

Abstract
Phosphate and parathyroid hormone related peptide (PTHrP) are required for normal growth plate maturation. Hypophosphatemia impairs hypertrophic chondrocyte apoptosis leading to rachitic expansion of the growth plate; however, the effect of phosphate restriction on chondrocyte differentiation during endochondral bone formation has not been examined. Investigations were, therefore, undertaken to address whether phosphate restriction alters the maturation of embryonic d15.5 murine metatarsal elements. Metatarsals cultured in low phosphate media exhibited impaired chondrocyte differentiation, analogous to that seen with PTHrP-treatment of metatarsals cultured in control media. Because phosphate restriction acutely increases PTHrP expression in cultured metatarsals, studies were undertaken to determine if this increase in PTHrP plays a pathogenic role in the impaired chondrocyte differentiation observed under low phosphate conditions. In contrast to what was observed with wild-type metatarsal elements, phosphate restriction did not impair the differentiation of metatarsals isolated from PTHrP heterozygous or PTHrP knockout mice. In vivo studies in postnatal mice demonstrated that PTHrP haploinsufficiency also prevents the impaired hypertrophic chondrocyte apoptosis observed with phosphate restriction.
To determine how signaling through the PTH/PTHrP receptor antagonizes the pro-apoptotic effects of phosphate, investigations were performed in primary murine hypertrophic chondrocytes. Receptor activation impaired phosphate-induced Erk1/2 phosphorylation specifically in the mitochondrial fraction and decreased levels of mitochondrial Bad, while increasing cytosolic phospho-Bad.
Thus, these data demonstrate that phosphate restriction attenuates chondrocyte differentiation as well as impairing hypertrophic chondrocyte apoptosis and implicate a functional role for the PTH/PTHrP signaling pathway in the abnormalities in chondrocyte differentiation and hypertrophic chondrocyte apoptosis observed under phosphate restricted conditions.
During normal growth plate maturation, chondrocytes differentiate from proliferative chondrocytes into prehypertrophic and hypertrophic chondrocytes. Terminally differentiated hypertrophic chondrocytes secrete signaling molecules that promote vascular invasion, undergo apoptosis, and are replaced by osteoblasts that give rise to the primary spongiosa of bone. In vivo analyses in growing mice have shown that hypophosphatemia impairs apoptosis of terminally differentiated hypertrophic chondrocytes, leading to rachitic expansion of the hypertrophic chondrocyte layer of the growth plate (1–3). The rachitic phenotype in mice with vitamin D receptor (VDR) ablation is prevented with a high calcium, high phosphate rescue diet, thus confirming that the growth plate abnormalities observed in these mice are not a direct consequence of impaired VDR action (1). In vitro studies demonstrate that induction of Erk1/2 phosphorylation by extracellular phosphate activates the mitochondrial apoptotic pathway in hypertrophic, but not proliferative, chondrocytes (2). Blocking Erk1/2 phosphorylation using MEK inhibitors impairs phosphate-induced apoptosis of hypertrophic chondrocytes in vitro and in vivo (2), demonstrating a critical role for phosphate induction of Erk1/2 phosphorylation in hypertrophic chondrocyte apoptosis.
Parathyroid hormone related peptide (PTHrP) is produced by periarticular chondrocytes and acts via the PTH/PTHrP receptor (PPR) to promote the proliferation and inhibit the differentiation of chondrocytes (4–7). The growth plates of PTHrP null mice exhibit accelerated differentiation with evidence of programmed cell death. In vitro analyses in chondrocytes isolated from these mice demonstrate increased susceptibility to TNFα-induced apoptosis (8, 9); however, the effect of PPR signaling on phosphate-induced hypertrophic chondrocyte apoptosis has not been examined. Based on the observation that phosphate restriction acutely increases PTHrP expression in murine metatarsals (2), studies were performed to determine whether phosphate restriction alters chondrocyte differentiation during endochondral bone formation and to address whether PPR signaling plays a role in the impaired hypertrophic chondrocyte apoptosis observed under low phosphate conditions.
