

Abstract
The mussel byssus is a remarkable attachment structure that is formed by injection molding and rapid in-situ hardening of concentrated solutions of proteins enriched in the catecholic amino acid 3,4-dihydroxy-L-phenylalanine (DOPA). Fe3+, found in high concentrations in the byssus, has been speculated to participate in redox reactions with DOPA that lead to protein polymerization, however direct evidence to support this hypothesis has been lacking. Using small molecule catechols, DOPA-containing peptides, and native mussel foot proteins, we report the first direct observation of catechol oxidation and polymerization accompanied by reduction of Fe3+ to Fe2+. In the case of the small molecule catechol, we identified two dominant dimer species and characterized their connectivities by nuclear magnetic resonance (NMR), with the C6-C6 and C5-C6 linked species as the major and minor products, respectively. For the DOPA-containing peptide, we studied the pH dependence of the reaction and demonstrated that catechol polymerization occurs readily at low pH, but is increasingly diminished in favor of metal-catechol coordination interactions at higher pH. Finally, we demonstrate that Fe3+ can induce cross-links in native byssal mussel proteins mefp-1 and mcfp-1 at acidic pH. Based on these findings, we discuss the potential implications to the chemistry of mussel adhesion.
Introduction
Mussels attach to underwater surfaces via a cluster of threads (the mussel byssus) that adhere strongly to solid surfaces via a terminal adhesive plaque (Figure 1a). Each thread of the mussel byssus is individually formed with an adhesive plaque in a fascinating manner akin to injection molding. Liquid protein secretions are extruded into the ventral groove and distal depression of the mussel foot, whereupon they solidify in only a few minutes to yield a newly formed byssal thread that, along with other threads, functions to tether the organism to the surface. Using this process, mussels are able to strongly bind to organic and inorganic surfaces in aqueous environments where many other glues fail.1–5
Figure 1.

Mussel byssus, model for pH-dependent Fe3+-catechol chemistry, and molecules studied. (a) Picture of mussel with labeled byssal structures. (b) Model for pH-dependent interactions between Fe3+ and catechol. At acidic pH, catechols and Fe3+ react to produce o-quinones, leading to catechol-catechol oligomerization. At basic pH, catechols form coordination bonds with Fe3+ that prevent oxidation of the catechol. In the context of the mussel byssus, Fe3+ could hypothetically induce covalent cross-linking during acidic processing of byssal protein secretions and enhance the mechanical properties of the byssal thread and adhesive by formation of mechanically active Fe3+-catechol coordination interactions upon equilibration to basic marine pH. Note that Fe3+-catechol coordination bonds would be expected to be mechanically reversible, whereas covalent catechol-catechol bonds would not.23 (c) Catechol-containing molecules studied: (1) DHPA and (2) Ac-Ser-DOPA-NH2 (dipeptide).
Previous studies revealed that 3,4-dihydroxy-L-phenylalanine (DOPA), an amino acid formed by post-translational modification of tyrosine, is distributed throughout the byssus with especially high concentrations in proteins located near the plaque-substrate interface.6–9 Although a comprehensive understanding of the role of DOPA in mussel adhesion has not been reached, existing experimental evidence points to both adhesive and cohesive functions. Strong interfacial interactions between DOPA or oxidized DOPA residues and surfaces likely contribute to adhesion between the mussel adhesive plaque and substrate,1–5 whereas, within the bulk of the adhesive plaque, DOPA-DOPA polymerization is thought to be a source of cross-linking that ultimately leads to solidification into a cohesive elastic solid.10–12
The concentration of Fe3+ in the mussel byssus has been measured at greater than 1 part per thousand - many orders of magnitude greater than the concentration of Fe3+ in seawater (typically 10 parts per billion).13–16 Through pulse 59Fe experiments, 59Fe has been found to be taken up by the mussel and redistribute into byssal thread.17 Fe3+ has been shown to form strong coordination bonds with catechols, in which one, two, or three catecholates can bind a single ferric ion.13, 18–21 Such noncovalent cross-links may result in a network of coordination bonds serving a mechanical role within adhesive plaques and byssal threads,22 as demonstrated recently using the surface forces apparatus and in biomimetic catechol polymer gels.4, 5, 23–27
Aside from these coordination interactions and literature reports of catechol oxidation to o-quinone by Fe3+,28–30 many aspects of catechol-Fe3+ interactions in the mussel byssal protein system remain unclear. Native oxide surfaces of steel have been reported to cure DOPA-containing polypeptides, which the authors suggest is due to the presence of soluble Fe3+ leading to the oxidation of DOPA.31 We recently demonstrated that catechol-modified poly(ethylene glycol) polymers could be covalently cross-linked at acidic pH in the presence of Fe3+.24 Others have speculated that Fe3+ is involved in redox-mediated covalent curing of mussel adhesives.15, 32–34 Though this type of metal-mediated dimerization has been known for decades to occur with phenols and naphthols,35, 36 to our knowledge, no direct evidence of similar reactions with catechols exists to support the hypothesis that Fe3+ could covalently cure mussel adhesives.
In an attempt to elucidate more details of Fe3+-catechol interactions relevant to mussel adhesion, we studied the reaction of small molecule catechols and DOPA-containing peptides (Figure 1b) with Fe3+ using spectroscopic and chromatographic methods that allow for conclusive detection of covalent catechol cross-linking, including the pH dependence of such reactions. In our initial studies, monofunctional catechols were selected over native mussel proteins and multivalent catechol polymers in order to more precisely characterize the reaction products.11 The reaction between a catechol-containing small molecule mimic of DOPA, 3-(3,4-dihydroxyphenyl)propionic acid (DHPA, 1, Figure 1c), and Fe3+ was studied as a function of time and metal:catechol stoichiometry by high performance liquid chromatography (HPLC), mass spectrometry (MS), and ultraviolet-visible spectroscopy (UV-Vis). At acidic pH, DHPA and Fe3+ mixtures formed catechol-catechol dimers within 1 min and higher oligomers at longer reaction times, along with the reduction of Fe3+ to Fe2+. The carbon-carbon connectivities of the main dimer reaction products were determined by nuclear magnetic resonance spectroscopy (NMR). The pH dependence of Fe3+-DOPA interactions was assessed with a DOPA-containing peptide (Ac-Ser-DOPA-NH2, 2, Figure 1c). Covalent cross-linking was demonstrated to occur at acidic pH but not at alkaline marine pH in the presence of Fe3+. Lastly, Fe3+ mediated cross-linking of the DOPA-rich mussel foot proteins mefp-1 and mcfp-1 was demonstrated at acidic pH.
