

Abstract
The first example of a sequential heterocycle formation/multicomponent reaction using an automated continuous flow microreactor assembly is reported. Consecutive Hantzsch thiazole synthesis, deketalization, and Biginelli multicomponent reaction provides rapid and efficient access to highly functionalized, pharmacologically significant 5-(thiazol-2-yl)-3,4-dihydropyrimidin-2(1H)-ones without isolation of intermediates. These complex small molecules are generated in reaction times less than 15 min and in high yields (39–46%) over three continuous chemical steps.
Keywords: flow chemistry, microreactors, multistep synthesis, Hantzsch thiazole synthesis, hydrobromic acid, Biginelli reaction
1. Introduction
Over the past decade, high-throughput chemical synthesis has become increasingly important because of its potential to accelerate drug discovery efforts [1]. Recently, multistep microfluidic chip-based processes have emerged as attractive methods for facilitating the rapid generation of small molecule libraries [2–7]. Specific advantages of this technology include optimal heat transfer, enhanced reagent mixing, precise reaction times, small reaction volumes, and the ability to conduct multistep reactions in a single, unbroken microreactor sequence [8]. Our group has initiated a research program focused on developing automated flow chemistry methods to rapidly access complex, drug-like compounds from readily available precursors. To this end, we have reported the continuous flow syntheses of bissubstituted 1,2,4-oxadiazoles [5], functionalized imidazo[1,2-a] heterocycles [6], and pyrrole-3-carboxylic acid derivatives [7]. We now report the first example of a consecutive heterocycle formation/multicomponent reaction using an uninterrupted continuous flow microreactor sequence. In this unique process, namely, sequential thiazole formation, deketalization, and Biginelli three-component reaction, provides rapid and efficient access to novel 5-(thiazol-2-yl)-3,4-dihydropyrimidin-2(1H)-one (DHPM) derivatives 1. These structures, which are generated from β-ketothiazoles 2 (Figure 1), are formed cleanly and without isolation of intermediates.
Figure 1.

(A) DHPMs 1 from intermediate β-ketothiazoles 2. (B) Biologically active thiazole MTEP, monastrol, and DHPM 1a
Thiazoles constitute a ubiquitous structural motif present in a broad range of biologically active small molecules and natural products. These heterocycles are present in approved and investigational drugs such as STX-107, a derivative of the potent and highly selective metabotropic glutamate subtype 5 (mGlu5) receptor negative allosteric modulator MTEP (Figure 1) [9a]. STX-107 is currently under clinical evaluation as a treatment for Fragile X syndrome [9b]. DHPM derivatives also constitute a large family of medicinally significant compounds displaying a wide range of pharmacological properties [10]. For example, monastrol has received much attention in recent years because of its activity as a mitotic kinesin-5 inhibitor (Figure 1) [11]. To date, the combination of thiazole and DHPM moieties in a single scaffold has not been explored to any significant extent. A single patent application discloses the synthesis of compounds such as 1a (Figure 1) with anti-HIV properties [12]. Thus, methods for joining thiazole and DHPM heterocycles into a combined structure as a novel drug-like scaffold are of potential interest.
2. Results and Discussion
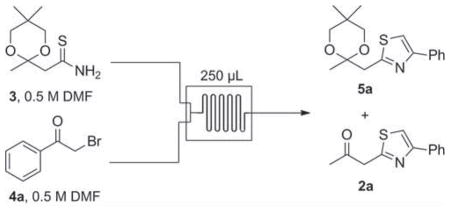
Thiazoles are typically prepared via condensation of thioamides with α-bromoketones using the Hantzsch thiazole synthesis [13]. Unlike oxazole formation, thiazole assembly proceeds under mild conditions without the need for base. In this process, molar equivalents of HBr and water are liberated in situ, serving as potentially useful by-products for continued chemistry. Knowing that the Biginelli reaction is promoted using acid [14], we envisioned taking advantage of this HBr in the context of a multistep continuous flow sequence. Thus, we imagined a flow process in which thiazole formation, with concomitant HBr release, would precede DHPM construction.
