‐to‐treat (ITT) basis: drop‐outs were always included in this analysis. Where participants had withdrawn from the trial before the endpoint, it was assumed they would have experienced the negative outcome by the end of the trial (e.g. failure to respond to treatment). When there were missing data and the method of "last observation carried forward" (LOCF) were been used to do an ITT analysis, then the LOCF data were used, with due consideration of the potential bias and uncertainty introduced. When dichotomous or continuous outcomes were not reported, trial authors were asked to supply these data.
When only the SE or t statistics or p values were reported, SDs were calculated according to Altman (Altman 1996). In the absence of supplemental data from the authors, the SDs of the HAM‐D (or any other depression scale) and response and remission rates were calculated according to validated methods (Furukawa 2005; Furukawa 2006). We examined the validity of these imputation in the sensitivity analyses.
Assessment of heterogeneity
We planned at protocol stage to present the skewed data and non‐quantitative data descriptively, however, no such relevant data were identified from the included studies. Should they be identified in future updates, any outcome whose minimum score is zero will be considered skewed when the mean is smaller than twice the SD.
Heterogeneity between studies was investigated by the I‐squared statistic (I‐squared equal to or more than 50% was considered indicative of heterogeneity) and the p value from the chi‐squared test (Higgins 2003), and by visual inspection of the forest plots.
Assessment of reporting biases
Where a sufficient number of trials were available, a funnel plot analysis was performed to check for existence of small study effects including publication bias.
Data synthesis
A random effects model was used to pool the results of single studies, because this model is more conservative than fixed effects model and incorporates both within‐study and between‐study variance. Further, a random effects model OR was used for the primary analysis rather than a random effect risk ratio (RR) because it has been shown that the highest generalisability in our empirical examination of summary effect measures for meta‐analyses (Furukawa 2002a). The robustness of this summary measure was routinely examined by checking the fixed effect model OR and the random effects model RR. Fixed effect analyses were done routinely for the continuous outcomes as well, to investigate the effect of the choice of method on the estimates. Material differences between the models were reported.
Subgroup analysis and investigation of heterogeneity
Subgroup analyses should be performed and interpreted with caution because multiple analyses will lead to false positive conclusions (Oxman 1992). However, we performed the following subgroup analyses, where possible, for the following reasons, which were stated a priori in our protocol.
Milnacipran dosing (fixed low dosage, fixed standard dosage, fixed high dosage; flexible low dosage, flexible standard dosage, flexible high dosage), because there was evidence to suspect that low dosage antidepressant might be associated with better outcomes both in terms of effectiveness and side effects than standard or high dosage antidepressants (Bollini 1999; Furukawa 2002b) and also because fixed versus flexible dosing schedule might affect estimates of treatment effectiveness (Khan 2003). In the case of milnacipran, based on previous reports (Lecrubier 1996; Lopez‐Ibor 1996; Okamura 2006), low dosage refers to <100, standard dosage to >=100 but <150, and high dosage to >=150 mg/day.
Comparator dosing (low effective range, medium to high effective range), as it is easy to imagine that there were greater chances of completing the study on the experimental drug than on the comparator drug that is increased to the maximum dosage.
Depression severity (severe major depression, moderate/mild major depression).
Treatment settings due to difference in severity of illness (psychiatric inpatients, psychiatric outpatients, primary care).
Elderly patients (>=65 years of age), separately from other adult patients
Sensitivity analysis
The following sensitivity analyses were planned a priori. By limiting the studies to be included to those with higher quality, we examined if the results changed, and checked for the robustness of the observed findings.
Excluding trials with unclear concealment of random allocation and/or unclear double blinding.
Excluding trials whose drop out rate is greater than 20%. Performing the worst case scenario ITT (all the patients in the experimental group experience the negative outcome and all those allocated to the comparison group experience the positive outcome) and the best case scenario ITT (all the patients in the experimental group experience the positive outcome and all those allocated to the comparison group experience the negative outcome).
Excluding trials for which the response rates had to be calculated based on the imputation method (Furukawa 2005) and those for which the SD had to be borrowed from other trials (Furukawa 2006).
Examination of "wish bias" by comparing milnacipran as investigational drug vs milnacipran as comparator, as there was evidence to suspect that a new antidepressant might perform worse when used as a comparator than when used as an experimental agent (Barbui 2004).
Excluding studies funded by the pharmaceutical company marketing milnacipran. This sensitivity analysis was particularly important in view of the recent repeated findings that funding strongly affects outcomes of research studies (Als‐Nielsen 2003; Bhandari 2004; Lexchin 2003; Montgomery 2004; Perlis 2005; Procyshyn 2004) and because industry sponsorship and authorship of clinical trials are increasing over the past 20 years (Buchkowsky 2004).
Our routine application of random effects models as well as our secondary outcomes of remission rates and continuous severity measures may be considered additional forms of sensitivity analyses. At protocol stage we planned (in the event of any of the subgroup or sensitivity analyses turning out to be significant) to run meta‐regression for exploratory analyses of their additive or multiplicative influences. However, it was impossible to run any analyses due to non‐significant results.
Results
Description of studies
See:Characteristics of included studies; Characteristics of excluded studies.
Results of the search
Twenty‐nine studies (38 references) were initially identified through an electronic search of the CCDAN register in May 2007 (see above). Seven additional studies were identified through hand search including contact with the manufacturing company of milnacipran (Pierre Fabre). Searches of the CCDAN register were rerun in August 2008 and a further two studies (two references) were identified. After looking over titles and abstracts, 25 studies were considered potentially relevant for further inspection. No ongoing studies were identified; one study currently awaits assessment and its data may appear in an update of this review (Yoshimura 2007).
Included studies
It was possible to include16 randomised controlled trials of milnacipran comparing other antidepressants in the meta‐analysis. In total, the studies included 2277 participants. The data reporting of most studies was incomplete even after supplementing the data provided by the two authors (Lee 2002b; Shinkai 2004). Therefore, with three exceptions (Tignol 1998; Sechter 2000; Shinkai 2004), the numbers of patients with response and remission were imputed. Except for Shinkai 2004, all the studies were sponsored by a pharmaceutical company.
Design
The median of number of participants per study was 120 (range: 41‐302), and the total participants of the entire study revealed to be 2277. The mean length of the trial was 7 weeks (SD 5.5). Most of the trials were conducted throughout the acute treatment phase (6 to12 weeks). However, six trials were limited to the early treatment phase (4 weeks: Annseau 1989a; Annseau 1989c; Annseau 1991c; Endo 1995; Shinkai 2004; Yamashita 1995). One trial had a longer length that ran up to 26 weeks (Leinonen 1997).
Milnacipran versus TCAs
Three studies were 4‐week trials (Annseau 1989a; Annseau 1989c; Yamashita 1995), two were 6‐week trials (Van Amerongen 2002; Lopez‐Ibor 2004), one was a 8‐week trial (Tignol 1998), and the remaining was a 26‐week trial (Leinonen 1997).
Milnacipran versus heterocyclics
A single study was a 4‐week trial (Endo 1995).
Milnacipran versus SSRIs
Two studies were 4‐week trials (Annseau 1991c;Shinkai 2004), four were 6‐week trials (Annseau 1994; Clerc 2001; Lee 2002b;Sechter 2000), one was a 8‐week trial (Yang 2003), and the remaining was a 12‐week trial (Guelfi 1998a).
Setting
Four studies enrolled out‐patients (Annseau 1994; Sechter 2000; Lee 2002b; Yang 2003), five both in‐ and out‐patients (Endo 1995; Yamashita 1995; Leinonen 1997; Tignol 1998; Clerc 2001), while the remaining studies were conducted in in‐patient facilities.
Milnacipran versus TCAs
Four studies were recruited in in‐patient settings (Annseau 1989a; Annseau 1989c; Van Amerongen 2002; Lopez‐Ibor 2004) and three were recruited in both in‐ and out‐patient settings (Leinonen 1997; Tignol 1998; Yamashita 1995).
Milnacipran versus heterocyclics
A single study was recruited in a both in‐ and out‐patient setting (Endo 1995).
Milnacipran versus SSRIs
Three studies were recruited in in‐patient settings (Annseau 1991c; Guelfi 1998a; Shinkai 2004), four were recruited in out‐patient settings (Annseau 1994; Lee 2002b; Sechter 2000; Yang 2003), and the remaining was recruited in a both in‐ and out‐patient setting (Clerc 2001).
Participants
Diagnosis
The majority of studies enrolled participants with pure unipolar major depression, whilst five studies enrolled participants with major depression that included bipolar depression (less than 20% of the participants) (Annseau 1994; Yamashita 1995; Leinonen 1997; Tignol 1998; Lopez‐Ibor 2004).
Milnacipran versus TCAs
Four studies enrolled patients with unipolar depression (Annseau 1989a; Annseau 1989c; Yamashita 1995; Van Amerongen 2002) while three studies enrolled patients with unipolar or bipolar depression (Leinonen 1997; Tignol 1998; Lopez‐Ibor 2004).
Milnacipran versus heterocyclics
Only one study enrolled patients with unipolar depression (Endo 1995).
Milnacipran versus SSRIs
Seven studies enrolled patients with unipolar depression (Annseau 1991c; Guelfi 1998a; Sechter 2000; Clerc 2001; Lee 2002b; Yang 2003; Shinkai 2004) while the remaining study enrolled patients with unipolar or bipolar depression (Annseau 1994).
Age
All participants were aged 18 or above and included some elderly participants (65 years or older). One study by Tignol 1998 was limited only to elderly participants and another study by Yang 2003 did not report age of the participants.
Interventions
Comparator intervention
There were seven studies comparing milnacipran with TCAs, one study with heterocyclics, and eight studies with SSRIs. We were not able to identify any study that compared milnacipran with newer antidepressants such as SNRIs, MAOIs or non‐conventional antidepressive agents. No study included a placebo arm.
Milnacipran versus TCAs
Four studies compared milnacipran with imipramine (Yamashita 1995; Tignol 1998; Van Amerongen 2002; Lopez‐Ibor 2004), two with amitriptyline (Annseau 1989a; Annseau 1989c), and the remaining one with clomipramine (Leinonen 1997). One study (Annseau 1989a) presented a comparison between three arms: milnacipran 50mg/day, milnacipran 100mg and amitriptyline 150mg/day.
Milnacipran versus heterocyclics
Only one study compared milnacipran with mianserin (Endo 1995).
Milnacipran versus SSRIs
Three studies compared milnacipran with fluoxetine (Annseau 1994; Guelfi 1998a; Lee 2002b), two with fluvoxamine (Annseau 1991c; Clerc 2001), two with paroxetine (Sechter 2000; Shinkai 2004), and the remaining one with sertraline (Yang 2003). One study (Guelfi 1998a) presented a comparison between three arms: milnacipran 100mg/day, milnacipran 200mg and fluoxetine 20mg/day, and other study (Annseau 1991c) presented a comparison between three arms: milnacipran 150‐300mg/day, milnacipran 200mg and fluvoxamine 200mg/day.
Dosage of the study drugs
In 8 out of the 16 studies, the dosage of milnacipran were within the standard therapeutic range (100‐150 mg/day), three within the higher dosage range (>150mg/day) (Annseau 1989cAnnseau 1991c; Leinonen 1997), three within the lower dosage range (<100mg/day) (Endo 1995; Yamashita 1995; Shinkai 2004), and the two had combined dosage range due to three arms. Of the combined dosage studies, one study (Annseau 1989a) had one arm within the standard therapeutic range and other in the lower dosage range, and another study (Guelfi 1998a) had one arm within the standard therapeutic range and other in the higher dosage range. On the other hand, the dosage of the comparator drug were within the standard therapeutic range for all the studies, except Clerc 2001 that had higher dosage range and Yamashita 1995 that had lower dosage range.
The use of a fixed‐ or a flexible‐dose regimen was consistent among comparisons within the same study in all of included trials. Six studies (Endo 1995; Yamashita 1995; Leinonen 1997; Tignol 1998; Yang 2003; Shinkai 2004) involved a flexible‐dose scheduling design, whereas the remainder of included trials involved a fixed‐dose scheduling design.
Outcomes
Outcome concerning efficacy during acute phase treatment (6‐12 weeks) were obtained from ten studies (n=1565). Of the ten studies, six studies were assessed at 6 weeks, 3 at 8 weeks and one at 12 weeks. Efficacy data during early phase were obtained from 13 studies (n=1934), and in 11 studies were assessed at two weeks. All studies used intention to treat analyses based on the last observation carried forward method for the efficacy outcome. Either 17, 21 or 24‐item HAM‐D were used to evaluate the efficacy data for all the studies included in the review.The data reporting of most studies were incomplete even after supplementing the provided data from contacted two authors (Lee 2002b; Shinkai 2004). Therefore, with three exceptions (Tignol 1998; Sechter 2000; Shinkai 2004), the number of patients with response and remission were imputed. In terms of acceptability, except for Leinonen 1997, all studies reported the total number of participants who dropped out prematurely during the trial. Yang 2003 did not provide the specific number of participants who dropped out during the study due to inefficacy or side effects. Annseau 1989a also did not provide the number of participants who dropped out during the study due to side effects. Outcome data concerning tolerability were extractable for the majority of studies but were not available for four (Annseau 1994; Leinonen 1997; Shinkai 2004; Yang 2003).
No data were obtained for social adjustment, social functioning, health‐related quality of life or costs to health care services from the included studies.
Excluded studies
Of the 25 studies considered for inclusion, 3 studies were excluded because they were additional publications of trials already included (Onodera 1992; Baek 2002b; Lee 2004). Two studies did not use other antidepressant as a comparator drug (Macher 1989; Kanemoto 2004). One study was not randomised (Wyeth 2006). Another study did not use relevant operational diagnostic criteria (Baek 2002a). One study looked at response to drugs by gender (Naito 2007). Finally, one study did not include acute phase treatment (Dardennes 1998). No study was excluded due to having more than 20% of the participants with bipolar depression as defined in our exclusion criteria. One study remains awaiting assessment (Yoshimura 2007).
Risk of bias in included studies
See Figure 1 and Figure 2 for a graphical summary of methodological quality for the 16 included studies, based on the six risk of bias domains.
1.
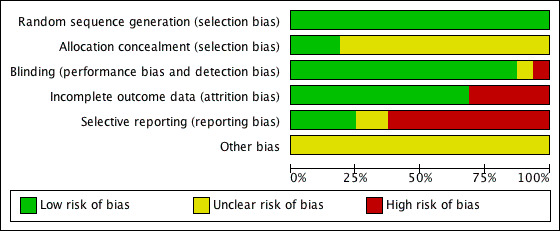
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
2.
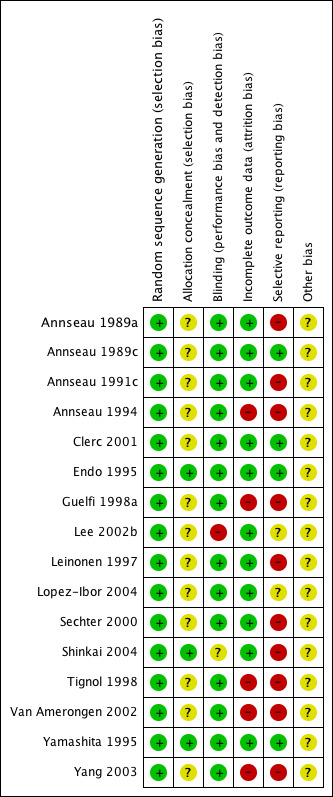
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
All trials were described as randomised. Using the Cochrane criteria which rate the adequacy of the random allocation concealment, most of the trials were rated as "unclear" or moderate risk of bias except Endo 1995, Shinkai 2004 and Yamashita 1995, in which risk of bias was rated as low.
Blinding
The outcome assessment was blind to treatment allocation in most of the studies except Shinkai 2004, in which the adequacy of the blinding was rated as "unclear" or moderate risk of bias, and Lee 2002b where the design was 'open label'.
Incomplete outcome data
Five studies were incomplete in outcome reporting (Annseau 1994;Guelfi 1998a;Tignol 1998; Van Amerongen 2002;Yang 2003).
Selective reporting
The study protocol was not available for all studies. Two studies lacked reporting of adverse events (Yang 2003; Shinkai 2004), one did not report the number of participants experiencing at least some side effects (Annseau 1994) and one (Tignol 1998) failed to report the MADRS scores indicated in the methods section of the published trial report. One study did not report the number of participants who dropped out from the trial due to any reason (Leinonen 1997) and other did not report the number of participants who dropped out from the trial due to side effects (Annseau 1989a).
Standard deviations were not reported In five studies (Annseau 1991c; Guelfi 1998a; Tignol 1998;Sechter 2000; Van Amerongen 2002). Two studies were rated as "unclear" due to insufficient information (Lee 2002b; Lopez‐Ibor 2004).
Other potential sources of bias
Except for Shinkai 2004, all the studies were sponsored by a pharmaceutical company marketing milnacipran.
Effects of interventions
The results are reported comparison by comparison (TCAs, Heterocyclics, SSRIs and newer antidepressants) and the forest plots are organised according to the relevance of outcomes, as reported in the review protocol. Some significant differences in efficacy, acceptability and tolerability were found and details are listed below.
1. Milnacipran versus TCAs
Efficacy outcomes were obtained from 7 studies (n=820) ([dichotomous outcomes] acute phase: 4 studies, n=537, early phase: 6 studies, n=802; [continuous outcomes] acute phase: 7 studies, n=820, early phase: 6 studies, n=765). Acceptability outcomes were obtained from 7 studies (n=902) (due to any reason: 6 studies (n=795), due to inefficacy: 7 studies (n=902), due to side effects: 6 studies (n=756)). Tolerability outcomes were obtained from 6 studies (n=795).
A. Milnacipran versus Imipramine
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was no evidence that milnacipran was more efficacious than imipramine (OR1.05, 95%CI: 0.71 to 1.54) (see Analysis 1.1, Figure 3).
1.1. Analysis.
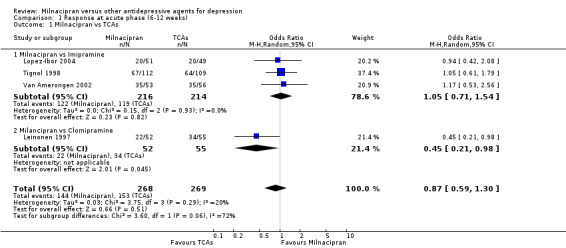
Comparison 1 Response at acute phase (6‐12 weeks), Outcome 1 Milnacipran vs TCAs.
3.
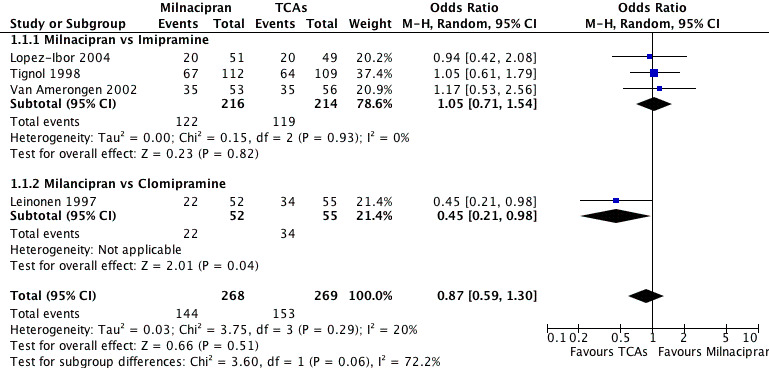
Forest plot of comparison: 1 Response at acute phase (6‐12 weeks), outcome: 1.1 Milnacipran vs TCAs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to imipramine (OR1.11, 95%CI: 0.73 to 1.69) (see Analysis 2.1, Figure 4).
