

Abstract
The adipokine, leptin (LEP), is a hormonal gateway, signaling energy stores to appetite-regulatory neurons, permitting reproduction when stores are sufficient. Dual-labeling for LEP receptors (LEPRs) and gonadotropins or GH revealed a 2-fold increase in LEPR during proestrus, some of which was seen in LH gonadotropes. We therefore investigated LEPR functions in gonadotropes with Cre-LoxP technology, deleting the signaling domain of the LEPR (Lepr-exon 17) with Cre-recombinase driven by the rat LH-β promoter (Lhβ-cre). Selectivity of the deletion was validated by organ genotyping and lack of LEPR and responses to LEP by mutant gonadotropes. The mutation had no impact on growth, body weight, the timing of puberty, or pregnancy. Mutant females took 36% longer to produce their first litter and had 50% fewer pups/litter. When the broad impact of the loss of gonadotrope LEPR on all pituitary hormones was studied, mutant diestrous females had reduced serum levels of LH (40%), FSH (70%), and GH (54%) and mRNA levels of Fshβ (59%) and inhibin/activin β A and β B (25%). Mutant males had reduced serum levels of GH (74%), TSH (31%), and prolactin (69%) and mRNA levels of Gh (31%), Ghrhr (30%), Fshβ (22%), and glycoprotein α-subunit (Cga) (22%). Serum levels of LEP and ACTH and mRNA levels of Gnrhr were unchanged. However, binding to GnRH receptors was reduced in LEPR-null LH or FSH gonadotropes by 82% or 89%, respectively, in females (P < .0001) and 27% or 53%, respectively, in males (P < .03). This correlated with reductions in GnRH receptor protein immunolabeling, suggesting that LEP's actions may be posttranscriptional. Collectively, these studies highlight the importance of LEP to gonadotropes with GnRH-binding sites and activin as potential targets. LEP may modulate population growth, adjusting the number of offspring to the availability of food supplies.
Leptin (LEP) is an adipocyte hormone that regulates appetite and signals levels of adiposity and nutritional status (1–7). Lep is also a critical signal for sufficient nutritional status leading to the optimal functioning of the hypothalamic-pituitary-gonadal axis. Humans deficient in LEP receptors (LEPRs) (8) or LEP (9, 10) are hypogonadal and infertile. In adult humans, low gonadotropin levels and functional hypothalamic amenorrhea occur when a relative energy deficit (from weight loss, excessive exercise, or eating disorders) disrupts the pituitary-gonadal axis (11–18).
LEP levels may rise normally to support reproductive events, including a preovulatory rise in serum LEP in women (19, 20), a proestrous LEP rise in rodents (21–25), and synchronous nocturnal LEP and LH pulses in women (26) and sheep (27). However, researchers have not yet fully identified functions for LEP in all of the LEP target cells. The importance of circulating LEP to the kisspeptin-neurokinin-dynorphin neuronal circuitry, which regulates GnRH neurons, is well established (28, 29). McMinn et al (30) used selective Cre-loxP technology to show that deletion of both alleles of the Lepr-exon 17 gene in neurons results in infertility, thus supporting LEP's role in the regulation of GnRH secretion (28, 29, 31, 32).
With respect to the pituitary, the significance of LEP to gonadotropes is less clear. Gonadotropes have been reported to respond to LEP (33–42), with changes in secretion of gonadotropins (22, 43–49). However, direct interactions between LEP and gonadotropes have been questioned because of early reports that gonadotropes lacked LEPRs (36, 37).
There is compelling systemic evidence for LEP's stimulatory effects on gonadotropes. Lep- or Lepr-deficient mice have reduced gonadotrope and somatotrope functions (6, 7, 41, 50) and are infertile. The fertility of LEP-deficient ob/ob mice is completely restored by exogenous LEP (51). LEP also restores LH secretion in fasted mice, rats, and monkeys (2, 4, 25, 27, 52–56). LEP therapy normalizes reproductive hormone levels in a LEP-deficient prepubertal child (57), as well as in adult men (58) and in women with functional amenorrhea (17, 18). In vitro evidence for LEP regulation of gonadotropes showed that LEP regulates gonadotropes via the nitric oxide pathway (49, 59). Our laboratory reported that the reduction in LH and FSH stores in gonadotropes after a 24-hour fast was restored by 100-pg/mL LEP (60).
The first objective of the present study was to determine whether gonadotropes express LEPR proteins and whether LEPR protein levels vary with the stage of the estrous cycle. After discovering that females have a 2-fold increase in LEPR during proestrus and estrus, we used Cre-LoxP technology to delete the Janus kinase-binding site of LEPR (exon 17) selectively in gonadotropes. This report validates the selectivity of the deletion and its broad impact on pituitary hormone mRNA, protein, and serum levels. We confirm that females are more sensitive to the loss of LEPR in gonadotropes with respect to reproduction, because they show a longer time to first pregnancy and reduced litter size. LEPR is less important for reproduction in mutant male gonadotropes, which show deficits that point to gonadotropes as metabolic sensors and paracrine regulators. Finally, we will report a reduction in 2 molecular targets for LEP-GnRH-binding sites (in male and female mutants) and activin mRNA (in female mutants). The data showing reduced GnRH receptor (GnRHR) proteins but not mRNA suggest a possible posttranscriptional pathway for LEP in the regulation of GnRHR.
Materials and Methods
Animal care and use
The Institutional Animal Care and Use Committee approved all animal care protocols. The early phases of the study, designed to detect LEPRs in gonadotropes, were done in male and cycling female Sprague-Dawley rats. Once this study transitioned to transgenic mice, the same detection protocols for LEPR were applied to control mice (FVB/NJ). Stages of the estrous cycle were detected by vaginal smears, as reported previously (61). Females were used after they exhibited 2 normal estrous cycles.
Male and female rats were housed 3/cage and acclimated for 10 days after receipt (Harlan Sprague-Dawley). They were exposed to a 12-hour light, 12-hour dark cycle, given food and water ad libitum, and fed the standard rodent chow (Rodent diet 8640; Harlan). Male and female mice were bred in house. After weaning, experimental and control mice were housed 4–5 mice/cage and fed Teklad 8640 diet (crude protein, 22%; crude fat, 5%; crude fiber, 4.5%; Harlan).
Production of the Lepr deletion mutant mouse model
Mice bearing 2 floxed alleles of Lepr (Lepr-exon 17loxP/loxP) were obtained from Dr Streamson Chua (30, 62). They were bred to mice bearing the Cre-recombinase gene driven by the bLH-β promoter (bLhβ-cre) (63), developed by Dr Sally Camper. The F1 generation of mice were hybrids of FVB/C57B/6 strains. Breeding was slow and sporadic, even among control mice bearing no Cre recombinase. Therefore, the bLhβ-cre transgene was moved to the more prolific FVB.129P2-Pde6b+ Tyrc-ch/AntJ background (FVB.129) through 4 generations to take advantage of the known higher breeding success in the FVB strain. The 129P2 variant of this FVB strain was used to avoid the confounding influence of retinal degeneration in adult FVB/NJ mice.
To produce the homozygous mutants, mice bearing 1 floxed allele of Lepr and LHβ-cre were backcrossed to mice bearing Lepr-exon 17loxP/loxP. The Cre recombinase was passed through the female line, because it was reported to be expressed in the testes (63). At weaning, tail snips were taken for genotyping with primers for Cre-recombinase or Lepr-exon 17loxP/loxP as in previous studies (64). Also, as previously described, (64), we took snips from 16 different organs, for organ genotyping to detect any extrapituitary Cre recombinase activity in mutants.
Collection of animals, pituitaries, and sera for immunoassay
Mice were anesthetized with isoflurane and then decapitated when fully unconscious. Trunk blood was collected and serum stored for assays of levels of LEP and LH, FSH, GH, TSH, prolactin (PRL), and ACTH. Pituitary hormones were assayed using the Luminex LX200 xPONENT 3.1 (Luminex Corp) with Millipore MAP Multiplex Mouse Pituitary hormone kits (Millipore Corp) (65). LEP levels were assayed by enzyme immunoassays (R&D Systems), as in previous reports (60).
Studies of growth and reproductive capacity
Mice were weaned at 21 days and weighed and measured (tail to nose) weekly. They were evaluated for signs of puberty, as described in our recent report of somatotrope-Lepr-exon 17-null mutants (64). After puberty, vaginal smears determined progression through the estrous cycle (61). To detect any changes in reproductive capacity, we set up breeding cages with deletion mutant males and/or females. The breeding study recorded the time to first pregnancy, time between litters, number of pups/litter, and survival of pups in each litter. The data were compared with those from 3 breeding cages containing control FVB.129P mice, which had delivered during the same season. At least 3 litters/cage were monitored. Unless otherwise noted, all mice in this study were 3–5 months of age.