β-Keto-oxadiazoles, and other heterocycles, have previously been identified as viable substrates for the batch synthesis of DHPMs [15]. By analogy, we hypothesized that β-ketothiazoles might be utilized in a similar fashion for the Biginelli multicomponent reaction. At the outset, we prepared the ketal-protected thioamide 3 (Table 1) from commercially available acetoacetamide in two steps (see Supporting Information for details). We next investigated the development of a flow method for the synthesis of β-ketothiazole derivatives 2 [16]. The microfluidic setup for the flow synthesis of 1-(thiazol-2-yl)propan-2-one derivatives (2) is outlined in Table 1. Our procedure is inspired by the reported microreactor syntheses of 2-aminothiazoles [17] and 4,5-disubstituted thiazoles [18].
Table 1.
Optimization of thiazole 2a synthesis
| ||||
|---|---|---|---|---|
| entry | time (min) | temp (°C) | H2O (equiv) | ratio (3:5a:2a)a |
| 1 | 2.5 | 50 | 0 | 0 : 8.8 : 1 |
| 2 | 5.0 | 50 | 0 | 8 : 8.6 : 1 |
| 3 | 2.5 | 100 | 0 | 0 : 1 : 1.7 |
| 4 | 5.0 | 100 | 0 | 0 : 1 : 2.9 |
| 5 | 2.5 | 150 | 0 | 0 : 1 : 8.7 |
| 6 | 5.0 | 150 | 0 | 0 : 1 : 6.8 |
| 7 | 5.0 | 150 | 1.0 | 0 : 1 : 11.7 |
| 8 | 5.0 | 150 | 5.0 | 0 : 1 : 104 |
| 9 | 5.0 | 150 | 10 | 1 : 0 : 3.5 |
An HPLC method was used to calculate the ratio of starting material 3 to ketal-protected thiazole 5a to the desired ketone 2a. See Supporting Information for selected HPLC data.
The optimization for the microfluidic synthesis of ketone 2a is also presented in Table 1. Initially, we were delighted to observe that in all cases (entries 1–6) the thioamide starting material 3 was consumed. As expected, the release of both HBr and water also triggered removal of the 1,3-dioxane protecting group at 150 °C (entries 5 and 6) yielding the best ratios of 2a to 5a. We then systematically varied the number of water equiv. (entries 7–9) used to prepare the stock solution of 4a in an effort to further accelerate removal of the ketal protecting group. Overall, using 5 equiv. of water (entry 8) yielded the largest product ratio in favor of 2a over 5a (104:1), while 10 equiv. (entry 9) precluded completion of the sequence with just under 25% of thioamide 3 remaining unreacted. Understanding that using 5 equiv. of water would likely hinder a subsequent Biginelli reaction, we opted for the conditions shown in entry 7 (1 equiv. of water, 2a:5a = 11.7:1) as a compromise between optimal 2a formation and suitable conditions for a two-chip sequence.
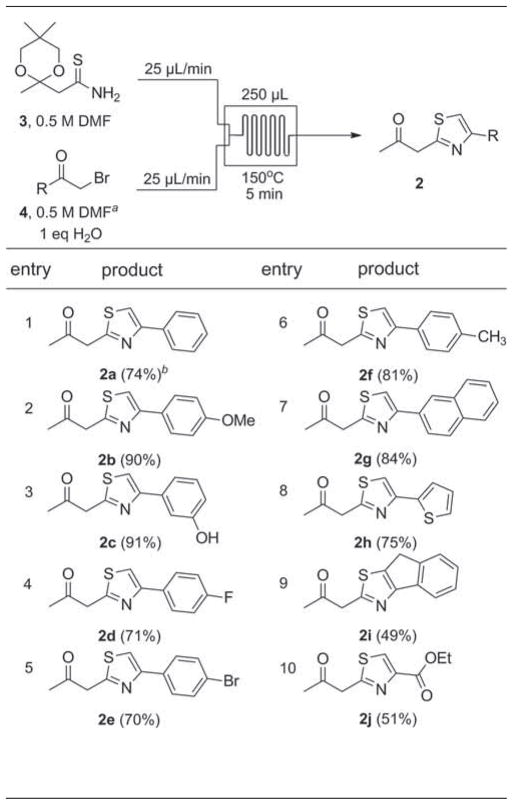
The scope for the continuous flow synthesis of β-ketothiazoles 2a–2j using the optimized conditions is shown in Table 2. Electron-rich α-bromoacetophenones (entries 2 and 3) performed the best with excellent isolated yields (90% for 2b and 91% for 2c). Unsubstituted phenyl (entry 1) and halogenated bromoacetophenones (entries 4–5) proceeded in high yield (70–74%) while the methyl-, naphthyl-, and thiophene-substituted thiazoles 2f–2h formed in 81%, 84%, and 75% yields, respectively. 2-Bromoindanone (entry 9) and ethyl bromopyruvate (entry 10) proved to be poorer substrates but still provided thiazole 2i and 2j in yields of 49% and 51%, respectively, over two chemical steps.
