

Cryo-EM and molecular dynamics simulations reveal unexpected flexibility in individual monomers and a stable interface between monomers in the spiral filaments formed by the ESCRT-III subunit Vps32/CHMP4B.
Abstract
The scission of biological membranes is facilitated by a variety of protein complexes that bind and manipulate lipid bilayers. ESCRT-III (endosomal sorting complex required for transport III) filaments mediate membrane scission during the ostensibly disparate processes of multivesicular endosome biogenesis, cytokinesis, and retroviral budding. However, mechanisms by which ESCRT-III subunits assemble into a polymer remain unknown. Using cryogenic electron microscopy (cryo-EM), we found that the full-length ESCRT-III subunit Vps32/CHMP4B spontaneously forms single-stranded spiral filaments. The resolution afforded by two-dimensional cryo-EM combined with molecular dynamics simulations revealed that individual Vps32/CHMP4B monomers within a filament are flexible and able to accommodate a range of bending angles. In contrast, the interface between monomers is stable and refractory to changes in conformation. We additionally found that the carboxyl terminus of Vps32/CHMP4B plays a key role in restricting the lateral association of filaments. Our findings highlight new mechanisms by which ESCRT-III filaments assemble to generate a unique polymer capable of membrane remodeling in multiple cellular contexts.
Introduction
Membrane bending and fission are required for numerous biological processes, including organelle biogenesis, the formation of vesicular transport carriers, and cellular division (Hurley and Hanson, 2010; Schmid and Frolov, 2011). Components of the endosomal complex required for transport (ESCRT) machinery facilitate scission events in cases in which membrane budding is oriented away from the cytoplasm (Henne et al., 2011). Specifically, ESCRT-III polymers have been implicated directly in the topological transition necessary for bilayers to rupture and reseal (Fabrikant et al., 2009; Guizetti and Gerlich, 2012). In vivo, membrane-associated ESCRT-III has been suggested to be composed primarily of polymerized Vps32 subunits (also known as Snf7p in yeast and homologous to three distinct isoforms of CHMP4 in mammals), which are nucleated by a complex of ESCRT-II and the ESCRT-III component Vps20 at multivesicular endosomes (MVEs) and through undefined mechanisms during cytokinetic abscission and viral budding (Teis et al., 2008; Pires et al., 2009; Fyfe et al., 2011). Vps32 recruits two additional ESCRT-III subunits, Vps24 and Vps2, which can also coassemble to form a distinct polymer (Lata et al., 2008a). At least four other ESCRT-III proteins have also been identified (Ist1, Did2, Vps60, and CHMP7), providing additional forms of regulation to ESCRT-III assembly and the membrane scission process.
In general, ESCRT-III subunits harbor a series of α helices that are joined by short linker sequences. Based on their related compositions, each is believed to exhibit an analogous conformation in solution (McCullough et al., 2013). Crystal structures solved for human isoforms of Vps24, Vps32, and Ist1 show that α1 and α2 form a helical hairpin (Muzioł et al., 2006; Bajorek et al., 2009; Martinelli et al., 2012). Additionally, α3 and α4 stack against the open end of the hairpin to generate an asymmetric four-helix bundle. The linker between α4 and α5 is unstructured but appears to exhibit a high degree of flexibility, enabling α5 to either be in tight apposition to the helical hairpin loop or isolated entirely away from the four-helix bundle. Current models suggest that these structures represent autoinhibited and active conformations, respectively. In a closed state, ESCRT-III subunits are unable to associate with one another and remain in solution as monomers. However, in an active configuration, many ESCRT-III subunits are able to coassemble (Lata et al., 2008b; Bajorek et al., 2009). For example, mutations at the interface between the helical hairpin loop and α5 stimulate human Vps24 polymerization in vitro (Bajorek et al., 2009). These data strongly suggest a critical role for displacing α5 to facilitate ESCRT-III polymerization.
Both Vps24 and Ist1 crystallize as dimers and exhibit multiple distinct crystal packing interactions (Muzioł et al., 2006; Bajorek et al., 2009). Although their biological significance is not clear, many of these associations involve the helical hairpin loop, further highlighting the contribution of this region to ESCRT-III coassembly. In addition, a wide variety of Vps32 filaments have been visualized, resembling rings, spirals, strings, and circular arrays (Ghazi-Tabatabai et al., 2008; Hanson et al., 2008; Pires et al., 2009; Fyfe et al., 2011; Henne et al., 2012; Cashikar et al., 2014). The variability in these structures has made their characterization challenging. Although overexpression of human Vps32 promotes the assembly of ∼5-nm-thick filaments, which appear as flat spirals on the inner leaflet of the plasma membrane, in vitro visualization of a mutant, activated form of yeast Vps32 revealed ∼9-nm-thick filaments that were composed of two unique intertwined strands (Hanson et al., 2008; Henne et al., 2012). These data are difficult to reconcile and underscore a need to better understand how ESCRT-III subunits associate. Our goal was to identify mechanisms by which ESCRT-III polymers adopt their spiral-like conformation using a combination of EM-based approaches and molecular dynamics (MD).
Results
Caenorhabditis elegans Vps32 spontaneously forms spiral filaments in the absence of activating mutations
We first identified a full-length isoform of Vps32 that was amenable to homopolymerization in vitro, without prerequisite truncations or other mutations. In contrast to orthologues examined previously (Fig. S1 A), we found that recombinant C. elegans Vps32, which exhibits the strongest similarity with human CHMP4B (61% identical and 76% similar along their entire lengths), formed both monomers and oligomers in solution at a low concentration (Fig. 1 A and Fig. S1 B). To determine whether the two discrete populations exhibited exchange with one another, we isolated monomeric Vps32 by size exclusion chromatography (∼500 nM) and subjected it to a second round of gel filtration. Immunoblot analysis of the fractions eluted using Vps32 antibodies demonstrated the formation of a new oligomer pool (Fig. 1 B). Similarly, when the oligomer pool was fractioned over a glycerol density gradient, Vps32 monomers were identified (Fig. 1 C). Our data indicate that C. elegans Vps32 exists in a monomer–oligomer equilibrium, even at nanomolar concentration. These findings highlight the intrinsic ability of Vps32 to homotypically associate and disassociate in a manner that does not require additional factors, a property that may be important for its dynamic assembly during membrane scission events. Importantly, depletion of Vps32 in C. elegans one-cell stage embryos resulted in a dramatic defect in the degradation of a previously characterized integral membrane, ubiquitin-modified cargo protein, consistent with it possessing a conserved role in the ESCRT-mediated lysosomal sorting pathway (Videos 1 and 2).
Figure 1.
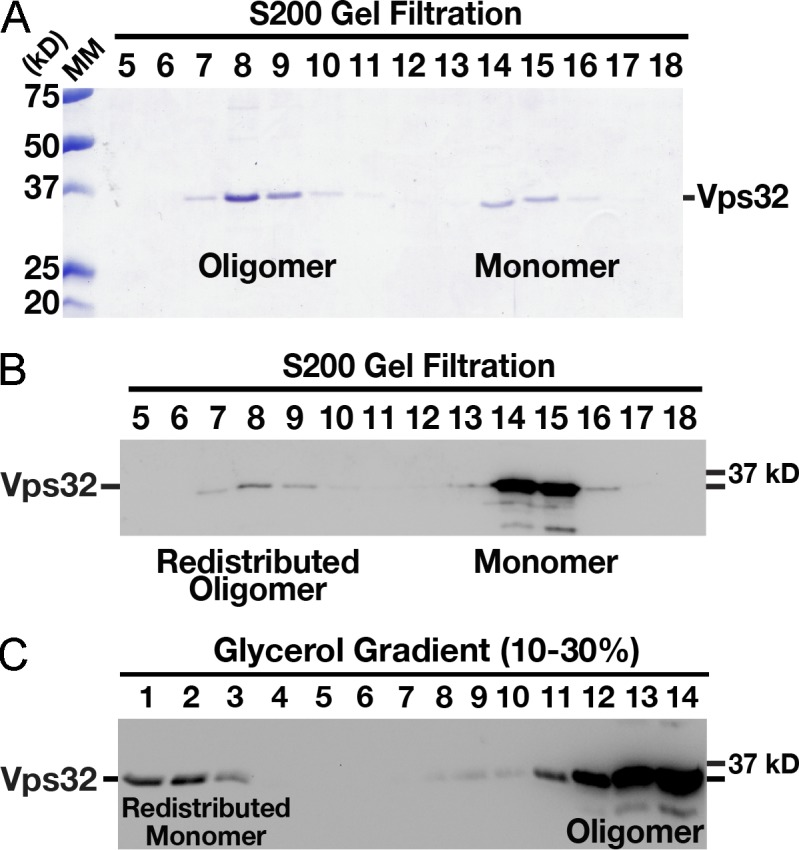
C. elegans Vps32 forms an exchangeable pool of monomers and oligomers in solution. (A) Coomassie-stained fractions of purified, recombinant C. elegans Vps32 after size exclusion chromatography and SDS-PAGE analysis. The monomer and oligomer peaks exhibit Stokes radii of 3.8 and 9.1 nm, respectively. (B) Immunoblot analysis using Vps32 antibodies demonstrating that a fraction of the monomer pool is redistributed to the oligomer pool after a second round of size exclusion chromatography. The monomer and oligomer peaks continue to exhibit Stokes radii of 3.8 and 9.1 nm, respectively. (C) Immunoblot analysis using Vps32 antibodies showing the distribution of recombinant Vps32 after fractionation over a glycerol gradient. The oligomer pool (fractions 7 and 8 recovered after size exclusion chromatography, as shown in A) was separated by velocity sedimentation. A redistributed monomer peak with a sedimentation value of 2.1 S is highlighted. Based on our hydrodynamic observations, the apparent molecular mass (MM) of Vps32 (monomer pool) is ∼33 kD (Siegel and Monty, 1966), similar to its predicted molecular mass of 25 kD, as determined by amino acid composition.