Pituitary cell dispersion and cytochemical protocols
The cell dispersion, culture, and LEP stimulation protocols were as described in previous studies (64). Immunocytochemical labeling for the long form of the LEPRr (LEPRb), for the phosphorylated signal transducer and activator of transcription 3 (pSTAT3), or for all pituitary hormones was done with antisera, controls, and protocols described previously (64). Table 1 includes all antibodies used in this study. We also immunolabeled cultured pituitary cells with antisera directed against amino acids 577–594 in the extracellular domain of LEPR, which is found in all isoforms of LEPR (Novus Biologicals, Inc). The immunoabsorption controls are reported in Figure 1.
Table 1.
Antibodies
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in, Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| LEPR | Amino acids 577–594 | LEPR-extracellular domain | Novus Biologicals, Inc | Rabbit, polyclonal | 1:10 000 |
| LEPRb | OBR13S | Alpha Diagnostics Int Services | Rabbit, polyclonal | 1:10 000 | |
| GH | Rat GH | A. F. Parlow, Hormone Distribution Program | Rabbit, polyclonal | 1:200 000 | |
| LH-β | β-Subunit | Bovine LH-β | J. G. Pierce | Rabbit, polyclonal | 1:200 000 |
| LH-β | β-Subunit | Human LH-β | A. F. Parlow, Hormone Distribution Program | Rabbit, polyclonal | 1:5000 |
| FSH-β | β-Subunit | Human FSH-β | A. F. Parlow, Hormone Distribution Program | Rabbit, polyclonal | 1:5 000 |
| ACTH | 17-39 ACTH | Anti-17-39 ACTH | Produced in laboratory | Rabbit, polyclonal | 1:40 000 |
| PRL | Anti-rPRL | A. F. Parlow, Hormone Distribution Program | Rabbit, polyclonal | 1:10 000 | |
| TSH | β-Subunit | Anti-rTSH-β | A. F. Parlow, Hormone Distribution Program | Rabbit, polyclonal | 1:30 000 |
| GnRHR | Amino acids 1–328 | Anti-GnRHR (Fl-328) | sc-13944; Santa Cruz Biotechnology, Inc | Rabbit, polyclonal | 1:300 |
| Anti-pSTAT3 | Anti-pSTAT3 | Cell Signaling | Rabbit, polyclonal | 1:10 000 |
Figure 1.

Immunocytochemical detection of the extracellular domain of LEPR in wild-type rats and mice. A, Counts of LEPR-labeled cells in male and cycling female mice. About half of the pituitary cell population expressed the extracellular domain of LEPR in males and metestrous or diestrous females. There is a significant increase in expression of LEPR in in proestrous and estrous females of both rodent species. B, Immunolabeled field, with dark field imaging, from a male mouse labeled with 1:10 000 dilution of the antibody produced against amino acids 577–594 of LEPR. This antibody detects all isoforms. C, Immunoabsorption controls showing almost complete neutralization of labeling after absorption with 5 μg/mL of the peptide used to make the antibody. In B and C, n = 4 male mice; in A, n = 4 male and 12 female rats; 4 male and 16 female mice. A star indicates significantly different from all other groups, ANOVA, Bonferroni's multiple comparison test; P < .001. Scale bar, 20 μm. D, Graph showing the proportion of each of 3 cell types that make up LEPR target cells. The proportion of somatotropes in the LEPR population is stable and thus somatotropes do not appear to contribute to the overall increased percentages of LEPR-bearing cells in proestrous or estrous mice seen in C. A star indicates increase in proportion of LH cells with LEPR from diestrus to proestrus (P = .05). E and F, Graphs showing the percentages of pituitary cells with LEPR and LH or FSH and the fact that most gonadotropes in the mouse express LEPR. The changes in gonadotropin stores with the stage of the cycle reflect the increase during diestrus to reach a peak on the AM of proestrus. (Highest values =closed star, P < .01) and the decrease seen in the morning of estrus (open star; lowest values, P < .0001)) because of secretion of LH or FSH the night before. This low value persists during metestrus (open star). The remaining figures depict dual-labeling for LEPR and GH (G and H), LH (I), or FSH (J) in male mice. The labeling for LEPR is black (arrows) and that for the pituitary hormone is orange-amber. Scale bar, 10 μm.
We detected biotinylated analogs of D-Lys6-GnRH (Bio-GnRH) in cultures grown for 24 hours, as described previously (66, 67). After a 24-hour incubation, cells were stimulated with 1nM–3nM Bio-GnRH for 10 minutes, fixed in glutaraldehyde; the biotin was detected by avidin-biotin peroxidase complexes (66, 67), and cells were dual-labeled for LH or FSH (67). Immunolabeling for GnRHRs used rabbit polyclonal anti-GnRHR (FL-328) at a 1:300 dilution, as recommended by the supplier (sc-13944; Santa Cruz Biotechnology, Inc). The epitope corresponds to amino acids 1–328, which represents the entire sequence of GnRHR. Cells were dual-labeled for LH, FSH, and GH, as described previously (64).
quantitative polymerase chain reaction (qPCR) assays for pituitary mRNA levels
Pituitary RNA was isolated using the Maxwell 16 LEV simplyRNA Tissue kit (AS1280; Promega). For quantitative reverse transcriptase polymerase chain reaction, cDNA samples and primers were added to Power SYBR Green PCR Master Mix (4367659; Applied Biosystems) and all reactions performed with the QuantStudio 12K Flex system (Applied Biosystems, Life Technologies) with the following protocol in 3 stages. Incubation/denaturation stage: 50°C for 2 minutes and 95°C for 10 minutes; PCR amplification stage (40 cycles): 95°C for 15 seconds, 55°C for 15 seconds, and 72°C for 1 second; and melt curve stage: 95°C for 15 seconds, 60°C for 1 second, and 95°C for 15 seconds. Samples were normalized to cyclophilin expression, and relative expression values were determined by the QuantStudio 12K Flex Software version 1.0 using the delta delta cycle threshold“ method. Supplemental Table 1 includes all qPCR primer sets for the following gene products: Cyclophilin, Fshβ, Lhβ, Gnrhr, proopiomelanocortin (Pomc), Gh, Ghrhr, glycoprotein hormone α-subunit (Cga), Prl, Tshβ, inhibin α-subunit (Ina), inhibin/activinβ-A subunit (Inhba), and Inhbb.
Statistics and power analyses
At least 5 animals were used for each test, and averages of in vitro tests were made with at least 3 repeats (n = 5–15). Cell counts and assay values were analyzed with Prism statistical software with ANOVA followed by Tukey or Bonferroni post hoc tests, as described previously (65). When 2 groups were compared, the Student's t test was run. A post hoc power analysis was done with pilot studies of gonadotrope Lepr-null male mutants. This allowed us to predict that a 50% reduction in serum FSH levels, from 90 ng/mL (controls) to 45 ng/mL (mutants) with an SD of 25 ng/mL, will have 81% statistical power with 5 replicates in a two-tailed, 0.05-level t test (see the following URL for the formula, http://www.dssresearch.com/KnowledgeCenter/toolkitcalculators/statisticalpowercalculators.aspx).
Results
Expression of LEPR by gonadotropes and somatotropes
The first objective of this study was to detect LEPR cytochemically in freshly dispersed, cultured pituitary cells during different reproductive states, with antisera directed against the extracellular LEPR domain. Figure 1A graphs the counts of LEPR target cells detected by this antibody, showing that 39%–50% of pituitary cells in male or metestrous or diestrous female rodents were immunolabeled. There was a significant increase (P < .001) in the percentages of immunolabeled cells in proestrous and estrous female mice Immunolabeling and specificity controls with cells are shown in Figure 1, B and C.
Most LEP-bearing cells store GH or gonadotropins as shown by dual-labeling (Figure 1D). LH cells with LEPR increase from diestrus to proestrus, indicating their contribution to the rise in LEPR-bearing cells. Neither GH cells nor FSH gonadotropes contribute to the overall increase in LEP target cells. Because 75%–90% of gonadotropes store both hormones, the percentages seen in this graph are not exactly additive. Also, the loss in gonadotropin stores during and after surge secretion prevents the detection of gonadotropes as target cells during proestrus and estrus.
The analyses show that most LH or FSH cells express LEPRs (Figure 1, E and F). The reduction in LH and FSH cells during estrus reflects secretion of stores during the proestrous LH surge or the estrous rise in FSH as described previously (68–70). Dual-labeling shows that labeling for LEPR is black and mostly at the cell periphery or in dark patches in the cell, labeling for pituitary hormones (Figure 1, G–J) is orange-amber.