Table 2.
Microfluidic synthesis of β-ketothiazoles 2
|
Refer to Supporting Information for structures of α-bromoketones 4b–4j.
Isolated yields based on 1000 μL collection volumes and following silica gel chromatography.
We next focused our efforts on joining this optimized transformation with a Biginelli reaction in a single continuous sequence. First, we designed a flow synthesis of DHPMs (see Supporting Information for a detailed example) [19]. For the multistep process, we envisioned that the liberated HBr utilized to unmask the ketone intermediates could be harnessed to catalyze the subsequent Biginelli reaction. Gratifyingly, we found that the two-chip flow synthesis of DHPMs 1 proceeded efficiently under the conditions shown in Scheme 1. When using the pump flow rate of the previously mentioned one-chip process (25 μL/min), we observed significant escape of HBr from solution, resulting in visible separations in flow leading to inefficient mixing. In order to prevent this, the flow rate of each pump was increased (32.5 μL/min), resulting in a reduced reaction time for the first microreactor (3.75 min) [20]. Overall, each continuous two-chip microfluidic sequence required less than 1 h for completion from start to finish (injection, reaction, and 1250 μL collection).
Scheme 1.

Final microfluidic setup for the synthesis of DHPMs 1
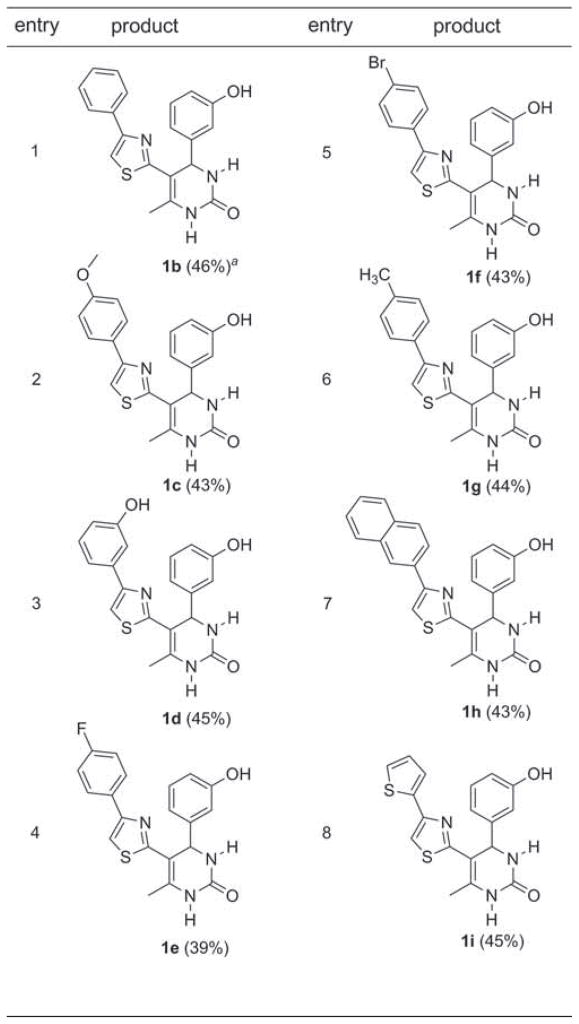
The multistep continuous flow synthesis of thiazolyl DHPMs 1 is presented in Table 3. For all entries (1–8), 3-hydroxybenz-aldehyde was pumped into the second chip to generate novel thiazole-substituted oxo-derivatives of monastrol (Figure 1) [11]. These structures are also putative analogues of the anti-HIV compound 1a (Figure 1). The overall yields for the three-step two-chip sequence were high (39–46%) when using aromatic α-bromoketones 4, averaging roughly 70% yield for each chemical step. Acetophenone substrates bearing either electron-donating (entries 2, 3, and 6) or electron-withdrawing substitution (entries 4 and 5) as well as 2-naphthyl (entry 7) and heteroaromatic (entry 8) were found to be suitable for this process. Importantly, these data largely represent the current supply of commercially available α-bromoketones, yielding complex drug-like compounds for extensive biological evaluation and/or continued chemistry.