Materials and Methods
Animal studies
Animal studies were approved by the institutional animal care committee. All mice were on a C57BL/6J background and were maintained in a virus and parasite-free barrier facility exposed to a 12-hour light-dark cycle. PTHrP ablation leads to embryonic lethality, thus postnatal studies were performed with PTHrP +/− mice (7). Both male and female mice were studied. Postnatal day 18, mice were weaned onto house chow (1% calcium, 0.6% phosphorus) or onto a low phosphate diet (0.6% calcium, 20% lactose, 0.02% phosphorus, Harlan TD 03486). Because phosphate restriction increases calcium absorption, the calcium content of the phosphate-restricted diet was decreased to provide equal bioavailable calcium to the house chow. Mice were treated with a single dose of PTH (1–34, 100 mcg/kg) on day 24 and killed 1 hour post injection.
Metatarsal cultures
Metatarsals were isolated from day 15.5pc (postcoitum) wild-type, PTHrP +/−, and PTHrP −/− litter mates and cultured in phosphate-free DMEM supplemented with 0.25% fetal bovine serum (FBS), 50 μg/mL ascorbic acid, antibiotic/antimycotic, sodium pyruvate, and sodium phosphate at a final concentration of 1.25 mM (control) or 0.05 mM (low phosphate). The two media have equivalent ionized calcium levels (control media: 1.08 mmol/L and low phosphate media: 1.12 mmol/L). To evaluate the effect of PPR signaling on endochondral bone formation, PTHrP was added to the media at a concentration of 10−7 M. Metatarsals were cultured at 37°C and 5% CO2 for 4 days.
Histology
Metatarsal morphology was evaluated by Toluidine blue staining of frozen sections. Morphology of postnatal bones was assessed by hematoxylin and eosin staining of paraffin embedded sections. Phospho-Erk1/2 (p-Erk1/2) immunohistochemistry was performed on frozen sections of metatarsals and paraffin sections of long bones as previously described (2, 10). VDR (Santa Cruz SC-1008) immunohistochemistry was performed on paraffin sections of long bones as previously described (11). Immunohistochemistry for Cyp27B1 (25-hydroxyvitamin D-1-α-hydroxylase, Millipore ABN182) was performed on paraffin sections of long bones; sections were treated with trypsin and subsequently blocked with 10% goat serum, 0.4% Triton X, 1% BSA in PBS and then incubated with primary antibody overnight at 4°C. Primary and secondary antibodies were diluted in 10% goat serum, 0.4% Triton X in PBS. In situ hybridization was performed on fixed frozen metatarsal sections using digoxigenin-labeled probes (3) and on paraffin sections of postnatal bones using 35S-UTP-labeled antisense RNA probes as previously reported (2, 12). The zones of Col X expression in metatarsals are expressed as the percent of metatarsal length. The terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay for apoptotic cells was performed using an in situ cell death detection kit (Roche Diagnostics) (1).
Cell culture
Primary chondrocytes were isolated from the rib cages of day 2 pups by sequential collagenase II digestions and plated onto gelatin-coated 6-well plates at density of 3 × 105/cm2 (1, 2). Cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 25 μg/mL ascorbic acid at 37°C, 5% CO2 for 2 weeks. To evaluate the effect of phosphate on Erk1/2 phosphorylation and apoptotic factors, chondrocytes were serum restricted (0.5% FBS) overnight prior to exposure to 7 mM sodium sulfate or sodium phosphate with or without PTH pretreatment (10−7 M PTH 1–34 for 30 minutes).
Western analysis and subcellular fractionation
Whole cell lysates of primary chondrocytes were prepared as previously described (2). To obtain subcellular fractions, cells were lysed in 250 mM sucrose, 20 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA. The lysate was passed through a 23-gauge needle 10 times and incubated on ice for 20 minutes. The nuclear fraction was pelleted at 600 g for 5 minutes, following which the mitochondrial fraction was pelleted at 10,000 g for 5 minutes. All buffers were supplemented with a cocktail of 1 mM sodium fluoride, 20 mM β-glycerol phosphate, 2 mM sodium orthovanadate, and protease inhibitor mixture (Roche Applied Science).
Protein concentration was calculated using the BCA protein assay (Pierce) and 7 mcg of protein was subjected to Western analysis. Membranes were blocked with 5% nonfat dry milk or BSA prior to incubation with primary antibodies against p-Erk1/2 (Cell Signaling 9101), Erk1/2 (Cell Signaling 9102), voltage-dependent anion-selective channel protein 1 (VDAC) (Millipore AB10527), HSP-90 (Cell Signaling 4875), Bad (Cell Signaling 9239), and phospho-Bad (Ser155) (Cell Signaling 9297). After incubation with horseradish peroxidase-conjugated secondary antibodies, signals were detected using ECL Plus (Amersham Biosciences).