To determine the conformation of Vps32 oligomers, we examined the mixed population by negative staining EM. Consistent with previous work (Ghazi-Tabatabai et al., 2008; Hanson et al., 2008; Pires et al., 2009; Fyfe et al., 2011; Henne et al., 2012; Cashikar et al., 2014), we observed the presence of ringlike structures, strings, and circular arrays (Fig. S1, C and D). The diameter of Vps32 rings ranged from ∼20 to 45 nm, whereas the circular arrays were often as large as 140 nm (Fig. S1 E). In addition, our findings indicated that all polymers exhibited a thickness of ∼4 nm, regardless of their conformation (Fig. S1 F). These data are consistent with Vps32 assembling as individual filaments that do not associate laterally, which contrasts with previous observations that used a mutant isoform of the yeast orthologue Snf7p (Henne et al., 2012). Although filament assembly occurred in a concentration-independent manner, their conformations varied from predominantly ringlike assemblies at 1 µM to mostly spirals at concentrations >10 µM (Fig. S1 G).
Some negative staining procedures used to study ESCRT-III structure include a desiccation step before the application of stain (Henne et al., 2012), which may incur structural rearrangements (Ohi et al., 2004). We therefore reexamined yeast Vps32 (Snf7p), using a method that did not involve desiccation of the sample before staining and further avoided the use of an epitope-tagged form of the protein, which might also impact subunit assembly. Consistent with previous work (Henne et al., 2012), yeast Snf7p that harbored the R52E mutation (shown to promote the formation of filaments) polymerized in solution (Fig. S2, A and B), but our measurements indicated that the vast majority of filaments were only ∼4 nm in width (Fig. S2 C), similar to that observed for C. elegans Vps32 polymers. In some cases (<20%), we observed ringlike assemblies that appeared to be thicker (Fig. S2 D). However, we were unable to identify cases in which filaments were intertwined, as was observed previously after desiccation of polyhistidine-tagged Snf7p (Henne et al., 2012). Collectively, these data continue to support the idea that Vps32 filaments are largely unable to associate laterally.
To increase our resolution of Vps32 polymers, we quickly froze the recombinant C. elegans protein and examined it using a low electron–dose EM imaging mode. Under these more native conditions and irrespective of concentration, we found that Vps32 formed mostly spirals, and only rarely were ringlike structures or strings observed (Fig. 2 A). The thickness of the ice was a critical parameter in visualizing Vps32 filaments. Only when the ice was appropriately thin could we obtain sufficient contrast to characterize them (Fig. S2 E). The spirals were typically paired, consisting of conjoined filaments that were mirror images of one another (Fig. S2 F). The width of filaments was highly uniform, exhibiting a thickness of 4.2 nm (Fig. 2 B), similar to that observed in situ for overexpressed human Vps32 after quick-freeze deep-etch EM (Hanson et al., 2008). Given that the asymmetric helical core domains of ESCRT-III monomers (α1–α4) are ∼3–4 nm wide, as determined previously by crystallographic studies (Muzioł et al., 2006; Bajorek et al., 2009), our results indicate that Vps32 subunits coassemble to generate single-stranded filaments that do not self-associate.
Figure 2.

Vps32 polymerizes to form curved, single-stranded filaments. (A) A representative cryo-EM image (inverted) of Vps32. Select spiral pairs (purple), hybrid spirals (purple/green), and ring structures (blue) are highlighted. Bar, 50 nm. (B) A straight filament (yellow boxed region in A) was chosen for FFT analysis and width measurement (intensity after background subtraction was used to calculate the width of filaments). Layer lines in the FFT analysis are highlighted with arrows. Bar, 5 nm. The data shown are from a single representative experiment, which was repeated six times (n = 15 for the experiment shown). (C) Distribution of spiral diameters. Both inner and outer diameters of spiral filaments were measured in two axes (x and y). Data shown are from multiple experiments, repeated at least six times independently. AU, arbitrary unit.
In contrast to their homogenous widths, the lengths of Vps32 filaments varied, averaging ∼110 nm for ringlike structures and ∼1.6 µm for spirals (Fig. S2 G). To determine the size of repeating units within each filament, we used a fast Fourier transform (FFT) algorithm. For these calculations, we specifically measured portions of spiral filaments that were linear. Our analysis revealed a clear layer line at 3.2 nm (Fig. 2 B). Based on these data, the largest filaments observed (∼2.5 µm) harbor nearly 800 repeating units. Nevertheless, spiral size was intrinsically limited, never exhibiting diameters >150 nm or <20 nm (Fig. 2 C). These findings indicate that Vps32 polymers assemble only within a specific range of curvatures.
Vps32 monomers associate in a head-to-tail orientation
Unlike other composite filaments, including actin, microtubules, and amyloid fibrils, Vps32 polymers lack helical symmetry, possess high curvature, and exhibit a very narrow width. The combination of these factors makes three-dimensional reconstruction unfeasible. Nevertheless, raw cryo-EM images suggest that Vps32 filaments are composed of a series of repeating globular densities. To improve their resolution, we treated spiral pairs as single particles and generated two-dimensional class averages. Because of their highly variable curvature, the use of currently available software packages proved to be impractical for this purpose. We therefore developed a new graphic user interface (GUI) for particle identification and trajectory tracking (Fig. S3 A). The GUI allows a user to manually define the path of spiral filaments. After two sequential rounds of interpolation, a best-fit model is generated, which provides a high level of accuracy. The GUI was validated using both simulated data and images acquired by cryo-EM (Fig. S3, B and C). Individual filaments were segmented using successive boxed areas with 90% overlap. Specifically, we followed the trajectory of filaments, starting from the center of one spiral that was oriented in a counterclockwise manner to its termination at the center of its paired spiral, which was oriented in a clockwise fashion (Fig. S3 C). Results from this analysis revealed the presence of distinct densities separated by narrow linkers along each filament (Fig. 3 A). The densities were also visible in the raw cryo-EM images (Fig. S3, D and E). The distance between the centers of each density was 3.2 nm, in agreement with our FFT analysis. Importantly, we found that the individual densities were not typically arrayed in a linear fashion but instead exhibited curvature. Overlaying multiple highly represented class averages suggested that the bent region corresponds to the linker between densities (Fig. 3 A).
Figure 3.

Stable interactions between neighboring α1 and α2 helices drive head-to-tail assembly of Vps32 filaments. (A) Two-dimensional class average analysis of vitrified Vps32 polymers. Representative class averages are overlaid to highlight flexibility observed in filaments. The distance between neighboring EM densities is indicated. The red line indicates the separation angle of three density maps with straight orientation. The blue line indicates the separation angle of three density maps with curved orientation. Bars, 5 nm. (B) Docking of Vps32 (top, pseudoatomic model) and human Vps24 (bottom, based on structure 2GD5; obtained from the Protein Data Bank) into an averaged EM isosurface. Helices of each monomer are labeled. (C and D) Homotypic association between Vps32 subunits is predicted to be mediated by their first two α helices. (C) The ZDOCK server was used to determine the relative positions of the helical hairpin from one Vps32 monomer and the asymmetric four-helix bundle of a neighboring Vps32 subunit. (top) The reference four-helix bundle is shown as a ribbon cartoon, and the top 500 predicted positions of the helical hairpin (from an adjacent Vps32 subunit) are shown as gray dots. (bottom) One highly probable position of the helical hairpin (cyan) is shown explicitly. (D) The Vps32 dimer interface is mediated by electrostatic interactions, which arise mainly from the positively charged residues within helices α1′ and α2′ from one monomer (blue) and the negatively charged residues in helices α1 and α2 of a neighboring Vps32 subunit (red). (E) Calculations of the RMSD of the α carbon atoms in the Vps32 dimer interface, relative to a reference structure (left), using an explicit solvent simulation demonstrates its stability. Representative snapshots of the structure at different time points are shown. The interface used for RMSD analysis is colored light red in the reference structure. Interface dynamics at selected time points are reflected by changes in the conformation of overlaid u-shaped lines (red). The black line indicates the RMSD relative to the time of the simulation. Snapshots of the structure at different time points are shown as well. (F) Coomassie-stained fractions of purified, recombinant C. elegans Vps32E94K after size exclusion chromatography and SDS-PAGE analysis. The monomer peak exhibits a Stokes radius of 3.8 nm. MM, molecular mass. (G) Circular dichroism spectroscopy was used to characterize 1 µM Vps32E94K. The data shown are from a single experiment, which was repeated twice. (H) Representative images showing the distribution of GFP-CPS in yeast cells lacking endogenous SNF7 and ectopically expressing either wild-type SNF7 (left), an empty vector (middle), or a mutant form of snf7 harboring an E95K substitution (right) from low copy plasmids. (bottom row) Merged images (fluorescence and differential interference contrast [DIC]) are also shown. Bar, 2 µm.