2.1. Analysis.
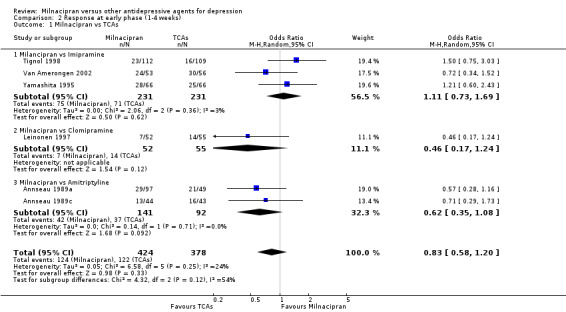
Comparison 2 Response at early phase (1‐4 weeks), Outcome 1 Milnacipran vs TCAs.
4.
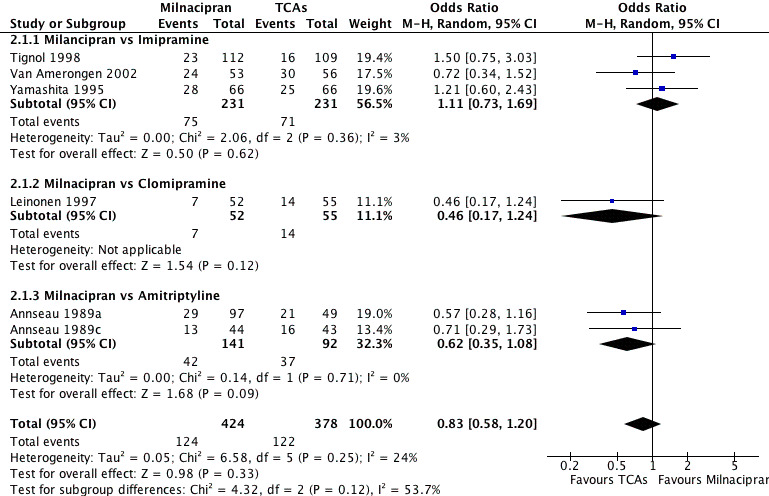
Forest plot of comparison: 2 Response at early phase (1‐4 weeks), outcome: 2.1 Milnacipran vs TCAs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to imipramine (see Analysis 4.1).
4.1. Analysis.
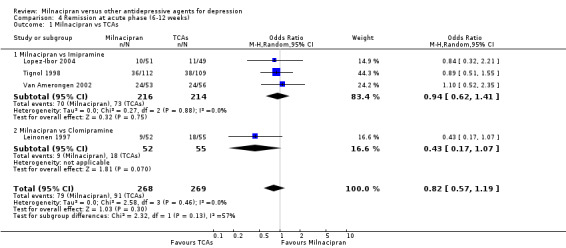
Comparison 4 Remission at acute phase (6‐12 weeks), Outcome 1 Milnacipran vs TCAs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to imipramine (see Analysis 5.1).
5.1. Analysis.
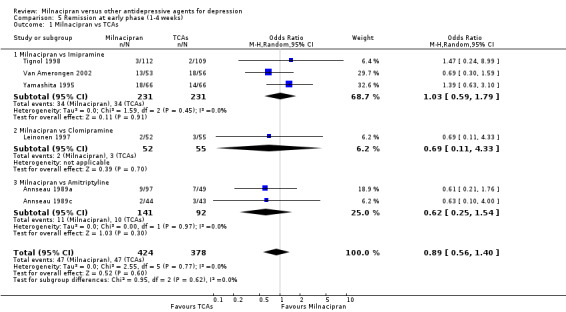
Comparison 5 Remission at early phase (1‐4 weeks), Outcome 1 Milnacipran vs TCAs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to imipramine (see Analysis 7.1).
7.1. Analysis.
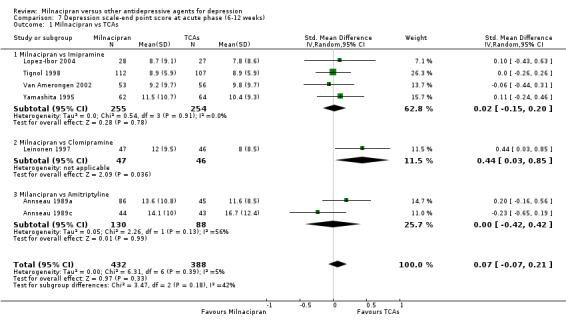
Comparison 7 Depression scale‐end point score at acute phase (6‐12 weeks), Outcome 1 Milnacipran vs TCAs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to imipramine (see Analysis 8.1).
8.1. Analysis.
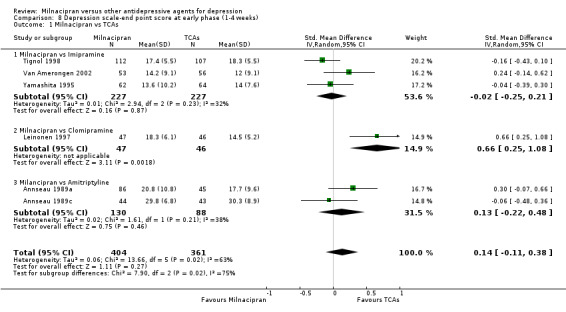
Comparison 8 Depression scale‐end point score at early phase (1‐4 weeks), Outcome 1 Milnacipran vs TCAs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to any cause compared to imipramine (see Analysis 9.1).
9.1. Analysis.
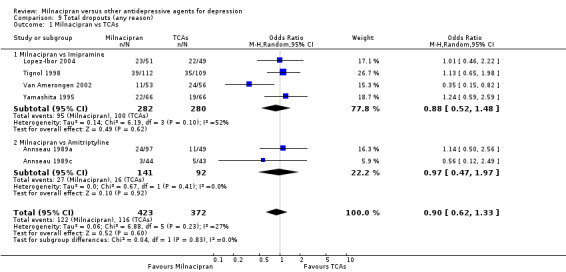
Comparison 9 Total dropouts (any reason), Outcome 1 Milnacipran vs TCAs.
b) Due to inefficacy
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to inefficacy compared to imipramine (see Analysis 10.1).
10.1. Analysis.
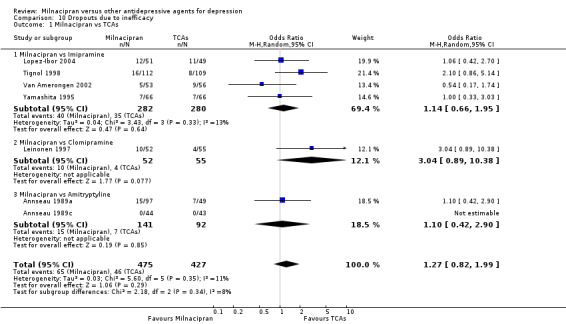
Comparison 10 Dropouts due to inefficacy, Outcome 1 Milnacipran vs TCAs.
c) Due to adverse events
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to adverse events compared to imipramine (see Analysis 11.1).
11.1. Analysis.
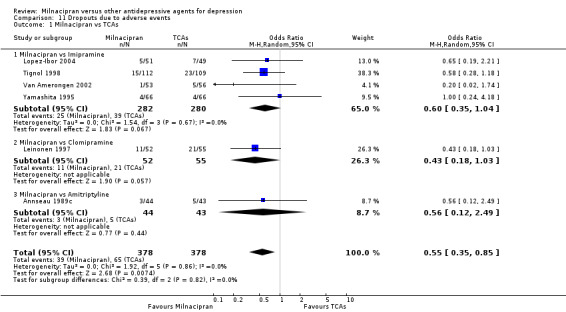
Comparison 11 Dropouts due to adverse events, Outcome 1 Milnacipran vs TCAs.
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was evidence that milnacipran was associated with a lower rate of patients experiencing adverse events than imipramine (OR 0.43, 95%CI 0.28 to 0.66) (see Analysis 12.1, Figure 5).
12.1. Analysis.
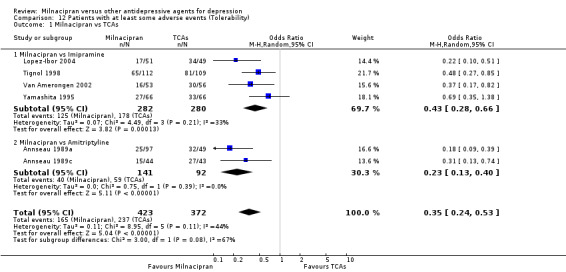
Comparison 12 Patients with at least some adverse events (Tolerability), Outcome 1 Milnacipran vs TCAs.
5.
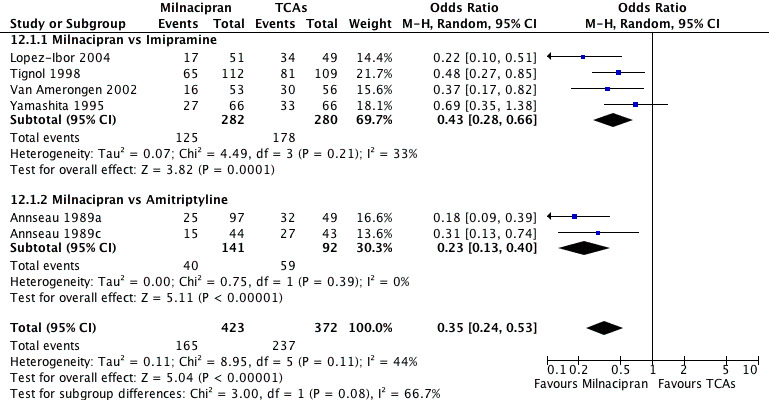
Forest plot of comparison: 12 Patients with at least some adverse events (Tolerability), outcome: 12.1 Milnacipran vs TCAs.
b) Total number of patients experiencing a specific adverse event
1. sleepiness/drowsiness
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing sleepiness/drowsiness than imipramine (see Analysis 13.1).
13.1. Analysis.
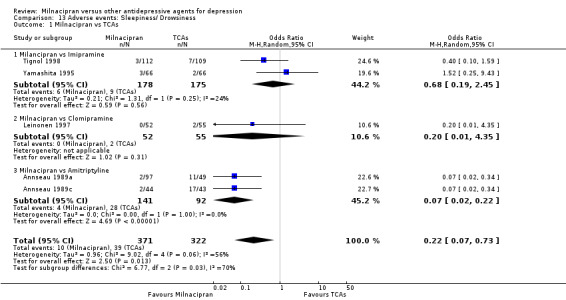
Comparison 13 Adverse events: Sleepiness/ Drowsiness, Outcome 1 Milnacipran vs TCAs.
2. insomnia
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than imipramine (see Analysis 14.1).
14.1. Analysis.
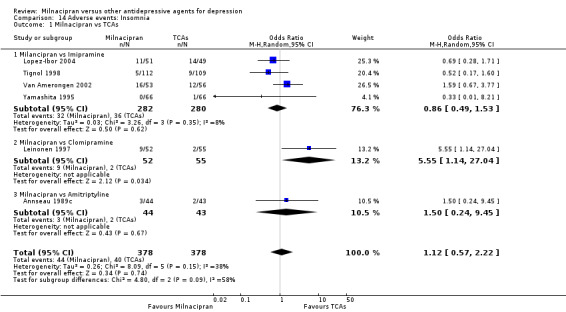
Comparison 14 Adverse events: Insomnia, Outcome 1 Milnacipran vs TCAs.
3. dry mouth
There was evidence that milnacipran was associated with a lower rate of participants experiencing dry mouth than imipramine (OR 0.57, 95%CI 0.37 to 0.86) (see Analysis 15.1).
15.1. Analysis.
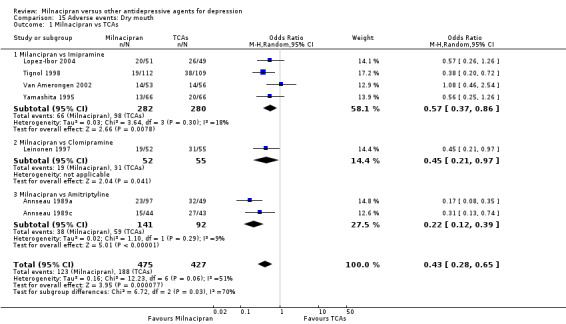
Comparison 15 Adverse events: Dry mouth, Outcome 1 Milnacipran vs TCAs.
4. constipation
There was evidence that milnacipran was associated with a lower rate of participants experiencing constipation than imipramine (OR 0.64, 95%CI 0.41 to 0.98) (see Analysis 16.1).
16.1. Analysis.
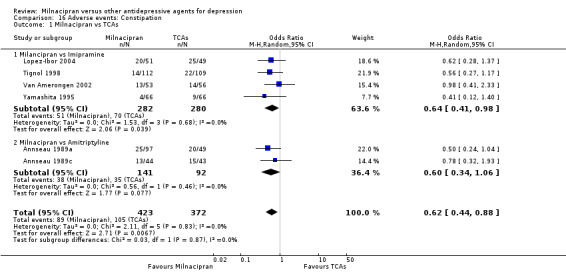
Comparison 16 Adverse events: Constipation, Outcome 1 Milnacipran vs TCAs.
5. urination problems
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing urination problems than imipramine (see Analysis 17.1).
17.1. Analysis.
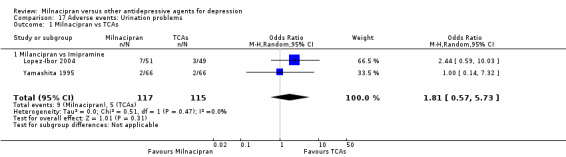
Comparison 17 Adverse events: Urination problems, Outcome 1 Milnacipran vs TCAs.
6. hypotension
There was no evidence that milnacipran was associated with a lower rate of participants experiencing hypotension than imipramine (see Analysis 18.1).
18.1. Analysis.
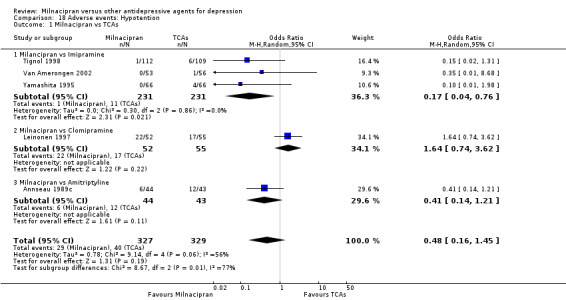
Comparison 18 Adverse events: Hypotention, Outcome 1 Milnacipran vs TCAs.
7. agitation/ anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/ anxiety than imipramine (see Analysis 19.1).
19.1. Analysis.
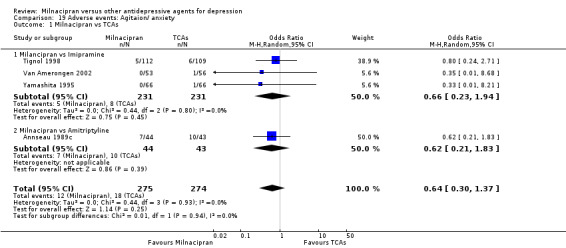
Comparison 19 Adverse events: Agitaion/ anxiety, Outcome 1 Milnacipran vs TCAs.
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/ nausea
There was evidence that milnacipran was associated with a higher rate of participants experiencing vomiting/ nausea than imipramine (OR 2.31, 95%CI 1.13 to 4.72) (see Analysis 22.1).
22.1. Analysis.
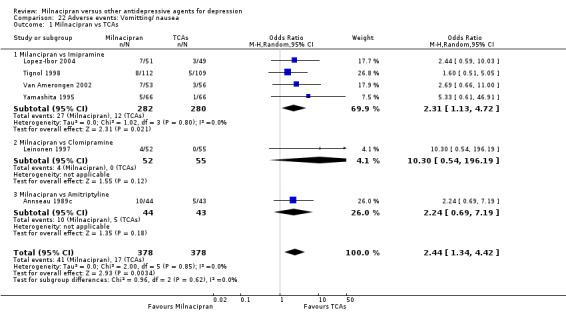
Comparison 22 Adverse events: Vomitting/ nausea, Outcome 1 Milnacipran vs TCAs.
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than imipramine (see Analysis 23.1).
23.1. Analysis.
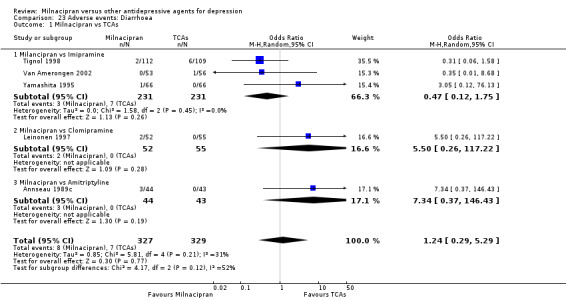
Comparison 23 Adverse events: Diarrhoea, Outcome 1 Milnacipran vs TCAs.
B. Milnacipran versus Clomipramine
Only Leinonen 1997 provided the data.
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was evidence that clomipramine was more efficacious than milnacipran (OR0.45, 95%CI: 0.21 to 0.98) (see Analysis 1.1, Figure 3).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to clomipramine (OR0.46, 95%CI: 0.17 to 1.24) (see Analysis 2.1, Figure 4).
c) Follow‐up phase treatment (16 to 24 weeks)
No substantial effect was found with milnacipran compared to clomipramine (OR0.72, 95%CI: 0.33 to 1.55) (see Analysis 3.1, Figure 6).
3.1. Analysis.
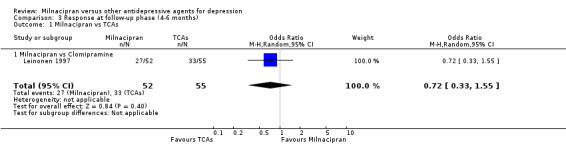
Comparison 3 Response at follow‐up phase (4‐6 months), Outcome 1 Milnacipran vs TCAs.
6.
Forest plot of comparison: 3 Response at follow‐up phase (4‐6 months), outcome: 3.1 Milnacipran vs TCAs.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to clomipramine (see Analysis 4.1).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to clomipramine (see Analysis 5.1).
c) Follow‐up phase treatment (16 to 24 weeks)
No substantial effect was found with milnacipran compared to clomipramine (see Analysis 6.1).
6.1. Analysis.
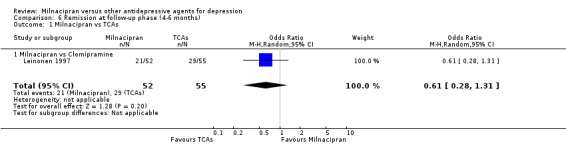
Comparison 6 Remission at follow‐up phase (4‐6 months), Outcome 1 Milnacipran vs TCAs.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
There was evidence that clomipramine was more efficacious than milnacipran (SMD 0.44, 95%C:I 0.03 to 0.85) (see Analysis 7.1).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to clomipramine (see Analysis 8.1).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to any cause compared to clomipramine (see Analysis 9.1).
b) Due to inefficacy
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to inefficacy compared to clomipramine (see Analysis 10.1).
c) Due to adverse events
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to adverse events compared to clomipramine (see Analysis 11.1).
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
No data available.
b) Total number of patients experiencing a specific adverse event
1. sleepiness/drowsiness
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing sleepiness/drowsiness than clomipramine (see Analysis 13.1).
2. insomnia
There was evidence that milnacipran was associated with a higher rate of participants experiencing insomnia than clomipramine (OR 5.55, 95%CI 1.14 to 27.04) (see Analysis 14.1).
3. dry mouth
There was evidence that milnacipran was associated with a lower rate of participants experiencing dry mouth than clomipramine (OR 0.45, 95%CI 0.21 to 0.97) (see Analysis 15.1).