Statistical analysis
Student's t test was used to analyze significance between two groups. P < .05 was considered significant.
Results
Phosphate restriction inhibits chondrocyte differentiation
The metatarsals of day 15.5 mouse embryos grown in culture recapitulate in vivo chondrocyte differentiation, thus, this culture system has been used extensively to investigate factors that regulate chondrocyte differentiation during endochondral bone formation (13–16). To determine if phosphate plays a role in embryonic endochondral bone formation, day 15.5pc metatarsals were cultured in control (1.25 mM) or low phosphate (0.05 mM) media for 4 days postharvest (Figure 1). Morphologic evaluation by toluidine blue staining demonstrated attenuation of the central hypertrophic chondrocyte region in metatarsals cultured in low phosphate media, relative to those cultured under control conditions. To further define the domains of proliferative and hypertrophic chondrocytes, in situ hybridization was performed for collagen type II (Col II; proliferative chondrocytes) and collagen type X (Col X; hypertrophic chondrocytes). Consistent with the morphological phenotype, wild-type metatarsals cultured in low phosphate media exhibited a marked decrease in the domain of Col X expressing hypertrophic chondrocytes relative to those cultured in control media as quantitated by percent of metatarsal length composed of Col X expressing cells (control media: 48.3 ± 3.2% vs low phosphate media: 31 ± 3.6%, P < .05), demonstrating that phosphate restriction impairs differentiation of proliferative into hypertrophic chondrocytes. Because Erk1/2 phosphorylation in cultured hypertrophic chondrocytes is induced by extracellular phosphate (2), immunohistochemical analyses were undertaken to examine the effect of low phosphate media on p-Erk1/2 in the cultured metatarsals. Whereas p-Erk1/2 immunoreactivity was present in the hypertrophic chondrocyte domain of metatarsals cultured in control media, it was not detectable in those cultured in low phosphate media.
Figure 1.
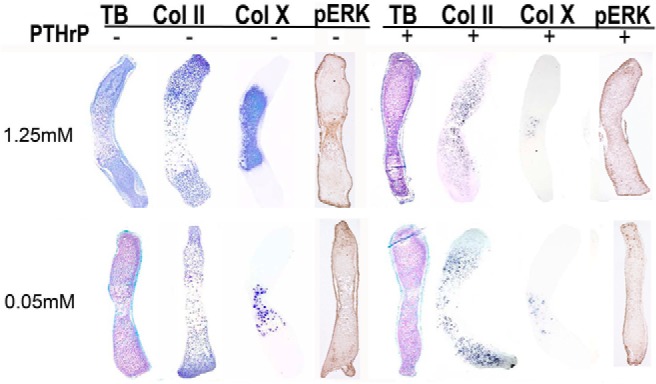
Phosphate restriction and PTHrP impair chondrocyte maturation. Toluidine blue staining (TB), in situ hybridization for collagen type II (Col II) and collagen type X (Col X), and immunohistochemistry for p-Erk1/2 (pERK) were performed on wild-type d15.5pc mouse metatarsals cultured with (+) or without (−) 10−7 M PTHrP in presence of control (1.25 mM sodium phosphate) or low phosphate (0.05 mM sodium phosphate) media for 4 days. Data are representative of 3 metatarsal elements for each condition.
Previous studies have reported that phosphate restriction of metatarsals induces PTHrP mRNA expression within 24 hours (2). To determine if PTHrP treatment leads to a phenotype similar to that observed with phosphate restriction, metatarsals were treated with 10−7 M PTHrP under normal and low phosphate conditions. PTHrP treatment resulted in a decrease in the hypertrophic chondrocyte region of the metatarsals, assessed by both toluidine blue staining and Col X mRNA expression (no treatment: 48.3 ± 3.2% vs PTHrP treatment: 12 ± 3.0%, P < .05), as well as a marked suppression of Erk1/2 phosphorylation (Figure 1) under control conditions. Phosphate-restriction did not appreciably alter the phenotype of the PTHrP-treated metatarsals, suggesting that PTHrP plays a pathogenic role in the impaired chondrocyte differentiation observed under low phosphate conditions.