Experimental Section
Materials
Acetonitrile, DHPA, ferric chloride hexahydrate, ferrous chloride tetrahydrate, formic acid, 1,10-phenanthroline hydrochloride monohydrate (phenanthroline), glacial acetic acid, sodium acetate trihydrate, bis-tris, bis-tris HCl, bicine, and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma Aldrich (Milwaukee, WI). All solutions of FeCl2 were made fresh so that spontaneous oxidation of Fe2+ to Fe3+ was minimized. All chemicals were used without further purification.
Oxidation of DHPA by Fe3+
Stock solutions of DHPA (75 mM) and FeCl3 (100 mM, 200 mM, 300 mM) were prepared in Nanopure H2O. Reactions were initiated by combining 800 μL of the DHPA stock solution and 200 μL of the appropriate FeCl3 solution. Fe3+:DHPA ratios of 1:3, 2:3, and 3:3 were accomplished by adding 200 μL of 100, 200, and 300 mM FeCl3, respectively. For reactions with Fe3+:DHPA ratios of 4:3 and 6:3, 800 μL of the DHPA stock solution was mixed with 400 μL of 200 mM FeCl3 and 300 mM FeCl3, respectively.
HPLC of small molecule reactions
The reaction products were separated by reversed-phase HPLC (Waters; Milford, MA) with a diphenyl column (Grace, Vydac # 219TP54). Separations were performed isocratically with a mobile phase of 90 % water with 0.1 % formic acid and 10 % acetonitrile and a flow rate of 1.0 mL/min. UV detection was performed at 220 nm and 280 nm.
MS of DHPA reaction
The reaction solutions were analyzed at Northwestern University’s Integrated Molecular Structure Education and Research Center (IMSERC) on an Agilent 1100 LC/MSD high performance ion trap mass spectrometer by direct injection in negative ion mode with water as the mobile phase.
HPLC-MS of DHPA reactions
The reaction solutions described above were diluted tenfold and the reaction products were separated by reversed-phase HPLC at IMSERC on an Agilent 1100 LC/MSD high performance ion trap mass spectrometer in negative ion mode equipped with a diphenyl column (Grace, Vydac # 219TP54). The isocratic solvent system used for separations was 90 % water with 0.1 % formic acid and 10 % acetonitrile at a flow rate of 1.0 mL/min. UV detection was performed at 220 nm and 280 nm.
NMR characterization of dimer products
HPLC was performed as described above, except that the solvent system was composed of 90 % water with 0.1 % trifluoroacetic acid (TFA) and 10 % acetonitrile with 0.1 % TFA. The products were collected, frozen, lyophilized, and dissolved in D2O. NMR spectra were acquired at IMSERC at 25 °C. 1H 1D and 2D-J-resolved spectra were acquired on a 500 MHz Varian Inova spectrometer equipped with a 5 mm Nalorac 13C{1H} dual broadband probe bearing z-axis pulsed field gradients. Data were acquired using VNMRJ 2.2D software. The 2D-J experiment employed Varian’s pulse sequence “hom2dj.c”. 1H, 13C, 1H-13C HSQC and HMBC spectra were acquired in automation on a 500 MHz Bruker Avance-III spectrometer equipped with a 5 mm 13C-detect DCH cryoprobe and Z-axis pulsed field gradients. The sample of Dimer 2 (the minor dimer product) was too dilute to obtain a 13C spectrum, but 13C resonance assignments were made using 2D HSQC and HMBC spectra.
UV-Vis characterization of Fe3+ reduction
Spectra of the following solutions were acquired on an Agilent HP8452 spectrometer at Northwestern University’s Keck Biophysics Facility: phenanthroline (20 μM); DHPA (30 μM) + phenanthroline (20 μM); FeCl3 (20 μM) + phenanthroline (20 μM); FeCl2 (20 μM) + phenanthroline (20 μM); DHPA (30 μM) + FeCl3 (20 μM) + phenanthroline (20 μM); DHPA (30 μM) + NaIO4 (15 μM) + phenanthroline (20 μM).
Synthesis of DOPA-containing dipeptide
Ac-Ser-DOPA-NH2 was prepared by standard Fmoc solid phase peptide synthesis methods (Supplementary Methods).37
pH-dependent oxidation of DOPA-containing dipeptide by Fe3+
Ac-Ser-DOPA-NH2 was dissolved in water or buffer. For 1× FeCl3 samples, 1× FeCl3 was added to the sample from a 100 mM FeCl3 stock within 1 min of dissolving the dipeptide in buffer. The final dipeptide concentration was 50 mM, and the final buffer concentration was 250 mM for all reactions. Reactions were studied at pH 5, 7, and 9, using acetate, bis-tris, and bicine buffers for each pH, respectively. After 1 h, 10× EDTA was added from a 0.100 M EDTA stock and the final peptide concentration was diluted to 1 mg/mL with water.
HPLC-MS of dipeptide reactions
The reaction solution from dipeptide reactions (10 μL) were injected onto an Agilent 6210 LC-TOF operated in negative ion mode and equipped with a Waters dC18 Atlantis column (#186001346) at a flow rate of 1.0 mL/min and column temperature of 45 °C at IMSERC. Mobile phases used were A, water with 0.1% formic acid, and B, acetonitrile with 0.1 % formic acid. A linear gradient method was employed: 0–5 min, 3 % B isocratic; 5–20 min, 3–50 % B; 20–25 min, 50–90 % B; 25–30 min, 90 % B. UV absorbance was collected at 280 and 220 nm. Approximately 50 % of the eluent was diverted after the column and UV detector, before reaching the MS. Extracted ion chromatograms were used to identify corresponding HPLC peaks.