Selectivity of Cre-recombinase excision in Lepr-exon 17-null deletion mutants
The findings for LEPR expression shown in Figure 1 fit well with the reported increases in serum LEP during the cycle (21–25) and suggested that females may be particularly dependent on gonadotrope LEPR for normal gonadotropin secretion. Cre-loxP technology was then used to selectively delete the signaling domain of LEPR in gonadotropes, and the model was validated as previously described (64).
The first validation step involved the use of organ genotyping to determine whether the pituitary was the sole tissue site of the selective excision of Lepr (Figure 2). Gels A and D show Cre-recombinase genotyping in female (or male) mouse pituitaries. Gels B and E show that all male and female mice expressed 2 alleles of floxed Lepr-exon 17 (249-bp band). Cre-positive mutant pituitaries also had a second 228-bp band indicating deletion of Lepr-exon 17 (LeprΔΔ). Gels in, C (females) and F (males), show the genotyping for Lepr in the remaining organs and no evidence of excised Lepr in females. The male shows LeprΔΔ in the testes (Figure 2F), in agreement with previous reports (63). For this reason, Cre-recombinase is passed down via the females in this line. Gels from tail snips from all offspring were scrutinized carefully for any evidence of extrapituitary Cre recombinase activity. Any evidence of a LeprΔΔ band would eliminate that pup from further study.
Figure 2.

Genotyping of organs from adult female (A–C) or male (D–F) deletion mutants and littermate controls. A, B, D, and E, Genotyping of the pituitaries from these mice. A and D, Mice 5–8 carry the Cre-recombinase transgene as a band at 166 bp. T-cell receptor δ (TCR-Δ) is a housekeeping gene seen in both controls (mice 1–4) and deletion mutants at 200 bp. Also included is DNA from wild-type (wt) and Cre+ animals on the other side of the ladder. The gels in B and F detect the presence of floxed Lepr exon 17 alleles (Lepr-exon17/loxP/loxP) in both controls and deletion mutants at 249 bp. No wt allele is detected (180 bp) except in the wt control (wt) on the other side of the ladder. B and E also show the presence of truncated Lepr (with exon 17 deleted) as a 228-bp band (LeprΔΔ) in the pituitaries of the Cre+ male or female mice (mice 5–8). C and F, Extrapituitary organ genotyping with primers for Lepr-exon 17/loxP/loxP. In females (C), there is no evidence for the presence of LeprΔΔ outside the pituitary, in deletion mutant mice (5–8). In contrast, organ genotyping of males (F) show LeprΔΔ in the testes from all deletion mutants (mice 5–8, arrow) and in the intestine from mouse 5 (arrow).
Cytochemical detection of selective loss of LEPR proteins in the pituitary
To determine whether the excision caused a significant reduction in LEPR protein levels, antisera raised against the cytoplasmic domain of LEPR were used in dual-labeling studies. It had been previously validated in studies of somatotrope-Lepr-exon 17-null mice (64). Figure 3A shows a significant overall decrease (P < .0001) in cells bearing LEPRb in mutant populations along with illustrations in Figure 3, B (control field) and C (mutant field). Dual-labeling showed the lack of LEPR immunolabeling in mutant gonadotropes (Figure 3, D–G). The mutants show significant 80% reductions (P < .0001) in LH and FSH cells with LEPRb, a slight, but significant 9% reduction in GH cells with LEPRb (P = .006), but no reductions in TSH, ACTH, or PRL cells with LEPRb (Figure 3H). Figure 3H also shows no changes in the percentages of any of the pituitary cell types in mutants.
Figure 3.

A, Overall reduction in the percentages of cells immunolabeled for LEPRb (the long form of the receptor) in cell populations from mutant males and diestrous females (n = 15–25). A star indicates significantly lower than controls, P < .0001. B, Field from a normal male mouse, showing the dense gray-black labeling for LEPRb (arrows). Unlabeled cells are pale gray. C, Reduced expression in the population from mutant male mice. Scale bar, 20 μm. D and F, Dual-labeling for LEPRb (black, arrows) and LH (orange) in fields from control (D) or mutant (E) mice. F and G, Dual-labeled LEPRb (black)-FSH (orange) cells from control (F) or mutant (G) male mice. In control and mutant fields, one must visualize the labeling in a through-focus series to detect the site of the peripheral labeling for LEPRb. In the case of the controls, the light micrographs in 1 focal plane do not depict peripheral labeling in several of the LH or FSH cells, which is out of the focal plane. Scale bar, 10 μm. H, Graph of percentages of each pituitary cells taken from counts of cells dual-labeled for LEPRb and each of the pituitary hormones (n = 9) in male mice. There are no changes in percentages of each cell type in these male mutants (Mut). Also, there are no changes in expression of LEPRb by TSH, Prl, and ACTH cells. There are 80% reductions (P < .0001) in LH or FSH cells with LEPRb and a 9% reduction (P = .006) in GH cells with LEPRb (stars). Con, controls.
Tests of gonadotrope responses to LEP, in vitro
To test for residual LEPR function in gonadotropes, we tested LEP's efficacy in stimulating gonadotropin stores and pSTAT3, in vitro. Littermate control mice responded to LEP by increasing numbers of gonadotropes storing LH (females P = .009; males P = .003) or FSH (females, P = .02; males, P = .005), responses that are similar to those reported for the rat (60). Mutant gonadotropes showed no significant responses to LEP (Figure 4, A–D).
Figure 4.

Validation of the loss of LEPR in gonadotropes by testing their responses to LEP. A–D, Counts of fields immunolabeled for LH or FSH and the fact that 100nM LEP stimulates an increase in percentage of LH- or FSH-bearing gonadotropes only in control mice, whereas mutants remain unresponsive. ANOVA followed by Bonferroni's multiple comparison test, n = 9/group. LH: males, P = .003; females, P = .009. FSH: males, P = .005; females, P = .02. E–H, Counts of gonadotropes that responded to LEP by increasing PSTAT3. Gonadotropes were dual-labeled for pSTAT3 (black) and LH or FSH (orange). The values are expressed as percentage of anterior pituitary cells that express both pSTAT3 and LH or FSH. LEP stimulated a significant 9-fold increase in percentages of cells with both pSTAT3 and LH or FSH, only in cells from littermate control males or females. The cells from mutants in which LEPR-exon 17 was ablated in gonadotropes were unresponsive to LEP. A star indicates the highest value; P < .0001, ANOVA followed by Bonferroni's multiple comparison test; n = 9/group. I–M, LEP-treated control and mutant populations from females showing orange/amber FSH cells (I–K) or LH cells (L and M) with pSTAT3 (black dots, arrow). There is little or no labeling for pSTAT3 in these mutant female LH or FSH cells. Scale bar, 10 μm.
Littermate control mice also showed increased pSTAT3 in LH and FSH cells (ANOVA, P < .0001), which indicates the presence of an intact, functional Janus Kinase binding site. In contrast, mutant gonadotropes showed no increased pSTAT3 after LEP treatment (Figure 4, E and F). Dual-labeling in Figure 4, I–M, show labeling for pSTAT3 in dark spots (black) in the cytoplasm of control fields (Figure 4, I, J, and L) and orange labeling for FSH (Figure 4, I–K) or LH (Figure 4, L and M). Fields from mutants had only orange labeling for LH or FSH in the gonadotropes.
Studies of puberty, growth, weight gain, and reproductive capacity
Once the selectivity of the deletion was established, we began an analysis of the overall development of the mutants, focusing mainly on the development and function of the reproductive system. Studies of hundreds of mice over a 5-year period showed that deletion of Lepr-exon 17 in gonadotropes did not affect either the time of onset or the progression of puberty, nor did it significantly affect cyclicity. We also found no differences in testicular or uterine weights in the mutants. We weighed homozygous and heterozygous deletion mutants beginning at 21 days and at 3- to 4-day intervals for 7–9 months. Body weights (grams) were not different when homozygous or heterozygous mutants were compared with littermate controls. Weights ± SEM for males are: 4 months controls 39 ± 1.8 (n = 9), mutants 38.6 ± 1.9 (n = 22); 7 months controls 36 ± 1 (n = 8), mutants 37 ± 1 (n = 8); and 9 months controls 37.5 ± 1.9 (n = 5), mutants 38 ± 2 (n = 8). Weights for females are: 4 months controls 28.5 ± 0.9 (n = 6), mutants 29 ± 1.3 (n = 16); 7 months controls 27.6 ± 1.6 (n = 6), mutants 28.6 ± 1 (n = 14); and 9 months controls 26 ± 2 (n = 4); mutants 26 ± 1.5 (n = 4).