PTHrP is required for the impaired chondrocyte differentiation observed with phosphate restriction
Because the phenotype of metatarsals cultured under phosphate restricted conditions is analogous to that observed with PTHrP treatment, studies were undertaken to determine if PTHrP plays a pathophysiologic role in the inhibition of chondrocyte differentiation and Erk1/2 phosphorylation observed. Metatarsals from day 15.5pc wild-type, PTHrP +/−, and PTHrP −/− littermates were cultured in control (1.25 mM) and low phosphate (0.05 mM) media (Figure 2). In contrast to what was observed in wild-type metatarsals, in PTHrP +/− metatarsals phosphate restriction did not decrease the hypertrophic chondrocyte region, assessed by toluidine blue staining and Col X mRNA expression (control media: 51.3 ± 3.1% vs low phosphate media: 52.7 ± 3.2%, P = .63), nor did it decrease p-Erk1/2 immunoreactivity (Figure 2). Metatarsals isolated from PTHrP −/− mice exhibited a dramatic acceleration of chondrocyte differentiation evidenced by the expanded domain of Col X mRNA expression and virtual absence of Col II expressing cells, accompanied by an increase in the region of p-Erk1/2 immunoreactivity. (Figure 2). These findings were not affected by phosphate restriction (zone of Col X expression, control media:87.3 ± 3.5% vs low phosphate media: 89.3 ± 3.1%, P = .50). Thus, PTHrP plays a pathogenic role in the impaired endochondral bone maturation observed under low phosphate conditions.
Figure 2.
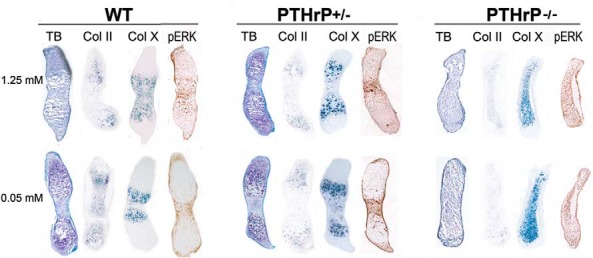
PTHrP is required for the impaired chondrocyte maturation observed with phosphate restriction. Toluidine blue staining (TB), in situ hybridization for collagen type II (Col II) and collagen type X (Col X), and immunohistochemistry for p-Erk1/2 (pERK) were performed on wild-type, PTHrP +/−, and PTHrP −/− d15.5pc mouse metatarsals cultured in control (1.25 mM sodium phosphate) media or low phosphate (0.05 mM sodium phosphate) media for 4 days. Data are representative of 3 metatarsal elements for each condition.
To determine whether PTHrP signaling can be implicated in the impaired hypertrophic chondrocyte apoptosis observed with phosphate restriction in vivo, wild-type and PTHrP +/− mice were weaned onto a low phosphate or control diet from day 18 to 24. The growth plates of wild-type mice fed the low phosphate diet exhibited an expansion of the hypertrophic chondrocyte layer, assessed by H and E staining and in situ hybridization for Col X (Figure 3, A and B). Decreased p-Erk1/2 immunoreactivity was also observed in the growth plate of wild-type mice on the low phosphate diet relative to that of wild-type mice fed the control diet (Figure 3C). In contrast, the low phosphate diet did not lead to appreciable growth plate abnormalities in the PTHrP +/− mice (Figure 3, A–C). However, PTH (1–34) acutely decreased p-Erk1/2 immunoreactivity in the growth plate of wild type and PTHrP +/− mice fed house chow (Figure 3C). To determine whether impaired VDR or Cyp27B1 expression contributes to the growth plate expansion observed with phosphate restriction, immunohistochemical analyses were performed (Figure 3, D and E). Neither VDR nor Cyp27B1 immunoreactivity was decreased in the growth plates of wild-type or PTHrP +/− mice fed a low phosphate diet.
Figure 3.

PTHrP haploinsufficiency attenuates the postnatal growth plate consequences of phosphate restriction. Hemotoxylin and eosin staining (A), in situ hybridization for collagen type X (B), and immunohistochemistry for p-Erk1/2 in phosphate-restricted mice and mice fed a control diet and killed 1 hour after PTH injection (C). Immunohistochemistry for VDR and Cyp27B1of growth plates of wild-type (D) and PTHrP +/− (E) mice fed a control (chow) or low phosphate diet (low Pi diet) from day 18 to 24. Data are representative of 3 mice for each genotype and diet.