Purification of mfp-1
Mfp-1 from Mytilus californianus (mcfp-1) was purified from mussels harvested off Goleta Pier, (Santa Barbara, CA), and held in circulation tanks before shucking the mussels and extracting the foot. Mfp-1 from Mytilus edulis (mefp-1) was purified from mussels feet supplied by Northeast Transport (Waldoboro, ME). Both proteins were purified as described previously,38 with some modifications. Briefly, the feet were frozen to −70 °C before flaying away the pigmented epithelium. Approximately 50 prepared feet were homogenized in four equivalents (w/v) of 5% acetic acid (v/v) with 10 μM leupeptin, 10 μM pepstatin, and 1 mM EDTA in a glass Kontes tissue grinder (Vineland, NJ) on ice and centrifuged at 20,000 × g, 4 °C for 40 min. The supernatant was acidified with 70 % perchloric acid to a final concentration of 1.5 % (v/v). After centrifugation at 20,000 × g, 4 °C for 40 min, the supernatant was dialyzed 4 × 4 L of 5 % acetic acid (v/v) for 4 h in 1,000 kDa molecular weight cutoff dialysis tubing (Spectrum Industries, Los Angeles, CA) before freeze drying. The lyophilized protein was resuspended in 200 μL of 5 % acetic acid (v/v) and 50 μL aliquots were run over a size exclusion column (Shodex KW-803 5 μm, 8 × 300 mm, New York, NY). The eluent was monitored at 280 nm and those fractions positive for protein were subjected to acid-urea polyacrylamide gel electrophoresis (7.5% acrylamide and 0.2% N,N-methylenebisacrylamide) containing 5% acetic acid and 8 M urea.39 After electrophoresis, gels were stained with Sigma-Aldrich Coomassie Blue R-250 (Brooklyn, NY). Pure mfp-1 fractions were pooled and aliquoted before freeze-drying and stored at −70 °C for future use.
Fe3+-mediated protein cross-linking
Protein (mefp-1 or mcfp-1) was resuspended to 50 μM in 0.1 M formic acid buffer (pH 3.0) (Fisher), mixed with ferric chloride adjusted to three different concentrations (0, 0.005, 0.05, 0.5 μM), and incubated for 5 min at room temperature. Solutions were diluted (1/5) with 0.1 M formic acid pH 3.0 with a small amount of tracking dye (0.3 μM methyl green, 5% acetic acid, 8M urea) and loaded onto an acid-urea polyacrylamide gel and ran for 1–2 h at 110 V and stained with Coomassie blue.
Results
Fe3+-induced cross-linking of a DOPA mimic
DHPA (Figure 1c, 1) was chosen as the catechol-containing small molecule because the presence of the carboxylic acid allowed the products to remain soluble throughout the reaction (other catechols studied formed precipitates). Initial reactions were carried out in unbuffered water, which was acidic (pH ~2) due to the presence of FeCl3. Upon mixing Fe3+ (orange) and DHPA (pale yellow) solutions, the mixture immediately turned deep green (Figure S1), a color associated previously with a noncovalent coordination interaction between one Fe3+ ion and one catechol.18, 28 As time progressed, the solution transitioned to yellow (~1 h) and brown (~2 d), reminiscent of catechol-quinone tanning.40 Ion trap mass spectrometric analysis of the reactant solutions revealed a mixture of unreacted DHPA, dimers and higher oligomers (Figure 2 and Figure S2). Several Fe3+:DHPA ratios ranging from iron-rich to iron-deficient were examined (Table 1) in order to determine the dependence of the reaction on reactant stoichiometry. At excess DHPA (Reaction 1, Fe3+:DHPA of 1:3), a large portion of DHPA remained, with limited dimer evident in the mass spectrum (Figure S2). With increasing Fe3+, the amount of unreacted DHPA decreased and the amount of higher mass products increased. At excess Fe3+ (Reaction 5, Fe3+:DHPA of 6:3), only a small portion of DHPA remained (Figure 2), in favor of dimers and trace quantities of trimers and tetramers. Similar results were obtained when the reaction was performed with 4-methylcatechol (4MC) (Supplementary Methods and Figure S3), indicating that the carboxylic acid of DHPA did not significantly influence the reaction.
Figure 2.
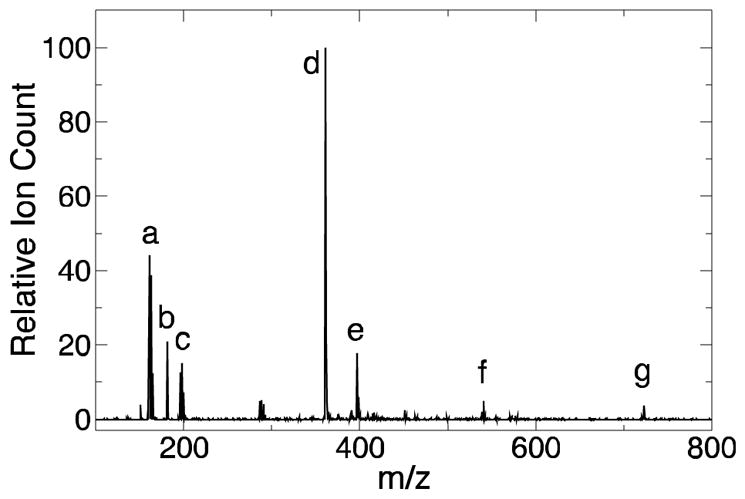
Fe3+ induces DHPA multimer formation. Representative negative ion MS spectrum of direct injection of reaction mixture (Reaction 5, Fe3+:DHPA of 6:3) after 7 d. The solution contained iron species (peaks a and c at m/z of 161 and 198, respectively), DHPA (peak b at m/z of 181), dimers (peak d at m/z of 361), trimers (peak f at m/z of 541), and tetramers (peak g at m/z of 721). Peak e represents dimers associating with a chloride ion (m/z of 397).
Table 1.
Fe3+-DHPA reactions and residual DHPA after 7 d
| Reaction | [Fe3+] (mM) | [DHPA] (mM) | [Fe3+]:[DHPA] | % DHPA unreacteda |
|---|---|---|---|---|
| 1 | 20 | 60 | 1:3 | 77 |
| 2 | 40 | 60 | 2:3 | 57 |
| 3 | 60 | 60 | 3:3 | 43 |
| 4 | 67 | 50 | 4:3 | 29 |
| 5 | 100 | 50 | 6:3 | 12 |
As determined by HPLC (Figure 3a)
In order to gain a more complete understanding of the reaction between Fe3+ and DHPA, HPLC was used to separate the products in the reaction mixture. Due to the aromatic character of DHPA and its dimers, a diphenyl column provided maximal separation of the reaction components. Initially, HPLC-MS was performed in order to identify the molecule associated with each HPLC peak (Figure S4). Two aspects of the HPLC-MS data were extremely interesting: the o-quinone form of DHPA (m/z = 179) could be specifically identified, and two different peaks associated with the dimer (m/z = 361) were detected, each separated by 3.5 min.