Cages containing a mutant female exhibited a 36% increase in time to first pregnancy (P = .04; n = 7) when compared with cages containing a mutant male (n = 4) (Figure 5A) or control male or female (P < .03) (data not shown). Mutant males produced litters consistently within the 21- to 22-day time range (Figure 5A). When pups were counted, litters from a mutant male had pup numbers as expected from the FVB/NJ background (n = 9, P = .01) (Figure 5B). However, litters from a mutant female had significantly lower numbers of pups (n = 12) compared with litters from control (n = 6, P = .004) or mutant male parents (n = 9; P = .02). Litters from 2 mutant parents were similar to those of the mutant female parent and significantly lower than those from control groups (n = 8, P = .01). Pup survival was greater than 98% for all groups. The deletion mutant females appeared to lactate normally; there was no difference in weight or length of weanlings from these litters.
Figure 5.
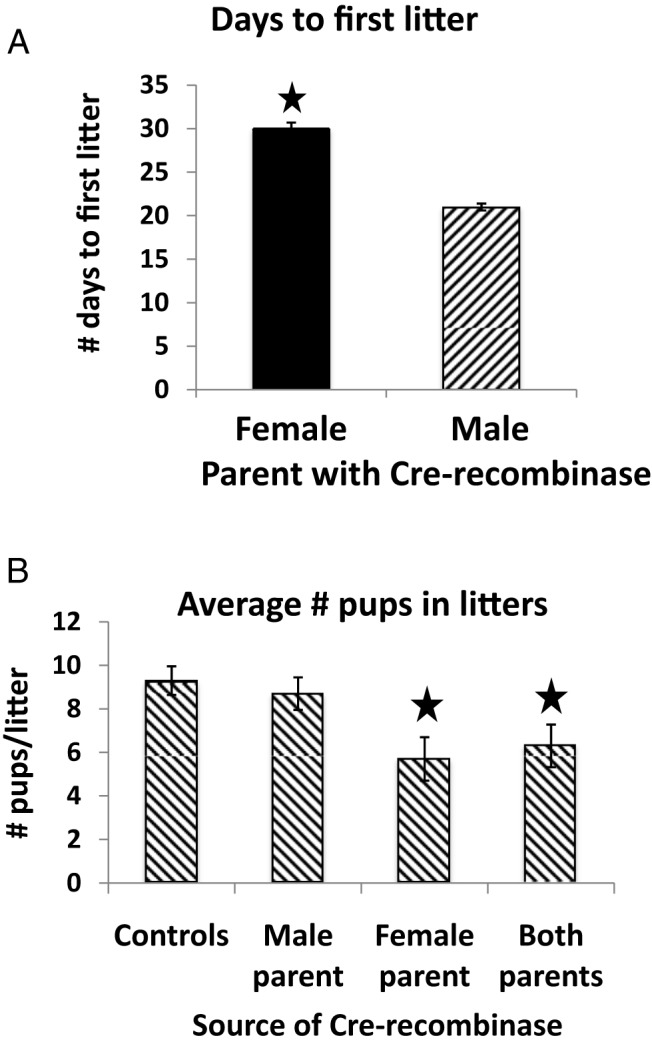
Phenotype of mutant mice-breeding and serum hormones. A, Results of the breeding study in which the numbers of pups/litter were monitored from at least 3 litters/cage containing control parents, and parents with either a mutant male or female, or 2 mutant parents. Time to first litter was monitored for 7 mutant females, and 4 of these females had significantly delayed litters, causing a 36% increase in numbers of days with the male before the first litter. No pregnancy was noted until the last 2 weeks. B, Significant reduction in the number of pups/litter in litters from parents that included a mutant female. A star indicates significantly or males; controls vs mutant female, P = .01 or vs 2 Cre+ male parent, P = .02; Student's t test.
Impact of loss of gonadotrope LEPR on females
We next determined the impact of loss of gonadotrope LEPR on expression of all pituitary hormones, GnRHR, GHRH receptor (GHRHR), and activin/inhibin subunits in females. Mutant diestrous females had a reduction in serum levels of LH (by 40%, P = .01) (Figure 6A), FSH (by 70%; P = .03) (Figure 6B), and GH (by 54%, P = .049) (Figure 6C) but no changes in serum LEP, TSH, PRL, or ACTH levels (Figure 6, D–G) or pituitary LEP-bearing cells (data not shown). Mutants had a significant reduction in mRNA levels of Fsh (by 54%, P = .01) but no changes in mRNA levels of Lh, Cga, or Gnrhr (Figure 6H), Gh or Ghrhr (Figure 6I), or Tsh, Prl, or Pomc (Figure 6J). However, both inhibin/activin β-subunit mRNA levels (A and B) were reduced by 25% (Figure 6K).
Figure 6.

Serum and mRNA levels of pituitary hormones in mutant females. A–C, Mutant diestrous females have significantly lower serum LH (P = .01, n = 15) (A), FSH (P = .03, n = 18) (B), and GH (P = .049; females, n = 21) (C). There were no significant changes in serum PRL (D), TSH (E), ACTH (n = 9) (F), or LEP (n = 8) (G). qPCR assays showed a 51% reduction in Fsh mRNA levels (P = .01, n = 5) (H) and 25% reductions in inhibin/activin β A and β B mRNA levels (P = .04, n = 5) (K). A star indicates significantly different from controls. There were no changes in mRNA levels for Lh, Cga, or Gnrhr (H), Gh or Ghrhr (I), and Tshβ, Prl, or Pomc (J). For the qPCR, the relative expression values were determined by the QuantStudio 12K Flex Software version 1.0 using the δ-δ-CT method.
Impact of loss of gonadotrope LEPR on males
Mutant males showed no significant reductions in serum LH and FSH (Figure 7, A and B). However, there were reductions in levels of serum GH (by 74%, P = .016) (Figure 7C), PRL (by 69%, P = .017) (Figure 7D), and TSH (by 31%, P = .015) (Figure 7E) but no reductions in levels of serum LEP (Figure 7G) or pituitary LEP-bearing cells (data not shown). Mutant males showed reductions in mRNA levels of Fsh and Cga (by 22%, P = .02 and P = .01, respectively) (Figure 7H), Gh (by 30%, P = .02), and Ghrhr (by 36%, P = .03) (Figure 7I). There were no changes in mRNA levels for Lh or Gnrhr (Figure 7H), inhibin/activin subunits (Figure 7J), or Tsh, Pomc, and Prl (Figure 7K).
Figure 7.

Serum and mRNA levels of pituitary hormones in mutant males. Mutant males show no change in LH (n = 19) (A) or FSH (n = 16) (B). However, mutants do show significantly reduced levels of serum GH (P = .016, n = 23) (C), PRL (P = .017, n = 9) (D), and TSH (P = .015, n = 9) (E). There were no reductions in serum ACTH (F) or LEP (G). qPCR assays showed no changes in Lh or Gnrhr mRNA levels, but there were 18% reductions in Fsh (P = .02, n = 5) and Cga (P = .01, n = 5) mRNA levels (H). There were 30%–40% reductions in mRNA levels of Gh (P = .02, n = 5) and Ghrhr (P = .03, n = 5) (I). There was no significant reduction in mRNA levels of inhibin/α-subunits (J) and Tsh, Pomc, or Prl (K). For the qPCR, the relative expression values were determined by the QuantStudio 12K Flex Software version 1.0 using the δ-δ-CT method.
Impact of loss of gonadotrope LEPR on GnRH-binding sites
Counts of gonadotropes dual-labeled for biotinylated GnRH and LH or FSH showed that 90% of LH cells and 92% of FSH cells bound this analog in control males. In contrast, cells from control females taken during the morning of diestrus showed lower levels of binding (70% of LH cells and 82% of FSH cells), which is similar to our previous studies of the rat, in which we showed that GnRH-binding sites in the female rise during the afternoon of diestrus to reach a peak on the morning of proestrus (71, 72). Figure 8, A and C, shows that diestrous mutant females exhibit a severe reduction in GnRH binding to only 13% of LH cells and 9.8% of FSH cells. In contrast, mutant males show a less severe reduction in GnRH binding to 43% of LH cells (Figure 8A) and 68% of FSH cells (Figure 8C). Figure 8, E and F, illustrates the black label for Bio-GnRH and orange label for LH in control (Figure 8E) females and the loss of Bio-GnRH labeling in mutant (Figure 8F) females.
Figure 8.

Expression of GnRH-binding sites in mutants. Counts of LH cells labeled for biotinylated GnRH-binding sites show more severe reductions in binding by LH cells (A) and FSH cells (C) of mutant females, when compared with mutant males. There are also 77%–78% reductions in the percentage of LH or FSH gonadotropes with GnRHR immunoreactivity in mutants (P < .0001, n = 6, ANOVA, Tukey's) (B or D). E and G, Dual-labeling for Bio-GnRH and LH in control (E) or mutant (F) females. The insets in E depict the control cells indicated by arrows, which show the black patch of label for Bio-GnRH and the orange label for LH. No black labeling is seen in the field from the mutant (F). G–J, Dual-labeling for GnRHR immunoreactivity (black, arrows) and LH (G, control; H, mutant) or FSH (I, control; J, mutant) in female populations. The immunoreactivity for GnRHR is peripheral (arrows) and cytoplasmic and sometimes difficult to show all of it in 1 focal plane. G and I, Arrows point to control cells that are depicted in the insets. Scale bars, 20 μm (E–J) and 10 μm (insets).