4. constipation
No data available.
5. urination problems
No data available.
6. hypotension
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing hypotension than clomipramine (see Analysis 18.1).
7. agitation/anxiety
No data available.
8. suicide wishes / gestures/ attempts
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing suicide wishes/ gestures/ attempts than clomipramine (see Analysis 20.1).
20.1. Analysis.
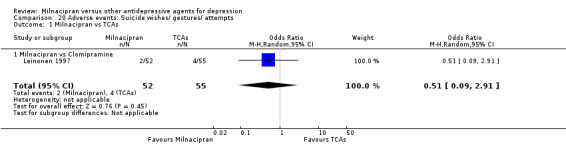
Comparison 20 Adverse events: Suicide wishes/ gestures/ attempts, Outcome 1 Milnacipran vs TCAs.
9. completed suicide
No data available.
10. vomiting/ nausea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing vomiting/ nausea than clomipramine (see Analysis 22.1).
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than clomipramine (see Analysis 23.1).
C. Milnacipran versus Amitriptyline
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
No data available.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to amitriptyline (OR 0.62, 95%CI: 0.35 to 1.08) (see Analysis 2.1, Figure 4).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No data available.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to amitriptyline (see Analysis 5.1).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to amitriptyline (see Analysis 7.1).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to amitriptyline (see Analysis 8.1).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with smaller or larger rate of drop out rate due to any cause compared to amitriptyline (see Analysis 9.1).
b) Due to inefficacy
There was no evidence that milnacipran was associated with smaller or larger rate of drop out rate due to inefficacy compared to amitriptyline (see Analysis 10.1).
c) Due to adverse events
There was no evidence that milnacipran was associated with smaller or larger rate of drop out rate due to adverse events compared to amitriptyline (see Analysis 11.1).
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was evidence that milnacipran was associated with smaller rate of patients experiencing adverse events than amitriptyline (OR 0.23, 95%CI 0.13 to 0.40) (see Analysis 12.1, Figure 5).
b) Total number of patients experiencing a specific adverse event
1. sleepiness/ drowsiness
There was evidence that milnacipran was associated with a lower rate of participants experiencing sleepiness/drowsiness than amitriptyline (OR 0.07, 95%CI 0.02 to 0.22) (see Analysis 13.1).
2. insomnia
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than amitriptyline (see Analysis 14.1).
3. dry mouth
There was evidence that milnacipran was associated with a lower rate of participants experiencing dry mouth than amitriptyline (OR 0.22, 95%CI 0.12 to 0.39) (see Analysis 15.1).
4. constipation
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than amitriptyline (see Analysis 16.1).
5. urination problems
No data available.
6. hypotension
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing hypotension than amitriptyline (see Analysis 18.1).
7. agitation/ anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/anxiety than amitriptyline (see Analysis 19.1).
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/ nausea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing vomiting/ nausea than amitriptyline (see Analysis 22.1).
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than amitriptyline (see Analysis 23.1).
2. Milnacipran versus Heterocyclics
Only Endo 1995 (n=179) that compared milnacipran with mianserin provided efficacy, acceptability and tolerability outcomes.
A. Milnacipran versus Mianserin
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
No data available.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to mianserin (OR1.10, 95%CI: 0.54 to 2.23) (see Analysis 2.3, Figure 7).
2.3. Analysis.
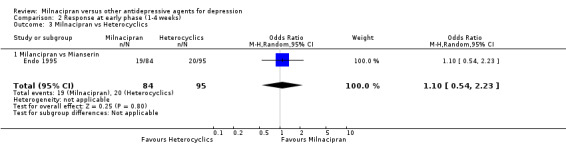
Comparison 2 Response at early phase (1‐4 weeks), Outcome 3 Milnacipran vs Heterocyclics.
7.
Forest plot of comparison: 2 Response at early phase (1‐4 weeks), outcome: 2.3 Milnacipran vs Hererocyclics.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No data available.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to mianserin (see Analysis 5.3).
5.3. Analysis.
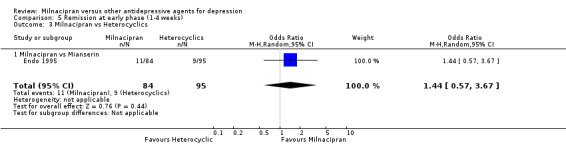
Comparison 5 Remission at early phase (1‐4 weeks), Outcome 3 Milnacipran vs Heterocyclics.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to mianserin (see Analysis 7.3).
7.3. Analysis.
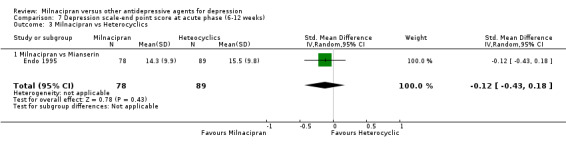
Comparison 7 Depression scale‐end point score at acute phase (6‐12 weeks), Outcome 3 Milnacipran vs Heterocyclics.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to mianserin (see Analysis 8.3).
8.3. Analysis.
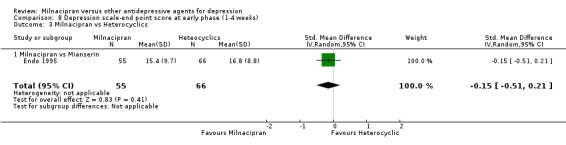
Comparison 8 Depression scale‐end point score at early phase (1‐4 weeks), Outcome 3 Milnacipran vs Heterocyclics.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with higher or lower rate of drop out rate due to any cause compared to mianserin (see Analysis 9.3).
9.3. Analysis.
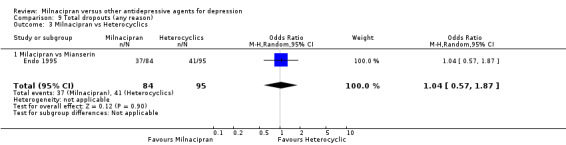
Comparison 9 Total dropouts (any reason), Outcome 3 Milnacipran vs Heterocyclics.
b) Due to inefficacy
There was no evidence that milnacipran was associated with higher or lower rate of drop out rate due to inefficacy compared to mianserin (see Analysis 10.3).
10.3. Analysis.
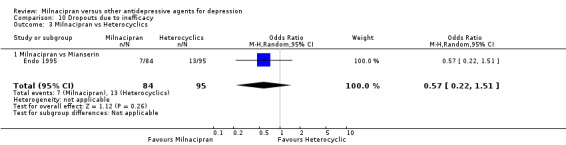
Comparison 10 Dropouts due to inefficacy, Outcome 3 Milnacipran vs Heterocyclics.
c) Due to adverse events
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to adverse events compared to mianserin (see Analysis 11.3).
11.3. Analysis.
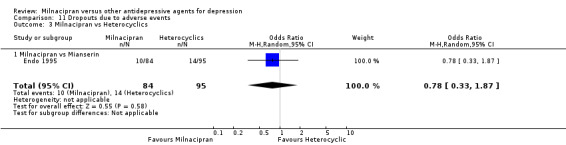
Comparison 11 Dropouts due to adverse events, Outcome 3 Milnacipran vs Heterocyclics.
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was no evidence that milnacipran was associated with higher or lower rate of patients experiencing adverse events than mianserin (see Analysis 12.3).
12.3. Analysis.
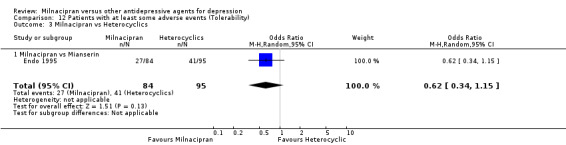
Comparison 12 Patients with at least some adverse events (Tolerability), Outcome 3 Milnacipran vs Heterocyclics.
b) Total number of patients experiencing a specific adverse event
1. sleepiness/ drowsiness
There was evidence that milnacipran was associated with a lower rate of participants experiencing sleepiness/drowsiness than mianserin (OR 0.21, 95%CI 0.08 to 0.58) (see Analysis 13.3).
13.3. Analysis.
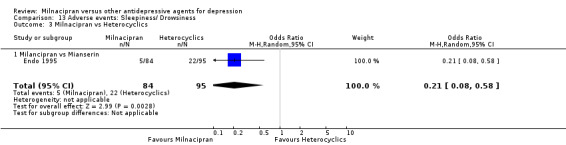
Comparison 13 Adverse events: Sleepiness/ Drowsiness, Outcome 3 Milnacipran vs Heterocyclics.
2. insomnia
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than mianserin (see Analysis 14.3).
14.3. Analysis.
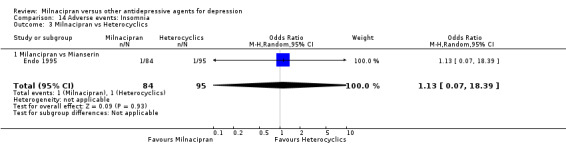
Comparison 14 Adverse events: Insomnia, Outcome 3 Milnacipran vs Heterocyclics.
3. dry mouth
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing dry mouth than mianserin (see Analysis 15.3).
15.3. Analysis.
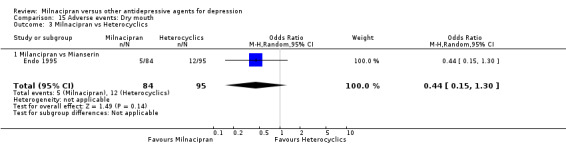
Comparison 15 Adverse events: Dry mouth, Outcome 3 Milnacipran vs Heterocyclics.
4. constipation
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than mianserin (see Analysis 16.3).
16.3. Analysis.
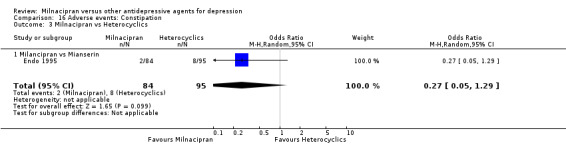
Comparison 16 Adverse events: Constipation, Outcome 3 Milnacipran vs Heterocyclics.
5. urination problems
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than mianserin (see Analysis 17.3).
17.3. Analysis.
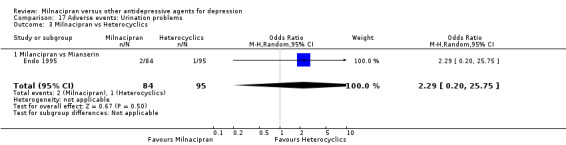
Comparison 17 Adverse events: Urination problems, Outcome 3 Milnacipran vs Heterocyclics.
6. hypotension
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing hypotension than mianserin (see Analysis 18.3).
18.3. Analysis.
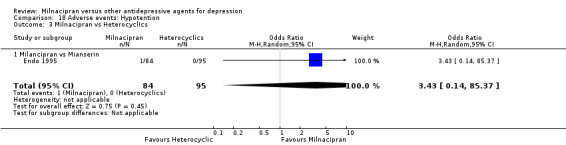
Comparison 18 Adverse events: Hypotention, Outcome 3 Milnacipran vs Heterocyclics.
7. agitation/ anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/anxiety than mianserin (see Analysis 19.3).
19.3. Analysis.
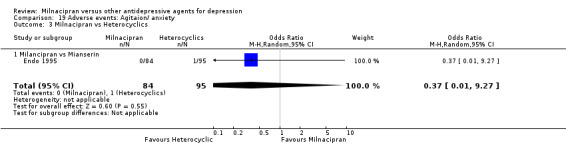
Comparison 19 Adverse events: Agitaion/ anxiety, Outcome 3 Milnacipran vs Heterocyclics.
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/ nausea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing vomiting/nausea than mianserin (see Analysis 22.3).
22.3. Analysis.
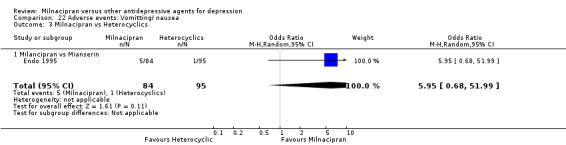
Comparison 22 Adverse events: Vomitting/ nausea, Outcome 3 Milnacipran vs Heterocyclics.
11. diarrhoea
No data reported.
3. Milnacipran versus SSRIs
Efficacy and acceptability outcomes were obtained from two studies comparing milnacipran with fluvoxamine (Annseau 1991c; Clerc 2001), three with fluoxetine (Annseau 1994; Guelfi 1998a; Lee 2002b), two with paroxetine (Sechter 2000; Shinkai 2004), and the remaining one with sertraline (Yang 2003). Outcome concerning tolerability were extractable from two studies comparing milnacipran with fluvoxamine (Annseau 1991c; Clerc 2001), two with fluoxetine (Guelfi 1998a; Lee 2002b), one with paroxetine (Sechter 2000) and none with sertraline.
A. Milnacipran versus Fluvoxamine
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was no evidence that milnacipran was more efficacious than fluvoxamine (OR1.76, 95%CI: 0.81 to 3.83) (see Analysis 1.2, Figure 8).
1.2. Analysis.
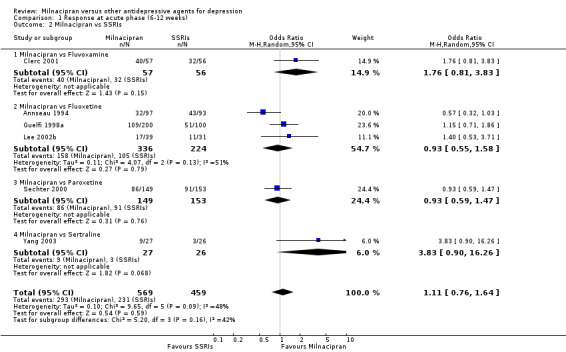
Comparison 1 Response at acute phase (6‐12 weeks), Outcome 2 Milnacipran vs SSRIs.
8.
Forest plot of comparison: 1 Response at acute phase (6‐12 weeks), outcome: 1.2 Milnacipran vs SSRIs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluvoxamine (OR1.54, 95%CI: 0.87 to 2.72) (see Analysis 2.2, Figure 9).
2.2. Analysis.
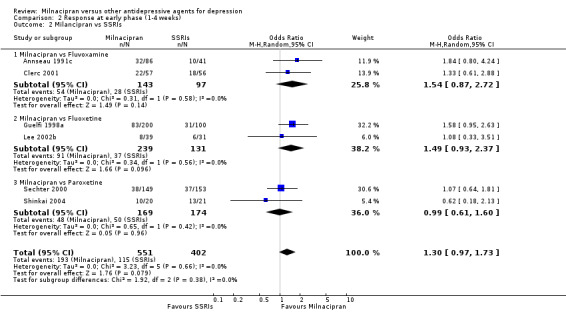
Comparison 2 Response at early phase (1‐4 weeks), Outcome 2 Milancipran vs SSRIs.
9.
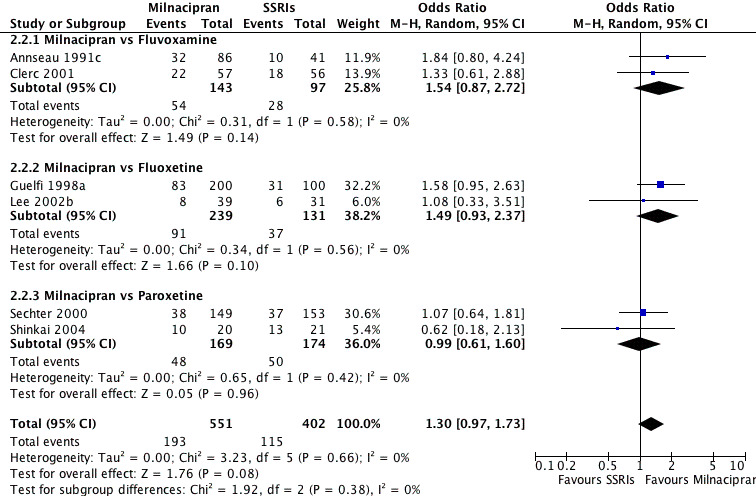
Forest plot of comparison: 2 Response at early phase (1‐4 weeks), outcome: 2.2 Milancipran vs SSRIs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to fluvoxamine (see Analysis 4.2).
4.2. Analysis.
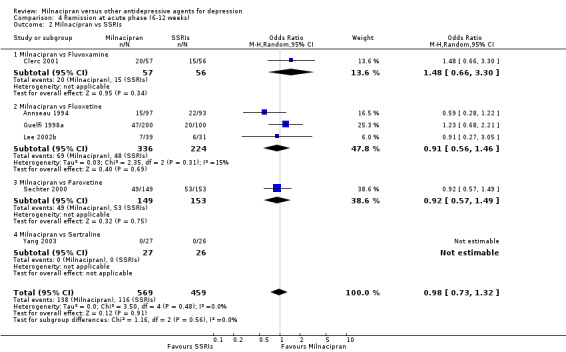
Comparison 4 Remission at acute phase (6‐12 weeks), Outcome 2 Milnacipran vs SSRIs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluvoxamine (see Analysis 5.2).
5.2. Analysis.
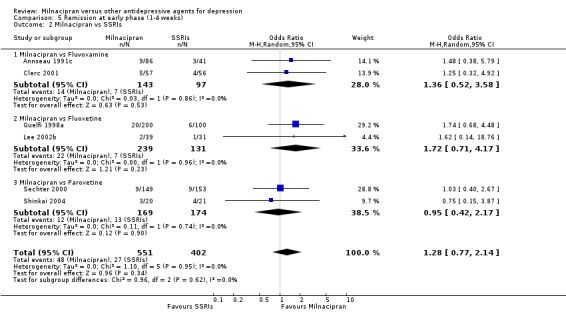
Comparison 5 Remission at early phase (1‐4 weeks), Outcome 2 Milnacipran vs SSRIs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to fluvoxamine (see Analysis 7.2).
7.2. Analysis.
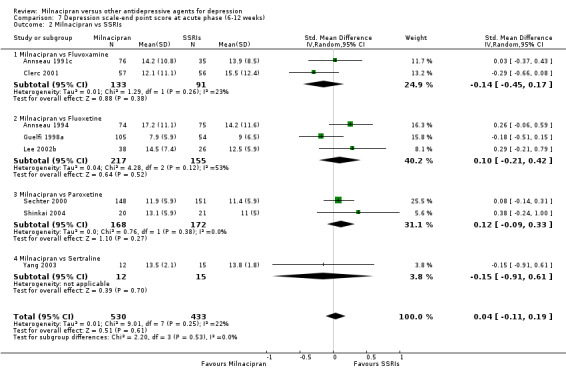
Comparison 7 Depression scale‐end point score at acute phase (6‐12 weeks), Outcome 2 Milnacipran vs SSRIs.
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluvoxamine (see Analysis 8.2).
8.2. Analysis.
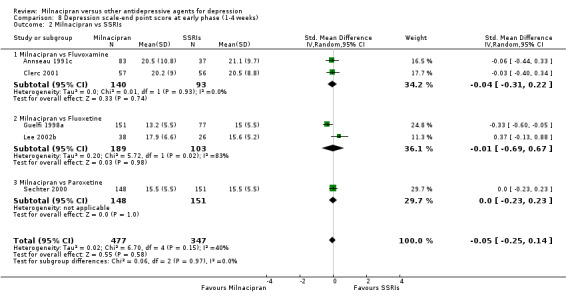
Comparison 8 Depression scale‐end point score at early phase (1‐4 weeks), Outcome 2 Milnacipran vs SSRIs.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to any cause compared to fluvoxamine (see Analysis 9.2).