Considering the relative sizes of ESCRT-III monomers and the width of Vps32 filaments, a limited number of orientations exist for Vps32 subunits to homopolymerize. Therefore, we used our class-averaged map to build an isosurface profile and docked onto it the previously solved structure for the ESCRT-III subunit Vps24 (helices α1–α4) and a pseudoatomic model of Vps32, which was based on crystal structures for human isoforms of Vps24 and Vps32 (Fig. 3 B and Fig. S3 F; Muzioł et al., 2006; Bajorek et al., 2009; Martinelli et al., 2012). The model of C. elegans Vps32, as determined using MD simulations, showed extensive similarity to both its human homologue and Vps24, including an overall length of ∼7 nm and electrostatic clusters for protein and lipid interactions (Fig. S3, G and H). Our docking results indicated that only one orientation was feasible, in which a single ESCRT-III monomer spanned a pair of EM densities (Video 3). For Vps32, the dimer interface consisted of the extended helical hairpin loop from one monomer and the asymmetric four-helix bundle from another, which filled the large EM densities (Fig. 3 B). More precisely, MD simulations identified a series of electrostatic interactions between neighboring Vps32 monomers that were largely restricted to the first two helices in each subunit (Fig. 3, C and D). To confirm the stability of the dimer interface, we calculated its root mean square deviation (RMSD) using MD simulations. These data indicated that the interface was stable and unlikely to tolerate dynamic changes in organization (Fig. 3 E).
To further validate this model, we generated a mutation within the dimer interface (E94K), which was predicted to disrupt key electrostatic interactions between subunits. When analyzed by size exclusion chromatography, the majority of Vps32E94K formed monomers in solution, with <5% of the total protein able to assemble into oligomers (Fig. 3 F). These data sharply contrasted the distribution of wild-type Vps32, which mostly formed oligomers subsequent to purification (Fig. 1 A). Importantly, analysis by circular dichroism spectroscopy indicated that Vps32E94K was folded, exhibiting a spectrum typical of an α-helical protein (Fig. 3 G). Given the degree of conservation of E94 in Vps32 isoforms (Fig. S1 A), we were able to test its physiological relevance using yeast as a model system. In contrast to wild-type yeast Vps32 (Snf7), the mutant form harboring the lysine substitution failed to support the normal trafficking of a model ESCRT cargo (GFP–carboxypeptidase S [CPS]) to the vacuole lumen (Fig. 3 H and Fig. S3, I and J). These data suggest that the mechanism of Vps32 filament assembly is functionally conserved.
A similar, but unique, orientation was observed for the human Vps24 dimer interface. In this case, MD simulations highlighted a series of hydrophobic residues, mainly restricted to the α2 helices of Vps24 monomers, which mediated their association (Fig. 4). Additional electrostatic interactions appeared to further contribute to the stability of the interface, which would reduce the flexibility needed to generate curved filaments. Collectively, our data argue that Vps32 and Vps24 assemble in a head-to-tail manner stably and that their helical hairpins must be pliable to accommodate changes in filament curvature during assembly.
Figure 4.

Stable interactions between neighboring α2 helices drive hVps24 dimer formation. (A) The ZDOCK server was used to determine the relative positions of the helical hairpin from one Vps24 monomer and the asymmetric four-helix bundle of a neighboring Vps24 subunit, as described in Fig. 3 C. (B) The human Vps24 dimer interface is dominated by hydrophobic interactions (white), with additional contributions by positively and negatively charged amino acids in the neighboring α2 helices (as described for Vps32). (C) Calculations of the RMSD of the α carbon atoms in the hVps24 dimer interface, relative to a reference structure (left), using an explicit solvent simulation that demonstrates its stability. Representative snapshots of the structure at different time points are shown as described for Fig. 3 E. The black line indicates the RMSD relative to the time of the simulation.
The crystal structures of Vps24 and the related ESCRT-III subunit Ist1 reveal dimeric crystal packing interfaces that are distinct from the orientation we determined (Muzioł et al., 2006; Bajorek et al., 2009; Martinelli et al., 2012). To address this discrepancy, we attempted to fit these previously solved structures onto the isosurface defined by our cryo-EM experiments. However, neither model was satisfactory, suggesting that these interfaces are unlikely to apply to Vps32 polymers (Fig. S4 A). Additionally, using MD simulations, we identified a key polar residue (Vps32Q64) within the hypothetical tip-to-tip interface predicted by the crystal structures, which should be required for the association of Vps32 monomers, if the model is accurate. However, mutation of this amino acid to leucine failed to impact Vps32 polymer formation or spiral filament assembly (Fig. S4 B). In addition, during the course of our docking attempts, we identified a single alternate configuration for Vps32 monomers that could partially fill the two-dimensional isosurface defined by our cryo-EM experiments. In this arrangement, an ESCRT-III monomer would occupy each EM density, and the interface between subunits would not rely on the asymmetric four-helix bundle. Moreover, the width of the filament would vary, depending on the angle at which it is viewed. To test this possibility, we analyzed negatively stained spiral filaments and determined their widths at multiple tilting angles. Our results demonstrated that the thickness of filaments does not vary significantly when rotated 60°, invalidating the model under scrutiny (Fig. S4, C–E). Collectively, our findings reveal the orientation of Vps32 monomers within a filament. Consistent with previous work (Bajorek et al., 2009), the interface between subunits requires them to exhibit an open, active conformation in which α5 is displaced away from the helical hairpin loop.
Vps32 monomers are intrinsically flexible
To investigate ESCRT-III subunit flexibility, we conducted MD simulations on our pseudoatomic model of the Vps32 helical core and helices α1–α4 of human Vps24 (Muzioł et al., 2006; Bajorek et al., 2009; Martinelli et al., 2012). Our observations demonstrated that helices α1 and α2 bend extensively in a single plane near their center, which we refer to as the hinge region, with angles ranging from 0 to 25° (Fig. 5 A). To characterize the free energy distribution for these conformations, we calculated the potential of mean force as a function of the bending angle in an explicit solvent simulation. This analysis revealed a free energy minimum at an angle of 9°, relative to the published crystal structures (Fig. 5 B). Based on our analysis of vitrified Vps32 spirals, we found that the bending angle between densities averaged 9.4 ± 0.7° at the innermost region of spirals, consistent with a minimal free energy state (Fig. 5, C and D). In contrast, the outermost portion of the spirals exhibited a mean bending angle of 3.8 ± 0.8°, which corresponds to an elevated energy state (Fig. 5 D). These data suggest that the spiral assembly pattern of Vps32 generates intrinsic free energy, which may contribute to the thermodynamically unfavorable pathway of membrane scission.
Figure 5.

Spiral assembly of Vps32 filaments harnesses intrinsic free energy. (A) Two views showing the bending flexibility of a Vps32 monomer (left) and a human Vps24 monomer (right) in MD simulations. In both cases, bending of the helical hairpin is restricted to a single plane. (B) In silico free energy analysis of Vps32 at different bending angles. The bending angle is defined in Fig. 6 B and normalized to 0°, as is observed in the pseudoatomic structure of Vps32 and the crystal structure of human Vps24. (right) A model showing the changes in free energy throughout the spiral is provided. (C) Bending angle analysis of a vitrified Vps32 spiral pair. The filament trajectory is labeled (red, black, and blue) and plotted (right). Linear treadlines that were fitted to each portion of the graph are shown. The termini of the spiral pair are indicated (S [starting point] and E [ending point]). Bar, 25 nm. The data shown are from a single representative experiment, which was repeated three times (n = 15 for the experiment shown). (D) Summary of bending angles within spirals (n = 25 spiral pairs). Error bars represent means ± SEM.
To directly test the importance of the helical hairpin “hinge” in spiral assembly, we used MD simulations and principal component analysis to identify mutations within this region that are predicted to impair bending. We focused our attention on a specific valine residue (V44) and found that substitution with tryptophan at this position rigidified the hairpin (Fig. 6, A and B). Importantly, purified Vps32V44W behaved identically to the wild-type protein in hydrodynamic experiments (Fig. 6 C), strongly suggesting that the mutation had little or no impact on the ability of Vps32 to polymerize. However, upon analysis using negative staining and cryo-EM, we found that spiral arrays and ring structures no longer assembled. Instead, Vps32V44W formed more linear filaments, consistent with a critical role for the hinge region in mediating spiral assembly (Fig. 6 D). Importantly, the mean thickness of the linear filaments was highly similar to that exhibited by wild-type spiral filaments, suggesting that the overall architecture of the polymer was otherwise intact (Fig. 6 E). Thus, by specifically reducing the ability of Vps32 monomers to bend at the hinge region, but not affecting the composition of the dimer interface, we could dramatically affect the curvature of ESCRT-III polymers that assemble.
Figure 6.