To determine whether the reduced binding reflected lower expression of GnRHR proteins, we immunolabeled for GnRHR and LH or FSH. Counts of labeled cells showed that labeling for GnRHR was reduced overall by 48% in females (from 20 ± 1 to 10.5 ± 1% of pituitary cells) and 53% in males (from 17 ± 0.8 to 8 ± 0.6% of pituitary cells) (ANOVA, Tukey's, P < .0001). Dual-labeling for GnRHR and LH or FSH showed that 80%–90% of gonadotropes show GnRHR labeling in control males and females. Diestrous females express more cells with GnRHR immunoreactivity than with GnRHR-binding sites (compare Figure 8, A–D).
In mutants, the percentages of LH cells with GnRHR labeling were reduced by 70% in males or 79% in females (ANOVA, Tukey's, P < .0001) (Figure 8B). Similarly, the percentages of FSH cells with GnRHR immunolabeling were reduced by 77% in mutant females and 82% in mutant males (ANOVA, Tukey's, P < .0001) (Figure 8D). Mutant somatotropes, which normally express GnRHR in 20% of their population (72, 73), showed no changes in GnRHR immunoreactivity (data not shown). Dual-labeling in control fields is illustrated in Figure 8, G (LH) or I (FSH), showing black label for GnRHR and orange label for LH or FSH. Figure 8, H and J, shows mutant fields where only orange label for LH or FSH is evident.
Discussion
This study is the first in a series that will characterize the impact of the loss of gonadotrope LEPR. This first study validated the model and determined whether the loss of LEPR had a broad impact on pituitary cell functions and reproductive competence. The broader view of pituitary function served secondary objectives, including proof for the selectivity of the loss of LEPR and the importance of gonadotropes as paracrine regulators. The discoveries presented in this study will form the basis for future studies that focus on basic mechanisms behind LEP actions on gonadotropes as well as the impact of the selective loss of LEPR on the reproductive cycle.
The development of the model itself was justified by the striking 2-fold increases in LEPR in proestrus and estrus, indicating that LEP may optimize gonadotrope function during the proestrous LH surge or the estrous rise in FSH. The timing of the increase in LEPR matches the midcycle serum LEP rise (21–25), thus supporting this hypothesis. This plasticity in LEPR expression in the female also suggests that females may be more dependent on LEP stimulation for gonadotrope and reproductive function than the males, a hypothesis that was indeed strengthened by the model.
Validation of the selectivity of the deletion of LEPR
Organ genotyping detected pituitary and testicular expression of Cre-recombinase by a second LeprΔΔ band indicating excised Lepr (Figure 3), which was expected, based on previous studies of this line (63). We did not detect extrapituitary Cre-recombinase expression in ovaries of female mice, and therefore, offspring came from cages in which the female carried the Cre-recombinase. We have not yet been able to identify the source of the extrapituitary Cre-recombinase seen in a subset of pups from most litters.
Dual-labeling for LEPR and all pituitary hormones showed that only cells known to produce LH (72–74) had significant losses in LEPR. Another discovery was that loss of LEPR did not affect hormone stores or percentages of any of the pituitary cell types (including LH and FSH cells), suggesting that LEP is not involved in differentiation of these cells, or their translation and storage of protein hormones.
Finally, our tests of gonadotrope responses to LEP showed no residual functional LEPR. These tests took advantage of 2 known LEP regulatory functions based on our previous studies of gonadotropes: the stimulation of LH and FSH stores (60) and the activation of the Janus Kinase STAT pathway, producing pSTAT3 (64).
The deletion of LEPR in gonadotropes impacted fertility in the female
When we examined the consequences of the loss of LEP signals to gonadotropes and the reproductive system, we found no impact on growth, weight gain, the timing of puberty, or on the overall capacity to produce pups. However, the formal breeding study showed a delayed time to first litter (by 36%) in cages with mutant females. Also, mutant females had reduced numbers of pups (compared with other females of the same strain), indicating that they are subfertile. We detected no impairment of fertility in males.
Lower GnRH-binding sites and activin/inhibin β-subunits contributed to subfertility in females
Diestrous females showed lower serum gonadotropins and GH, which could contribute to their subfertility. The absence of changes in percentages of LH, FSH, or GH cells suggests no role for gonadotrope LEPR on hormone stores. Our tests of regulators for these cells led to the discovery of 2 important gene products that could explain the lower gonadotropins and the subfertility. The most striking is the severe reduction in binding sites for biotinylated GnRH. In addition, females showed significant reductions in mRNA levels for inhibin/activin β-subunits A and B. These subunits dimerize to form activin, which is an important autocrine/paracrine regulator for FSH (75–78). The fact that inhibin α-subunit mRNA levels are unchanged suggests that the deficiency may lie in the production of activin. A significant reduction in activin mRNA correlates well with lower levels of FSH, GnRH-binding sites, and GH secretion, because we have shown that activin stimulates GnRH-binding sites by gonadotropes and somatotropes (79).
The fact that GnRH-binding sites are low in diestrous mutants is important, because there is a well-established normal cyclic increase in GnRHR expression during diestrus reaching a peak on the morning of proestrus and which is needed for the LH surge (71, 72). This rise parallels the rise in LEPR (Figure 1) and the rise in serum LEP (21–25). It is preceded by a midcycle rise in pituitary LEP, which is mainly in GH cells (80). Thus, the lowered GnRH binding resulting from the deletion of LEPR indicates that LEP from either paracrine or endocrine sources may be needed to facilitate the production and/or trafficking of GnRHR to the surface.
This validation study of mutant diestrous females is a snapshot in time of the events that may be ongoing during the estrous cycle. A complete study of GnRH binding and activin expression throughout the cycle will be needed to determine the impact of loss of LEPR on the reproductive cycle. In addition, future studies are needed to determine whether GnRHR and activin are direct targets. With respect to GnRHR, is interesting to note that the deficiency appears to be in GnRHR protein expression and not in GnRHR mRNA, suggesting that LEP's actions on this target molecule may be posttranscriptional. This finding suggests a potential novel mechanism behind LEP actions on gonadotropes, which will also be explored in future studies.
These findings have translational relevance, because therapeutic approaches for infertility (clomiphene) rely on active, functional GnRHRs. This might explain why overweight and obese women, which are LEP resistant, have poor responses to fertility treatments like clomiphene (81–83). A study of obese pubertal girls reported that hyperleptinemia correlated inversely with responses to exogenous GnRH, which was detected by reduced serum levels of LH and FSH when the girls were given GnRH (84). Thus, if normal GnRHR expression depends on LEP, females that are LEP deficient or LEP resistant may not be able to respond as well to infertility treatments.
Finally, in mutant females, the reduced serum GH was not correlated with reductions in stores of GH in the somatotropes, or Gh or Ghrhr mRNA levels. Further studies of GHRH protein levels may detect deficiencies that would explain the reduced serum levels. Also, it is important to note that, because GH is considered a cogonadotropin, which stimulates the development of preantral follicles (73), the lower serum GH might have contributed to the lower number of pups from mutant females.
Loss of LEPR in male gonadotropes impacts the pituitary specific positive transcription factor-1 (Pit-1 line of cells
The findings in the male correlate well with the fact that the male mutants remain fully fertile. The reduction in GnRH-binding sites is much less severe than that in the mutant females. Gonadotropes have been reported to have spare receptors, and the actual number of GnRHRs is considered a weak determinant of the amount of LH that is secreted (85, 86). In fact, only 20% of the GnRHRs need to be occupied for LH secretion (86). Thus, the male gonadotropes appear to have sufficient GnRHR for responses needed in reproduction. There is a slight but significant reduction in Fsh and Cga mRNA levels, with no apparent impact on protein stores or secretion. The trend towards reduced serum LH and FSH did not reach significance, even when we increased the n to 16–19 mice.
Gonadotropes are metabolic sensors, receiving direct signals from serum glucose, for example (87, 88). They also produce paracrine factors that may regulate other cell types (89, 90). The loss of LEPR in male gonadotropes shows a potential paracrine impact on the cells most involved in metabolic regulation: TSH, GH, and PRL cells. Because hormone storage in these cells is not deficient, the low serum levels in the mutants suggest a deficiency in 1 or more regulators that may come from gonadotropes. A recent comprehensive review by Denef (89) reported over 100 regulatory compounds in pituitary cells and identified 22 produced by gonadotropes. Among these peptides are known regulators for somatotropes, thyrotropes, and lactotropes. A number of studies have reported severe reductions in PRL cell functions in animals in which gonadotropes are ablated, thus further supporting the lactotrope dependency on gonadotropes (89–94).