9.2. Analysis.
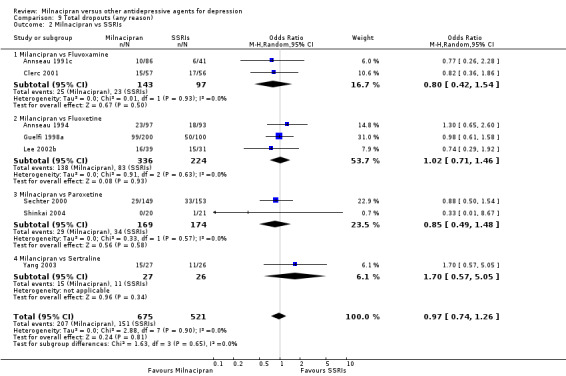
Comparison 9 Total dropouts (any reason), Outcome 2 Milnacipran vs SSRIs.
b) Due to inefficacy
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to inefficacy compared to fluvoxamine (see Analysis 10.2).
10.2. Analysis.
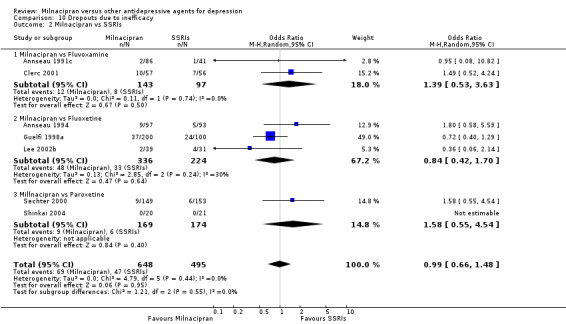
Comparison 10 Dropouts due to inefficacy, Outcome 2 Milnacipran vs SSRIs.
c) Due to adverse events
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to adverse events compared to fluvoxamine (see Analysis 11.2).
11.2. Analysis.
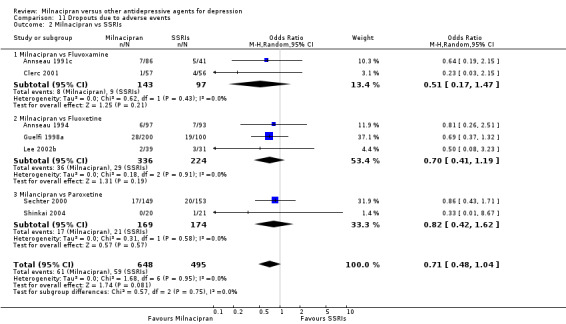
Comparison 11 Dropouts due to adverse events, Outcome 2 Milnacipran vs SSRIs.
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was evidence that milnacipran was associated with lower rate of patients experiencing adverse events than fluvoxamine (OR 0.51, 95%CI 0.28 to 0.94) (see Analysis 12.2).
12.2. Analysis.
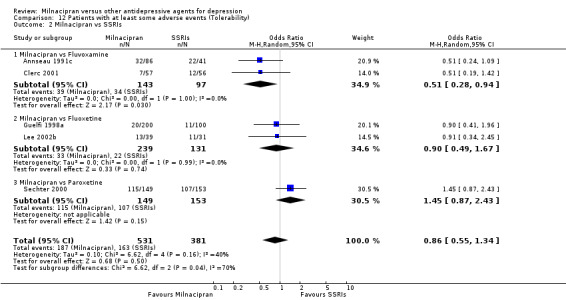
Comparison 12 Patients with at least some adverse events (Tolerability), Outcome 2 Milnacipran vs SSRIs.
b) Total number of patients experiencing a specific adverse event
1. sleepiness/drowsiness
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing sleepiness/drowsiness than fluvoxamine (see Analysis 13.2).
13.2. Analysis.
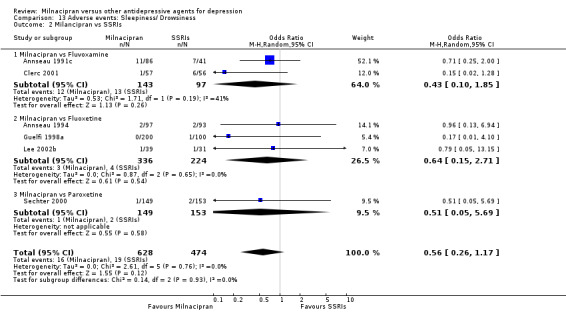
Comparison 13 Adverse events: Sleepiness/ Drowsiness, Outcome 2 Milancipran vs SSRIs.
2. insomnia
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than fluvoxamine (see Analysis 14.2).
14.2. Analysis.
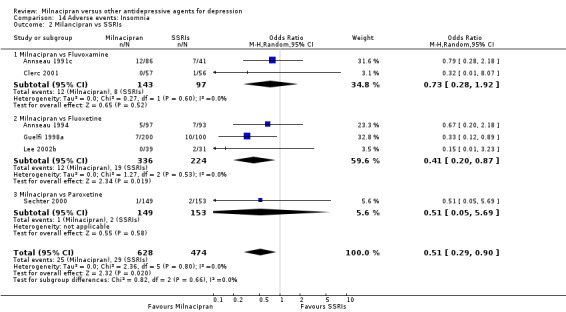
Comparison 14 Adverse events: Insomnia, Outcome 2 Milancipran vs SSRIs.
3. dry mouth
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing dry mouth than fluvoxamine (see Analysis 15.2).
15.2. Analysis.
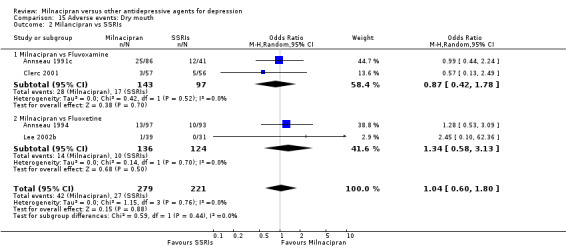
Comparison 15 Adverse events: Dry mouth, Outcome 2 Milancipran vs SSRIs.
4. constipation
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing constipation than fluvoxamine (see Analysis 16.2).
16.2. Analysis.
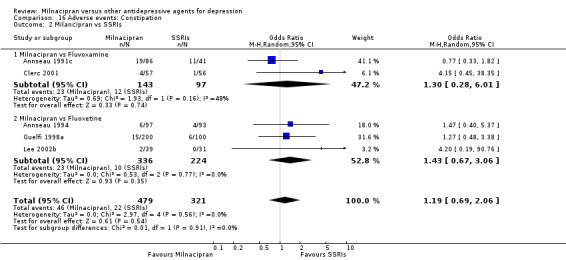
Comparison 16 Adverse events: Constipation, Outcome 2 Milancipran vs SSRIs.
5. urination problems
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing urination problems than fluvoxamine (see Analysis 17.2).
17.2. Analysis.
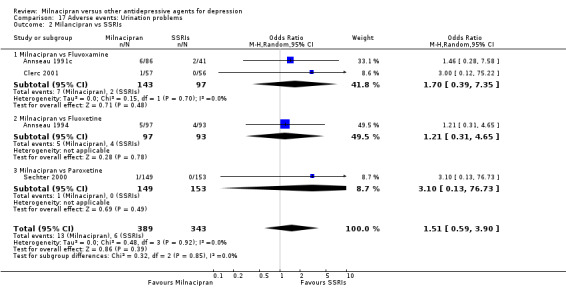
Comparison 17 Adverse events: Urination problems, Outcome 2 Milancipran vs SSRIs.
6. hypotension
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing hypotension than fluvoxamine (see Analysis 18.2).
18.2. Analysis.
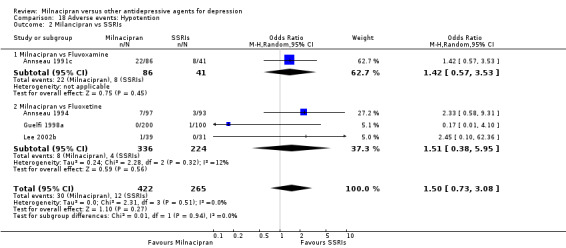
Comparison 18 Adverse events: Hypotention, Outcome 2 Milancipran vs SSRIs.
7. agitation/anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/anxiety than fluvoxamine (see Analysis 19.2).
19.2. Analysis.
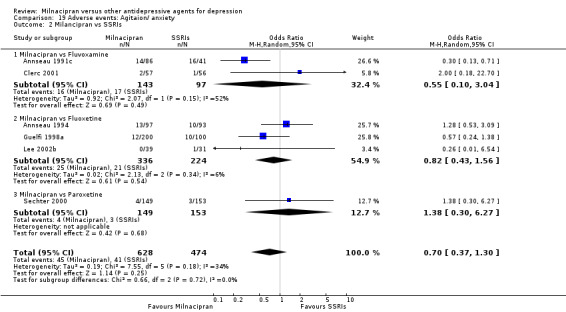
Comparison 19 Adverse events: Agitaion/ anxiety, Outcome 2 Milancipran vs SSRIs.
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/ nausea
There was evidence that milnacipran was associated with a lower rate of participants experiencing vomiting/nausea than fluvoxamine (OR 0.51, 95%CI 0.28 to 0.94)(see Analysis 22.2).
22.2. Analysis.
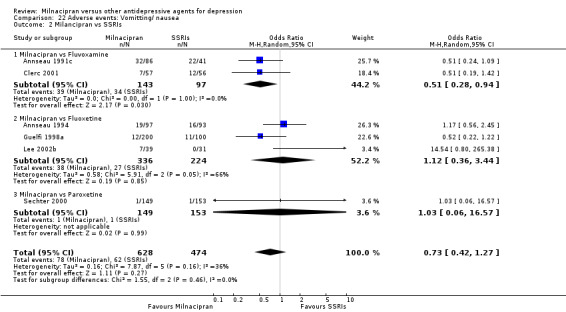
Comparison 22 Adverse events: Vomitting/ nausea, Outcome 2 Milancipran vs SSRIs.
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than fluvoxamine (see Analysis 23.2).
23.2. Analysis.
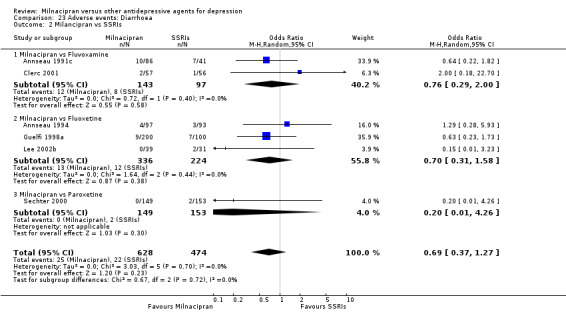
Comparison 23 Adverse events: Diarrhoea, Outcome 2 Milancipran vs SSRIs.
B. Milnacipran versus Fluoxetine
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was no evidence that milnacipran was more efficacious than fluoxetine (OR0.93, 95%CI: 0.55 to 1.58) (see Analysis 1.2, Figure 8).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluoxetine (OR1.49, 95%CI: 0.93 to 2.37) (see Analysis 2.2, Figure 9).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to fluoxetine (see Analysis 4.2).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluoxetine (see Analysis 5.2).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to fluoxetine (see Analysis 7.2).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to fluoxetine (see Analysis 8.2).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to any cause compared to fluoxetine (see Analysis 9.2).
b) Due to inefficacy
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to inefficacy compared to fluoxetine (see Analysis 10.2).
c) Due to adverse events
There was no evidence that milnacipran was associated with higher or lower rate of drop out due to adverse events compared to fluoxetine (see Analysis 11.2).
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was no evidence that milnacipran was associated with higher or lower rate of patients experiencing adverse events than fluoxetine (see Analysis 12.2).
b) Total number of patients experiencing a specific adverse event
1. sleepiness/drowsiness
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing sleepiness/drowsiness than fluoxetine (see Analysis 13.2).
2. insomnia
There was evidence that milnacipran was associated with a lower rate of participants experiencing insomnia than fluoxetine (OR 0.41, 95%CI: 0.20 to 0.87) (see Analysis 14.2).
3. dry mouth
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing dry mouth than fluoxetine (see Analysis 15.2).
4. constipation
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing constipation than fluoxetine (see Analysis 16.2).
5. urination problems
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing urination problems than fluoxetine (see Analysis 17.2).
6. hypotension
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing hypotension than fluoxetine (see Analysis 18.2).
7. agitation/ anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/ anxiety than fluoxetine (see Analysis 19.2).
8. suicide wishes/ gestures/ attempts
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing suicide wishes/ gestures/ attempts than fluoxetine (see Analysis 20.2).
20.2. Analysis.
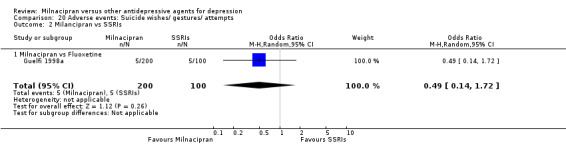
Comparison 20 Adverse events: Suicide wishes/ gestures/ attempts, Outcome 2 Milancipran vs SSRIs.
9. completed suicide
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing completed suicide than fluoxetine (see Analysis 21.1).
21.1. Analysis.
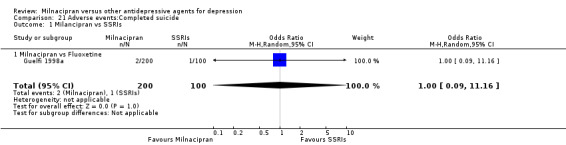
Comparison 21 Adverse events:Completed suicide, Outcome 1 Milancipran vs SSRIs.
10. vomiting/ nausea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing vomiting/nausea than fluoxetine (see Analysis 22.2).
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than fluoxetine (see Analysis 23.2).
C. Milnacipran versus Paroxetine
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was no evidence that milnacipran was more efficacious than paroxetine (OR0.93, 95%CI: 0.59 to 1.47) (see Analysis 1.2, Figure 8).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to paroxetine (OR 0.99, 95%CI: 0.61 to 1.60) (see Analysis 2.2, Figure 9).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to paroxetine (see Analysis 4.2).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to paroxetine (see Analysis 5.2).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to paroxetine (see Analysis 7.2).
b) Early phase treatment (1 to 4 weeks)
No substantial effect was found with milnacipran compared to paroxetine (see Analysis 8.2).
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to any cause compared to paroxetine (see Analysis 9.2).
b) Due to inefficacy
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to inefficacy compared to paroxetine (see Analysis 10.2).
c) Due to adverse events
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to adverse events compared to paroxetine (see Analysis 11.2).
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
There was no evidence that milnacipran was associated with a higher or lower rate of patients experiencing adverse events than paroxetine (see Analysis 12.2).
b) Total number of patients experiencing a specific adverse event
1. sleepiness/ drowsiness
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing sleepiness/drowsiness than paroxetine (see Analysis 13.2).
2. insomnia
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing insomnia than paroxetine (see Analysis 14.2).
3. dry mouth
No data available.
4. constipation
No data available.
5. urination problems
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing urination problem than paroxetine (see Analysis 17.2).
6. hypotension
No data available.
7. agitation/ anxiety
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing agitation/ anxiety than paroxetine (see Analysis 19.2).
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/ nausea
There was evidence that milnacipran was associated with a higher or lower rate of participants experiencing vomiting/nausea than paroxetine (see Analysis 22.2).
11. diarrhoea
There was no evidence that milnacipran was associated with a higher or lower rate of participants experiencing diarrhoea than paroxetine (see Analysis 23.2).
D. Milnacipran versus Sertraline
1.PRIMARY OUTCOME
1‐1. EFFICACY ‐ Number of patients who responded to treatment
a) Acute phase treatment (6 to 12 weeks)
There was no evidence that milnacipran was more efficacious than sertraline (OR 3.83, 95%CI: 0.90 to 16.26) (see Analysis 1.2, Figure 8).
b) Early phase treatment (1 to 4 weeks)
No data available.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2. SECONDARY OUTCOMES (only figures for substantial differences were reported in the text)
2‐1. EFFICACY ‐ Number of patients who achieved remission
a) Acute phase treatment (6 to 12 weeks)
Not estimable.
b) Early phase treatment (1 to 4 weeks)
No data available.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐2. EFFICACY ‐ Severity of depression at treatment phase
a) Acute phase treatment (6 to 12 weeks)
No substantial effect was found with milnacipran compared to sertraline (see Analysis 7.2).
b) Early phase treatment (1 to 4 weeks)
No data available.
c) Follow‐up phase treatment (16 to 24 weeks)
No data available.
2‐3 to ‐5. EFFICACY‐ Social adjustment, social functioning, health‐related quality of life, costs to health care services
No data available.
2‐6. ACCEPTABILITY ‐ Drop out rate
a) Due to any cause
There was no evidence that milnacipran was associated with a higher or lower rate of drop out due to any cause compared to sertraline (see Analysis 9.2).
b) Due to inefficacy
No data available.
c) Due to adverse events
No data available.
2‐7. TOLERABILITY
a) Total number of patients experiencing at least one adverse event
No data available.
b) Total number of patients experiencing a specific adverse event
1. sleepiness/drowsiness
No data available.
2. insomnia
No data available.
3. dry mouth
No data available.
4. constipation
No data available.
5. urination problems
No data available.
6. hypotension
No data available.
7. agitation/anxiety
No data available.
8. suicide wishes/ gestures/ attempts
No data available.
9. completed suicide
No data available.
10. vomiting/nausea
No data available.
11. diarrhoea
No data available.
Subgroup analysis
1. Milnacipran dosing
When we limited to participants treated with high dose milnacipran, no difference was found for response at early phase compared with TCAs (see Analysis 24.1). Response at acute phase was not analysed because only Leinonen 1997 provided relevant data. In terms of SSRIs, all studies were within the therapeutic range with the exception of Annseau 1991c, in which a higher dose was used. Due to the small number of trials without the therapeutic range, it was not considered meaningful to carry out this pre‐planned subgroup analysis.
24.1. Analysis.
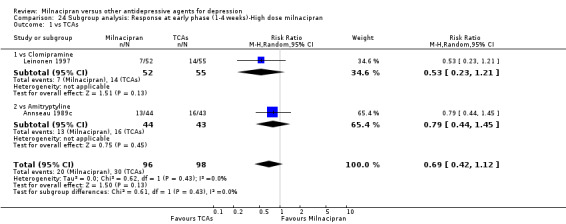
Comparison 24 Subgroup analysis: Response at early phase (1‐4 weeks)‐High dose milnacipran, Outcome 1 vs TCAs.
Participants treated with low dose (<100mg/day) milnacipran were compared with TCAs (Yamashita 1995), with heterocyclics (Endo 1995) and with SSRIs (Shinkai 2004). Due to the small number of trials without the therapeutic range for each comparison, it was not considered meaningful to carry out this pre‐planned subgroup analysis.
Four studies (Yamashita 1995, Endo 1995, Leinonen 1997, Tignol 1998) involved a flexible‐dose scheduling design. When we limited to studies involving a flexible‐dose scheduling design, no difference was found for response compared with TCAs (see Analysis 25.1, Analysis 26.1).
25.1. Analysis.
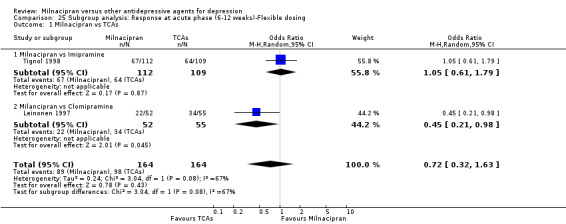
Comparison 25 Subgroup analysis: Response at acute phase (6‐12 weeks)‐Flexible dosing, Outcome 1 Milnacipran vs TCAs.