Increasing the rigidity of the Vps32 helical hairpin inhibits spiral filament assembly. (A) Principal component analysis was used to compare the flexibilities of wild-type C. elegans Vps32, human Vps24, and a mutant isoform of C. elegans Vps32 (Vps32V44W), predicted to exhibit a more rigid architecture. Green arrows indicate the direction of movement, and the length of the arrows indicates the magnitude of motion. The degree of structural flexibility is highlighted with different colors, with red indicating regions of high flexibility and blue indicating regions of low flexibility. The hinge region and the tip of the helical hairpin are circled for comparison. (B) The bending angle can be defined as the change in a specific dihedral angle in Vps32. The dihedral is composed of the four geometric centers of α carbon atoms in three motifs (residues 18–21, 29–33, and 52–55) in helix α1 and a single motif (residues 79–82) in helix α2. The position of valine 44 is highlighted. (C) A comparison of the elution profiles of wild-type (FL, full length) and mutant (V44W) Vps32 after gel filtration chromatography indicates only minor differences in their hydrodynamic properties. The data shown are from single representative experiments, which were repeated independently at least two times for each protein. (D) Representative negatively stained EM images of purified, recombinant Vps32V44W, which continues to form filaments but fails to assemble into spirals or ring structures. Bar, 40 nm. (E) Width analysis of a Vps32V44W filament that is highlighted in D (intensity after background subtraction was used to calculate the width of filaments). The data shown are from a single representative experiment, which was repeated independently three times (n = 15 for the experiment shown). AU, arbitrary unit.
The carboxyl terminus of Vps32 inhibits lateral filament association
Although thermodynamic parameters predict that the hinge region should maintain a bending angle close to 9°, the spirals of wild-type Vps32 are not tightly packed. Instead, filaments are consistently separated from one another (on average, 6.84 ± 2.16 nm from their centers), suggesting the presence of a steric barrier that restricts lateral interactions (Fig. 7 A and Fig. S5, A–C). Because it was not feasible to place the carboxyl-terminal helices (α5 and α6) of Vps32 onto our EM density map, we considered the possibility that these helices protrude into the space between filaments within spirals. Consistent with this idea, we sometimes observe weak densities that extend from filaments in our two-dimensional class averages, suggesting the presence of an additional region within Vps32 monomers that could act as a spacer (Fig. 3 A). However, in most cases, these densities are averaged out, suggesting a highly flexible character, which is consistent with previous structural data (Bajorek et al., 2009). Although deletion of helices α5 and α6 leads to protein aggregation, an isoform of Vps32 lacking α6 and its adjacent MIM2 (MIT-interacting motif 2) domain, which binds to the Vps4 ATPase (Kieffer et al., 2008), exhibited similar hydrodynamic properties as the wild-type protein (Fig. S5 D). These data indicate that the carboxyl-terminal 31 amino acids of Vps32 are dispensable for homopolymerization, consistent with previous work (Martinelli et al., 2012). When examined by negative staining EM, the truncated protein formed thick ringlike structures, similar to that reported previously for yeast Vps24p (Ghazi-Tabatabai et al., 2008). However, by conducting electron tomography, we determined that these structures were composed of single-stranded spirals with substantially compacted conformations relative to those composed of full-length Vps32 (Fig. S5, E and F; and Videos 4 and 5). Specifically, deletion of the MIM2 domain and helix α6 reduced the spacing between filaments within the spirals by ∼2 nm to 4.81 ± 1.29 nm (from their centers), suggesting that the carboxyl terminus of Vps32 plays an important role in regulating lateral associations between individual single-stranded polymers (Fig. 7 B and Fig. S5 G). Importantly, the thickness of filaments composed on Vps321–190 was ∼4.2 nm on average, highly similar to that observed for wild-type Vps32 (Fig. S5 H). These data are consistent with our docking experiments and MD simulations, which places the carboxyl terminus of each Vps32 subunit outside of the confines of conjoined densities observed by cryo-EM. Collectively, our findings suggest a new function for the carboxyl terminus of Vps32 in regulating filament assembly.
Figure 7.

The carboxyl terminus of Vps32 inhibits the lateral association of filaments within spirals. (A) Analysis of the spacing between filaments in Vps32 spirals. (left) A representative cryopreserved Vps32 spiral with its trajectory highlighted in purple is shown. The ends of the spiral are indicated (S [starting point] and E [ending point]). Bar, 25 nm. (middle) A portion of this spiral was straightened, and its distance to the adjacent filament within the same structure was calculated along its entire length. (right) The average distance between the centers of each filament is plotted. Error bars represent means ± SEM. (B) Analysis of the spacing between filaments in a negatively stained spiral composed of a truncated form of Vps32 (amino acids 1–190) was conducted using electron tomography. Representative raw images, tomograms, and trajectories are shown for a spiral pair (top) and a large-diameter spiral (bottom). (right) The average distance between the centers of each filament is plotted. Boxed regions highlight the areas that were magnified (shown in the images to the right). Bars: (23,000× magnification) 50 nm; (59,000× magnification, tomogram, and trajectory) 20 nm. The differences in distances are statistically significant (P < 0.001, compared with wild type, one-tail t test). Error bars represent means ± SEM.
The Vps4 ATPase promotes a conformational change in Vps32 spirals that is independent of ATP hydrolysis
To test directly whether displacement of the Vps32 carboxyl terminus could account for the restructuring of spiral filaments, we investigated the impact of incubating them with Vps4, the ATPase that binds to the Vps32 carboxyl-terminal MIM2 domain. Because Vps4 disassembles ESCRT-III polymers in an ATP-dependent manner (Lata et al., 2008a), we used a mutant form of the enzyme that harbors a substitution within its Walker B motif (Vps4E219Q) and cannot hydrolyze ATP. Based on size exclusion chromatography coupled to multiangle light scattering (MALS), we found that C. elegans Vps4E219Q exhibits a molecular mass of 52.6 kD (±3.5%) in the absence of nucleotides, similar to that predicted by its amino acid sequence (48.9 kD), indicating that it is a monomer is solution (Fig. 8 A). Upon incubation with ATP, our analysis by size exclusion chromatography coupled to MALS indicated that a substantial pool of Vps4E219Q assembled into monodisperse, hexameric complexes with a molecular mass of 283.3 kD (±1.4%; Fig. 8, A and B). Negative staining EM of Vps4E219Q in the presence of ATP further highlighted the formation of ringlike structures exhibiting a diameter of ∼14 nm (Fig. 8 C). Upon mixing Vps32 with a 2.5-fold molar excess of Vps4E219Q (10 µM), we observed numerous compacted spirals and several discrete strands of Vps32 after negative staining EM (Fig. 8 D). Furthermore, immunogold labeling of Vps4E219Q indicated that it was typically associated with compacted spirals (Fig. 8 D, inset). Although the width of individual Vps32 filaments was not affected by the presence of Vps4E219Q (Fig. 8 E), the distance between Vps32 filaments within spiral arrays was reduced significantly, relative to the spacing observed in the absence of Vps4 (Fig. 8, F and G). Strikingly, the interfilament distance observed (∼5.0 nm) was similar to that seen in spirals composed on Vps321–190. These data suggest that Vps4 can remodel and constrict Vps32 spiral filaments in a manner that does not require its ATPase activity.
Figure 8.

Vps4 promotes the compaction of Vps32 spirals independently of its ATPase activity. (A) Coomassie-stained fractions of purified, recombinant C. elegans Vps4E219Q after size exclusion chromatography and SDS-PAGE analysis in the presence (bottom) or absence (top) of 10 mM ATP. MM, molecular mass. (B) A comparison of the elution profiles of Vps4E219Q in the absence and presence of 10 mM ATP after gel filtration chromatography highlights the ability of Vps4 to oligomerize in the presence of ATP. The data shown are from single representative experiments, which were repeated independently at least two times for each condition. (C) A representative EM image of recombinant C. elegans Vps4E219Q (10 µM), purified in the presence of 10 mM ATP. On the right are typical Vps4E219Q structures (end-on views) that were selected after low pass filtering. Bars: (left) 50 nm; (right) 10 nm. (D) A representative EM image of purified, recombinant C. elegans Vps4E219Q (10 µM), purified in the presence of 10 mM ATP, mixed with 4 µM Vps32. Bar, 50 nm. The inset shows a 2× zoomed image of a compacted Vps32 spiral after labeling using 1.8 nm Ni-NTA-Nanogold, which recognizes the polyhistidine tag on Vps4E219Q. A gold particle is highlighted (yellow arrowhead). The blue boxed regions highlight typical examples of Vps32 spirals that are compacted in the presence of Vps4E219Q. The red boxed region highlights a single-stranded ringlike structure, which is not affected by the presence of Vps4E219Q. Bar, 25 nm. (E) A single-stranded Vps32 ring (red boxed region in C) was straightened, and its width was measured (intensity after background subtraction was used to calculate the width of filaments). Bar, 5 nm. The data shown are from a single representative experiment, which was repeated independently two times (n = 15 for the experiment shown). (F and G) Analysis of the spacing between filaments (marked blue and red) in a negatively stained Vps32 spiral exposed to oligomerized Vps4E219Q was conducted. The average spacing between neighboring filaments was calculated to be 5.0 nm (G). Error bars represent means ± SEM. The difference in spacing between Vps32 filaments in the presence or absence of Vps4E219Q was statistically significant (P < 0.01, one-tail t test). The data shown are from a single representative experiment, which was repeated independently two times (n = 15 for the experiment shown). AU, arbitrary unit.