These 3 cell types have in common the fact that they all share the transcription factor, Pit-1, as a regulator, especially during early development (95–98). Pit-1 also regulates the expression of GHRHR mRNA (99), which is reduced in the mutant males and could cause the reduction in serum GH in these mutants. Thus, at this point, the impact of LEPR on gonadotropes in the male appears to focus on the gonadotropes' role as metabolic sensors and paracrine regulators of cells of the Pit-1 lineage. Future studies will be needed to identify the paracrine regulators that are also dependent on LEP. The heteogeneity in gonadotropes correlates well with previous studies of this dynamic population (100).
Summary
To summarize, this study of mice, in which the signaling domain of LEPR was deleted selectively in gonadotropes, showed that LEP signals to gonadotropes are not needed for the maintenance of optimal numbers of gonadotropes. The female gonadotrope-LEPR-null model shows clearly the importance of the rise in LEPR during proestrus as the deletion of LEPR in gonadotropes reduces GnRH-binding sites and activin mRNA. This reduction correlates well with the lowered serum FSH and LH and ultimately with delays in pregnancy and reduced numbers of pups/litter.
It is clear from these studies of the male LEPR-null gonadotrope model that LEPRs are not needed for normal fertility. Rather, this model may highlight the male gonadotrope's role as a metabolic sensor and paracrine regulator. The male mutant model will thus elucidate LEP molecular targets that may serve as paracrine regulators of the Pit-1 line of cells.
The model revealed translationally relevant information when it identified GnRH-binding sites as potential LEP targets. Infertility treatments that use clomiphene rely on responses to GnRH. These treatments may be compromised in cases of LEP resistance (obesity) or deficiency (anorexia nervosa). The findings might explain why overweight and obese women, who are LEP resistant, have poor responses to clomiphene (81–83).
The permissive regulatory function for LEP in females could reflect its role as a metabolic signal, to promote adaptation of the reproductive system to available energy supplies. Because LEP declines during nutritional deprivation, this loss of signal is detected by gonadotropes, and the females will adapt by producing fewer offspring. Thus, LEP may serve as a gateway that optimizes reproductive outcomes to fit the environment. This might benefit a population faced with food shortages, without preventing reproduction altogether.
Acknowledgments
We thank Dr Sally Camper (University of Michigan) for the generous gift of the mice bearing Cre-recombinase driven by the bLhβ promoter and Dr Streamson Chua (Albert Einstein College of Medicine) for the generous gift of mice bearing Lepr-exon 17loxP/loxP. We also thank Dr A. Parlow and the National Institute of Health Hormone Pituitary Distribution program for the antisera to GH, FSH, and LH. A portion of this study was presented at 95th Annual Meeting of The Endocrine Society, June 15–18, 2013, San Francisco, CA, during oral session (OR04-4), and another portion was presented at the 16th Annual Meeting of the International Congress of Endocrinologists, June 21–24, 2014, Chicago, IL in a poster session.
This work was supported by bridging funds from the University of Arkansas for Medical Sciences Research Council and by National Institute of Health Grants R03 HD059066 and 1R01HD059056 (to G.V.C.). Core facilities were funded by the National Institute of Health National Center for Research Resources Grant P20 RR020146 and the National Institue of Health Grant P30 NS047546 at University of Arkansas for Medical Sciences.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Bio-GnRH
- biotinylated analogs of D-Lys6-GnRH
- Cga
- glycoprotein hormone α-subunit
- GHRHR
- GHRH receptor
- GnRHR
- GnRH receptor
- LEP
- leptin
- LEPR
- LEP receptor
- Pit-1
- pituitary specific positive transcription factor-1
- Pomc
- proopiomelanocortin
- PRL
- prolactin
- pSTAT3
- phosphorylated signal transducer and activator of transcription 3
- qPCR
- quantitative polymerase chain reaction.
References
- 1. Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. J Clin Invest. 1997;99:391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252 [DOI] [PubMed] [Google Scholar]
- 3. Barash IA, Cheung CC, Weigle DS, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147 [DOI] [PubMed] [Google Scholar]
- 4. Finn PD, Cunningham MJ, Pau KY, Spies HG, Clifton DK, Steiner RA. The stimulatory effect of leptin on the neuroendocrine reproductive axis of the monkey. Endocrinology. 1998;139:4652–4662 [DOI] [PubMed] [Google Scholar]
- 5. Ishii S, Shibasaki T, Murakami T, Shima K, Wakabayashi I. Response of leptin mRNA to 24-h food deprivation and refeeding is influenced by age in rats. Regul Pept. 2000;92:45–50 [DOI] [PubMed] [Google Scholar]
- 6. Mann DR, Plant TM. Leptin and pubertal development. Semin Reprod Med. 2002;20:93–102 [DOI] [PubMed] [Google Scholar]
- 7. Urbanski HF. Leptin and puberty. Trends Endocrinol Metab. 2001;12:428–429 [DOI] [PubMed] [Google Scholar]
- 8. Clément K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401 [DOI] [PubMed] [Google Scholar]
- 9. Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908 [DOI] [PubMed] [Google Scholar]
- 10. Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–215 [DOI] [PubMed] [Google Scholar]
- 11. Kaufman BA, Warren MP, Dominguez JE, Wang J, Heymsfield SB, Pierson RN. Bone density and amenorrhea in ballet dancers are related to a decreased resting metabolic rate and lower leptin levels. J Clin Endocrinol Metab. 2002;87:2777–2783 [DOI] [PubMed] [Google Scholar]
- 12. Laughlin GA, Yen SS. Hypoleptinemia in women athletes: absence of a diurnal rhythm with amenorrhea. J Clin Endocrinol Metab. 1997;82:318–321 [DOI] [PubMed] [Google Scholar]
- 13. Miller KK, Parulekar MS, Schoenfeld E, et al. Decreased leptin levels in normal weight women with hypothalamic amenorrhea: the effects of body composition and nutritional intake. J Clin Endocrinol Metab. 1998;83:2309–2312 [DOI] [PubMed] [Google Scholar]
- 14. Thong FS, Graham TE. Leptin and reproduction: is it a critical link between adipose tissue, nutrition, and reproduction? Can J Appl Physiol. 1999;24:317–336 [DOI] [PubMed] [Google Scholar]
- 15. Thong FS, McLean C, Graham TE. Plasma leptin in female athletes: relationship with body fat, reproductive, nutritional, and endocrine factors. J Appl Physiol. 2000;88:2037–2044 [DOI] [PubMed] [Google Scholar]
- 16. Warren MP, Voussoughian F, Geer EB, Hyle EP, Adberg CL, Ramos RH. Functional hypothalamic amenorrhea: hypoleptinemia and disordered eating. J Clin Endocrinol Metab. 1999;84:873–877 [DOI] [PubMed] [Google Scholar]
- 17. Welt CK. Will leptin become the treatment of choice for functional hypothalamic amenorrhea? Nat Clin Pract Endocrinol Metab. 2007;3:556–557 [DOI] [PubMed] [Google Scholar]
- 18. Welt CK, Chan JL, Bullen J, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997 [DOI] [PubMed] [Google Scholar]
- 19. Cella F, Giordano G, Cordera R. Serum leptin concentrations during the menstrual cycle in normal-weight women: effects of an oral triphasic estrogen-progestin medication. Eur J Endocrinol. 2000;142:174–178 [DOI] [PubMed] [Google Scholar]
- 20. Riad-Gabriel MG, Jinagouda SD, Sharma A, Boyadjian R, Saad MF. Changes in plasma leptin during the menstrual cycle. Eur J Endocrinol. 1998;139:528–531 [DOI] [PubMed] [Google Scholar]
- 21. Carro E, Pinilla L, Seoane LM, et al. Influence of endogenous leptin tone on the estrous cycle and luteinizing hormone pulsatility in female rats. Neuroendocrinology. 1997;66:375–377 [DOI] [PubMed] [Google Scholar]
- 22. De Biasi SN, Apfelbaum LI, Apfelbaum ME. In vitro effect of leptin on LH release by anterior pituitary glands from female rats at the time of spontaneous and steroid-induced LH surge. Eur J Endocrinol. 2001;145:659–665 [DOI] [PubMed] [Google Scholar]