Table 3.
Synthesis of thiazole-substituted DHPMs 1
|
Isolated yields based on 1250 μL collection volumes and following silica gel chromatography.
Several advantages of this methodology compared to the standard batch synthesis of analogue libraries should be noted. First, optimization of each reaction step can be conducted very rapidly. For example, the flow conditions shown in Table 1 leading to optimal reaction parameters (entry 7) were investigated in less than 1 day (see Supporting Information for additional details). Second, fully automated multistep flow technology allows for swift construction of compound libraries. In this case, the highly functionalized DHPMs (1b–1i) presented in Table 3 can also be assembled in less than 1 day. Furthermore, because the process is fully automated, unsupervised (e.g., overnight) high-throughput synthesis is an option. We conservatively estimate that approximately 100 derivatives of 1 could be synthesized, purified (using preparative HPLC), and prepared for biological testing within 1 week.
3. Conclusions
In summary, we have developed the first example of a sequential heterocycle formation/multicomponent reaction using an automated continuous flow process that allows efficient access to a novel class of DHPMs (1). These complex structures can be constructed rapidly and in high yield. We have also demonstrated the utility of released by-products within a flow sequence, harnessing in situ generated acid for both protecting group removal and promotion of a multicomponent reaction [7]. Further applications of 2, biological evaluation of 1b–1i, and expansion of our methodology to access highly functionalized DHPMs 1 are in progress.
Supplementary Material
Acknowledgments
NP dedicates this manuscript in loving memory of his brother, CPT Gregory T. Dalessio. This work was supported by National Institutes of Health Grant Nos. HG005033, GM079590, and GM081261.
Footnotes
Supporting Information. Experimental procedures for the batch synthesis of thioamide 3, selected HPLC data for Table 1, structures of α-bromoketones 4b–4j, experimental procedure for the flow synthesis of β-ketothiazole derivatives 2, example of one-chip Biginelli flow synthesis, experimental procedure for the flow synthesis of DHPM derivatives 1, and characterization data for all new compounds including NMR spectra and MS data. This material is available free of charge on the journal’s homepage at www.akademiai.com.
References
- 1.(a) Chighine A, Sechi G, Bradley M. Drug Discov Today. 2007;12:459–464. doi: 10.1016/j.drudis.2007.04.004. [DOI] [PubMed] [Google Scholar]; (b) Huryn DM, Cosford NDP. Annu Rep Med Chem. 2007;42:401–416. doi: 10.1016/S0065-7743(07)42026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Baxendale IR, Hayward JJ, Lanners S, Ley SV, Smith CD. In: Microreactors in Organic Synthesis and Catalysis. Wirth T, editor. Wiley-VCH; Weinheim: 2008. [Google Scholar]; (b) Ehrfeld W, Hessel V, Lowe H. Microreactors: New Technology for Modern Chemistry. Wiley-VCH; Weinheim: 2000. [Google Scholar]
- 3.For recent selected reviews, see: Glasnov TN, Kappe CO. J Heterocycl Chem. 2011;48:11–30.Webb D, Jamison TF. Chem Sci. 2010;1:675–680.Geyer K, Gustafsson T, Seeberger PH. Synlett. 2009;15:2382–2391.Mak XY, Laurino P, Seeberger PH. Beilstein J Org Chem. 2009;5:19–29. doi: 10.3762/bjoc.5.19.Weiler A, Junkers M. Pharm Technol. 2009;S6:S10–S11.Baxendale IR, Hayward JJ, Ley SV. Comb Chem High Throughput Screen. 2007;10:802–836. doi: 10.2174/138620707783220374.Watts P. Curr Opin Drug Discov Dev. 2004;7:807–812.