Discussion
ESCRT-III subunits function in a nonredundant manner
Although all ESCRT-III subunits are predicted to share a similar overall structure (Babst et al., 2002; Shim et al., 2007), their lack of redundancy indicates that they operate via distinct mechanisms. During MVE biogenesis, the ESCRT-III subunit Vps20, together with ESCRT-II, appears to function as a recruitment factor to facilitate Vps32 polymerization specifically at regions of elevated membrane curvature (e.g., nascent vesicle bud necks; Teis et al., 2008; Fyfe et al., 2011). In contrast, alternative regulators of Vps32 must operate during cytokinetic abscission and retroviral budding, as ESCRT-II and Vps20 are largely dispensable for these events (Bajorek et al., 2009; Morita et al., 2010, 2011). Our new findings provide mechanisms by which spiral arrays of Vps32 form, irrespective of their site of action. Additionally, our results suggest that an inherent property of these spirals is their ability to harness free energy in the varying curvature of the polymer during filament formation. These data support a model in which the spiral assembly of Vps32 contributes to membrane scission by providing a key source of energy to help overcome the barriers to lipid bilayer restructuring.
Vps32 has also been suggested to recruit the Vps24–Vps2 subcomplex, which can generate ∼40-nm helical tubes that narrow and close at one end (Lata et al., 2008a). The domelike configuration observed at the termination of these helical tubes could support membrane scission in ways that Vps32 spiral filaments cannot. Strikingly, at an energy minimum, Vps32 spirals exhibit a diameter of ∼35 nm, similar to that of a Vps24–Vps2 helical tube. Thus, the innermost portion of the Vps32 spiral may act as an adaptor to assemble Vps24–Vps2 domes. Alternatively, Vps24 may bind and change the conformation of Vps32 flat spirals, creating tubelike assemblies that can narrow in diameter (Henne et al., 2012). Irrespective of the precise mechanism of action, it is likely that a combination of nonredundant ESCRT-III subunit activities is necessary to complete the membrane scission process.
A new function for the Vps32 carboxyl terminus
Similar to Vps24, the Vps4 ATPase has also been shown to restructure Vps32 spirals (Hanson et al., 2008). Our data suggest a mechanism by which Vps4 binding could alter their compaction and thereby enable free energy release that contributes to membrane scission. Specifically, our results place the Vps4-interacting MIM2 domain of Vps32 at the interface between adjacent filaments within a spiral. Binding of Vps4, in a manner that does not require ATP hydrolysis, appears to displace these domains and facilitate the constriction of spirals, which would release energy stored within them (Fig. 9). Consistent with this idea, Vps4 binding to Vps32 spiral arrays has been suggested to promote their contraction and concomitant reorganization in vivo (Hanson et al., 2008; Adell et al., 2014). In this scenario, Vps24–Vps2 would act as adaptors in the recruitment and local concentration of Vps4. Indeed, the Vps2 MIM1 domain exhibits a significantly tighter binding affinity for Vps4 as compared with the MIM2 domain in Vps32 (Stuchell-Brereton et al., 2007; Kieffer et al., 2008). An alternative possibility, which is most consistent with available genetic data, is that a combination of Vps24–Vps2- and Vps4-mediated remodeling events on Vps32 spirals function during ESCRT-mediated membrane constriction, which ultimately facilitates scission.
Figure 9.

A model illustrating ESCRT-III spiral assembly and constriction during membrane scission. Based on our findings, ESCRT-III subunits adopt a head-to-tail interacting mode to assemble into single-stranded filaments. Flexibility of the hinge region within each monomer facilitates the formation of curved spiral polymers. (top left) The carboxyl-terminal helices function as spacers between neighboring filaments. We propose that displacement of the carboxyl-terminal helices in ESCRT-III subunits away from the interface between individual filaments within spirals results in their compaction and a concomitant release of free energy that may contribute to membrane scission.
Although our data are consistent with a role for the Vps32 carboxyl terminus in restricting lateral association of filaments, another explanation is that this region actually promotes transient contacts with adjacent subunits within a spiral filament to help regulate curvature. To date, such an interface has yet to be defined. However, our previous analysis of C. elegans Vps32 harboring a carboxyl-terminal hexahistidine tag demonstrated that it was not able to generate polymers spontaneously (Fyfe et al., 2011), in contrast to the untagged form of the protein used in this study. Thus, the presence of a short, charged epitope on the carboxyl terminus impedes filament assembly, potentially by inhibiting intermolecular associations. Nevertheless, Vps32 fused to a hexahistidine tag could still undergo polymerization in the presence of ESCRT-II, Vps20, and highly bent membranes, suggesting that proper nucleation can bypass the inhibitory effect of a carboxyl-terminal tag.
The structure of a Vps32 filament
There is currently a wide discrepancy in the experimentally calculated width of Vps32 filaments. Our data are most consistent with studies that examined Vps32 spirals assembled in cells, which are composed of individual filaments that do not associate constitutively with one another (Hanson et al., 2008; Cashikar et al., 2014). Moreover, our findings demonstrate a mechanism by which Vps32 filaments remain separated from one another. However, a study using a mutant form of yeast Vps32 (Snf7R52E), tagged at its amino terminus with a hexahistidine tag, indicates they assemble as protofilaments that are composed of two intertwined subfilaments (Henne et al., 2012). Using untagged yeast Snf7R52E, we were unable to reproduce these data. The discrepancy in our results may be caused by differences in the preparation of samples for EM. Alternatively, the presence of a polyhistidine tag on Snf7 in the previous study may have altered its oligomeric properties to promote stable, lateral associations between filaments, especially in the context of using a mutant (R52E) form of the protein at a high (>30 µM) concentration (Henne et al., 2012). In contrast, our experiments take advantage of a wild-type form of Vps32 that polymerizes at a physiologically relevant concentration to generate a single-stranded spiral filament. In cells, ESCRT-II and Vps20 have been suggested to nucleate and/or stabilize these filaments (Teis et al., 2008, 2010). Notably, each ESCRT-II complex is capable of seeding up to two ESCRT-III filaments (Hierro et al., 2004; Teo et al., 2004). As our previous work suggested that Vps32 filaments are typically oriented in opposing directions away from a single ESCRT-II complex (Fyfe et al., 2011), it is feasible that multiple spirals form during membrane scission events, which may act in concert to accelerate the process. Alternatively, the presence of multiple nucleation sites may simply improve the likelihood that a single Vps32 polymer is able to assemble. Ultimately, the further characterization of native ESCRT-III polymers is necessary to resolve this discrepancy. To date, only a single electron tomography–based study has resolved a spiral filament that depends on the presence of the ESCRT machinery to assemble in wild-type metazoan cells. These ∼17-nm-thick filaments were observed at constriction sites within the intracellular bridge of postmitotic cells, where ESCRT-III has been shown previously to facilitate cytokinetic abscission (Guizetti et al., 2011). However, it remains unclear whether this filament actually consists of ESCRT subunits or represents an alternative filamentous system that forms during cytokinesis. A similar study in archaea highlights the assembly of ∼3.5-nm filaments during cytokinesis, which likely correspond to ESCRT-III and are much more similar to the size of Vps32 filaments that we define in this study (Dobro et al., 2013). In Vps4-depleted human cells, ESCRT-III filaments that accumulate on the cell surface appear to range in width from 4 nm (consistent with our work) to ∼15 nm (Cashikar et al., 2014). However, because a major regulator of ESCRT-III polymer dynamics is absent in these studies, the physiological relevance of these filaments is challenging to determine. Future improvements in EM-based imaging in wild-type mammalian cells will reveal the true nature of the ESCRT-III complex, not only at the cytokinetic furrow but also at MVEs and sites of retroviral budding, which will aid in our further understanding of its mechanism of action.
Materials and methods
Protein sequence alignment
Protein sequences including C. elegans Vps32 (NP_495337; accession numbers were obtained from the NCBI Protein database), Saccharomyces cerevisiae Snf7p (NP_013125), Homo sapiens CHMP4A (NP_054888), H. sapiens CHMP4B (NP_789782), H. sapiens CHMP4C (NP_689497), Arabidopsis thaliana Vps32.2 (NP_001078468), and H. sapiens Vps24 (NP_057163) were downloaded from PubMed in FASTA format. COBALT was used to align the sequences (Papadopoulos and Agarwala, 2007).
Protein purification, immunoblotting, and live-cell imaging
Recombinant protein expression was performed using BL21-T1R (DE3) Escherichia coli. Wild-type and mutant isoforms of C. elegans Vps32 were cloned into pGEX6P-1, which encodes a cleavable, amino-terminal GST tag. Initial purifications from bacterial extracts were conducted using glutathione agarose beads. Bound proteins were supplemented with PreScission protease to remove the tag, and eluted Vps32 isoforms were applied to a S200 size exclusion column equilibrated in cryo-EM buffer (50 mM Hepes, pH 7.6, 100 mM NaCl, and 1 mM DTT). 1-ml fractions were collected, and the Stokes radius of each protein or protein complex was calculated from its elution volume based on the elution profiles of characterized globular standards. To determine the sedimentation values for each fraction eluted from the S200 column, samples were applied to 4-ml glycerol gradients (10–30%), which were fractionated serially by hand (200 µl each). Sedimentation values were calculated by comparing the position of the peak with that of characterized standards run on a separate gradient in parallel.