26.1. Analysis.
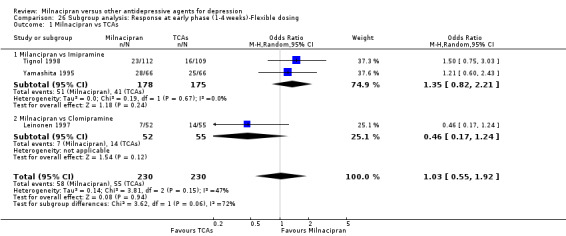
Comparison 26 Subgroup analysis: Response at early phase (1‐4 weeks)‐Flexible dosing, Outcome 1 Milnacipran vs TCAs.
2. Comparator dosing
All comparator doses were within the therapeutic range, with the exception of Clerc 2001, which used a higher dose, and Yamashita 1995, which used a lower dose. Due to the small number of trials without the therapeutic range, it was not considered meaningful to carry out this pre‐planned subgroup analysis.
3. Depression severity
All studies reported a mean baseline score corresponding to moderate major depression, with the exception of Guelfi 1998a where the mean baseline score corresponded to a severe major depression. Therefore, it was not meaningful to carry out this pre‐planned subgroup analysis.
4. Treatment settings
Among subgroups by the study settings, we did not find difference for response between milnacipran and TCAs or SSRIs based on seven studies for inpatients (comparing TCAs: Annseau 1989a; Annseau 1989c; Van Amerongen 2002; Lopez‐Ibor 2004, comparing SSRIs: Annseau 1991c; Guelfi 1998a; Shinkai 2004) and four studies for outpatients (comparing SSRIs only:Annseau 1994; Sechter 2000; Lee 2002b; Yang 2003). Among subgroups by the study settings, we did not find difference for response in each settings (see Analysis 27.1, Analysis 28.1, Analysis 29.1, Analysis 29.2, Analysis 30.1).
27.1. Analysis.
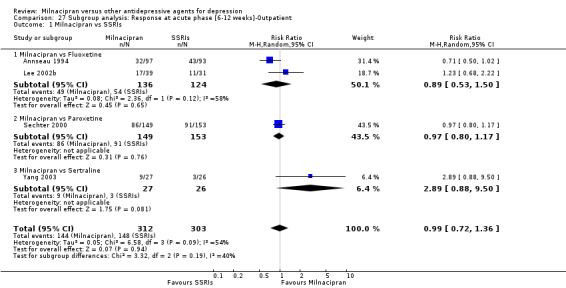
Comparison 27 Subgroup analysis: Response at acute phase [6‐12 weeks]‐Outpatient, Outcome 1 Milnacipran vs SSRIs.
28.1. Analysis.
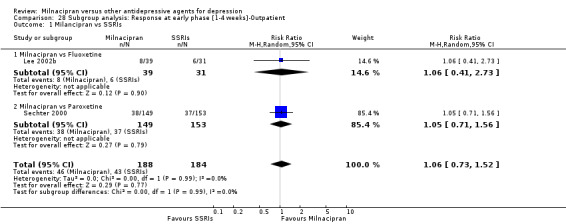
Comparison 28 Subgroup analysis: Response at early phase [1‐4 weeks]‐Outpatient, Outcome 1 Milancipran vs SSRIs.
29.1. Analysis.
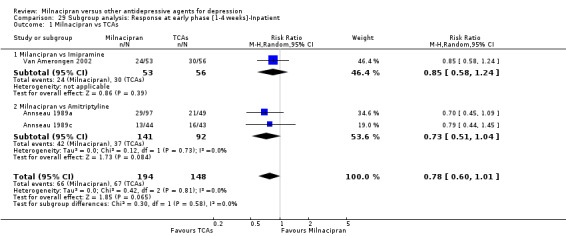
Comparison 29 Subgroup analysis: Response at early phase [1‐4 weeks]‐Inpatient, Outcome 1 Milnacipran vs TCAs.
29.2. Analysis.
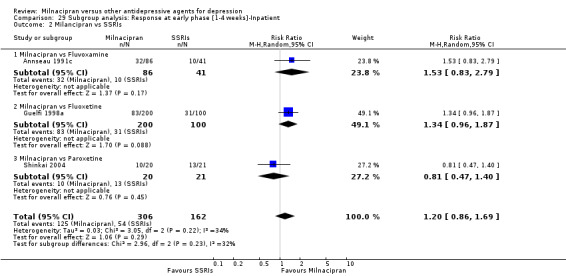
Comparison 29 Subgroup analysis: Response at early phase [1‐4 weeks]‐Inpatient, Outcome 2 Milancipran vs SSRIs.
30.1. Analysis.
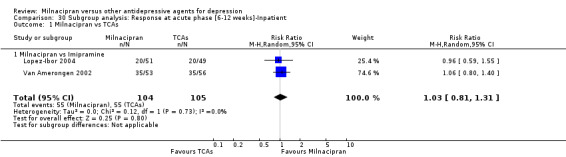
Comparison 30 Subgroup analysis: Response at acute phase [6‐12 weeks]‐Inpatient, Outcome 1 Milnacipran vs TCAs.
5. Elderly patients
As only one study specifically recruited elderly patients (Tignol 1998), it was not meaningful to carry out this pre‐planned sub‐group analysis.
Sensitivity analysis
1. Excluding trials with unclear concealment of random allocation and/or unclear double blinding
Although technically possible to carry out these sensitivity analyses, they were not performed, because they would not have contributed useful information due to the small number of studies (only three trials) reporting clear details on concealment of random allocation (Endo 1995, Yamashita 1995, Shinkai 2004).
2. Excluding trials whose dropout rate was greater than 20%
Referring to TCAs, a dropout rate greater than 20% was found for four studies comparing milnacipran with imipramine (Yamashita 1995, Tignol 1998, Van Amerongen 2002, Lopez‐Ibor 2004) and one with amitriptyline (Annseau 1989a). In terms of heterocyclics, the only study (Endo 1995) comparing milnacipran with mianserin reported a dropout rate greater than 20%. Among SSRIs, a dropout rate greater than 20% was found for two studies comparing milnacipran with fluoxetine (Guelfi 1998a, Lee 2002b), one with fluvoxamine (Clerc 2001), one with paroxetine (Sechter 2000) and one with sertraline (Yang 2003). Therefore, these pre‐planned sensitivity analyses were not carried out because there were insufficient trials to allow meaningful formal assessment.
3. Performing the worst and best‐case scenario analysis
Results from these sensitivity analyses did not materially change the main findings (full details available on request from authors)
4. Excluding trials for which the imputation methods were used
a) Imputed response rate
Excluding trials for which the response rate had to be calculated based on the imputation method, results for all comparisons did not materially change the main findings.
b) Borrowed SDs
Excluding trials for which the SD had to be borrowed from other trials, results from these sensitivity analyses did not materially change the main findings.
5. Examination of "wish bias" and exclusion of studies funded by the pharmaceutical company marketing milnacipran
These pre‐planned sensitivity analyses were not carried out because there were insufficient trials run by manufacturers other than the pharmaceutical company marketing milnacipran to allow meaningful formal assessment. All the studies were sponsored by a pharmaceutical company marketing milnacipran except for Shinkai 2004.
Funnel plot analysis
There was no evidence of publication bias or other small study effects based on visual inspections of the funnel‐plots with regard to the outcome variables.
Discussion
Summary of main results
A total of 16 randomised controlled trials (n=2277) were included in this review. Milnacipran does not seem to provide a significant advantage in efficacy over other antidepressive agents for the acute phase treatment of major depression. However, the data from one trial suggest that milnacipran may be inferior in terms of response compared to clomipramine (OR0.45, 95%CI: 0.21 to 0.98). Further, compared with TCAs, intervention groups including patients taking milnacipran were associated with fewer patients leaving the trial early due to adverse events as compared to patients taking TCAs (OR 0.55; 95%CI 0.35 to 0.85) (Analysis 11.1). There was also small amount of evidence that patients taking milnacipran experienced fewer adverse events of sleepiness/ drowsiness, dry mouth or constipation, as compared with those taking TCAs.
The included studies did not report on all the outcomes that were pre‐specified in the protocol of this review. Outcomes of clear relevance to patients and clinicians, in particular, patients' and their carers' attitudes to treatment, their ability to return to work and resume normal social functioning, were not reported in the included studies. Also ,only a small number of trials per comparison were found for most of the antidepressants. This limits the power of the review to detect moderate but clinically meaningful differences between the antidepressive agents.
Overall completeness and applicability of evidence
It has long been argued that placebo controlled trials are required to adequately demonstrate the efficacy of novel antidepressant drugs (Kupfer 2002), however, in the present review we focused only on the comparison between milnacipran and other active treatments for two reasons. First, in this review we focused on the following important clinical matter: "When an milnacipran is to be prescribed, would it constitute the better choice ?"
To answer this question within a clinically sound perspective, we included studies with active treatment comparisons. Second, placebo‐controlled studies are different from active comparator trials in terms of design, conduct and population, and furthermore it has been shown that placebo response in published trials of antidepressant drug for major depressive disorder is highly variable and often substantial (Walsh 2002). Retrieved randomised evidence compared milnacipran with a small selection of possible comparator antidepressants and no trials comparing milnacipran with escitalopram or citalopram (amongst SSRIs), or with venlafaxine, duloxetine, mirtazapine, bupropion, reboxetine or hypericum (amongst the newer antidepressive agents), or with some of the first generation antidepressants (such as MAOIs) were found. Although the search was comprehensive and thorough, it is still possible that there are unpublished studies that have not been identified but the small number of trials identified per comparison hinders the detection of any publication bias.
As in all systematic reviews and meta‐analyses, in the present study the main concern is about assessing the studies which were identified. The more information that is pooled together, the more precise and accurate is the estimate (Higgins 2005). We are realistically aware that a possibly significant piece of information has not been published and thus is not contributing to the true treatment estimate we were seeking. Although we did our very best to retrieve as many data as possible, through asking pharmaceutical companies and study authors to supply all available information, we can assume that data from some trials are still lacking, most of which are likely to be studies with negative findings. We are also aware of the possibility that a number of further randomised controlled trials comparing milnacipran with other antidepressant drugs are currently being conducted and these will be included in future updates of the review.
Potential biases in the review process
Some possible limitations of this review should be noted. Firstly, we had to impute the response and remission rates, our primary outcome, for most of the included trials. However, we consider that this is hardly rare, since incomplete reporting of outcome (i.e. an outcome reporting bias) is common within published articles of randomised trials (Chan 2004, Chan 2005). Further, imputation of response and remission rates by a validated statistical method (Furukawa 2006) in our review should minimize those biases. Nevertheless, we regret that we were unable to do a sensitivity analysis excluding trials with imputed response rates. As we update this review and assemble more trials involving milnacipran, we hope to conduct such a sensitivity analysis and be able to examine if our conclusions are robust.
Secondly, high dropout rates reduce the reliability of the assessment of other outcomes. Further, by making multiple comparisons we might have committed a type 1 error, that is, identifying and reporting a spurious association. Thirdly, all but one of the included trials had been funded by the drug company marketing milnacipran. There is nothing inherently wrong or biasing in this but, in view of the overwhelming evidence that sponsorship bias exists not only in psychiatry (Heres 2006) but also in medicine overall (Bekelman 2003), we should pay special attention that we may not inadvertently fall prey to such a bias. Therefore, these associations should be made clear to let anyone judge the relevance of the current findings.
Agreements and disagreements with other studies or reviews
Venlafaxine, another dual serotonin‐norepinephrine reuptake inhibitor, has been the first new generation antidepressant to be claimed to have differential effectiveness vis‐a‐vis the other antidepressants. The superiority of venlafaxine was first demonstrated in a drug company sponsored meta‐analysis (Thase 2001), and subsequently confirmed in an independently conducted meta‐analysis (Smith 2002). In addition, duloxetine, another SNRI, is also reported to be efficacious for the treatment of major depressive disorder and is well tolerated, safe and effective for the treatment of core depressive symptoms (Goldstein 2002, Cowen 2005, Frampton 2007). In keeping with these findings, milnacipran has been reported to be superior to SSRIs and equal to TCAs in two drug company sponsored meta‐analyses (Puech 1997, Lopez‐Ibor 1996). However, this superiority was not replicated in a subsequent independently‐conducted meta‐analysis (Papakostas 2007a). Our current review, which has included two additional trials involving SSRIs (Shinkai 2004, Yang 2003), and seven TCAs trials (Tignol 1998, Van Amerongen 2002, Lopez‐Ibor 2004, Leinonen 1997, Annseau 1989a, Annseau 1989c, Yamashita 1995) and one heterocyclic trial (Endo 1995) since Papakostas 2007a, confirms its findings.
The methodological limitation of standard systematic reviews is that they can rely only on evidence from direct comparisons. However, given the wide spectrum of available comparisons for the treatment of major depression, the use of the methodology of multiple treatments meta‐analysis (MTM, also known as network meta‐analysis) may help overcome this limitation (Lumley 2002; Lu 2004: Lu 2006; Salanti 2008). MTM is a statistical method that enables to integrate data from direct comparisons (when treatments are compared within a randomised trial) and indirect comparisons (when treatments are compared between trials by combining results on how effective they are against a common comparator treatment) involving diverse regimens, and to assess the strength and consistency of the evidence. MTM has already been used in other fields of medicine (Psaty 2003; Elliott 2007), and these comparisons may provide a clinically useful summary that can be used to guide treatment decisions. In the field of major depression, we have recently published a MTM including our data for milnacipran to compare both direct and indirect effects of 12 new‐generation antidepressants (Cipriani 2009a). The corresponding OR with 95%CI for efficacy (response rate) and acceptability (total dropout rate) are shown in Table 1. All of the confidence intervals overlap widely between MTM and direct comparisons, mainly because the confidence intervals of the direct comparisons are wide, generally indicating that the network of evidence is consistent. When the relative ratio of ORs was smaller than 0.59 (vs fluvoxamine and vs sertraline for response), the ORs of the MTM were revealed to be more conservative (i.e. closer to the null result) than the direct comparisons. It is possible that including indirect evidence may have 'cancelled out' the potential biases such as sponsorship and publication biases. The value of MTMs are increasingly acknowledged (Santaguida 2005) along with their pitfalls (Ioannidis 2006). Further methodological work is needed to confirm the relevance and validity of the review.
1. Comparative efficacy and acceptability of milnacipran for acute major depression.
| MTM (Cipriani 2009a) | Current review | ||
| OR (95%CI) | OR (95%CI) | Relative ratio of ORsa | |
| Efficacy (response rate) | |||
| Fluvoxamine | 1.03 (0.73‐1.47) | 1.76 (0.81‐3.83) | 0.59 |
| Fluoxetine | 1.01 (0.76‐1.35) | 0.93 (0.55‐1.58) | 1.09 |
| Paroxetine | 1.00 (0.74‐1.33) | 0.93 (0.59‐1.47) | 1.08 |
| Sertraline | 0.81 (0.60‐1.11) | 3.83 (0.90‐16.26) | 0.21 |
| Acceptability (total dropout rate) | |||
| Fluvoxamine | 0.85 (0.57‐1.32) | 0.82 (0.36‐1.86)b | 1.03 |
| Fluoxetine | 1.03 (0.76‐1.45) | 1.02 (0.71‐1.46) | 1.01 |
| Paroxetine | 0.94 (0.68‐1.31) | 0.88 (0.50‐1.54) | 1.07 |
| Sertraline | 1.17 (0.84‐1.72) | 1.70 (0.57‐5.05)b | 0.69 |
For efficacy, OR higher than 1 favour milnacipran. For acceptability, OR lower than 1 favour milnacipran .
aORs of the current review as reference.
bTwo trials comparing milnacipran with fluvoxamine (Annseau 1991c) and with sertraline (Shinkai 2004) were excluded because these 4‐week trials were not included in MTM.
Abbreviations: MTM=multiple‐treatments meta‐analysis, OR=odds ratio, CI=confidence interval
Authors' conclusions
Implications for practice.
The results of this review suggest that milnacipran is no more or less effective than other antidepressants in the acute phase treatment of major depression. There is also inadequate evidence to detect a substantial difference between milnacipran and other antidepressive agents in terms of acceptability and tolerability. However, there is some evidence in favour of milnacipran over TCAs in terms of dropouts due to adverse events and the rates of experiencing adverse events.
Implications for research.
More randomised controlled trials comparing milnacipran with other antidepressants are needed to generate more precise and accurate information about the drug. Also, randomised controlled trials comparing milnacipran with other comparator such as escitalopram, venlafaxine, duloxetine, mirtazapine, or hypericum are needed. Furthermore, future studies should focus to a greater extent on outcomes of clear relevance to patients and clinicians, in particular, patients' and their carers' attitudes to treatment, their ability to return to work and resume normal social functioning. Cost‐effectiveness also need to be assessed.
History
Protocol first published: Issue 2, 2007 Review first published: Issue 3, 2009
| Date | Event | Description |
|---|---|---|
| 20 April 2009 | Amended | Changed title from 'Milnacipran versus types of pharmacotherapy for depression' to 'Milnacipran versus other antidepressive agents for depression' |
| 8 October 2008 | Amended | Converted to new review format. |
| 20 September 2008 | Amended | Substantive amendment |
Acknowledgements
This review is one publication of the Meta‐Analyses of New Generation Antidepressants (MANGA) project in which a group of researchers within the Cochrane Collaboration Depression, Anxiety and Neurosis Group agreed to conduct a systematic review of all available evidence for 12 new generation antidepressants to inform clinical practice and mental health policies.
As of April 2009, we have completed an individual review for fluoxetine (Cipriani 2005), sertraline (Cipriani 2009b) and escitalopram (Cipriani 2009c), and published the protocols for fluvoxamine (Omori 2006), citalopram (Imperadore 2007), paroxetine (Cipriani 2007a), venlafaxine (Cipriani 2007b), duloxetine (Nose 2007) and mirtazapine (Watanabe 2006).