Because expansion of the hypertrophic chondrocyte layer in hypophosphatemic mice has been shown to be secondary to impaired apoptosis of these cells, TUNEL assays were performed. As previously reported, the low phosphate diet markedly impaired apoptosis of the late hypertrophic chondrocytes in wild-type mice; however, phosphate-restriction did not significantly impair hypertrophic chondrocyte apoptosis in PTHrP +/− mice (Figure 4, A and B).
Figure 4.
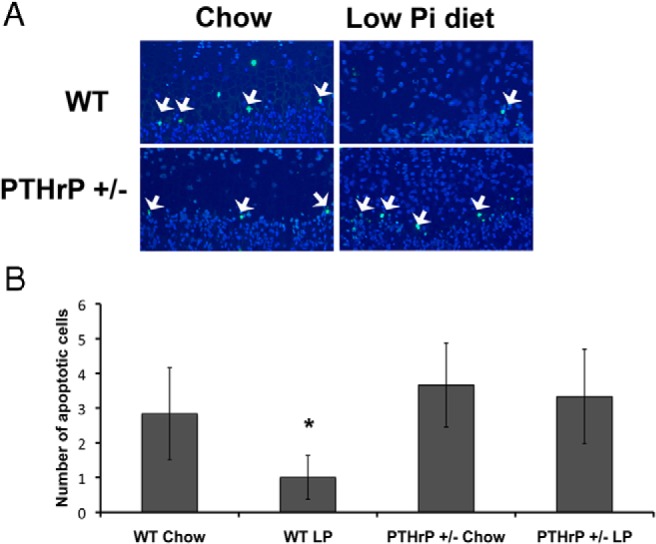
PTHrP haploinsufficiency circumvents the effects of phosphate restriction on hypertrophic chondrocyte apoptosis in vivo. A, TUNEL labeling was performed on wild-type and PTHrP +/− mice fed a control (chow) or low phosphate diet (low Pi diet) from days 18 to 24. B, TUNEL positive cells were counted in the distal 2 rows of the hypertrophic chondrocyte columns. Data are representative of 3 mice for each genotype and diet. *P < .05, significantly different from wild-type mice fed control diet. Arrowheads indicate TUNEL positive hypertrophic chondrocytes.
PPR activation modulates mitochondrial Erk1/2 and apoptotic factors in vitro
Based on these data that demonstrate a role for PTHrP in the impaired hypertrophic chondrocyte apoptosis observed during phosphate restriction, investigations were undertaken to determine if PPR activation alters the induction, activation or subcellular distribution of apoptotic factors during phosphate-induced hypertrophic chondrocyte apoptosis. Because phosphate induction of Erk1/2 phosphorylation is required for hypertrophic chondrocyte apoptosis in vivo and in vitro (2), hypertrophic chondrocytes were treated, or not, with PTH at 10−6 M, 10−7 M, and 10−8 M for 30 minutes prior to the addition of 7 mM sodium sulfate (control) or sodium phosphate (Figure 5A). These studies demonstrated that PTH pretreatment impairs phosphate-induced Erk1/2 phosphorylation at all doses examined. To determine whether this PTH-mediated decrease in p-Erk1/2 was cellular compartment specific, subcellular fractionation was performed. Whereas p-Erk1/2 was induced in both the cytosolic and mitochondrial fractions one hour after treatment with sodium phosphate, PTH impaired phosphate-induced Erk1/2 phosphorylation only in the mitochondria (Figure 5B). PTH also attenuated the increase in mitochondrial total Erk1/2, normalized for VDAC, observed 1 hour post phosphate treatment (Figure 5B). By four hours post treatment, the phosphate-mediated changes in p-Erk1/2 and total Erk1/2 had waned significantly (Figure 5B).
Figure 5.