C. elegans Vps4E219Q was cloned into pET3aTR with a polyhistidine tag appended onto its carboxyl terminus to enable purification using Ni-nitrilotriacetic acid (NTA) agarose. Alternatively, Vps4E219Q was purified as a GST fusion protein using glutathione agarose beads, and the tag was removed using PreScission protease. Irrespective of its method of purification, Vps4E219Q was further subjected to size exclusion chromatography using a S200 gel filtration column (in the presence or absence of ATP) before analysis by EM or light scattering. For MALS experiments, purified Vps4E219Q was applied onto a size exclusion chromatography column (WTC-030S5; Wyatt Technology Corporation), which was coupled to a three-angle light scattering detector (miniDAWN TREOS; Wyatt Technology Corporation). Data were collected every second at a flow rate of 0.7 ml/min and analyzed using ASTRA software (Wyatt Technology Corporation), which determines molecule weight and mass distribution of samples based on light scattering. Yeast Snf7R52E was cloned into pGEX6P-1 and cleaved from glutathione agarose resin using PreScission protease to remove the tag before EM and size exclusion chromatography experiments.
For circular dichroism experiments, Vps32E94K was analyzed using a 0.1-cm path length quartz cell. The protein was dialyzed overnight into 25 mM sodium phosphate, pH 7.2, and spectra were collected using a circular dichroism spectrophotometer (model 202SF; Aviv Biomedical) at 25°C. Antibodies directed against C. elegans Vps32 (C56C10.3) were raised in rabbits by immunization with a GST fusion to the full-length protein produced in E. coli. The antibodies were affinity purified from serum by binding to a column of the same antigen after removal of the GST tag. Immunoblotting was performed as described previously (Audhya et al., 2005). For live imaging of C. elegans, animals expressing a GFP fusion to Cav1 were immobilized on agarose pads (Wang and Audhya, 2014) and imaged on a swept field confocal microscope (Ti-E; Nikon) equipped with a charge-coupled device camera (CoolSNAP HQ2; Roper Scientific) using a Nikon 60×, 1.4 NA Plan Apochromat oil objective lens. Acquisition parameters were controlled by Elements software (Nikon), and image analysis was conducted using MetaMorph software (Molecular Devices). Imaging of yeast was conducted similarly. A plasmid encoding GFP-CPS (a gift of D. Katzmann, Mayo Clinic, Rochester, MN) was cotransformed with pRS415-based plasmids expressing either wild-type or mutant SNF7 (under its endogenous regulatory sequences) into yeast harboring a deletion of endogenous SNF7 (SEY6210; snf7Δ::HIS3).
EM and tomography
Wild-type and mutant isoforms of C. elegans Vps32, C. elegans Vps4E219Q, and yeast Snf7R52E (3 µl) were negatively stained using a 1% (wt/vol) uranyl formate solution on glow-discharged continuous carbon-coated copper grids (easiGlow; PELCO) or directly frozen on Quantifoil grids using a vitrification device (Vitrobot Mark IV; FEI). Both negatively stained and vitrified samples were examined under an electron microscope (Tecnai G2 F30; FEI) with a field emission gun operated at 300 kV and recorded on a charge-coupled device camera (UltraScan 1000XP, 2k × 2k; Gatan). The negatively stained samples were captured at magnifications of 23,000, 39,000, or 59,000×. The frozen hydrated samples were examined in low-dose mode to avoid irradiation damage. Magnifications were set at 39,000 or 59,000×, with final pixel sizes of 2.8 or 1.8 Å, respectively. The defocus ranged from −2 to −5 µm. Cryo-EM images were selected for further trajectory and 2D class average analysis. In cases in which Vps4E219Q was mixed with Vps32, the proteins were incubated together on glow-discharged continuous carbon-coated copper grids for 15 min at 4°C, washed twice with buffer, and stained as described for the individual proteins.
A subset of negatively stained Vps32 samples was examined using electron tomography. Samples were placed on a tomography holder (FEI) with maximum tilt angles of ±60°. Data were collected using Xplore3D software (FEI) at a magnification of 59,000×, with the defocus set at −3 µm. Tilting step sizes were set at 4° between 0 and 50° and 2° between 50 and 60°. The tomography data were saved in a single .mrc format and subsequently converted into a .st format with a bin size of 2 using the IMOD software package (Kremer et al., 1996). Regions of interest were centered and aligned in a fiducialless mode. Three-dimensional models were subjected to a low pass filter to increase contrast. Model building was conducted using the IMOD software package, and only distinct trajectories were marked at different tilting angles. The spacing between neighboring filaments in models was calculated using the straightening script (described in the Filament straightening section), taking advantage of a similar strategy to that used in measuring raw images.
For immunogold labeling, 4 µM Vps32 was incubated with polyhistidine-tagged Vps4E219Q (10 µM) at 4°C for 30 min. The mixture (3 µl) was placed on EM grids for 1 min, and excess solution was removed by filter paper. Immediately after blotting, a droplet of 1.8 nm Ni-NTA-Nanogold particles (5 nM) was added to the grids and kept at room temperature for 10 min. The mixtures were then subjected to negative staining EM, as described for other purified proteins.
Trajectory analysis
We developed a new user-friendly GUI in MATLAB (MathWorks, Inc.) for trajectory tracking. The GUI interface incorporates a wide range of functions, including the ability to invert, scale, and threshold images and adjust contrast and color points. The GUI can accept grayscale image files in either .mrc or .tif format. By manually marking several points along any filament, the script tracks the path of filaments using two iterations of Bezier interpolation. First, several user-defined points are inserted between neighboring marks on filaments. An equipartition script subsequently divides the path evenly, after calculating the total filament length. A second round of Bezier interpolation repeats this process and provides more evenly spaced points. If necessary, a straightening script can be used before the second interpolation.
Filament straightening
As a result of the inherently inaccurate process of marking filaments manually, small deviations may arise after the first round of Bezier interpolation. To address this problem, we developed a peak finding script for automated correction. At each interpolated point, the normal direction is defined, and the image is rotated to vertical using this point as its center. The area between this point and the next is boxed with a rectangular region, the dimensions of which are determined by filament thickness and pixel size. A one-dimensional projection of this boxed area is calculated, and the maximum is defined. Because the previously identified interpolated points were already close to the peak, searching is limited to a small area. The coordinate of this new peak position then replaces the previous one with improved accuracy.
The aforementioned peak finding script is also used to straighten curved filaments to define the spacing between neighboring polymers. A relatively larger boxed area (larger than the estimated filament space) is used to cover adjacent filaments. Serial rotated rectangles are stacked in a new image, and both the straightened and neighboring filaments are easily defined. The GUI system follows the trajectories of neighboring filaments, calculates the distance at specific points, and averages them over their lengths.
Two-dimensional class average analysis
Considering that Vps32 single-stranded filaments are highly curved with a narrow width (4.2 nm), cryo-EM images were taken at a magnification of 59,000× for two-dimensional class average analysis. We used a 90% overlap strategy to box regions of the Vps32 filaments, after trajectory analysis. To do so, Vps32 filament trajectory was interpolated linearly. The interpolated points were spaced at 0.9-nm intervals and subsequently used as the centers of individual boxed regions. The Batchboxer program in EMAN (Electron Micrograph Analysis) then defined specific regions with the box size of 9 nm (Ludtke et al., 1999). Image stacks were subjected to reference-free two-dimensional alignment and classification using IMAGIC software (Image Science Software GmbH; van Heel et al., 1996). In total, 8,286 particles were classified into 50 unique, but highly related, classes. The center density maps in each class were selected using the EMAN Boxer program and averaged in IMAGIC to define an isosurface. The averaged isosurface was used to replace all three densities in a single box manually, after 3.2-nm intervals without bending. One representative straight Vps32 filament was boxed using EMAN Helixboxer, and an FFT algorithm was performed in the same software package. A one-dimensional projection profile was processed using a script we wrote in MATLAB (Supplemental material).
MD simulation protocols
Classical MD simulations were used to probe the structural flexibility of both human Vps24 and C. elegans Vps32 monomers as well as the binding interface of dimers. All MD simulations were run with the AMBER (Assisted Model Building with Energy Refinement) MD package on graphics processing units (Götz et al., 2012; Salomon-Ferrer et al., 2013). The ff99SBnmr1 force field (Li and Brüschweiler, 2010) was used to describe the system because recent benchmark calculations showed that this force field provides reliable structural properties with both explicit and implicit solvent simulations (Beauchamp et al., 2012). Both explicit solvent and implicit solvent MD simulations were performed to confirm the robustness of the results; the length of the simulation ranged from 400 to 600 ns for explicit solvent simulations and from 400 ns to 3 µs for implicit solvent simulations.
For explicit solvent simulations, the TIP3P water model (Price and Brooks, 2004) was used to solvate the protein with periodic boundary conditions. The system size was ∼33,000 atoms for monomer simulations and 83,000 atoms for dimer simulations. The system was neutralized with counterions, and electrostatic interactions were calculated with the particle mesh Ewald method using a grid spacing of ∼1.0 Å. The nonbonded cutoff for van der Waals interactions was set at 10 Å. The SHAKE algorithm (Ryckaert et al., 1977) was applied to all bonds involving hydrogen, allowing an integration time step of 2 fs. Simulations were run with the NVT (constant number, volume, and temperature) ensemble at 300 K using the weak-coupling algorithm (Berendsen et al., 1984).