Data and analyses
Comparison 1. Response at acute phase (6‐12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 4 | 537 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.59, 1.30] |
| 1.1 Milnacipran vs Imipramine | 3 | 430 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.71, 1.54] |
| 1.2 Milancipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.21, 0.98] |
| 2 Milnacipran vs SSRIs | 6 | 1028 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.76, 1.64] |
| 2.1 Milnacipran vs Fluvoxamine | 1 | 113 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [0.81, 3.83] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.55, 1.58] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.59, 1.47] |
| 2.4 Milnacipran vs Sertraline | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 3.83 [0.90, 16.26] |
Comparison 2. Response at early phase (1‐4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 802 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.20] |
| 1.1 Milancipran vs Imipramine | 3 | 462 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.73, 1.69] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.17, 1.24] |
| 1.3 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.35, 1.08] |
| 2 Milancipran vs SSRIs | 6 | 953 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.97, 1.73] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.87, 2.72] |
| 2.2 Milnacipran vs Fluoxetine | 2 | 370 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.93, 2.37] |
| 2.3 Milnacipran vs Paroxetine | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.61, 1.60] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.54, 2.23] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.54, 2.23] |
Comparison 3. Response at follow‐up phase (4‐6 months).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.33, 1.55] |
| 1.1 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.33, 1.55] |
Comparison 4. Remission at acute phase (6‐12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 4 | 537 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.57, 1.19] |
| 1.1 Milnacipran vs Imipramine | 3 | 430 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.62, 1.41] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.17, 1.07] |
| 2 Milnacipran vs SSRIs | 6 | 1028 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.73, 1.32] |
| 2.1 Milnacipran vs Fluvoxamine | 1 | 113 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [0.66, 3.30] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.56, 1.46] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.57, 1.49] |
| 2.4 Milnacipran vs Sertraline | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Remission at early phase (1‐4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 802 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.56, 1.40] |
| 1.1 Milnacipran vs Imipramine | 3 | 462 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.59, 1.79] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.11, 4.33] |
| 1.3 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.25, 1.54] |
| 2 Milnacipran vs SSRIs | 6 | 953 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.77, 2.14] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.52, 3.58] |
| 2.2 Milnacipran vs Fluoxetine | 2 | 370 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.71, 4.17] |
| 2.3 Milnacipran vs Paroxetine | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.42, 2.17] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.57, 3.67] |
| 3.1 Milnacipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.57, 3.67] |
Comparison 6. Remission at follow‐up phase (4‐6 months).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.28, 1.31] |
| 1.1 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.28, 1.31] |
Comparison 7. Depression scale‐end point score at acute phase (6‐12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 7 | 820 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.07, 0.21] |
| 1.1 Milnacipran vs Imipramine | 4 | 509 | Std. Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.15, 0.20] |
| 1.2 Milnacipran vs Clomipramine | 1 | 93 | Std. Mean Difference (IV, Random, 95% CI) | 0.44 [0.03, 0.85] |
| 1.3 Milancipran vs Amitriptyline | 2 | 218 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.00 [‐0.42, 0.42] |
| 2 Milnacipran vs SSRIs | 8 | 963 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.11, 0.19] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 224 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.45, 0.17] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 372 | Std. Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.21, 0.42] |
| 2.3 Milnacipran vs Paroxetine | 2 | 340 | Std. Mean Difference (IV, Random, 95% CI) | 0.12 [‐0.09, 0.33] |
| 2.4 Milnacipran vs Sertraline | 1 | 27 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.91, 0.61] |
| 3 Milnacipran vs Heterocyclics | 1 | 167 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.43, 0.18] |
| 3.1 Milnacipran vs Mianserin | 1 | 167 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.43, 0.18] |
Comparison 8. Depression scale‐end point score at early phase (1‐4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 765 | Std. Mean Difference (IV, Random, 95% CI) | 0.14 [‐0.11, 0.38] |
| 1.1 Milnacipran vs Imipramine | 3 | 454 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.25, 0.21] |
| 1.2 Milnacipran vs Clomipramine | 1 | 93 | Std. Mean Difference (IV, Random, 95% CI) | 0.66 [0.25, 1.08] |
| 1.3 Milancipran vs Amitriptyline | 2 | 218 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.22, 0.48] |
| 2 Milnacipran vs SSRIs | 5 | 824 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.25, 0.14] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 233 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.31, 0.22] |
| 2.2 Milnacipran vs Fluoxetine | 2 | 292 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.69, 0.67] |
| 2.3 Milnacipran vs Paroxetine | 1 | 299 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.23, 0.23] |
| 3 Milnacipran vs Heterocyclics | 1 | 121 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.51, 0.21] |
| 3.1 Milnacipran vs Mianserin | 1 | 121 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.51, 0.21] |
Comparison 9. Total dropouts (any reason).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 795 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.62, 1.33] |
| 1.1 Milnacipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.52, 1.48] |
| 1.2 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.47, 1.97] |
| 2 Milnacipran vs SSRIs | 8 | 1196 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.74, 1.26] |
| 2.1 Milancipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.42, 1.54] |
| 2.2 Milancipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.71, 1.46] |
| 2.3 Milnacipran vs Paroxetine | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.49, 1.48] |
| 2.4 Milancipran vs Sertraline | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [0.57, 5.05] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.57, 1.87] |
| 3.1 Milacipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.57, 1.87] |
Comparison 10. Dropouts due to inefficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 7 | 902 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.82, 1.99] |
| 1.1 Milnacipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.66, 1.95] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 3.04 [0.89, 10.38] |
| 1.3 Milnacipran vs Amitryptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.42, 2.90] |
| 2 Milnacipran vs SSRIs | 7 | 1143 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.66, 1.48] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.53, 3.63] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.42, 1.70] |
| 2.3 Millnacipran vs Paroxetine | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.55, 4.54] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.22, 1.51] |
| 3.1 Milnacipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.22, 1.51] |
Comparison 11. Dropouts due to adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 756 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.35, 0.85] |
| 1.1 Milnacipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.35, 1.04] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.18, 1.03] |
| 1.3 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.12, 2.49] |
| 2 Milnacipran vs SSRIs | 7 | 1143 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.48, 1.04] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.17, 1.47] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.41, 1.19] |
| 2.3 Milancipran vs Paroxetine | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.42, 1.62] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.33, 1.87] |
| 3.1 Milnacipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.33, 1.87] |
Comparison 12. Patients with at least some adverse events (Tolerability).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 795 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.24, 0.53] |
| 1.1 Milnacipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.28, 0.66] |
| 1.2 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.23 [0.13, 0.40] |
| 2 Milnacipran vs SSRIs | 5 | 912 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.55, 1.34] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.28, 0.94] |
| 2.2 Milnacipran vs Fluoxetine | 2 | 370 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.49, 1.67] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.87, 2.43] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.34, 1.15] |
| 3.1 Milnacipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.34, 1.15] |
Comparison 13. Adverse events: Sleepiness/ Drowsiness.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 5 | 693 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.07, 0.73] |
| 1.1 Milancipran vs Imipramine | 2 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.19, 2.45] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.35] |
| 1.3 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.07 [0.02, 0.22] |
| 2 Milancipran vs SSRIs | 6 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.26, 1.17] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.10, 1.85] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.15, 2.71] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.05, 5.69] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.08, 0.58] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.08, 0.58] |
Comparison 14. Adverse events: Insomnia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 756 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.57, 2.22] |
| 1.1 Milancipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.49, 1.53] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 5.55 [1.14, 27.04] |
| 1.3 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 1.5 [0.24, 9.45] |
| 2 Milancipran vs SSRIs | 6 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.29, 0.90] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.28, 1.92] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.41 [0.20, 0.87] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.05, 5.69] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.07, 18.39] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.07, 18.39] |
Comparison 15. Adverse events: Dry mouth.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 7 | 902 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.28, 0.65] |
| 1.1 Milancipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.37, 0.86] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.21, 0.97] |
| 1.3 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.12, 0.39] |
| 2 Milancipran vs SSRIs | 4 | 500 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.60, 1.80] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.42, 1.78] |
| 2.2 Milnacipran vs Fluoxetine | 2 | 260 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.58, 3.13] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.15, 1.30] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.15, 1.30] |
Comparison 16. Adverse events: Constipation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 795 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.44, 0.88] |
| 1.1 Milancipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.41, 0.98] |
| 1.2 Milnacipran vs Amitriptyline | 2 | 233 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.34, 1.06] |
| 2 Milancipran vs SSRIs | 5 | 800 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.69, 2.06] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.28, 6.01] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.67, 3.06] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.05, 1.29] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.05, 1.29] |
Comparison 17. Adverse events: Urination problems.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 1.81 [0.57, 5.73] |
| 1.1 Milancipran vs Imipramine | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 1.81 [0.57, 5.73] |
| 2 Milancipran vs SSRIs | 4 | 732 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [0.59, 3.90] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [0.39, 7.35] |
| 2.2 Milnacipran vs Fluoxetine | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.31, 4.65] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 3.10 [0.13, 76.73] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 2.29 [0.20, 25.75] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 2.29 [0.20, 25.75] |
Comparison 18. Adverse events: Hypotention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 5 | 656 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.16, 1.45] |
| 1.1 Milancipran vs Imipramine | 3 | 462 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.04, 0.76] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 1.64 [0.74, 3.62] |
| 1.3 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 0.41 [0.14, 1.21] |
| 2 Milancipran vs SSRIs | 4 | 687 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.73, 3.08] |
| 2.1 Milnacipran vs Fluvoxamine | 1 | 127 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.57, 3.53] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [0.38, 5.95] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 3.43 [0.14, 85.37] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 3.43 [0.14, 85.37] |
Comparison 19. Adverse events: Agitaion/ anxiety.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 4 | 549 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.30, 1.37] |
| 1.1 Milancipran vs Imipramine | 3 | 462 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.23, 1.94] |
| 1.2 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.21, 1.83] |
| 2 Milancipran vs SSRIs | 6 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.37, 1.30] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.10, 3.04] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.43, 1.56] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.30, 6.27] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.01, 9.27] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.01, 9.27] |
Comparison 20. Adverse events: Suicide wishes/ gestures/ attempts.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.09, 2.91] |
| 1.1 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.09, 2.91] |
| 2 Milancipran vs SSRIs | 1 | 300 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.14, 1.72] |
| 2.1 Milnacipran vs Fluoxetine | 1 | 300 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.14, 1.72] |
Comparison 21. Adverse events:Completed suicide.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milancipran vs SSRIs | 1 | 300 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.09, 11.16] |
| 1.1 Milnacipran vs Fluoxetine | 1 | 300 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.09, 11.16] |
Comparison 22. Adverse events: Vomitting/ nausea.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 6 | 756 | Odds Ratio (M‐H, Random, 95% CI) | 2.44 [1.34, 4.42] |
| 1.1 Milancipran vs Imipramine | 4 | 562 | Odds Ratio (M‐H, Random, 95% CI) | 2.31 [1.13, 4.72] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 10.30 [0.54, 196.19] |
| 1.3 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 2.24 [0.69, 7.19] |
| 2 Milancipran vs SSRIs | 6 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.42, 1.27] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.28, 0.94] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.36, 3.44] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.06, 16.57] |
| 3 Milnacipran vs Heterocyclics | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 5.95 [0.68, 51.99] |
| 3.1 Milancipran vs Mianserin | 1 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 5.95 [0.68, 51.99] |
Comparison 23. Adverse events: Diarrhoea.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 5 | 656 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.29, 5.29] |
| 1.1 Milancipran vs Imipramine | 3 | 462 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.12, 1.75] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 5.50 [0.26, 117.22] |
| 1.3 Milnacipran vs Amitriptyline | 1 | 87 | Odds Ratio (M‐H, Random, 95% CI) | 7.34 [0.37, 146.43] |
| 2 Milancipran vs SSRIs | 6 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.37, 1.27] |
| 2.1 Milnacipran vs Fluvoxamine | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.29, 2.00] |
| 2.2 Milnacipran vs Fluoxetine | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.31, 1.58] |
| 2.3 Milnacipran vs Paroxetine | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.26] |
Comparison 24. Subgroup analysis: Response at early phase (1‐4 weeks)‐High dose milnacipran.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 vs TCAs | 2 | 194 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.42, 1.12] |
| 1.1 vs Clomipramine | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.23, 1.21] |
| 1.2 vs Amitryptyline | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.44, 1.45] |
Comparison 25. Subgroup analysis: Response at acute phase (6‐12 weeks)‐Flexible dosing.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 2 | 328 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.32, 1.63] |
| 1.1 Milnacipran vs Imipramine | 1 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.61, 1.79] |
| 1.2 Milancipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.21, 0.98] |
Comparison 26. Subgroup analysis: Response at early phase (1‐4 weeks)‐Flexible dosing.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 3 | 460 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.55, 1.92] |
| 1.1 Milancipran vs Imipramine | 2 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.82, 2.21] |
| 1.2 Milnacipran vs Clomipramine | 1 | 107 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.17, 1.24] |
Comparison 27. Subgroup analysis: Response at acute phase [6‐12 weeks]‐Outpatient.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs SSRIs | 4 | 615 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.72, 1.36] |
| 1.1 Milnacipran vs Fluoxetine | 2 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.53, 1.50] |
| 1.2 Milnacipran vs Paroxetine | 1 | 302 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.80, 1.17] |
| 1.3 Milnacipran vs Sertraline | 1 | 53 | Risk Ratio (M‐H, Random, 95% CI) | 2.89 [0.88, 9.50] |
Comparison 28. Subgroup analysis: Response at early phase [1‐4 weeks]‐Outpatient.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milancipran vs SSRIs | 2 | 372 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.73, 1.52] |
| 1.1 Milnacipran vs Fluoxetine | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.41, 2.73] |
| 1.2 Milnacipran vs Paroxetine | 1 | 302 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.71, 1.56] |
Comparison 29. Subgroup analysis: Response at early phase [1‐4 weeks]‐Inpatient.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 3 | 342 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.60, 1.01] |
| 1.1 Milancipran vs Imipramine | 1 | 109 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.58, 1.24] |
| 1.2 Milnacipran vs Amitriptyline | 2 | 233 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.51, 1.04] |
| 2 Milancipran vs SSRIs | 3 | 468 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.86, 1.69] |
| 2.1 Milnacipran vs Fluvoxamine | 1 | 127 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.83, 2.79] |
| 2.2 Milnacipran vs Fluoxetine | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.96, 1.87] |
| 2.3 Milnacipran vs Paroxetine | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.47, 1.40] |
Comparison 30. Subgroup analysis: Response at acute phase [6‐12 weeks]‐Inpatient.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Milnacipran vs TCAs | 2 | 209 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.81, 1.31] |
| 1.1 Milnacipran vs Imipramine | 2 | 209 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.81, 1.31] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Annseau 1989a.
| Methods | 4 week randomised double blind study. | |
| Participants | Diagnosis: Inpatients with RDC major depressive disorder Male and Female. Threshold of baseline severity: MADRS>=25, CGI<=4, Raskin Scale for Depression>Covi Anxiety Scale Total number of all allocated participants: N=146 Age: mean 48.6 (SD 10.8) y, range 20‐70y. |
|
| Interventions | Milnacipran 50/100mg: N=97 (50mg: N=47, 100mg: N=48)
Amitriptyline 150mg: N=49 Fixed dosing schedule |
|
| Outcomes | Hamilton Depression Rating Scale‐24 item, MADRS,CGI‐I, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind". Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | High risk | Did not provide number of participants who dropped out during the study due to side effects. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Annseau 1989c.
| Methods | 4 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with RDC major depressive disorder Male and Female. Threshold of baseline severity: MADRS>=25, CGI<=5, Raskin Scale for Depression>Covi Anxiety Scale Total number of all allocated participants: N=87 Age: mean 49.6 (SD 11.6) y, range 23‐68y. |
|
| Interventions | Milnacipran 200mg: N=44
Amitriptyline 150mg: N=43 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐24 item, MADRS,CGI‐I, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Annseau 1991c.
| Methods | 4 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with RDC major depressive disorder Male and Female. Threshold of baseline severity: MADRS>=25, CGI<=5, Raskin Scale for Depression>Covi Anxiety Scale Total number of all allocated participants: N=127 Age: mean 43.7 (SD 12.4)y, range 20‐70y. |
|
| Interventions | Milnacipran 150‐300/200mg: N=86 (150‐300mg:N=42, 200mg:N=44)
Fluvoxamine 200mg: N=41 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐24 item, MADRS,CGI‐I, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | High risk | Some missing standard deviations. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Annseau 1994.
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Outpatients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: MADRS>=25, CGI‐S<=4, Raskin Scale for Depression>Covi Anxiety Scale Total number of all allocated participants: N=190 Age: mean 44.9 (SD 11.2)y, range 19‐68y. |
|
| Interventions | Milnacipran 100mg: N=97
Fluoxetine 20mg: N=93 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐24 item, MADRS,CGI‐I, CGI‐S, CGI‐E, 100mm VAS for depressed mood, psychomotor retardation, anxiety and insomnia | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Some of actual figures of outcome data are missing. Incoherence between denominators. |
| Selective reporting (reporting bias) | High risk | No data of participants who experienced at least some side effects. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Clerc 2001.
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Outpatients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: MADRS>=25, Raskin Scale for Depression Total number of all allocated participants: N=113 Age: mean 48.7 (SD 15.1)y for milnacipran, mean 51.2 (SD 12.6)y for fluvoxamine |
|
| Interventions | Milnacipran 100mg: N=57
Fluvoxamine 200mg: N=56 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐24 item, MADRS,CGI‐I, CGI‐S, CGI‐E | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Endo 1995.
| Methods | 4 week randomised double blind study | |
| Participants | Diagnosis: In‐ and Out‐patients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: Not reported. Total number of all allocated participants: N=179 Age: range 20‐65y |
|
| Interventions | Milnacipran 50‐150mg (mean: 68.6mg): N=84
Mianserine 30‐ 60mg : N=95 Flexible dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐21 item, CGI‐I, CPRG | |
| Notes | Funding: by industry Article in Japanese. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Low risk | Central allocation used. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Guelfi 1998a.
| Methods | 12 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with DSM‐III‐R major depression Male and Female. Threshold of baseline severity: HDRS‐17>=22, Newcastle scale>=6, HDRS specific endogenous subscale>=8 Total number of all allocated participants: N=300 Age: mean 45.6 (SD 12.8)y for milnacipran 100mg, mean 45.2 (SD 12.5)y for milnacipran 200mg, mean 45.8 (SD 12.8)y for fluoxetine; range 18‐70y |
|
| Interventions | Milnacipran 100/200mg: N=200 (100mg:N=100, 200mg:N=100)
Fluoxetine 20mg: N=100 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing primary outcome data at early phase. |
| Selective reporting (reporting bias) | High risk | Some missing standard deviations. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Lee 2002b.
| Methods | 6 week randomised open‐label study | |
| Participants | Diagnosis: Outpatients with DSM‐IV major depressive disorder Male and Female. Threshold of baseline severity: HDRS‐17>=17, MADRS>=21 Total number of all allocated participants: N=70 Age: mean 49 (SD 15)y for milnacipran , mean 51 (SD 12)y for fluoxetine; range 17‐70y |
|
| Interventions | Milnacipran 100mg: N=39
Fluoxetine 20mg: N=31 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐I, Covi Anxiety Scale | |
| Notes | Funding: by industry Article in Korean. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Leinonen 1997.
| Methods | 26 week randomised double blind study | |
| Participants | Diagnosis: In‐ and Out‐patients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: HDRS‐17>=18, CGI>=moderately ill Total number of all allocated participants: N=107 Age: mean 49.2 (SD 9.8)y for milnacipran, mean 47.1 (SD 10.6)y for clomipramine; range 18‐70y |
|
| Interventions | Milnacipran 100‐200mg: N=52
Clomipramine 75‐150mg: N=55 Flexible dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome. |
| Selective reporting (reporting bias) | High risk | Number of participants who dropped out the trial is missing. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Lopez‐Ibor 2004.
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: MADRS>=25 Total number of all allocated participants: N=100 Age: range 18‐70y |
|
| Interventions | Milnacipran 100mg: N=51
Imipramine 150mg: N=49 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐21 item, MADRS, 100mm VAS for subjective depression | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised" |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Unclear risk | Number of patient receiving each intervention at the early phase is unclear. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Sechter 2000.
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Outpatients with DSM‐IV major depressive disorder Male and Female. Threshold of baseline severity: MADRS>=20 Total number of all allocated participants: N=302 Age: mean 44.8 (SD 11.6)y for milnacipran, mean 42.8 (SD 11.2)y for paroxetine; range 18‐70y |
|
| Interventions | Milnacipran 100mg: N=149
Imipramine 20mg: N=153 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐I, CGI‐S | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised" |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome. |
| Selective reporting (reporting bias) | High risk | Missing standard deviations. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Shinkai 2004.
| Methods | 4 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with DSM‐IV major depressive disorder without psychotic features Male and Female. Threshold of baseline severity: HDRS‐17>=15 Total number of all allocated participants: N=41 Age: mean 53 (SD 17)y; range 20‐78y |
|
| Interventions | Milnacipran mean 80.25mg: N=20
Paroxetine mean 34.28mg: N=21 Flexible dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item | |
| Notes | Funding: independent from industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly" |
| Allocation concealment (selection bias) | Low risk | Quote: "randomly divided into either milnacipran or the paroxetine group using StatView, a computerized statistical package" |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome. |
| Selective reporting (reporting bias) | High risk | Adverse events were not reported so that could not be entered in to the meta‐analysis. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Tignol 1998.