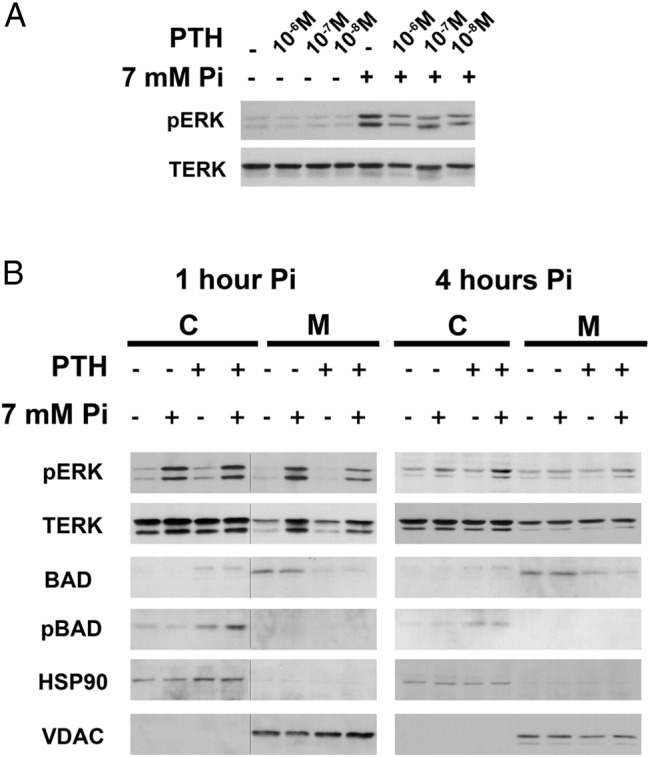
PTH suppresses phosphate-induced mitochondrial Erk1/2 phosphorylation and mitochondrial Bad. A, Hypertrophic chondrocytes were pretreated for 30 minutes with (+) or without (−) 10−6 M, 10−7 M, and 10−8 M PTH prior to a 30-minute incubation with 7 mM sodium sulfate (−) or sodium phosphate (+). Western analyses were performed to evaluate phospho-Erk1/2 (pERK) and total Erk1/2 (TERK). B, Subcellular fractionation of hypertrophic chondrocytes pretreated with (+) or without (−) 10−7 M PTH prior to incubation with 7 mM sodium sulfate (−) or phosphate (+) for 1 or 4 hours. Western analyses were performed on cytosolic (C) and mitochondrial (M) fractions. VDAC (mitochondrial control) and HSP90 (cytosolic control). Data are representative of those obtained from three independent chondrocyte preparations. The line in B (1 hour data) indicates that the two panels were not contiguous on the same gel.
The Bcl-2-associated death associated promoter (Bad) protein promotes apoptosis by binding to and blocking the action of Bcl-2/Bcl-XL (17, 18). Upon phosphorylation, Bad releases Bcl-2/Bcl-XL and moves from the mitochondria to the cytosol, inhibiting apoptosis (19). Phosphate treatment of hypertrophic chondrocytes failed to alter the levels or subcellular distribution of Bcl-2 (data not shown) phospho-Bad or total Bad (Figure 5B). However, 30 minutes pretreatment with PTH led to a marked decrease in mitochondrial Bad, accompanied by a corresponding increase in cytosolic phospho-Bad 1 and 4 hours post phosphate treatment (Figure 5B).
Discussion
The importance of phosphate in skeletal mineralization has long been appreciated. However, studies addressing the role of extracellular phosphate in chondrocyte differentiation during embryonic endochondral bone formation have been limited by the absence of models of fetal hypophosphatemia. To circumvent this problem, investigations were undertaken using the mouse embryonic metatarsal culture system, a model that has been shown to recapitulate embryonic endochondral bone formation ex vivo. Acute phosphate restriction has been shown to increase PTHrP expression in cultured embryonic metatarsals. In the current studies, phosphate restriction impaired the differentiation of proliferative into hypertrophic chondrocytes in a PTHrP-dependent fashion. Thus, low phosphate conditions lead to an increase in PTHrP expression in metatarsals that is required for the impaired chondrocyte differentiation observed with phosphate restriction.
The importance of PPR signaling in chondrocyte differentiation is well established. Chondrocyte-specific overexpression of PTHrP and constitutive activation of the PPR delay chondrocyte maturation (6, 20), whereas homozygous inactivation of the PPR in humans and mice results in accelerated chondrocyte differentiation and embryonic lethality (7, 21, 22). Neither humans nor mice with PTHrP haploinsufficiency manifest skeletal abnormalities during development. In humans, PTHrP haploinsufficiency is characterized by short stature due to growth arrest in the early teens associated with premature fusion of growth plates. In mice, skeletal abnormalities have been reported at 3 months (23, 24). This difference in phenotype may be a consequence of normal lack of growth plate closure in rodents. Mice haploinsufficient for PTHrP have a significant reduction in renal PTHrP mRNA levels (24), thus, PTHrP heterozygous mice, and metatarsals derived from them, provide a suitable model for studying the interactions of phosphate and PTHrP on chondrocyte maturation during development and postnatal growth plate maturation.