- 23. Fungfuang W, Nakada T, Nakao N, et al. Serum leptin concentrations, leptin mRNA expression, and food intake during the estrous cycle in rats. Lab Anim Res. 2013;29:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fungfuang W, Terada M, Komatsu N, Moon C, Saito TR. Effects of estrogen on food intake, serum leptin levels and leptin mRNA expression in adipose tissue of female rats. Lab Anim Res. 2013;29:168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider JE, Goldman MD, Tang S, Bean B, Ji H, Friedman MI. Leptin indirectly affects estrous cycles by increasing metabolic fuel oxidation. Horm Behav. 1998;33:217–228 [DOI] [PubMed] [Google Scholar]
- 26. Licinio J, Negrão AB, Mantzoros C, et al. Synchronicity of frequently sampled, 24-h concentrations of circulating leptin, luteinizing hormone, and estradiol in healthy women. Proc Natl Acad Sci USA. 1998;95:2541–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagatani S, Zeng Y, Keisler DH, Foster DL, Jaffe CA. Leptin regulates pulsatile luteinizing hormone and growth hormone secretion in the sheep. Endocrinology. 2000;141:3965–3975 [DOI] [PubMed] [Google Scholar]
- 28. Lehman MN, Merkley CM, Coolen LM, Goodman RL. Anatomy of the kisspeptin neural network in mammals. Brain Res. 2010;1364:90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lehman MN, Coolen LM, Goodman RL. Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology. 2010;151:3479–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMinn JE, Liu SM, Liu H, et al. Neuronal deletion of Lepr elicits diabesity in mice without affecting cold tolerance or fertility. Am J Physiol Endocrinol Metab. 2005;289:E403–E411 [DOI] [PubMed] [Google Scholar]
- 31. Elias CF. Leptin action in pubertal development: recent advances and unanswered questions. Trends Endocrinol Metab. 2012;23:9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elias CF, Purohit D. Leptin signaling and circuits in puberty and fertility. Cell Mol Life Sci. 2013;70:841–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cai A, Hyde JF. Upregulation of leptin receptor gene expression in the anterior pituitary of human growth hormone-releasing hormone transgenic mice. Endocrinology. 1998;139:420–423 [DOI] [PubMed] [Google Scholar]
- 34. Cai A, Hyde JF. The human growth hormone-releasing hormone transgenic mouse as a model of modest obesity: differential changes in leptin receptor (OBR) gene expression in the anterior pituitary and hypothalamus after fasting and OBR localization in somatotrophs. Endocrinology. 1999;140:3609–3614 [DOI] [PubMed] [Google Scholar]
- 35. Iqbal J, Pompolo S, Considine RV, Clarke IJ. Localization of leptin receptor-like immunoreactivity in the corticotropes, somatotropes, and gonadotropes in the ovine anterior pituitary. Endocrinology. 2000;141:1515–1520 [DOI] [PubMed] [Google Scholar]
- 36. Sone M, Nagata H, Takekoshi S, Osamura RY. Expression and localization of leptin receptor in the normal rat pituitary gland. Cell Tissue Res. 2001;305:351–356 [DOI] [PubMed] [Google Scholar]
- 37. Sone M, Osamura RY. Leptin and the pituitary. Pituitary. 2001;4:15–23 [DOI] [PubMed] [Google Scholar]
- 38. Jin L, Zhang S, Burguera BG, et al. Leptin and leptin receptor expression in rat and mouse pituitary cells. Endocrinology. 2000;141:333–339 [DOI] [PubMed] [Google Scholar]
- 39. Giusti M, Bocca L, Florio T, et al. In vitro effect of human recombinant leptin and expression of leptin receptors on growth hormone-secreting human pituitary adenomas. Clin Endocrinol (Oxf). 2002;57:449–455 [DOI] [PubMed] [Google Scholar]
- 40. Jin L, Burguera BG, Couce ME, et al. Leptin and leptin receptor expression in normal and neoplastic human pituitary: evidence of a regulatory role for leptin on pituitary cell proliferation. J Clin Endocrinol Metab. 1999;84:2903–2911 [DOI] [PubMed] [Google Scholar]
- 41. Lloyd RV, Jin L, Tsumanuma I, et al. Leptin and leptin receptor in anterior pituitary function. Pituitary. 2001;4:33–47 [DOI] [PubMed] [Google Scholar]
- 42. Shimon I, Yan X, Magoffin DA, Friedman TC, Melmed S. Intact leptin receptor is selectively expressed in human fetal pituitary and pituitary adenomas and signals human fetal pituitary growth hormone secretion. J Clin Endocrinol Metab. 1998;83:4059–4064 [DOI] [PubMed] [Google Scholar]
- 43. Chan JL, Mantzoros CS. Leptin and the hypothalamic-pituitary regulation of the gonadotropin-gonadal axis. Pituitary. 2001;4:87–92 [DOI] [PubMed] [Google Scholar]
- 44. Ogura K, Irahara M, Kiyokawa M, et al. Effects of leptin on secretion of LH and FSH from primary cultured female rat pituitary cells. Eur J Endocrinol. 2001;144:653–658 [DOI] [PubMed] [Google Scholar]
- 45. Spicer LJ. Leptin: a possible metabolic signal affecting reproduction. Domest Anim Endocrinol. 2001;21:251–270 [DOI] [PubMed] [Google Scholar]
- 46. Tezuka M, Irahara M, Ogura K, et al. Effects of leptin on gonadotropin secretion in juvenile female rat pituitary cells. Eur J Endocrinol. 2002;146:261–266 [DOI] [PubMed] [Google Scholar]
- 47. Swerdloff RS, Batt RA, Bray GA. Reproductive hormonal function in the genetically obese (ob/ob) mouse. Endocrinology. 1976;98:1359–1364 [DOI] [PubMed] [Google Scholar]
- 48. Swerdloff RS, Peterson M, Vera A, Batt RA, Heber D, Bray GA. The hypothalamic-pituitary axis in genetically obese (ob/ob) mice: response to luteinizing hormone-releasing hormone. Endocrinology. 1978;103:542–547 [DOI] [PubMed] [Google Scholar]
- 49. Yu WH, Kimura M, Walczewska A, Karanth S, McCann SM. Role of leptin in hypothalamic-pituitary function. Proc Natl Acad Sci USA. 1997;94:1023–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yura S, Ogawa Y, Sagawa N, et al. Accelerated puberty and late-onset hypothalamic hypogonadism in female transgenic skinny mice overexpressing leptin. J Clin Invest. 2000;105:749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320 [DOI] [PubMed] [Google Scholar]
- 52. Gonzalez LC, Pinilla L, Tena-Sempere M, Aguilar E. Leptin(116–130) stimulates prolactin and luteinizing hormone secretion in fasted adult male rats. Neuroendocrinology. 1999;70:213–220 [DOI] [PubMed] [Google Scholar]
- 53. Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL. Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. 1998;67:370–376 [DOI] [PubMed] [Google Scholar]
- 54. Schneider JE, Blum RM, Wade GN. Metabolic control of food intake and estrous cycles in syrian hamsters. I. Plasma insulin and leptin. Am J Physiol Regul Integr Comp Physiol. 2000;278:R476–R485 [DOI] [PubMed] [Google Scholar]
- 55. Schneider JE, Buckley CA, Blum RM, et al. Metabolic signals, hormones and neuropeptides involved in control of energy balance and reproductive success in hamsters. Eur J Neurosci. 2002;16:377–379 [DOI] [PubMed] [Google Scholar]
- 56. Schneider JE, Zhou D. Interactive effects of central leptin and peripheral fuel oxidation on estrous cyclicity. Am J Physiol. 1999;277:R1020–R1024 [DOI] [PubMed] [Google Scholar]
- 57. Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884 [DOI] [PubMed] [Google Scholar]
- 58. Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu WH, Walczewska A, Karanth S, McCann SM. Nitric oxide mediates leptin-induced luteinizing hormone-releasing hormone (LHRH) and LHRH and leptin-induced LH release from the pituitary gland. Endocrinology. 1997;138:5055–5058 [DOI] [PubMed] [Google Scholar]
- 60. Crane C, Akhter N, Johnson BW, et al. Fasting and glucose effects on pituitary leptin expression: is leptin a local signal for nutrient status? J Histochem Cytochem. 2007;55:1059–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Akhter N, Johnson BW, Crane C, et al. Anterior pituitary leptin expression changes in different reproductive states: in vitro stimulation by gonadotropin-releasing hormone. J Histochem Cytochem. 2007;55:151–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McMinn JE, Liu SM, Dragatsis I, et al. An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome. 2004;15:677–685 [DOI] [PubMed] [Google Scholar]