- 4.Kang L, Chung BG, Langer R, Khademhosseini A. Drug Discov Today. 2008;13:1–13. doi: 10.1016/j.drudis.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grant D, Dahl R, Cosford NDP. J Org Chem. 2008;73:7219–7223. doi: 10.1021/jo801152c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herath A, Dahl R, Cosford NDP. Org Lett. 2010;12:412–415. doi: 10.1021/ol902433a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herath A, Cosford NDP. Org Lett. 2010;12:5182–5185. doi: 10.1021/ol102216x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Razzaq T, Kappe CO. Chem Asian J. 2010;5:1274–1289. doi: 10.1002/asia.201000010. [DOI] [PubMed] [Google Scholar]
- 9a.Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, MacDonald I, Rao S, Washburn M, Varney MA. J Med Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 9b.Cosford ND, Seiders TJ, Payne J, Roppe JR, Huang D, Smith ND, Poon SF, King C, Eastman BW, Wang B, Arruda JM, Vernier J-M, Zhao X. 7,879,882 B2. US Patent. 2011
- 10.Kappe CO. Eur J Med Chem. 2000;35:1043–1052. doi: 10.1016/s0223-5234(00)01189-2. [DOI] [PubMed] [Google Scholar]
- 11.Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Cechetto J, No Z, Christophe T, Kim T, Taehee N, Nam JY, So W, Jo M, Ok T, Park C, Seo MJ, Sohn J-H, Sommer P, Boese AS, Han S-J, Park YS, Kim HP. 2010046780. WO. 2010
- 13.(a) Hantzch AR, Weber JH. Ber. 1887;20:3118–3132. [Google Scholar]; (b) Metzger JV. Comprehensive Heterocyclic Chemistry. Vol. 6. Pergamon; Oxford: 1984. p. 235. [Google Scholar]
- 14.(a) Biginelli P. Ber. 1891;24:2962–2967. [Google Scholar]; (b) Biginelli P. Gazz Chim Ital. 1893;23 [Google Scholar]; (c) Kürti L, Czakó B. In: Strategic Applications of Named Reactions in Organic Synthesis. Hayhurst J, editor. Elsevier; Burlington, VT: 2005. pp. 58–59. [Google Scholar]
- 15.(a) Kharchenko JV, Detistov OS, Orlov VD. J Comb Chem. 2009;11:216–219. doi: 10.1021/cc800111h. [DOI] [PubMed] [Google Scholar]; (b) Dzvinchuk IB, Makitruk TV, Lozinskii MO. Chem Heterocycl Comp. 2003;39:455–460. [Google Scholar]; (c) Dzvinchuk IB, Makitruk TV, Lozinskii MO. Chem Heterocycl Comp. 2002;38:1000–1007. [Google Scholar]
- 16.All microfluidic syntheses were conducted on a Syrris AFRICA synthesis station using Syrris etched glass chips.
- 17.Garcia-Egido E, Wong SYF, Warrington BH. Lab Chip. 2002;2:31–33. doi: 10.1039/b109360f. [DOI] [PubMed] [Google Scholar]
- 18.Baxendale IR, Ley SV, Smith CD, Tamborini L, Voica A-F. J Comb Chem. 2008;10:851–857. doi: 10.1021/cc800070a. [DOI] [PubMed] [Google Scholar]
- 19.Glasnov TN, Vugts DJ, Konigstein MM, Desai B, Fabian WMF, Orru RVA, Kappe CO. QSAR Comb Sci. 2006;5:509–518.For microwave assisted Biginelli reactions, see: Stadler A, Kappe CO. J Comb Chem. 2001;3:624–630. doi: 10.1021/cc010044j.(c) Complete details of our study will be published in due course.
- 20.It should be noted that no improvement in yield was observed with addition of 1 equiv water to the α-bromoketone stock solution. Thus, water was not incorporated into the two-chip sequences.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.