Using the assignments from HPLC-MS, the effect of Fe3+:DHPA on the reaction products was further studied (Figure 3a). By determining the area under the DHPA peak relative to the total area under the peaks of DHPA and dimers, the amount of DHPA consumed in the reaction could be estimated (Table 1). At low Fe3+:DHPA (1:3), over 77 % of the DHPA remained unreacted. However, the amount of unreacted DHPA decreased to 57 %, 43 %, 29 %, and 12 % at Fe3+:DHPA of 2:3, 3:3, 4:3, and 6:3, respectively. An interesting trend was noted in the relative abundance of the two dimer peaks appearing at 7.5 and 10.5 min (Table S1). The ratio of the peak areas of major (7.5 min; Dimer 1) to minor (10.5 min; Dimer 2) dimer products increased from 2.5 at Fe3+:DHPA of 1:3 to 5.8 at Fe3+:DHPA of 6:3. Thus, increasing the amount of Fe3+ relative to DHPA favors production of Dimer 1.
Figure 3.
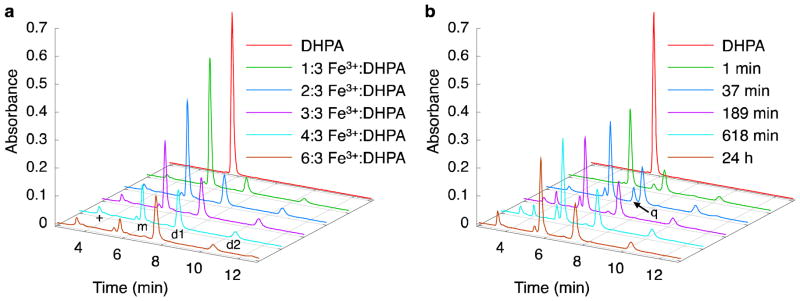
Two DHPA-dimer species are produced after Fe3+-coupled oxidation. HPLC chromatograms (280 nm) of reactions between DHPA and Fe3+. (a) Effect of stoichiometry on the reaction between DHPA and Fe3+ after 7 d. The peaks at ~3.5 min (+), ~5.5 min (m), ~7.5 min (d1), and ~10.5 min (d2) correspond to soluble Fe3+ and Cl− ions, DHPA, Dimer 1, and Dimer 2; these results were determined by HPLC-MS (Figure S4). (b) Time dependence of the reaction between DHPA and Fe3+ (1:1 molar equiv.). The peaks are identified as in (a), with the addition of a small peak at ~7 min (q) that corresponds to the o-quinone form of DHPA.
The time dependence of the reaction between Fe3+ and DHPA was also studied (Figure 3b). Using Fe3+:DHPA equal to 3:3 as a representative reaction condition, the reaction mixture was analyzed by HPLC after 1 min, 37 min, 189 min, 618 min, and 24 h. A general observation about this chemical transformation is that a significant amount of DHPA is consumed within the first minute, yielding dimer products (Figure 3b and Table S1). Between the two forms of dimers, Dimer 1 was the dominant species at all time points and was roughly 3.2–3.6 times more abundant than Dimer 2. An interesting feature of the time-dependent HPLC is the transient presence of the o-quinone form of DHPA. This species was observed after 1 min of reaction time, peaked at 37 min, decreased significantly thereafter, and was not observed at 7 days.
Structural determination of DHPA dimers by NMR
Evidence from HPLC-MS pointing to the presence of two distinct dimer reaction products prompted us to undertake structural analysis. HPLC fractions containing Dimer 1 and Dimer 2 were collected, frozen, lyophilized, and characterized by NMR with D2O as the solvent (Figs. S5 – S16). Assuming C-C bond formation, DHPA dimers potentially contain an element of symmetry that would make the four 1H atoms on the two aromatic rings appear at two frequencies instead of four. The relative position (ortho, meta, or para) of the two 1H’s on each ring can therefore be determined by the 1H-1H scalar coupling constant.41
Dimer 1, the compound isolated from the major dimer peak at ~7.5 min, presented two resonances in the aromatic portion of the 1H spectrum (Figure 4a), indicating both rings had the same substitution pattern. The 1H-1H scalar coupling constant between these nuclei was determined to be 0.3 Hz by analysis of the 2D-J-resolved NMR spectrum (Figure S11), indicating that the 1H atoms have a para relationship to one another.41 Thus, Dimer 1 was concluded to contain a C6-C6 linkage between the aromatic rings (Figure 4b and Figure S5).
Figure 4.
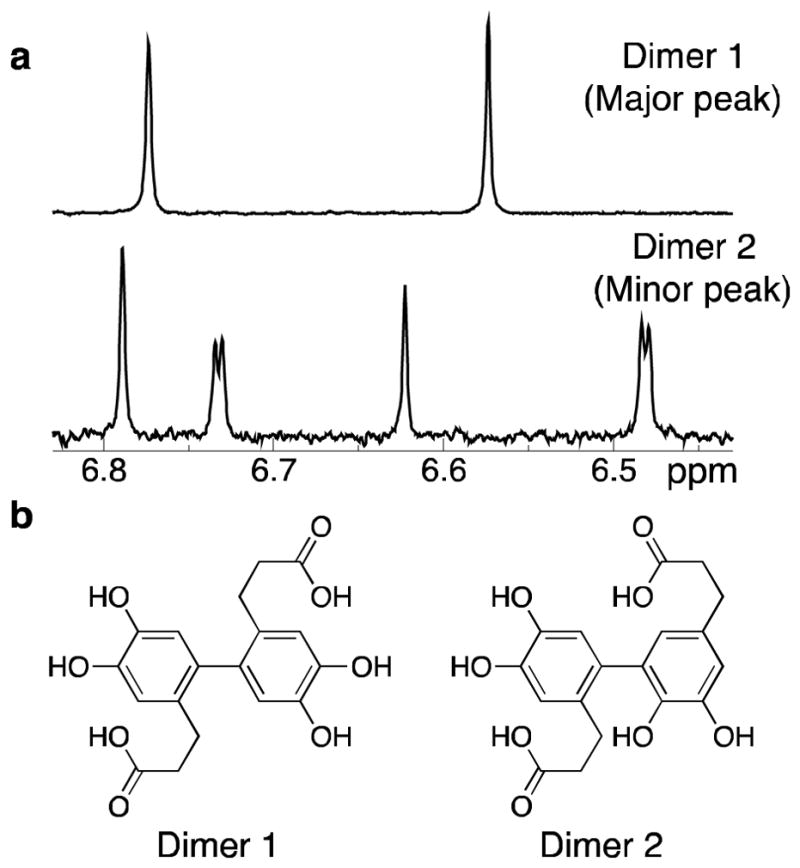
Connectivity of DHPA dimers determined by NMR. Structural analysis of dimers formed by reaction of DHPA and Fe3+. (a) Aromatic regions of the 500 MHz 1H 1D NMR spectra of dimers eluting at ~7.5 min (Dimer 1) and ~10.5 min (Dimer 2) from Figure 3. Dimer 1 exhibits two signals, indicating a symmetric species; the 1H-1H scalar coupling is smaller than the signals’ linewidth (~1.0 Hz), indicating the hydrogens are para to one another. Dimer 2 exhibits four signals, indicating an asymmetric species; the two scalar couplings are <1.0 Hz and 2.2 Hz, indicating para and meta 1H-1H relationships, respectively. (b) Dimer 1 is connected in a C6-C6 fashion, resulting in a symmetric molecule with hydrogens that are para to one another. Dimer 2 has a C5–C6 linkage, producing an asymmetric molecule with two hydrogens para to one another and two hydrogens meta to one another.