Based on the data demonstrating that haploinsufficiency of PTHrP impairs the effects of phosphate restriction on embryonic metatarsals, studies were undertaken in growing mice. These studies demonstrated that, unlike what is observed in wild-type mice, the growth plates of mice heterozygous for PTHrP ablation do not demonstrate impaired hypertrophic chondrocyte apoptosis when fed a low phosphate diet during a period of rapid growth. These data demonstrate that PPR signaling is required for the impaired hypertrophic chondrocyte apoptosis observed with phosphate restriction. However, previous studies have demonstrated that a low phosphate diet partially rescues the premature closure of the growth plate observed with postnatal PPR ablation. Whereas incomplete cre-mediated PPR ablation could result in this phenotype, signaling by the intact receptor in cells that did not express the inducible Col II-Cre transgene, or activation of compensatory pathways in this model could also account for the observation (25–28).
Activation of the PPR has previously been reported to have both pro- and anti-apoptotic effects in other cell types. PTH has been shown to exert significant anti-apoptotic effects on osteoblasts and osteocytes in vivo in mice, as well as cause resistance to dexamethasone-induced apoptosis of these cells (29). Similarly, in mesenchymal cell culture models, overexpression of the PPR attenuates dexamethasone and serum deprivation-induced apoptosis, as does treatment of preconfluent cells with PPR ligands (30). In contrast, in postconfluent mesenchymal cells, PPR activation induces apoptosis, associated with Akt dephosphorylation (30). PPR activation in Caco-2 cells also leads to dephosphorylation of Akt and Bad, decreasing cell survival (31). These data contrast with our studies demonstrating that PTH treatment of primary hypertrophic chondrocytes increases phospho-Bad and prevents phosphate-induced mitochondrial Erk1/2 phosphorylation. Taken together, these studies suggest that the actions of PPR on apoptosis are cell type and differentiation stage-specific.
Thus, the current investigations demonstrate that a low phosphate environment attenuates chondrocyte differentiation during embryonic endochondral bone formation by a PPR-dependent mechanism. They also show that PPR activation modulates the effects of extracellular phosphate on hypertrophic chondrocyte apoptosis during postnatal growth plate maturation.
Acknowledgments
Current address for F.G.: Harvard School of Dental Medicine, Boston, MA 02115.
This work was supported by Grants R01 AR061376, T32 DK007529, and F32 AR065386 from the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Bad
- Bcl-2-associated death
- Col II
- collagen type II
- Col X
- collagen type X
- FBS
- fetal bovine serum
- pc
- postcoitum
- p-Erk1/2
- Phospho-Erk1/2
- PPR
- PTH/PTHrP receptor
- PTHrP
- parathyroid hormone related peptide
- TUNEL
- terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- VDAC
- voltage-dependent anion-selective channel protein 1
- VDR
- vitamin D receptor.