- 63. Charles MA, Mortensen AH, Potok MA, Camper SA. Pitx2 deletion in pituitary gonadotropes is compatible with gonadal development, puberty, and fertility. Genesis. 2008;46:507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Childs GV, Akhter N, Haney A, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology. 2011;152:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Syed M, Cozart M, Haney AC, et al. Ghrelin restoration of function in vitro in somatotropes from male mice lacking the Janus kinase (JAK)-binding site of the leptin receptor. Endocrinology. 2013;154:1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Childs GV, Naor Z, Hazum E, Tibolt R, Westlund KN, Hancock MB. Localization of biotinylated gonadotropin releasing hormone on pituitary monolayer cells with avidin-biotin-peroxidase complexes. J Histochem Cytochem. 1983;31:1422–1425 [DOI] [PubMed] [Google Scholar]
- 67. Childs GV, Naor Z, Hazum E, Tibolt R, Westlund KN, Hancock MB. Cytochemical characterization of pituitary target cells for biotinylated gonadotropin releasing hormone. Peptides. 1983;4:549–555 [DOI] [PubMed] [Google Scholar]
- 68. Childs GV, Unabia G, Tibolt R, Lloyd JM. Cytological factors that support nonparallel secretion of luteinizing hormone and follicle-stimulating hormone during the estrous cycle. Endocrinology. 1987;121:1801–1813 [DOI] [PubMed] [Google Scholar]
- 69. Childs GV. Division of labor among gonadotropes. Vitam Horm. 1995;50:215–286 [DOI] [PubMed] [Google Scholar]
- 70. Childs GV. Gonadotropes and lactotropes. In: Physiology of Reproduction. Neill J, Knobil E, eds. New York, NY: Elsevier Press; 2006:1483–1579 [Google Scholar]
- 71. Lloyd JM, Childs GV. Changes in the number of GnRH-receptive cells during the rat estrous cycle: biphasic effects of estradiol. Neuroendocrinology. 1988;48:138–146 [DOI] [PubMed] [Google Scholar]
- 72. Childs GV, Unabia G, Miller BT. Cytochemical detection of gonadotropin-releasing hormone-binding sites on rat pituitary cells with luteinizing hormone, follicle-stimulating hormone, and growth hormone antigens during diestrous up-regulation. Endocrinology. 1994;134:1943–1951 [DOI] [PubMed] [Google Scholar]
- 73. Childs GV. Growth hormone cells as co-gonadotropes: partners in the regulation of the reproductive system. Trends Endocrinol Metab. 2000;11:168–175 [DOI] [PubMed] [Google Scholar]
- 74. Childs GV, Unabia G, Rougeau D. Cells that express luteinizing hormone (LH) and follicle-stimulating hormone (FSH) β-subunit messenger ribonucleic acids during the estrous cycle: the major contributors contain LH β, FSH β, and/or growth hormone. Endocrinology. 1994;134:990–997 [DOI] [PubMed] [Google Scholar]
- 75. Bilezikjian LM, Vale WW. The local control of the pituitary by activin signaling and modulation. Open Neuroendocrinol J. 2011;4:90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bilezikjian LM, Blount AL, Leal AM, Donaldson CJ, Fischer WH, Vale WW. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol Cell Endocrinol. 2004;225:29–36 [DOI] [PubMed] [Google Scholar]
- 77. Popovics P, Rekasi Z, Stewart AJ, Kovacs M. Regulation of pituitary inhibin/activin subunits and follistatin gene expression by GnRH in female rats. J Endocrinol. 2011;210:71–79 [DOI] [PubMed] [Google Scholar]
- 78. Cheng GF, Yuen CW, Ge W. Evidence for the existence of a local activin follistatin negative feedback loop in the goldfish pituitary and its regulation by activin and gonadal steroids. J Endocrinol. 2007;195:373–384 [DOI] [PubMed] [Google Scholar]
- 79. Childs GV, Unabia G. Cytochemical studies of the effects of activin on gonadotropin-releasing hormone (GnRH) binding by pituitary gonadotropes and growth hormone cells. J Histochem Cytochem. 1997;45:1603–1610 [DOI] [PubMed] [Google Scholar]
- 80. Akhter N, Crane C, Childs GV. Pituitary Leptin-A Paracrine Regulator of Gonadotropes: A Review. The Open Neuroendocrinology Journal. [published online May 6, 2011] 4:25.42 doi:10.2174/1876528901104010025 [Google Scholar]
- 81. Koning AM, Kuchenbecker WK, Groen H, et al. Economic consequences of overweight and obesity in infertility: a framework for evaluating the costs and outcomes of fertility care. Hum Reprod Update. 2010;16:246–254 [DOI] [PubMed] [Google Scholar]
- 82. Pandey S, Pandey S, Maheshwari A, Bhattacharya S. The impact of female obesity on the outcome of fertility treatment. J Hum Reprod Sci. 2010;3:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Metwally M, Li TC, Ledger WL. The impact of obesity on female reproductive function. Obes Rev. 2007;8:515–523 [DOI] [PubMed] [Google Scholar]
- 84. Bouvattier C, Lahlou N, Roger M, Bougnères P. Hyperleptinaemia is associated with impaired gonadotrophin response to GnRH during late puberty in obese girls, not boys. Eur J Endocrinol. 1998;138:653–658 [DOI] [PubMed] [Google Scholar]
- 85. Smith PF, Neill JD. Simultaneous measurement of hormone release and secretagogue binding by individual pituitary cells. Proc Natl Acad Sci USA. 1987;84:5501–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Naor Z, Clayton RN, Catt KJ. Characterization of gonadotropin-releasing hormone receptors in cultured rat pituitary cells. Endocrinology. 1980;107:1144–1152 [DOI] [PubMed] [Google Scholar]
- 87. Sorenson RL, Stout LE, Brelje TC, Jetton TL, Matschinsky FM. Immunohistochemical evidence for the presence of glucokinase in the gonadotropes and thyrotropes of the anterior pituitary gland of rat and monkey. J Histochem Cytochem. 2007;55:555–566 [DOI] [PubMed] [Google Scholar]
- 88. Zelent D, Golson ML, Koeberlein B, et al. A glucose sensor role for glucokinase in anterior pituitary cells. Diabetes. 2006;55:1923–1929 [DOI] [PubMed] [Google Scholar]
- 89. Denef C. Paracrinicity: the story of 30 years of cellular pituitary crosstalk. J Neuroendocrinol. 2008;20:1–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Denef C, Andries M. Evidence for paracrine interaction between gonadotrophs and lactotrophs in pituitary cell aggregates. Endocrinology. 1983;112:813–822 [DOI] [PubMed] [Google Scholar]
- 91. Kendall SK, Saunders TL, Jin L, et al. Targeted ablation of pituitary gonadotropes in transgenic mice. Mol Endocrinol. 1991;5:2025–2036 [DOI] [PubMed] [Google Scholar]
- 92. Seuntjens E, Vankelecom H, Quaegebeur A, Vande Vijver V, Denef C. Targeted ablation of gonadotrophs in transgenic mice affects embryonic development of lactotrophs. Mol Cell Endocrinol. 1999;150:129–139 [DOI] [PubMed] [Google Scholar]
- 93. Van Bael A, Huygen R, Himpens B, Denef C. In vitro evidence that LHRH stimulates the recruitment of prolactin mRNA-expressing cells during the postnatal period in the rat. J Mol Endocrinol. 1994;12:107–118 [DOI] [PubMed] [Google Scholar]
- 94. Vankelecom H, Seuntjens E, Hauspie A, Denef C. Targeted ablation of gonadotrophs in transgenic mice depresses prolactin but not growth hormone gene expression at birth as measured by quantitative mRNA detection. J Biomed Sci. 2003;10:805–812 [DOI] [PubMed] [Google Scholar]
- 95. Ingraham HA, Chen RP, Mangalam HJ, et al. A tissue-specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell. 1988;55:519–529 [DOI] [PubMed] [Google Scholar]
- 96. Mangalam HJ, Albert VR, Ingraham HA, et al. A pituitary POU domain protein, Pit-1, activates both growth hormone and prolactin promoters transcriptionally. Genes Dev. 1989;3:946–958 [DOI] [PubMed] [Google Scholar]
- 97. Nelson C, Albert VR, Elsholtz HP, Lu LI, Rosenfeld MG. Activation of cell-specific expression of rat growth hormone and prolactin genes by a common transcription factor. Science. 1988;239:1400–1405 [DOI] [PubMed] [Google Scholar]
- 98. Steinfelder HJ, Hauser P, Nakayama Y, et al. Thyrotropin-releasing hormone regulation of human TSHB expression: role of a pituitary-specific transcription factor (Pit-1/GHF-1) and potential interaction with a thyroid hormone-inhibitory element. Proc Natl Acad Sci USA. 1991;88:3130–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Salvatori R, Fan X, Mullis PE, Haile A, Levine MA. Decreased expression of the GHRH receptor gene due to a mutation in a Pit-1 binding site. Mol Endocrinol. 2002;16:450–458 [DOI] [PubMed] [Google Scholar]
- 100. Childs GV, Unabia G, Lloyd J. Recruitment and maturation of small subsets of luteinizing hormone gonadotropes during the estrous cycle. Endocrinology. 1992;130:335–344 [DOI] [PubMed] [Google Scholar]