In contrast, Dimer 2, the compound isolated from the minor dimer peak at ~10.5 min, presented four resonances in the aromatic portion of the 1H spectrum (Figure 4a), indicating that the two rings had different substitution patterns. Analysis of the 2D-J-resolved NMR spectrum (Figure S16) revealed that one pair of resonances possessed a similar coupling constant to that of Dimer 1 (0.3 Hz), indicating that one ring has two hydrogens para to one another. The other pair of resonances exhibited a scalar coupling constant of 2.1 Hz, indicating a meta relationship between them. Thus, Dimer 2, was assigned a C5-C6 bond between the aromatic rings (Figure 4b and Figure S5).
Reduction of Fe3+ to Fe2+
A UV-Vis assay based on phenanthroline28 was employed in order to demonstrate that oxidation of the catechol to the o-quinone was accompanied by reduction of Fe3+ to Fe2+. In this simple colorimetric assay, addition of phenanthroline to a Fe2+ solution produces a deep red color, whereas addition of phenanthroline to a Fe3+ solution (orange) produces no visible color change.28 Therefore, the UV-Vis spectra of dilute solutions containing phenanthroline combined with DHPA, Fe3+, Fe2+, Fe3++DHPA, and IO4-+DHPA were recorded (Figure S17). Notably, the reaction mixture of DHPA and Fe3+ yielded a strong absorbance between ~350 nm and ~550 nm in the presence of phenanthroline, identical to the spectrum obtained from a control mixture of phenanthroline and Fe2+ but different from that arising from the reaction of DHPA with NaIO4. These data show that Fe3+ is reduced to Fe2+ during the dimerization of DHPA, likely through a quinone-mediated process.
Fe3+-induced cross-linking of a DOPA-containing peptide
The reactions between DHPA and Fe3+ described above were performed under unbuffered acidic conditions (pH ~2) as a consequence of the acidity of FeCl3. Recognizing that mussel byssus formation occurs at higher pH, we investigated the effect of pH on the oxidation and cross-linking of catechols by Fe3+. Analysis of DHPA was complicated by the formation of insoluble precipitates at pH >4, presumably due to metal carboxylate salt formation. Instead, pH-dependent reactions were investigated using the DOPA-containing dipeptide Ac-Ser-DOPA-NH2 (2, Figure 1c), in unbuffered water (pH ~2 due to FeCl3 acidity) and at pH 5, 7, and 9 buffered with acetate, bis-tris, and bicine, respectively. Ac-Ser-DOPA-NH2 is based on the repeating decapeptide motif of the mussel adhesive protein mfp-1.7 The buffers for these experiments were carefully chosen in order to eliminate unwanted side reactions, precipitation, and chelation of Fe3+. Figure 5 shows HPLC-MS chromatograms of the reactions between Ac-Ser-DOPA-NH2 and Fe3+. Extracted ion chromatograms (EIC) were used to identify peaks (Supplemental Figure S18). In the presence of Fe3+, the dimer of the dipeptide is seen to form at pH ≤5, but significantly less so at higher pHs. Four dimers of the dipeptide peaks were identified (+), three of which appear to be present in significant quantities at pH 5 with 1× Fe3+. The dimer peak seen at ~6 min in the EIC (Figure S18) at pH 5 with 1× Fe3+, only appears to be significant in acidic unbuffered water with 1× Fe3+, along with many small unidentified peaks (seen in the chromatogram baseline). It appears that reactions of DOPA with 1× Fe3+ in unbuffered water are more promiscuous than in the buffered systems. At pH 9 in the absence of Fe3+, a peak at ~9 min was seen, however it could not be identified by HPLC-MS. It is likely attributable to an autoxidation product, as the solution was noted to develop a light pink color at this basic pH that remained even after acidification with EDTA. In the presence of Fe3+ at pH 9, this peak is not seen, supporting the idea that coordination of Fe3+ protects DOPA from autoxidation at high pH. The color of the reaction solutions in the pH 5–9 range was consistent with the formation of Fe-catechol coordination interactions.13, 18, 20
Figure 5.
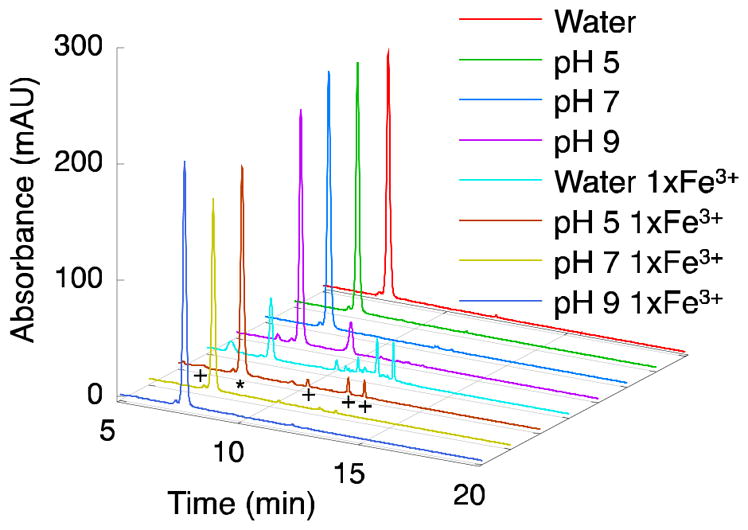
DOPA-containing dipeptide forms dimers at acidic pH in the presence of Fe3+. DOPA-containing dipeptide (Ac-Ser-DOPA-NH2) reaction with and without Fe3+ as studied by HPLC-MS (280 nm shown) at different reaction pHs. Peaks were identified as: ~7 min, dipeptide (*); dimers of dipeptide (+) were detected at ~6, ~10, ~11, and ~12 min (seen prominently in pH 5 and water 1× Fe3+ samples); peak in pH 9 sample at ~9 min, unidentified. In unbuffered water (pH ~2) many unidentified products are produced, while at pH 5 three dimers are the dominant species formed after the reactions with Fe3+. Dimer at ~6 min is seen prominently only in water 1× Fe3+ sample. At higher pH in the presence of Fe3+, the dipeptide appears to be protected from oxidation by the presence of Fe3+.