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: In‐ and Out‐patients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: HDRS‐17>=17, MADRS>=25, improvement during washout phase less than 25% of the initial score, MMSE>=20 Total number of all allocated participants: N=221 Age: mean 74.0 (SD 6.2)y for milnacipran, mean 74.2 (SD 6.8)y for imipramine; range 65‐93y |
|
| Interventions | Milnacipran 75‐100mg N=112
Imipramine 75‐100mg N=109 Flexible dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐I, CGI‐S, Covi Anxiety Scale, WAIS, Digit Symbol Substitution Test (DSST), Word‐paired test, MMSE, Functional Status Questionnaire (FSQ) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing primary outcome at early phase. |
| Selective reporting (reporting bias) | High risk | MADRS scores were reported incompletely. Missing standard deviations. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Van Amerongen 2002.
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Inpatients with DSM‐III major depression Male and Female. Threshold of baseline severity: HDRS‐17>=17, MADRS>=25, improvement during washout phase less than 25% of the initial score, MMSE>=20 Total number of all allocated participants: N=109 Age: mean 46.7y (range 23‐70y) for milnacipran, mean 45.9 y (range 20‐71y) for imipramine; range 18‐70y for total sample |
|
| Interventions | Milnacipran 100mg N=53
Imipramine 150mg N=56 Fixed dosing schedule. |
|
| Outcomes | Hamilton Depression Rating Scale‐21 item, MADRS, CGI‐3, 100mm VAS for subjective depression | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing primary outcome at early phase. |
| Selective reporting (reporting bias) | High risk | Missing standard deviations. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Yamashita 1995.
| Methods | 4 week randomised double blind study | |
| Participants | Diagnosis: In‐ and Out‐ patients with DSM‐III‐R major depressive episode Male and Female. Threshold of baseline severity: Not reported. Total number of all allocated participants: N=132 Age: range 20‐65y for total sample |
|
| Interventions | Milnacipran 50‐150mg (mean:77.2mg) N=66
Imipramine 50‐150mg (mean:89.1mg) N=66 Flexible dosing schedule |
|
| Outcomes | Hamilton Depression Rating Scale‐21 item, CGI‐I, CPRG | |
| Notes | Funding: by industry Article in Japanese. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Low risk | Cental allocation used. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing primary outcome data. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Yang 2003.
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: Outpatients with DSM‐IV major depressive disorder Male and Female. Threshold of baseline severity: HDRS‐17>=17 Total number of all allocated participants: N=53 Age: not shown |
|
| Interventions | Milnacipran 100mg N=27
Sertraline 100mg N=26 Flexible dosing schedule |
|
| Outcomes | Hamilton Depression Rating Scale‐17 item, MADRS, CGI‐I, CGI‐S | |
| Notes | Funding: by industry Article in Korean. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "double‐blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary outcome data missing. |
| Selective reporting (reporting bias) | High risk | Adverse events are not reported so that could not be entered in a meta‐analysis. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Baek 2002a | Did not use relevant operational diagnostic criteria. |
| Baek 2002b | Additional publication of trial already included. |
| Dardennes 1998 | Did not include acute phase treatment. |
| Kanemoto 2004 | Did not use other antidepressant as a comparator drug. |
| Lee 2004 | Additional publication of trial already included. |
| Macher 1989 | Did not use other antidepressant as a comparator drug. |
| Naito 2007 | Secondary analysis of separate two studies (Ito 2002; Yoshida 2002), thus not a concurrent comparison study |
| Onodera 1992 | Additional publication of trial already included. |
| Wyeth 2006 | Method of allocation was not randomised (e.g. controlled clinical trial). |
Characteristics of studies awaiting assessment [ordered by study ID]
Yoshimura 2007.
| Methods | RCT |
| Participants | 42 Japanese adults |
| Interventions | paroxetine vs milnacipran |
| Outcomes | Serum BDNF levels, response and remission rates |
| Notes |
Differences between protocol and review
None.
Contributions of authors
AN, NW, TAF, AC, CB, RC and HMG conceived and designed the review. AN, NW and HMG identified and acquired reports of trials,and contacted authors of trials and pharmaceutical industries for additional information. AN and NW extracted data. AN, NW and TAF analysed and interpreted the data. AC, CB and HMG contributed to the interpretation of the data. AN wrote the first draft of the manuscript. All authors contributed to make critical revision of the manuscript for important intellectual content and have approved the final version of the manuscript.
Declarations of interest
AN, NW, AC, CB, HMG, RC: none declared TAF has received several research grants and fees for speaking from some pharmaceutical companies, which market antidepressants (paroxetine, fluvoxamine, milnacipran, trazodone, mianserin), antipsychotics (risperidone, olanzapine, quetiapine, paliperidone, asenapine), nootropics (donepezil) and anxiolytics (loflazepate, tandospirone).
Edited (no change to conclusions)
References
References to studies included in this review
Annseau 1989a {published data only}
- Ansseau M, Frenckell R, Mertens C, Wilde J, Botte L, Devoitille JM, Evrard JL, Nayer A, Darimont P, Dejaiffe G, Mirel J, Meurice E, Parent M, Couzinier JP, Demarez JP, Serre C. Controlled comparison of two doses of milnacipran (F 2207) and amitriptyline in major depressive inpatients. Psychopharmacology 1989;98(2):163‐8. [DOI] [PubMed] [Google Scholar]
- Frenckell R, Ansseau M, Serre C, Sutet P. Pooling two controlled comparisons of milnacipran (F2207) and amitriptyline in endogenous inpatients. A new approach in dose ranging studies. International Clinical Psychopharmacology 1990;5:49‐56. [DOI] [PubMed] [Google Scholar]
Annseau 1989c {published data only}
- Ansseau M, Frenckell R, Papart P, Mertens C, Wilde J, Botte L, Devoitille JM, Evrard JL, Nayer A, Koch‐Bourdouxhe S, Darimont P, Lecoq A, Mirel J, Couzinier JP, Demarez JP, Serre C. Controlled comparison of milnacipran (F2207) 200 mg and amitriptyline in endogenous depressive inpatients. Human Psychopharmacology 1989;4:221‐7. [Google Scholar]
- Ansseau M, Frenckell R, Serre C, Demarez JP. Controlled comparison of milnacipran (F2207) 200 mg and amitriptyline in endogenous depressive inpatients. Therapie 1990;45(3):292. [Google Scholar]
Annseau 1991c {published data only}
- Ansseau M, Frenckell R, Gerard MA, Mertens C, Wilde J, Botte L, Devoitille JM, Evrard JL, Nayer A, Darimont P, Mirel J, Troisfontaines B, Toussaint C, Couzinier JP, Demarez JP, Serre C. Interest of a loading dose of milnacipran in endogenous depressive inpatients. Comparison with the standard regimen and with fluvoxamine. European Neuropsychopharmacology 1991;1(2):113‐21. [DOI] [PubMed] [Google Scholar]
Annseau 1994 {published data only}
- Ansseau M, Papart P, Troisfontaines B, Bartholome F, Bataille M, Charles G, Schittecatte M, Darimont P, Devoitille JM, Wilde J, Dufrasne M, Gilson H, Evrard J‐L, Nayer A, Kremer P, Mertens C, Serre C. Controlled comparison of milnacipran and fluoxetine in major depression. Psychopharmacology 1994;114(1):131‐7. [DOI] [PubMed] [Google Scholar]
Clerc 2001 {published data only}
- Clerc G, Assicot M, Bouchard JM, Cottin M, Guibert M, Liegaut D, Engel P, May JP, Oules J, Pagot R, Patris MF, Ruimy P, Sombret A, Amerongen AP, Tournoux A. Antidepressant efficacy and tolerability of milnacipran, a dual serotonin and noradrenaline reuptake inhibitor: A comparison with fluvoxamine. International Clinical Psychopharmacology 2001;16(3):145‐51. [DOI] [PubMed] [Google Scholar]
Endo 1995 {published data only}
- Endo S, Miura S, Miyasaka M, Yamauchi T, Asai M, Ushijima S, Kamijima K, Hasegawa K, Ogura T. Clinical Evaluation of Milnacipran Hydrochloride, a New Antidepressant for Depression and Depressive State Phase III Clinical Trial with Mianserin Hydrochloride as a Control Drug. Rinsho Hyoka 1995;23(1):39‐64. [Google Scholar]
Guelfi 1998a {published data only}
- Guelfi JD, Ansseau M, Corruble E, Samuelian JC, Tonelli I, Tournoux A, Pletan Y. A double‐blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatients. International Clinical Psychopharmacology 1998;13(3):121‐8. [DOI] [PubMed] [Google Scholar]
Lee 2002b {published data only}
- Lee MS, Ham BJ, Kee BS, Kim JB, Yeon BK, Oh KS, Oh BH, Lee C, Jung HY, Chee IS, Choe BM, Paik IH. A comparison of the efficacy and safety between milnacipran and fluoxetine in the treatment of patients with depression. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):205. [Google Scholar]
- Lee MS, Ham BJ, Kee BS, Kim JB, Yeon BK, Oh KS, Oh BH, Lee C, Jung HY, Chee IS, Choe BM, Paik IH. Comparison of efficacy and safety of milnacipran and fluoxetine in Korean patients with major depression. Current Medical Research & Opinion 2005;21(9):1369‐75. [DOI] [PubMed] [Google Scholar]
Leinonen 1997 {published data only}
- Leinonen E, Lepola U, Koponen H, Mehtonen OP, Rimon R. Long‐term efficacy and safety of milnacipran compared to clomipramine inpatients with major depression. Acta Psychiatrica Scandinavica 1997;96(6):497‐504. [DOI] [PubMed] [Google Scholar]
Lopez‐Ibor 2004 {published data only}
- Lopez‐Ibor JJ, Conesa A, Spanish Milnacipran/Imipramine Study. A comparative study of milnacipran and imipramine in the treatment of major depressive disorder. Current Medical Research and Opinion 2004;20(6):855‐60. [DOI] [PubMed] [Google Scholar]
Sechter 2000 {published data only}
- Sechter D, Vandel P, Weiller E, Pezous N, Cabanac F, Tournoux A. A comparative study of milnacipran and paroxetine in outpatients with major depression. Journal of Affective Disorders 2004;83(2‐3):233‐6. [DOI] [PubMed] [Google Scholar]
- Sechter D, Weiller E, Pezous N, Bisserbe JC. Psychomotor retardation is a predictive factor of milnacipran response. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):190. [Google Scholar]
- Vandel P, Sechter D, Weiller E, Pezous N, Cabanac F, Tournoux A, Panconi E. Post‐treatment emergent adverse events in depressed patients following treatment with milnacipran and paroxetine. Human Psychopharmacology 2004;19(8):585‐6. [DOI] [PubMed] [Google Scholar]
Shinkai 2004 {published data only}
- Shinkai K, Yoshimura R, Ueda N, Okamoto K, Nakamura J. Associations between baseline plasma MHPG (3‐methoxy‐4‐hydroxyphenylglycol) levels and clinical responses with respect to milnacipran versus paroxetine treatment. Journal of Clinical Psychopharmacology 2004;24(1):11‐17. [DOI] [PubMed] [Google Scholar]
Tignol 1998 {published data only}
- Tignol J, Pujol Domenech J, Chartres JP, Leger JM, Pletan Y, Tonelli I, Tournoux A, Pezous N. Double‐blind study of the efficacy and safety of milnacipran and imipramine in elderly patients with major depressive episode. Acta Psychiatrica Scandinavica 1998;97(2):157‐65. [DOI] [PubMed] [Google Scholar]
Van Amerongen 2002 {published data only}
- Amerongen AP, Ferrey G, Tournoux A. A randomised, double‐blind comparison of milnacipran and imipramine in the treatment of depression. Journal of Affective Disorders 2002;72(1):21‐31. [DOI] [PubMed] [Google Scholar]
Yamashita 1995 {published data only}
- Matsubara R, Onodera I, Ito K, Okada F, Asano Y. A double‐blind comparison of milnacipran and imipramine in depressive patients. XXth Collegium Internationale Neuro psychopharmacologicum, Melbourne, Australia. 1996.
- Yamashita I, Matubara R, Onodera I, Ito K, Okada K, Asano Y. Clinical evaluation of milnacipran hydrochloride (tn‐912) on depression and depressive states ‐phase iii clinical trial with imipramine hydrochloride as a control drug. Rinsyoiyaku 1995;11(4):819‐42. [Google Scholar]
Yang 2003 {published data only}
- Yang JC, Kim SW, Yu BH. Milnacipran versus Sertraline in Major Depressive Disorder: A Double‐Blind Randomized Comparative Study on the Treatment Effect and cbeta‐Adrenergic Receptor Responsiveness. Korean Journal of Psychopharmacology 2003;14(4):387‐96. [Google Scholar]
References to studies excluded from this review
Baek 2002a {published data only}
- Baek HK, Kim EJ, Yu BH. The Effect of Antidepressant Treatment on Beta‐Adrenergic Receptor Responsiveness in Patients with Major Depression. Korean Journal of Psychopharmacology 2002;13(3):154‐63. [Google Scholar]
Baek 2002b {published data only}
- Baek HK, Yu BH. The effect of antidepressant treatment on beta‐adrenergic receptor responsiveness in patients with major depression. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):148. [Google Scholar]
Dardennes 1998 {published data only}
- Dardennes R, Berdeaux G, Bisserbe JC, Lafuma A, Pribil C, Fagnani F. Cost‐utility of milnacipran (Ixel(r)) in the prevention of recurrent depression. 11th European College of Neuropsychopharmacology Congress. Paris, France. 31st October 4th November 1998.
- Dardennes RM, Lafuma A, Fagnani F, Pribil C, Bisserbe JC, Berdeaux G. Economic assessment of a maintenance treatment strategy in prevention of recurrent depressive disorder. Value in Health 2000;3(1):40‐7. [DOI] [PubMed] [Google Scholar]
- Lafuma A, Dardennes R, Fagnani F, Pribil C, Bisserbe JC, Berdeaux G. Clinico‐economic assessment of milnacipran in the prevention of depressive episodes. Encephale 1999;25(5):401‐7. [PubMed] [Google Scholar]
- Rouillon F, Berdeaux G, Bisserbe JC, Warner B, Mesbah M, Smadja C, Chwalow J. Prevention of recurrent depressive episodes with milnacipran: consequences on quality of life. Journal of Affective Disorders 2000;58(3):171‐80. [DOI] [PubMed] [Google Scholar]
- Rouillon F, Berdeaux G, Chwalow J, Mesbah M. The efficacy of milnacipran (IxelR) in the prevention of recurrent depression: effects on quality of life. 11th European College of Neuropsychopharmacology Congress. Paris, France. 31st October 4th November 1998.
- Rouillon F, Warner B, Pezous N, Bisserbe JC. Milnacipran efficacy in the prevention of recurrent depression: a 12‐month placebo‐controlled study. International Clinical Psychopharmacology 2000;15(3):133‐40. [DOI] [PubMed] [Google Scholar]
Kanemoto 2004 {published data only}
- Kanemoto K, Matsubara M, Yamashita K, Tarao Y, Inada E, Sekine T. Controlled comparison of two different doses of milnacipran in major depressive outpatients. International Clinical Psychopharmacology 2004;19(6):343‐6. [DOI] [PubMed] [Google Scholar]
Lee 2004 {published data only}
- Lee MS, Ham BJ, Kee BS, Kim JB, Yeon BK, Oh KS, Oh BH, Lee C, Jung HY, Chee IS, Choe BM, Paik IH. The Efficacy and Safety of Milnacipran in Patients with Major Depression: A comparison with Fluoxetine. Journal of the Korean Neuropsychiatric Association 2004;43(4):415‐24. [Google Scholar]
Macher 1989 {published data only}
- Macher JP, Sichel JP, Serre C, Frenckell R, Huck JC, Demarez JP. Double‐blind placebo‐controlled study of milnacipran in hospitalized patients with major depressive disorders. Neuropsychobiology 1989;22(2):77‐82. [DOI] [PubMed] [Google Scholar]
Naito 2007 {published data only}
- Shingo N, Kazuhiro S, Keizo Y, Hisashi H, Hitoshi T, Mitsuhiro K, Kenichi I, Tadashi O, Tetsuo S. Gender differences in the clinical effects of fluvoxamine and milnacipran in Japanese major depressive patients. Psychiatry and Clinical Neurosciences 2007;61(4):421‐7. [DOI] [PubMed] [Google Scholar]
Onodera 1992 {published data only}
- Onodera I, Asano Y, Ito K, Matsubara R, Okada F. Double‐blind controlled comparison of two doses of milnacipran in depressive patients. Clinical Neuropharmacology 1992;15(1 Pt B):418. [Google Scholar]
Wyeth 2006 {published data only}
- Wyeth. Phase III multi‐center, double‐blind, comparative study of Effexor XR for the treatment of depression. controlled‐trials.com 2006.