For implicit solvent simulations, the GB7 generalized Born model (Mongan et al., 2007) was used, and nonbonded interactions were calculated without any cutoff. A salt concentration of 0.15 M was applied using a modified generalized Born model based on the Debye–Hückel limiting law for ionic screening of electrostatic interactions (Srinivasan et al., 1999). The temperature of 300 K was controlled through Langevin dynamics with a collision frequency of 20 ps−1.
Initial monomer structure preparation and flexibility analysis using MD simulations
We used the I-TASSER (Iterative Threading Assembly Refinement) server (Zhang, 2008; Roy et al., 2010) to build the structural model of C. elegans Vps32. Seven other Vps32 models were also built with MODELLER (Eswar et al., 2006) using different strategies, but each led to a less stable structure during subsequent MD simulations. The structure building process of I-TASSER used the crystal structures of both CHMP4B helices (α1 and α2) and human Vps24 (α1–α4). The final Vps32 model was of similar conformation to human Vps24 and showed a stable RMSD of ∼3 Å during 3 µs of GB7 simulation, supporting the validity of the I-TASSER model. This model structure, together with the human Vps24 crystal structure, was chosen as the starting configurations for further molecular dynamic simulations on their flexibilities.
To quantify the structural flexibility of the protein, MD trajectories were analyzed with a principal component analysis using the ProDy package (Bakan et al., 2011). The bending angle of the helical hairpin (α1 and α2) was defined as the dihedral angle spanned by the two ends of helix α1 with respect to the hinge in the middle of the structure. The hinge region was predicted using the DynDom protein domain motion analysis program (Hayward and Lee, 2002) with representative conformations from the MD trajectories after clustering analysis. Following a best fit of a pair of conformations, the DynDom program partitions a protein into dynamic domains based on a clustering analysis of rotational vectors associated with short main-chain segments. Interdomain screw axes are then used to identify residues that serve as mechanical hinges between the dynamic domains. The bending potential of the mean force was directly inverted from the bending angle distribution, P(θ), from the MD trajectories W(θ) = −kBTlnP(θ), in which kB is the Boltzmann constant, and T is the absolute temperature (300 K).
MD dimeric interface simulation and its stability analysis
Our EM docking results indicate that the four-helix bundle in one Vps32 monomer interacts with an adjacent monomer (through its helical hairpin) to assemble into single-stranded filaments. A Vps32 molecule was split into a “head” region (amino acids 12–33 and 77–137) and a “tail” region (amino acids 34–76) for interface prediction. The head and tail regions were submitted to the ZDOCK server as receptor and ligand structures, respectively (Chen et al., 2003). No residue was specifically selected or blocked in either the receptor or the ligand during the prediction.
Next, to identify the interface shared by two interacting ESCRT-III molecules and explore the structural stability of the interface, we subjected the crystal structure of human Vps24 (helices α1–α4) to two different protein docking servers: ZDOCK (Chen et al., 2003) and HADDOCK (High Ambiguity Driven Biomolecular Docking; Dominguez et al., 2003; de Vries et al., 2007). With ZDOCK, one set of calculations did not include any specific constraints, whereas the other two sets included spatial constraints to impose proximity between R46-E129 and K54/R69-E129/E133 across the interface. In HADDOCK, the same sets of restraints were included. Docking, using the conformational ensemble collected from MD simulations of human Vps24, was also attempted. 635 top-ranked dimer models from these calculations were screened according to the EM density map and whether the dimer exhibited a uniform interface that potentially binds to a lipid bilayer. Further refinement of selected dimer models with MD simulations led to a stable complex in which the two monomers were arranged in a head to tail manner with helix α2 contributing most to the binding interface. This stable complex was also predicted by ZDOCK without any constraints and had an initial rank of 25 among the top 200 predictions. The dimer model of C. elegans Vps32 was based upon the structure of the human Vps24 dimer, which we defined and subjected to further MD refinement. The resulting model of the C. elegans Vps32 dimer showed a similar conformation as compared with human Vps24. However, both helices α1 and α2 contributed to the binding interface. In each case, the dimer complexes remain structurally stable in both implicit and explicit solvent simulations. Based on the crystal structures of human Vps24 and Ist1 (Muzioł et al., 2006; Bajorek et al., 2009; Martinelli et al., 2012), MD simulations were also used to generate a model for the hypothetical tip-to-tip association of Vps32 subunits. The residues contributing most to the stability of dimer interfaces were predicted using the KFC2 server (Darnell et al., 2007; Zhu and Mitchell, 2011).
Structural analysis
The structural analysis including surface electrostatic distribution and structural superimposition was fulfilled in the University of California, San Francisco Chimera package (Pettersen et al., 2004). The principal component analysis results were visualized in VMD (Visual Molecular Dynamics; Humphrey et al., 1996).
Online supplemental material
Fig. S1 highlights the sequence conversation exhibited by ESCRT-III subunits and demonstrates that recombinant C. elegans Vps32 forms two distinct populations in solution, one of which assembles into polymers (both rings and spirals) that can be observed and characterized by negative staining EM. Fig. S2 shows that a yeast isoform of Vps32 also forms two distinct populations in solution and forms single-stranded polymers of similar thickness to that observed for C. elegans Vps32. Fig. S3 provides an overview of the computational approaches developed to analyze Vps32 spiral filaments and shows results from two-dimensional class averaging. Fig. S4 shows that alternative modes of docking Vps32 subunits within the density map defined by cryo-EM fail to satisfy their constraints. Fig. S5 describes the approach used to calculate the distance between filaments within Vps32 spirals and highlights the key role of the Vps32 carboxyl terminus in restricting the lateral association of filaments. Video 1 shows caveolin transport during oocyte ovulation and early embryonic development. Video 2 shows caveolin degradation is impaired in embryos depleted of Vps32. Video 3 illustrates that only a single docking solution is compatible with the density map defined by cryo-EM analysis of Vps32 polymers. Video 4 shows a tomogram of an individual spiral composed of truncated Vps321–190. Video 5 shows a tomogram of a spiral pair composed of truncated Vps321–190. A ZIP file containing the source code for MATLAB scripts used to characterize Vps32 spirals is also provided. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201403108/DC1.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM088151 to A. Audhya) and the National Science Foundation (MCB1157824 to M.S. Otegui). Y. Zheng is supported by a grant from the National Science Foundation (DMS-1160360 to J.C. Mitchell and Q. Cui). Core facility support was provided by the National Science Foundation (BIR-9512577 and 1126441), the National Institutes of Health (S10 RR13790), and the Army Research Office (W911NF-11-1-0327).
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- CPS
- carboxypeptidase S
- ESCRT
- endosomal complex required for transport
- FFT
- fast Fourier transform
- GUI
- graphic user interface
- MALS
- multiangle light scattering
- MD
- molecular dynamics
- MVE
- multivesicular endosome
- NTA
- nitrilotriacetic acid
- RMSD
- root mean square deviation
References
- Adell, M.A., Vogel G.F., Pakdel M., Müller M., Lindner H., Hess M.W., and Teis D.. 2014. Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J. Cell Biol. 205:33–49 10.1083/jcb.201310114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya, A., Hyndman F., McLeod I.X., Maddox A.S., Yates J.R. III, Desai A., and Oegema K.. 2005. A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J. Cell Biol. 171:267–279 10.1083/jcb.200506124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst, M., Katzmann D.J., Estepa-Sabal E.J., Meerloo T., and Emr S.D.. 2002. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell. 3:271–282 10.1016/S1534-5807(02)00220-4 [DOI] [PubMed] [Google Scholar]
- Bajorek, M., Schubert H.L., McCullough J., Langelier C., Eckert D.M., Stubblefield W.M., Uter N.T., Myszka D.G., Hill C.P., and Sundquist W.I.. 2009. Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16:754–762 10.1038/nsmb.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan, A., Meireles L.M., and Bahar I.. 2011. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 27:1575–1577 10.1093/bioinformatics/btr168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp, K.A., Lin Y.S., Das R., and Pande V.S.. 2012. Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput. 8:1409–1414 10.1021/ct2007814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., and Haak J.R.. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690 [Google Scholar]
- Cashikar, A.G., Shim S., Roth R., Maldazys M.R., Heuser J.E., and Hanson P.I.. 2014. Structure of cellular ESCRT-III spirals and their relationship to HIV budding. eLife. 3:e02184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R., Li L., and Weng Z.. 2003. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 52:80–87 10.1002/prot.10389 [DOI] [PubMed] [Google Scholar]
- Darnell, S.J., Page D., and Mitchell J.C.. 2007. An automated decision-tree approach to predicting protein interaction hot spots. Proteins. 68:813–823 10.1002/prot.21474 [DOI] [PubMed] [Google Scholar]