Fe3+-induced cross-linking of mefp-3 and mcfp-3
Purified DOPA-rich mefp-1 and mcfp-1, MW ~110 kDa 19, 42 and ~92 kDa,43 respectively, were studied to determine whether Fe3+ could induce covalent cross-links in native proteins. Mefp-1 has a DOPA content of approximately 11 % (~62 DOPA residues/protein),7, 43 while mcfp-1 has an approximate DOPA content of 13 % (~95 DOPA residues/protein).43 In a 0.1 M formic acid buffer solution (pH 3), 50 μM mefp-1 or mcfp-1 was incubated with increasing concentrations of Fe3+, up to 0.5 μM. As shown in Figure 6, the mefp-1 and mcfp-1 monomer bands are seen to decrease, while a new, higher MW band is seen to appear with increasing concentrations of Fe3+. The higher MW band is absent from the Fe3+ free experiment, indicating that Fe3+ is able to cross-link mefp-1 and mcfp-1, likely through DOPA-DOPA covalent cross-links. Given the acidity of the solution, Fe3+-DOPA coordination cross-links would not be expected to occur.18
Figure 6.
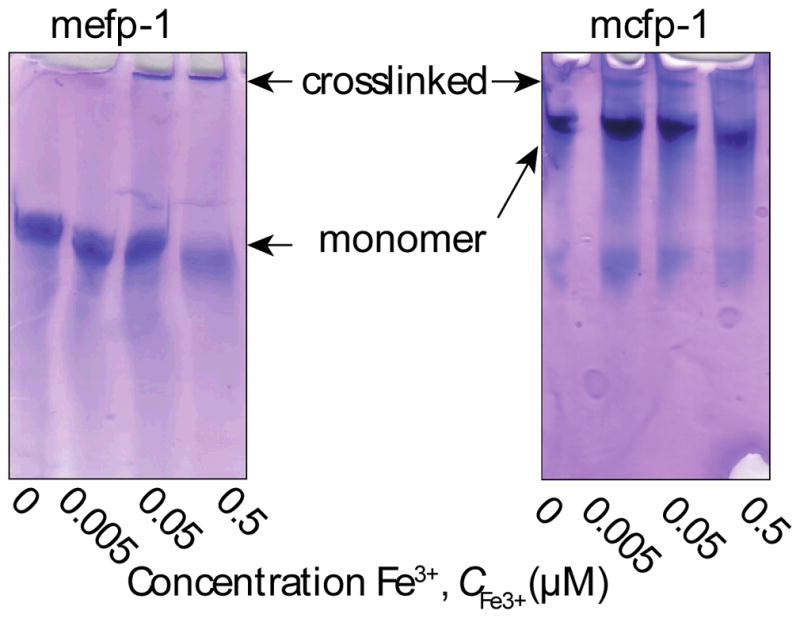
Effect of Fe3+ on protein crosslinking. Purified protein (mefp-1 and mcfp-1) in 0.1 M formic acid pH 3.0 was incubated at room temperature with varying iron concentrations (0–0.5 μM) and run on an acid-urea polyacrylamide gel. Higher molecular weight bands represent dimer or higher oligomer formation. Note that the mefp-1 gel was run for 2 h while the mcfp-1 gel was run for 1 h.
Discussion
Using catechol-containing small molecules and peptides, our results provide the first direct characterization of the covalent cross-linking of catechols induced by redox coupling with Fe3+. For the reaction between Fe3+ and DHPA under acidic conditions, analysis of the reaction solutions revealed that formation of the o-quinone form of DHPA was accompanied by the reduction of Fe3+ to Fe2+ and the formation of dimers, trimers, and tetramers. One- and two-dimensional NMR analyses of HPLC fractions containing DHPA dimers unequivocally identified the major reaction product formed under these conditions to be coupled via a C6-C6 linkage between aromatic rings, with the remainder coupled via a C5-C6 linkage. Covalent multimer formation was also observed for reactions between Fe3+ and a DOPA-containing peptide at low pH (pH 2–3). However, the formation of covalent multimers in the presence of Fe3+ decreased as the pH was increased, and became nearly undetectable at pH 9.
These findings show a clear pH dependence of the interactions between Fe3+ and catechols, suggesting a refinement of existing views regarding the role of Fe3+ in mussel adhesion. To place our results into context, it is useful to consider the mechanism and conditions under which mussel byssal proteins are secreted.44 Byssal threads and adhesive plaques are formed by injection molding of liquid precursors within the distal depression and ventral groove of the mussel foot, followed by release of the newly formed thread and adhesive plaque after several minutes of ‘curing’. Recent unpublished work has determined that the initial pH of secreted byssal proteins is ~4,45 while published work from the same group had earlier indicated an initial pH of ~5.8 ± 0.3.46 After release of the newly formed thread, rapid neutralization to marine pH (8–8.5) presumably occurs upon exposure to seawater. Therefore, the interactions between Fe3+ and catechols that are most relevant to mussel adhesion are likely to occur in the mildly acidic to mildly alkaline pH range (pH 4–8.5).
The formation of coordination complexes between Fe3+ and catechols within this pH range is well documented in the literature.13, 18–21 At catechol:Fe3+ ratios greater than 3, the bis Fe(DOPA)2 complex is favored in the moderately acidic and neutral pH range, whereas tris coordination is favored at alkaline pH.20 Tris Fe3+(DOPA)3 coordination interactions may be responsible for the unique physical properties of the byssal thread cuticle,22, 46 which is rich in Fe3+ and the DOPA-containing protein mfp-1. The mechanical behavior of biomimetic polymer hydrogels formed through tris Fe3+-catechol coordination interactions leaves little doubt that such interactions are mechanically active and can contribute to solidification of the mussel byssus.23–26 Although covalent DOPA-DOPA cross-links have been detected in the mussel byssus,12 the reaction pathway has not been well characterized. Given our results, one would expect that either Fe3+ is stored separately from the DOPA-containing mfps or an additional factor inhibits the oxidation of DOPA by Fe3+ observed in our studies. If Fe3+ was stored together with mfps premature cross-linking in byssal secretions would likely result, leading to a more viscous liquid or even a solid-like gel, making byssal formation difficult or even intractable.