References to studies awaiting assessment
Yoshimura 2007 {published data only}
- Yoshimura R, Mitoma M, Sugita A, Hori H, Okamoto T, Umene W, Ueda N, Nakamura J. Effects of paroxetine or milnacipran on serum brain‐derived neurotrophic factor in depressed patients. Progress in Neuropsychopharmacology and Biological Psychiatry 2007;31:1034–7. [DOI] [PubMed] [Google Scholar]
Additional references
Als‐Nielsen 2003
- Als‐Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials: a reflection of treatment effect or adverse events?. JAMA 2003 Aug 20;290(7):921‐8. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996 Nov 9;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 2000a
- Anderson IM, Nutt DJ, Deakin JF. Evidence‐based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines.British Association for Psychopharmacology. Journal of Psychopharmacology 2000 Mar;14(1):3‐20. [DOI] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III). 3rd Edition. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III‐R). 3rd Edition. American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. American Journal of Psychiatry 2000 Apr;157(4 Suppl):1‐45. [PubMed] [Google Scholar]
Appleton 2006
- Appleton KM, Hayward RC, Gunnell D, Peters TJ, Rogers PJ, Kessler D, Ness AR. Effects of n‐3 long‐chain polyunsaturated fatty acids on depressed mood: systematic review of published trials. American Journal of Clinical Nutrition 2006;84(6):1308‐16. [DOI] [PubMed] [Google Scholar]
Arroll 2005
- Arroll B, Macgillivray S, Ogston S, Reid I, Sullivan F, Williams B, et al. Efficacy and tolerability of tricyclic antidepressants and SSRIs compared with placebo for treatment of depression in primary care: a meta‐analysis. Annals of Family Medicine 2005 Sep‐Oct;3(5):449‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barbui 2004
- Barbui C, Cipriani A, Brambilla P, Hotopf M. "Wish bias" in antidepressant drug trials?. Journal of Clinical Psychopharmacology 2004 Apr;24(2):126‐30. [DOI] [PubMed] [Google Scholar]
Barbui 2007
- Barbui C, Hotopf M, Freemantle N, Boynton J, Churchill R, Eccles MP, Geddes JR, Hardy R, Lewis G, Mason JM. Selective serotonin reuptake inhibitors versus tricyclic and heterocyclic antidepressants: comparison of drug adherence. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: ] [DOI] [PubMed] [Google Scholar]
Bekelman 2003
- Bekelman JE, Li Y, Gross CP. Scope and impact of financial conflicts of interest in biomedical research: a systematic review. JAMA 2003;289(4):454‐65. [DOI] [PubMed] [Google Scholar]
Bhandari 2004
- Bhandari M, Busse JW, Jackowski D, Montori VM, Schunemann H, Sprague S, Mears D, Schemitsch EH, Heels‐Ansdell D, Devereaux PJ. Association between industry funding and statistically significant pro‐industry findings in medical and surgical randomised trials. Canadian Medical Association Journal 2004 Feb 17;170(4):477‐80. [PMC free article] [PubMed] [Google Scholar]
Bollini 1999
- Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta‐analysis of dose‐effect relationships in randomised clinical trials. British Journal of Psychiatry 1999 Apr;174:297‐303. [DOI] [PubMed] [Google Scholar]
Briley 1996
- Briley M, Prost J, Moret C. Preclinical pharmacology of milnacipran. International Clinical Psychopharmacology 1996;11(Suppl. 4):9‐14. [DOI] [PubMed] [Google Scholar]
Buchkowsky 2004
- Buchkowsky SS, Jewesson PJ. Industry sponsorship and authorship of clinical trials over 20 years. Annals of Pharmacotherapy 2004 Apr;38(4):579‐85. [DOI] [PubMed] [Google Scholar]
Chan 2004
- Chan AW, Hrobjartsson A, Haahr MT, Gotzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291(20):2457‐65. [DOI] [PubMed] [Google Scholar]
Chan 2005
- Chan AW, Altman DG. Identifying outcome reporting bias in randomised trials on PubMed: review of publications and survey of authors. BMJ 330;7494:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2005
- Cipriani A, Brambilla P, Furukawa T, Geddes J, Gregis M, Hotopf M, Malvini L, Barbui C. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2005 Oct 19, Issue 4. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2007a
- Cipriani A, Furukawa TA, Veronese A, Watanabe N, Churchill R, McGuire HF and Barbui C for the Meta‐Analysis of New Generation Antidepressants (MANGA) Study Group. Paroxetine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2007b
- Cipriani A, Signoretti A, Furukawa TA, Churchill R, Tomelleri S, Omori IM, McGuire HF and Barbui C for the Meta‐Analysis of New Generation Antidepressants (MANGA) Study Group. Venlafaxine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2009a
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, Watanabe N, Nakagawa A, Omori IM, McGuire H, Tansella M, Barbui C. Comparative efficacy and acceptability of 12 new‐generation antidepressants: a multiple‐treatments meta‐analysis. Lancet 2009;373(9665):746‐58. [DOI] [PubMed] [Google Scholar]
Cipriani 2009b
- Cipriani A, Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, McGuire H, Barbui C. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2:CD006117. [DOI] [PubMed] [Google Scholar]
Cipriani 2009c
- Cipriani A, Santilli C, Furukawa TA, Signoretti A, Nakagawa A, McGuire H, Churchill R, Barbui C. Escitalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2:CD006532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ciuna 2004
- Ciuna A, Andretta M, Corbari L, Levi D, Mirandola M, Sorio A, et al. Are we going to increase the use of antidepressants up to that of benzodiazepines?. European Journal of Clinical Pharmacology 2004 Nov;60(9):629‐34. [DOI] [PubMed] [Google Scholar]
Cowen 2005
- Cowen PJ, Ogilvie AD, Gama J. Efficacy, safety and tolerability of duloxetine 60 mg once daily in major depression. Current Medical Research and Opinion 2005;21(3):345‐56. [DOI] [PubMed] [Google Scholar]
Coyne 1994
- Coyne JC, Fechner‐Bates S, Schwenk TL. Prevalence, nature, and comorbidity of depressive disorders in primary care. General Hospital Psychiatry 1994 Jul;16(4):267‐76. [DOI] [PubMed] [Google Scholar]
Elliott 2007
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta‐analysis. Lancet 2007;369(9557):201‐7. [DOI] [PubMed] [Google Scholar]
Ellis 2004
- Ellis P. Australian and New Zealand clinical practice guidelines for the treatment of depression. Australian and New Zealand Journal of Psychiatry 2004 Jun;38(6):389‐407. [DOI] [PubMed] [Google Scholar]
Frampton 2007
- Frampton JE, Plosker GL. Duloxetine : a review of its use in the treatment of major depressive disorder. CNS Drugs 2007;21(7):581‐609. [DOI] [PubMed] [Google Scholar]
Furukawa 2002a
- Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the 'number needed to treat'? An empirical study of summary effect measures in meta‐analyses. International Journal of Epidemiology 2002 Feb;31(1):72‐6. [DOI] [PubMed] [Google Scholar]
Furukawa 2002b
- Furukawa TA, McGuire H, Barbui C. Meta‐analysis of effects and side effects of low dosage tricyclic antidepressants in depression: systematic review. BMJ 2002 Nov 2;325(7371):991‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta‐analysis. International Clinical Psychopharmacology 2005;20(1):49‐52. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006 Jan;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Geddes 2000
- Geddes JR, Freemantle N, Mason J, Eccles MP, Boynton J. SSRIs versus other antidepressants for depressive disorder. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: ] [DOI] [PubMed] [Google Scholar]
Goldstein 2002
- Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack MA. Duloxetine in the treatment of major depressive disorder: a double‐blind clinical trial. Journal of Clinical Psychiatry 2002;63(6):225‐31. [DOI] [PubMed] [Google Scholar]
Greenberg 2003
- Greenberg PE, Kessler RC, Birnbaum HG, Leong SA, Lowe SW, Berglund PA, Corey‐Lisle PK. The economic burden of depression in the United States: how did it change between 1990 and 2000?. Journal of Clinical Psychiatry 2003 Dec;64(12):1465‐75. [DOI] [PubMed] [Google Scholar]
Greenberg 2005
- Greenberg PE, Birnbaum HG. The economic burden of depression in the US: societal and patient perspectives. Expert Opinion on Pharmacotherapy 2005 Mar;6(3):369‐76. [DOI] [PubMed] [Google Scholar]
Guaiana 2005
- Guaiana G, Andretta M, Corbari L, Mirandola M, Sorio A, D'Avanzo B, Barbui C. Antidepressant drug consumption and public health indicators in Italy, 1955 to 2000. Journal of Clinical Psychiatry 2005 Jun;66(6):750‐5. [DOI] [PubMed] [Google Scholar]
Guaiana 2007
- Guaiana G, Barbui C, Hotopf M. Amitriptyline versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: ] [DOI] [PubMed] [Google Scholar]
Guelfi 1998
- Guelfi JD, Ansseau M, Corruble E, Samuelian JC, Tonelli I, Tournoux A, Pletan Y. A double‐blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatients. International Clinical Psychopharmacology 1998 May;13(3):121‐8. [DOI] [PubMed] [Google Scholar]
Guy 1970
- Guy W, Bonato RR. Manual for the ECDEU Assessment Battery.2. Chevy Chase, MD: National Institute ofMental Health, 1970.. Manual for the ECDEU Assessment Battery 2. Chevy Chase, MD: National Institute of Mental Health, 1970. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hansen 2005
- Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second‐generation antidepressants in the treatment of major depressive disorder. Annals of Internal Medicine 2005 Sep 20;143(6):415‐26. [DOI] [PubMed] [Google Scholar]
Heres 2006
- Heres S, Davis J, Maino K, Jetzinger E, Kissling W, Leucht S. Why olanzapine beats risperidone, risperidone beats quetiapine, and quetiapine beats olanzapine: an exploratory analysis of head‐to‐head comparison studies of second‐generation antipsychotics. American Journal of Psychiatry 2006;163(2):185‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003 Sep 6;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5. Chichester, UK: John Wiley & Sons, Ltd., May 2005. [Google Scholar]
Higgins 2008a
- Higgins JPT, Altman DG. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 5.01 (updated Sept 2008) www.cochrane‐handbook.org. The Cochrane Collaboration, 2008. [Google Scholar]
Higgins 2008b
- Higgins JPT, Altman DG. Section 8. Assessing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 5.01 (updated Sept 2008) www.cochrane‐handbook.org. The Cochrane Collaboration, 2008. [Google Scholar]
Imperadore 2007
- Imperadore G, Cipriani A, Signoretti A, Furukawa TA, Watanabe N, Churchill R, McGuire HF, Barbui C for the Meta‐Analysis of New Generation Antidepressants (MANGA) Study Group. Citalopram versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. [Google Scholar]
Ioannidis 2006
- Ioannidis JP. Indirect comparisons: the mesh and mess of clinical trials. Lancet 2006;368(9546):1470‐2. [DOI] [PubMed] [Google Scholar]
Ito 2002
- Ito K, Yoshida K, Sato K, Takahashi H, Kamata M, Higuchi H, Shimizu T, Itoh K, Inoue K, Tezuka T, Suzuki T, Ohkubo T, Sugawara K, Otani K. A variable number of tandem repeats in the serotonin transporter gene does not affect the antidepressant response to fluvoxamine. Psychiatry Research 2002;111(2‐3):235‐9. [DOI] [PubMed] [Google Scholar]
Kessler 2003
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. National Comorbidity Survey Replication. JAMA 2003 Jun 18;289(23):3095‐105. [DOI] [PubMed] [Google Scholar]
Khan 2003
- Khan A, Khan SR, Walens G, Kolts R, Giller EL. Frequency of positive studies among fixed and flexible dose antidepressant clinical trials: an analysis of the food and drug administration summary basis of approval reports. Neuropsychopharmacology 2003 Mar;28(3):552‐7. [DOI] [PubMed] [Google Scholar]
Kupfer 2002
- Kupfer DJ, Frank E. Placebo in clinical trials for depression: complexity and necessity. JAMA 2002;287:1853‐54. [DOI] [PubMed] [Google Scholar]
Lecrubier 1996
- Lecrubier Y, Pletan Y, Solles A, Tournoux A, Magne V. Clinical efficacy of milnacipran: placebo‐controlled trials. International Clinical Psychopharmacology 1996 Sep;11(Suppl 4):29‐33. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003 May 31;326(7400):1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Linde 2008
- Linde K, Berner MM, Kriston L. St John's wort for major depression. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lopez‐Ibor 1996
- Lopez‐Ibor J, Guelfi JD, Pletan Y, Tournoux A, Prost JF. Milnacipran and selective serotonin reuptake inhibitors in major depression. International Clinical Psychopharmacology 1996 Sep;11(Suppl 4):41‐6. [DOI] [PubMed] [Google Scholar]
Lu 2004
- Lu M, Ades T. Combination of direct and indirect evidence in mixed treatment comparison. Statistics in Medicine 2004;23:3105‐24. [DOI] [PubMed] [Google Scholar]
Lu 2006
- Lu M, Ades T. Assessing evidence consistency in mixed treatment comparisons. Journal of the American Statistical Association 2006;101:447‐59. [Google Scholar]
Luborsky 1962
- Luborsky L. Clinician's judgments of mental health. Archives of General Psychiatry 1962;7:407‐17. [DOI] [PubMed] [Google Scholar]
Lumley 2002
- Lumley S. Network meta‐analysis for indirect treatment comparisons. Statistics in Medicine 2002;21:2313‐2324. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Montgomery 2004
- Montgomery JH, Byerly M, Carmody T, Li B, Miller DR, Varghese F, Holland R. An analysis of the effect of funding source in randomized clinical trials of second generation antipsychotics for the treatment of schizophrenia. Controlled Clinical Trials 2004 Dec;25(6):598‐612. [DOI] [PubMed] [Google Scholar]
Moret 1985
- Moret C, Charveron M, Finberg JPM, Couzinier JP, Briley M. Biochemical profile of milnacipran (F 2207), 1‐phenyl‐1‐diethyl‐aminocarbonyl‐2‐aminomethyl‐cyclopropane (Z) hydrochloride, a potential fourth generation antidepressant drug. Neuropharmacology 1985;24:1211‐19. [DOI] [PubMed] [Google Scholar]
NICE 2007
- NICE. Depression: management of depression in primary and secondary care (amended). London: National Institute for Health and Clinical Excellence, 2007. [Google Scholar]
Nose 2007
- Nose M, Cipriani A, Furukawa TA, Omori IM, Churchill R, McGuire HF, Barbui C for the Meta‐Analysis of New Generation Antidepressants (MANGA) Study Group. Duloxetine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. [Google Scholar]
Okamura 2006
- Okamura K, Furukawa TA. Remission rates with milnacipran 100mg/day and 150 mg/day in the long‐term treatment of major depression. Clinical Drug Investigation 2006;26(3):135‐42. [DOI] [PubMed] [Google Scholar]
Omori 2006
- Omori IM, Watanabe N, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA for the MANGA Study. Fluvoxamine versus other anti‐depressive agents for depression. (Protocol). Cochrane Database of Systematic Reviews 2006, Issue 3: CD006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Oxman 1992
- Oxman AD, Guyatt GH. A consumer's guide to subgroup analyses. Annals of Internal Medicine 1992 Jan 1;116(1):78‐84. [DOI] [PubMed] [Google Scholar]
Papakostas 2007a
- Papakostas GI, Fava M. A meta‐analysis of clinical trials comparing milnacipran, a serotonin‐norepinephrine reuptake inhibitor, with a selective serotonin reuptake inhibitor for the treatment of major depressive disorder. European Neuropsychopharmacology 2007;17(1):32‐6. [DOI] [PubMed] [Google Scholar]
Perlis 2005
- Perlis RH, Perlis CS, Wu Y, Hwang C, Joseph M, Nierenberg AA. Industry sponsorship and financial conflict of interest in the reporting of clinical trials in psychiatry. American Journal of Psychiatry 2005 Oct;162(10):1957‐60. [DOI] [PubMed] [Google Scholar]
Procyshyn 2004
- Procyshyn RM, Chau A, Fortin P, Jenkins W. Prevalence and outcomes of pharmaceutical industry‐sponsored clinical trials involving clozapine, risperidone, or olanzapine. Canadian Journal of Psychiatry 2004 Sep;49(9):601‐6. [DOI] [PubMed] [Google Scholar]
Psaty 2003
- Psaty BM, Lumley T, Furberg CD, Schellenbaum G, Pahor M, Alderman MH, Weiss NS. Health outcomes associated with various antihypertensive therapies used as first‐line agents: a network meta‐analysis. JAMA 2003;289(19):2533‐44. [DOI] [PubMed] [Google Scholar]
Puech 1997
- Puech A, Montgomery SA, Prost JF, Solles A, Briley M. Milnacipran, a new serotonin and noradrenaline reuptake inhibitor: an overview of its antidepressant activity and clinical tolerability. International Clinical Psychopharmacology 1997 Mar;12(2):99‐108. [DOI] [PubMed] [Google Scholar]
Puozzo 1996
- Puozzo C, Leonard BE. Pharmacokinetics of milnacipran in comparison with other antidepressants. International Clinical Psychopharmacology 1996 Sep;11(Suppl 4):15‐27. [DOI] [PubMed] [Google Scholar]
Puozzo 2005
- Puozzo C, Lens S, Reh C, Michaelis K, Rosillon D, Deroubaix X, Deprez D. Lack of interaction of milnacipran with the cytochrome P450 isoenzymes frequently involved in antidepressant metabolism. Clinical Pharmacokinetics 2005;44:977‐8. [DOI] [PubMed] [Google Scholar]
Salanti 2008
- Salanti G, Higgins JP, Ades A, Ioannidis JP. Evaluation of networks of randomized trials. Statistical Methods in Medical Research 2008;17:279‐301. [DOI] [PubMed] [Google Scholar]
Santaguida 2005
- Santaguida PL, Helfand M, Raina P. Challenges in systematic reviews that evaluate drug efficacy or effectiveness. Annals of Internal Medicine 2005;142(12 Pt 2):1066‐72. [DOI] [PubMed] [Google Scholar]
Sawada 2001
- Sawada Y, Ohtani H. Pharmacokinetics and drug interactions of antidepressive agents. Nippon Rinsho 2001;59:1539‐1545. [PubMed] [Google Scholar]
Sechter 2004
- Sechter D, Vandel P, Weiller E, Pezous N, Cabanac F, Tournoux A, study co‐coordinators. A comparative study of milnacipran and paroxetine in outpatients with major depression. Journal of Affective Disorders 2004 Dec;83(2‐3):233‐6. [DOI] [PubMed] [Google Scholar]
Smith 2002
- Smith D, Dempster C, Glanville J, Freemantle N, Anderson I. Efficacy and tolerability of venlafaxine compared with selective serotonin reuptake inhibitors and other antidepressants: a meta‐analysis. British Journal of Psychiatry 2002 May;180:396‐404. [DOI] [PubMed] [Google Scholar]
Spencer 1998
- Spencer CM, Wilde MI. Milnacipran. A review of its use in depression. Drugs 1998 Sep;56(3):405‐27. [DOI] [PubMed] [Google Scholar]
Steen 1997
- Steen A, Boer JA. A double‐blind six months comparative study of milnacipran and clomipramine in major depressive disorder. International Clinical Psychopharmacology 1997 Sep;12(5):269‐81. [DOI] [PubMed] [Google Scholar]
Thase 2001
- Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. British Journal of Psychiatry 2001;178:234‐41. [DOI] [PubMed] [Google Scholar]
Walsh 2002
- Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA 2002 Apr 10;287(14):1840‐7. [DOI] [PubMed] [Google Scholar]
Ware 1993
- Ware JE, Snow KK, Kosinski M, Gandek B. SF‐36 Health Survey Manual and Interpretation Guide. Boston,MA: New England Medical Centre, 1993. [Google Scholar]
Watanabe 2006
- Watanabe N, Barbui C, Churchill R, Cipriani A, Furukawa TA, McGuire HF, Omori IM for the MANGA Study. Mirtazapine versus other anti‐depressive agents for depression (Protocol).. Cochrane Database of Systematic Reviews 2006, Issue 3: CD006528. [Google Scholar]
WHO 1978
- World Health Organization. The Ninth Revision of the International Classification of Disease and Related Health Problems (ICD‐9). World Health Organization, 1978. [Google Scholar]
WHO 1992
- World Health Organization. The Tenth Revision of the International Classification of Disease and Related Health Problems (ICD‐10). World Health Organization, 1992. [Google Scholar]
WHO 2004
- World Health Organization. The Global Burden of Disease 2004 Update. Geneva: World Health Organization, 2004. [http://www.who.int/entity/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf] [Google Scholar]
WHOQOL Group 1998
- WHOQOL Group. The World Health Organization quality of life assessment (WHOQOL): Development and general psychometric properties. Social Science and Medicine 1998;46(12):1569‐65. [DOI] [PubMed] [Google Scholar]
Williams 2000
- Williams JW, Jr, Mulrow CD, Chiquette E, Noel PH, Aguilar C, Cornell J. A systematic review of newer pharmacotherapies for depression in adults: evidence report summary. Annals of Internal Medicine 2000 May 2;132(9):743‐56. [DOI] [PubMed] [Google Scholar]
Williams 2001
- Williams JB. Standardizing the Hamilton Depression Rating Scale: past, present, and future. European Archives of Psychiatry and Clinical Neuroscience 2001;251(Suppl 2):II16‐12. [DOI] [PubMed] [Google Scholar]
Wing 1994
- Wing J. Measuring mental health outcomes: a perspective from the Royal College of Psychiatrists. Outcomes into Clinical Practice. London, London: BMJ Publishing, 1994. [Google Scholar]
Yoshida 2002
- Yoshida K, Ito K, Sato K, Takahashi H, Kamata M, Higuchi H, Shimizu T, Itoh K, Inoue K, Tezuka T, Suzuki T, Ohkubo T, Sugawara K, Otani K. Influence of the serotonin transporter gene‐linked polymorphic region on the antidepressant response to fluvoxamine in Japanese depressed patients. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2002;26(2):383‐6. [DOI] [PubMed] [Google Scholar]
Zimmerman 2004
- Zimmerman M, Posternak MA, Chelminski I. Derivation of a definition of remission on Montgomery‐Asberg depression rating scale corresponding to the definition of remission on the Hamilton rating scale for depression. Journal of Psychiatric Research 2004;38:577‐82. [DOI] [PubMed] [Google Scholar]