- de Vries, S.J., van Dijk A.D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A.M.. 2007. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 69:726–733 10.1002/prot.21723 [DOI] [PubMed] [Google Scholar]
- Dobro, M.J., Samson R.Y., Yu Z., McCullough J., Ding H.J., Chong P.L., Bell S.D., and Jensen G.J.. 2013. Electron cryotomography of ESCRT assemblies and dividing Sulfolobus cells suggests that spiraling filaments are involved in membrane scission. Mol. Biol. Cell. 24:2319–2327 10.1091/mbc.E12-11-0785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, C., Boelens R., and Bonvin A.M.. 2003. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125:1731–1737 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- Eswar, N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., and Sali A.. 2006. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. Chapter 5:Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrikant, G., Lata S., Riches J.D., Briggs J.A., Weissenhorn W., and Kozlov M.M.. 2009. Computational model of membrane fission catalyzed by ESCRT-III. PLOS Comput. Biol. 5:e1000575 10.1371/journal.pcbi.1000575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe, I., Schuh A.L., Edwardson J.M., and Audhya A.. 2011. Association of the endosomal sorting complex ESCRT-II with the Vps20 subunit of ESCRT-III generates a curvature-sensitive complex capable of nucleating ESCRT-III filaments. J. Biol. Chem. 286:34262–34270 10.1074/jbc.M111.266411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi-Tabatabai, S., Saksena S., Short J.M., Pobbati A.V., Veprintsev D.B., Crowther R.A., Emr S.D., Egelman E.H., and Williams R.L.. 2008. Structure and disassembly of filaments formed by the ESCRT-III subunit Vps24. Structure. 16:1345–1356 10.1016/j.str.2008.06.010 [DOI] [PubMed] [Google Scholar]
- Götz, A.W., Williamson M.J., Xu D., Poole D., Le Grand S., and Walker R.C.. 2012. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 8:1542–1555 10.1021/ct200909j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizetti, J., and Gerlich D.W.. 2012. ESCRT-III polymers in membrane neck constriction. Trends Cell Biol. 22:133–140 10.1016/j.tcb.2011.11.007 [DOI] [PubMed] [Google Scholar]
- Guizetti, J., Schermelleh L., Mäntler J., Maar S., Poser I., Leonhardt H., Müller-Reichert T., and Gerlich D.W.. 2011. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 331:1616–1620 10.1126/science.1201847 [DOI] [PubMed] [Google Scholar]
- Hanson, P.I., Roth R., Lin Y., and Heuser J.E.. 2008. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 180:389–402 10.1083/jcb.200707031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, S., and Lee R.A.. 2002. Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J. Mol. Graph. Model. 21:181–183 [DOI] [PubMed] [Google Scholar]
- Henne, W.M., Buchkovich N.J., and Emr S.D.. 2011. The ESCRT pathway. Dev. Cell. 21:77–91 10.1016/j.devcel.2011.05.015 [DOI] [PubMed] [Google Scholar]
- Henne, W.M., Buchkovich N.J., Zhao Y., and Emr S.D.. 2012. The endosomal sorting complex ESCRT-II mediates the assembly and architecture of ESCRT-III helices. Cell. 151:356–371 10.1016/j.cell.2012.08.039 [DOI] [PubMed] [Google Scholar]
- Hierro, A., Sun J., Rusnak A.S., Kim J., Prag G., Emr S.D., and Hurley J.H.. 2004. Structure of the ESCRT-II endosomal trafficking complex. Nature. 431:221–225 10.1038/nature02914 [DOI] [PubMed] [Google Scholar]
- Humphrey, W., Dalke A., and Schulten K.. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Hurley, J.H., and Hanson P.I.. 2010. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat. Rev. Mol. Cell Biol. 11:556–566 10.1038/nrm2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer, C., Skalicky J.J., Morita E., De Domenico I., Ward D.M., Kaplan J., and Sundquist W.I.. 2008. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell. 15:62–73 10.1016/j.devcel.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Kremer, J.R., Mastronarde D.N., and McIntosh J.R.. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76 10.1006/jsbi.1996.0013 [DOI] [PubMed] [Google Scholar]
- Lata, S., Schoehn G., Jain A., Pires R., Piehler J., Gottlinger H.G., and Weissenhorn W.. 2008a. Helical structures of ESCRT-III are disassembled by VPS4. Science. 321:1354–1357 10.1126/science.1161070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata, S., Roessle M., Solomons J., Jamin M., Gottlinger H.G., Svergun D.I., and Weissenhorn W.. 2008b. Structural basis for autoinhibition of ESCRT-III CHMP3. J. Mol. Biol. 378:818–827 10.1016/j.jmb.2008.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D.W., and Brüschweiler R.. 2010. NMR-based protein potentials. Angew. Chem. Int. Ed. Engl. 49:6778–6780 10.1002/anie.201001898 [DOI] [PubMed] [Google Scholar]
- Ludtke, S.J., Baldwin P.R., and Chiu W.. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]
- Martinelli, N., Hartlieb B., Usami Y., Sabin C., Dordor A., Miguet N., Avilov S.V., Ribeiro E.A. Jr, Göttlinger H., and Weissenhorn W.. 2012. CC2D1A is a regulator of ESCRT-III CHMP4B. J. Mol. Biol. 419:75–88 10.1016/j.jmb.2012.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough, J., Colf L.A., and Sundquist W.I.. 2013. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82:663–692 10.1146/annurev-biochem-072909-101058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongan, J., Simmerling C., McCammon J.A., Case D.A., and Onufriev A.. 2007. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 3:156–169 10.1021/ct600085e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, E., Colf L.A., Karren M.A., Sandrin V., Rodesch C.K., and Sundquist W.I.. 2010. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc. Natl. Acad. Sci. USA. 107:12889–12894 10.1073/pnas.1005938107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, E., Sandrin V., McCullough J., Katsuyama A., Baci Hamilton I., and Sundquist W.I.. 2011. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe. 9:235–242 10.1016/j.chom.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzioł, T., Pineda-Molina E., Ravelli R.B., Zamborlini A., Usami Y., Göttlinger H., and Weissenhorn W.. 2006. Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell. 10:821–830 10.1016/j.devcel.2006.03.013 [DOI] [PubMed] [Google Scholar]
- Ohi, M., Li Y., Cheng Y., and Walz T.. 2004. Negative staining and image classification - powerful tools in modern electron microscopy. Biol. Proced. Online. 6:23–34 10.1251/bpo70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, J.S., and Agarwala R.. 2007. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 23:1073–1079 10.1093/bioinformatics/btm076 [DOI] [PubMed] [Google Scholar]
- Pettersen, E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., and Ferrin T.E.. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pires, R., Hartlieb B., Signor L., Schoehn G., Lata S., Roessle M., Moriscot C., Popov S., Hinz A., Jamin M., et al. . 2009. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure. 17:843–856 10.1016/j.str.2009.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, D.J., and Brooks C.L. III. 2004. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121:10096–10103 10.1063/1.1808117 [DOI] [PubMed] [Google Scholar]
- Roy, A., Kucukural A., and Zhang Y.. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J.-P., Ciccotti G., and Berendsen H.J.C.. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-Alkanes. J. Comput. Phys. 23:327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- Salomon-Ferrer, R., Götz A.W., Poole D., Le Grand S., and Walker R.C.. 2013. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 9:3878–3888 10.1021/ct400314y [DOI] [PubMed] [Google Scholar]
- Schmid, S.L., and Frolov V.A.. 2011. Dynamin: functional design of a membrane fission catalyst. Annu. Rev. Cell Dev. Biol. 27:79–105 10.1146/annurev-cellbio-100109-104016 [DOI] [PubMed] [Google Scholar]
- Shim, S., Kimpler L.A., and Hanson P.I.. 2007. Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic. 8:1068–1079 10.1111/j.1600-0854.2007.00584.x [DOI] [PubMed] [Google Scholar]
- Siegel, L.M., and Monty K.J.. 1966. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta. 112:346–362 10.1016/0926-6585(66)90333-5 [DOI] [PubMed] [Google Scholar]
- Srinivasan, J., Trevathan M.W., Beroza P., and Case D.A.. 1999. Application of a pairwise generalized born model to proteins and nucleic acids: inclusion of salt effects. Theor. Chem. Acc. 101:426–434 10.1007/s002140050460 [DOI] [Google Scholar]
- Stuchell-Brereton, M.D., Skalicky J.J., Kieffer C., Karren M.A., Ghaffarian S., and Sundquist W.I.. 2007. ESCRT-III recognition by VPS4 ATPases. Nature. 449:740–744 10.1038/nature06172 [DOI] [PubMed] [Google Scholar]
- Teis, D., Saksena S., and Emr S.D.. 2008. Ordered assembly of the ESCRT-III complex on endosomes is required to sequester cargo during MVB formation. Dev. Cell. 15:578–589 10.1016/j.devcel.2008.08.013 [DOI] [PubMed] [Google Scholar]
- Teis, D., Saksena S., Judson B.L., and Emr S.D.. 2010. ESCRT-II coordinates the assembly of ESCRT-III filaments for cargo sorting and multivesicular body vesicle formation. EMBO J. 29:871–883 10.1038/emboj.2009.408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo, H., Perisic O., González B., and Williams R.L.. 2004. ESCRT-II, an endosome-associated complex required for protein sorting: crystal structure and interactions with ESCRT-III and membranes. Dev. Cell. 7:559–569 10.1016/j.devcel.2004.09.003 [DOI] [PubMed] [Google Scholar]
- van Heel, M., Harauz G., Orlova E.V., Schmidt R., and Schatz M.. 1996. A new generation of the IMAGIC image processing system. J. Struct. Biol. 116:17–24 10.1006/jsbi.1996.0004 [DOI] [PubMed] [Google Scholar]
- Wang, L., and Audhya A.. 2014. In vivo imaging of C. elegans endocytosis. Methods. 68:518–528 10.1016/j.ymeth.2014.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9:40 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X., and Mitchell J.C.. 2011. KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins. 79:2671–2683 10.1002/prot.23094 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.