A primary role for covalent cross-linking of mussel adhesive proteins by Fe3+ has been suggested,15, 32 however, the subtleties of byssal precursor storage and processing by the mussel are not well understood. The polyphenolic gland has been identified and demonstrated to contain o-diphenols.47, 48 If dilute FeCl3 is incubated with the foot, this structure turns grey-green in color, and if Na2CO3 is subsequently added, the structure turns red.47 This, together with research suggesting iron is secreted from the foot into the thread,17 provides limited support for the idea that Fe3+ and the DOPA-rich mfps are stored separately in the glandular mussel foot. If this case were true, our results suggest that the mussel may use Fe3+ as an oxidant for DOPA during the initial (acidic) stages of byssal secretion to induce covalent DOPA-DOPA crosslinks. DOPA may then coordinate Fe3+ after the byssus is released into the basic marine environment to enhance the thread’s mechanical properties (Figure 1b).
In a synthetic catechol-modified poly(ethylene glycol) system, we recently demonstrated that the mechanical properties of hydrogels could be dramatically enhanced by applying a similar pH switch in the presence of Fe3+.24 In that system, Fe3+ was first used to covalently cross-link the hydrogel at acidic pH and then the pH was adjusted to a basic pH to allow for Fe3+-catechol coordination bonds to form. At higher pH, gels had an increased modulus and an improved energy dissipating capacity, mechanical features that are important for the byssal thread. Similarly, a recent DOPA-modified peptide amphiphile network demonstrated increased modulus and self-healing properties in the presence of Fe3+ at basic pH.27
While further work is needed to definitively elucidate the interactions between Fe3+ and the various mfps before, during, and after byssus formation, we hypothesize that a complex role for Fe3+ may exist – one that includes both covalent and coordination interactions that are strongly modulated by pH. The mechanical implications of covalent versus coordination cross-links for the performance of the mussel byssus cannot be ignored. Evidence at the single molecule3 and macroscopic gel23–26 levels indicate that fundamentally different mechanical behavior emerges when catechol polymers are cross-linked via coordination and covalent interactions. It may be that mussels achieve the optimal balance between these two types of cross-links through modulation of pH during byssus formation. While an oxidative role for Fe3+ in the mussel adhesive remains hypothetical at this point, these results are highly relevant to the many bioinspired systems where catechols and iron are present.4, 5, 23–27
Conclusions
Herein, we have characterized the products of Fe3+-induced oxidation of the catechol DHPA. Through HPLC-MS and NMR, we have shown that the dominant products are the C6-C6 and C5-C6 connected dimers. HPLC-MS experiments involving a DOPA-containing peptide showed that at acidic pH Fe3+ induces covalent bond formation, whereas at basic pH, DOPA is protected through coordination bond formation and covalent bonds are not formed. Using the DOPA-rich mussel foot proteins, mefp-1 and mcfp-1, we have shown that Fe3+ can induce covalent bond formation between the proteins at acidic pH. We have discussed the implications of this chemistry to byssal thread and mussel adhesive formation, hypothesizing that Fe3+ may play both an oxidant role to induce covalent DOPA-DOPA bond formation and act as a coordination center for DOPA, and that these roles may be switched with the temporal acidic to basic pH change that occurs during byssal secretion and release. We further discuss the mechanical implications of such chemistry and the possible advantages.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH. DEF and DGB were partially supported by a NIH National Research Service Award from the National Heart, Lung, and Blood Institute (F30HL096292) and the IBNAM-Baxter Early Career Development Award in Bioengineering, respectively. The authors thank Dr. J. Herbert Waite, Dr. Thomas V. O’Halloran, Dr. Jennifer Seymour, Dr. Jose G. Rivera, Dr. Sungjin Park, Dr. Niels Holten-Andersen, Saman Shafaie, and Leah Russel for insightful discussions. HPLC-MS and NMR experiments were performed at Northwestern IMSERC.
Footnotes
Supporting Information. Supplementary methods for the oxidation of 4MC and characterization of products by MS and synthesis of Ac-Ser-DOPA-NH2 are available. Figure S1, DHPA oxidation of by Fe3+ digital images; Figure S2, MS DHPA oxidation; Figure S3, MS 4MC oxidation; Figure S4, HPLC-MS with EICs of DHPA oxidation; Figure S5, labeled DHPA dimer structures; Figure S6, Dimer 1 1H NMR; Figure S7, Dimer 1 13C NMR; Figure S8, Dimer 1 1H-1H COSY; Figure S9, Dimer 1 1H-13C HSQC; Figure S10, Dimer 1 1H-13C HMBC; Figure S11, Dimer 1 2D-J NMR; Figure S12, Dimer 2 1H NMR; Figure S13, Dimer 2 1H-1H COSY; Figure S14, Dimer 2 1H-13C HSQC; Figure S15, Dimer 2 1H-13C HMBC; Figure S16, Dimer 2 2D-J NMR; Figure S17, UV-Vis demonstrating Fe3+ reduction to Fe2+ with phenanthroline; Figure S18, HPLC-MS with EICs of dipeptide oxidation. Table S1, Relative abundance of DHPA, o-quinone, and dimers under different reaction conditions.