References
- 1. Sabbagh Y, Carpenter TO, Demay M. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA. 2005;102:9637–9642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miedlich SU, Zalutskaya A, Zhu ED, Demay MB. Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J Biol Chem. 2010;285:18270–18275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zalutskaya AA, Cox MK, Demay MB. Phosphate regulates embryonic endochondral bone development. J Cell Biochem. 2009;108:668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666 [DOI] [PubMed] [Google Scholar]
- 5. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336 [DOI] [PubMed] [Google Scholar]
- 6. Schipani E, Lanske B, Hunzelman J, et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA. 1997;94:13689–13694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289 [DOI] [PubMed] [Google Scholar]
- 8. Amizuka N, Henderson JE, Hoshi K, et al. Programmed cell death of chondrocytes and aberrant chondrogenesis in mice homozygous for parathyroid hormone-related peptide gene deletion. Endocrinology. 1996;137:5055–5067 [DOI] [PubMed] [Google Scholar]
- 9. Okoumassoun LE, Russo C, Denizeau F, Averill-Bates D, Henderson JE. Parathyroid hormone-related protein (PTHrP) inhibits mitochondrial-dependent apoptosis through CK2. J Cell Physiol. 2007;212:591–599 [DOI] [PubMed] [Google Scholar]
- 10. Provot S, Nachtrab G, Paruch J, Chen AP, Silva A, Kronenberg HM. A-raf and B-raf are dispensable for normal endochondral bone development, and parathyroid hormone-related peptide suppresses extracellular signal-regulated kinase activation in hypertrophic chondrocytes. Mol Cell Biol. 2008;28:344–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luderer HF, Nazarian RM, Zhu ED, Demay MB. Ligand-dependent actions of the vitamin D receptor are required for activation of TGF-β signaling during the inflammatory response to cutaneous injury. Endocrinology. 2013;154:16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. 2002;143:3691–3694 [DOI] [PubMed] [Google Scholar]
- 13. Klement BJ, Spooner BS. Embryonic mouse pre-metatarsal development in organ culture. J Exp Zool. 1993;265:285–294 [DOI] [PubMed] [Google Scholar]
- 14. Serra R, Karaplis A, Sohn P. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor beta (TGF-beta) on endochondral bone formation. J Cell Biol. 1999;145:783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao H, Minkin C. The effects of 1,25-dihydroxyvitamin D3 on osteoclast formation in fetal mouse metatarsal organ cultures. Bone. 1994;15:217–223 [DOI] [PubMed] [Google Scholar]
- 16. Minkin C, St James S, Tao HH, et al. Skeletal development and formation of osteoclast-like cells from in situ progenitors in fetal mouse metatarsals cultured in chemically defined medium. Bone Miner. 1991;12:141–155 [DOI] [PubMed] [Google Scholar]
- 17. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291 [DOI] [PubMed] [Google Scholar]
- 18. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628 [DOI] [PubMed] [Google Scholar]
- 19. Zhou XM, Liu Y, Payne G, Lutz RJ, Chittenden T. Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155. J Biol Chem. 2000;275:25046–25051 [DOI] [PubMed] [Google Scholar]
- 20. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. 1996;93:10240–10245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jobert AS, Zhang P, Couvineau A, et al. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang P, Jobert AS, Couvineau A, Silve C. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab. 1998;83:3365–3368 [DOI] [PubMed] [Google Scholar]
- 23. Klopocki E, Hennig BP, Dathe K, et al. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Human Genet. 2010;86:434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amizuka N, Karaplis AC, Henderson JE, et al. Haploinsufficiency of parathyroid hormone-related peptide (PTHrP) results in abnormal postnatal bone development. Dev Biol. 1996;175:166–176 [DOI] [PubMed] [Google Scholar]
- 25. Hirai T, Chagin AS, Kobayashi T, Mackem S, Kronenberg HM. Parathyroid hormone/parathyroid hormone-related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc Natl Acad Sci USA. 2011;108:191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee K, Deeds JD, Segre GV. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology. 1995;136:453–463 [DOI] [PubMed] [Google Scholar]
- 27. Farquharson C, Jefferies D, Seawright E, Houston B. Regulation of chondrocyte terminal differentiation in the postembryonic growth plate: the role of the PTHrP-Indian hedgehog axis. Endocrinology. 2001;142:4131–4140 [DOI] [PubMed] [Google Scholar]
- 28. Kobayashi T, Chung UI, Schipani E, et al. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986 [DOI] [PubMed] [Google Scholar]
- 29. Weinstein RS, Jilka RL, Almeida M, Roberson PK, Manolagas SC. Intermittent parathyroid hormone administration counteracts the adverse effects of glucocorticoids on osteoblast and osteocyte viability, bone formation, and strength in mice. Endocrinology. 2010;151:2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen HL, Demiralp B, Schneider A, et al. Parathyroid hormone and parathyroid hormone-related protein exert both pro- and anti-apoptotic effects in mesenchymal cells. J Biol Chem. 2002;277:19374–19381 [DOI] [PubMed] [Google Scholar]
- 31. Calvo NG, Gentili CR, de Boland AR. The early phase of programmed cell death in Caco-2 intestinal cells exposed to PTH. J Cell Biochem. 2008;105:989–997 [DOI] [PubMed] [Google Scholar]