References
- 1.Waite JH. Int J Adhes Adhes. 1987;7:9–14. [Google Scholar]
- 2.Deming TJ. Curr Opin Chem Biol. 1999;3:100–105. doi: 10.1016/s1367-5931(99)80018-0. [DOI] [PubMed] [Google Scholar]
- 3.Lee H, Scherer NF, Messersmith PB. Proceedings of the National Academy of Sciences. 2006;103:12999–13003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson TH, Yu J, Estrada A, Hammer MU, Waite JH, Israelachvili JN. Advanced Functional Materials. 2010;20:4196–4205. doi: 10.1002/adfm.201000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng H, Hwang DS, Israelachvili JN, Waite JH. Proceedings of the National Academy of Sciences. 2010;107:12850–12853. doi: 10.1073/pnas.1007416107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waite JH, Tanzer ML. Science. 1981;212:1038–1040. doi: 10.1126/science.212.4498.1038. [DOI] [PubMed] [Google Scholar]
- 7.Waite JH. J Biol Chem. 1983;258:2911–2915. [PubMed] [Google Scholar]
- 8.Papov VV, Diamond TV, Biemann K, Waite JH. J Biol Chem. 1995;270:20183–20192. doi: 10.1074/jbc.270.34.20183. [DOI] [PubMed] [Google Scholar]
- 9.Waite JH, Qin XX. Biochemistry. 2001;40:2887–2893. doi: 10.1021/bi002718x. [DOI] [PubMed] [Google Scholar]
- 10.Burzio LA, Waite JH. Biochemistry. 2000;39:11147–11153. doi: 10.1021/bi0002434. [DOI] [PubMed] [Google Scholar]
- 11.Yu M, Hwang J, Deming TJ. J Am Chem Soc. 1999;121:5825–5826. [Google Scholar]
- 12.McDowell LM, Burzio LA, Waite JH, Schaefer J. J Biol Chem. 1999;274:20293–20295. doi: 10.1074/jbc.274.29.20293. [DOI] [PubMed] [Google Scholar]
- 13.Taylor SW, Luther GW, Waite JH. Inorganic Chemistry. 1994;33:5819–5824. [Google Scholar]
- 14.Monahan J, Wilker JJ. Chem Commun. 2003:1672–1673. doi: 10.1039/b301052j. [DOI] [PubMed] [Google Scholar]
- 15.Wilker JJ. Angewandte Chemie-International Edition. 2010;49:8076–8078. doi: 10.1002/anie.201003171. [DOI] [PubMed] [Google Scholar]
- 16.Coombs TL, Keller PJ. Aquatic Toxicology. 1981;1:291–300. [Google Scholar]
- 17.George SG, Pirie BJS, Coombs TL. Journal of Experimental Marine Biology and Ecology. 1976;23:71–84. [Google Scholar]
- 18.Avdeef A, Sofen SR, Bregante TL, Raymond KN. J Am Chem Soc. 1978;100:5362–5370. [Google Scholar]
- 19.Taylor SW, Chase DB, Emptage MH, Nelson MJ, Waite JH. Inorganic Chemistry. 1996;35:7572–7577. [Google Scholar]
- 20.Sever MJ, Wilker JJ. Dalton Transactions. 2004:1061–1072. doi: 10.1039/b315811j. [DOI] [PubMed] [Google Scholar]
- 21.Sever MJ, Wilker JJ. Dalton Transactions. 2006:813–822. doi: 10.1039/b509586g. [DOI] [PubMed] [Google Scholar]
- 22.Harrington MJ, Masic A, Holten-Andersen N, Waite JH, Fratzl P. Science. 2010;328:216–220. doi: 10.1126/science.1181044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH. Proceedings of the National Academy of Sciences. 2011;108:2651–2655. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett DG, Fullenkamp DE, He L, Holten-Andersen N, Lee KYC, Messersmith PB. Advanced Functional Materials. 2013;23:1111–1119. doi: 10.1002/adfm.201201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menyo MS, Hawker CJ, Waite JH. Soft Matter. 2013;9:10314–10323. doi: 10.1039/C3SM51824H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holten-Andersen N, Jaishankar A, Harrington MJ, Fullenkamp DE, DiMarco G, He L, McKinley GH, Messersmith PB, Lee KYC. Journal of Materials Chemistry B. 2014;2:2467–2472. doi: 10.1039/C3TB21374A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ceylan H, Urel M, Erkal TS, Tekinay AB, Dana A, Guler MO. Advanced Functional Materials. 2013;23:2081–2090. [Google Scholar]
- 28.Mentasti E, Pelizzetti E. Journal of the Chemical Society, Dalton Transactions. 1973:2605–2608. [Google Scholar]
- 29.Mentasti E, Pelizzetti E, Saini G. Journal of the Chemical Society, Dalton Transactions. 1973:2609–2614.
- 30.Powell H, Taylor M. Australian Journal of Chemistry. 1982;35:739–756. [Google Scholar]
- 31.Yu M. PhD Thesis. University of California; Santa Barbara: 2000. [Google Scholar]
- 32.Wilker JJ. Curr Opin Chem Biol. 2010;14:276–283. doi: 10.1016/j.cbpa.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Sever MJ, Weisser JT, Monahan J, Srinivasan S, Wilker JJ. Angewandte Chemie-International Edition. 2004;43:448–450. doi: 10.1002/anie.200352759. [DOI] [PubMed] [Google Scholar]
- 34.Weisser JT, Nilges MJ, Sever MJ, Wilker JJ. Inorganic Chemistry. 2006;45:7736–7747. doi: 10.1021/ic060685p. [DOI] [PubMed] [Google Scholar]
- 35.Cook C, English E, Wilson B. The Journal of Organic Chemistry. 1958;23:755–756. [Google Scholar]
- 36.Dewar MJS, Nakaya T. J Am Chem Soc. 1968;90:7134–7135. [Google Scholar]
- 37.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; USA: 2000. [Google Scholar]
- 38.Zhao H, Waite JH. J Biol Chem. 2006;281:26150–26158. doi: 10.1074/jbc.M604357200. [DOI] [PubMed] [Google Scholar]
- 39.Waite JH, Benedict CV. In: Methods in Enzymology. Finn Wold KM, editor. Vol. 107. Academic Press; 1984. pp. 397–413. [DOI] [PubMed] [Google Scholar]
- 40.Bittner S. Amino Acids. 2006;30:205–224. doi: 10.1007/s00726-005-0298-2. [DOI] [PubMed] [Google Scholar]
- 41.Becker ED. High Resolution NMR: Theory and Chemical Applications. 2. Academic Press; San Francisco: 1980. [Google Scholar]
- 42.Deacon MP, Davis SS, Waite JH, Harding SE. Biochemistry. 1998;37:14108–14112. doi: 10.1021/bi9806192. [DOI] [PubMed] [Google Scholar]
- 43.Holten-Andersen N, Zhao H, Waite JH. Biochemistry. 2009;48:2752–2759. doi: 10.1021/bi900018m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverman HG, Roberto FF. In: Biological Adhesive Systems. von Byern J, Grunwald I, editors. ch 18. Springer-Verlag; Vienna: 2010. pp. 273–283. [Google Scholar]
- 45.Waite JH. personal communication.
- 46.Yu J, Wei W, Danner E, Ashley RK, Israelachvili JN, Waite JH. Nature Chemical Biology. 2011;7:588–590. doi: 10.1038/nchembio.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown CH. Quarterly Journal of Microscopical Science. 1952;s3–93:487–502. [Google Scholar]
- 48.Tamarin A, Keller PJ. Journal of Ultrastructure Research. 1972;40:401–416. doi: 10.1016/s0022-5320(72)90110-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.