

Abstract
The voltage-gated Nav1.5 channel is essential for the propagation of action potentials in the heart. Malfunctions of this channel are known to cause hereditary diseases. It is a prime target for class 1 antiarrhythmic drugs and a number of antidepressants. Our study investigated the Nav1.5 blocking properties of fluoxetine, a selective serotonin reuptake inhibitor. Nav1.5 channels were expressed in HEK-293 cells, and Na+ currents were recorded using the patch-clamp technique. Dose-response curves of racemic fluoxetine (IC50 = 39 μM) and its optical isomers had a similar IC50 [40 and 47 μM for the (+) and (−) isomers, respectively]. Norfluoxetine, a fluoxetine metabolite, had a higher affinity than fluoxetine, with an IC50 of 29 μM. Fluoxetine inhibited currents in a frequency-dependent manner, shifted steady-state inactivation to more hyperpolarized potentials, and slowed the recovery of Nav1.5 from inactivation. Mutating a phenylalanine (F1760) and a tyrosine (Y1767) in the S6 segment of domain (D) IV (DIVS6) significantly reduced the affinity of fluoxetine and its frequency-dependent inhibition. We used a noninactivating Nav1.5 mutant to show that fluoxetine displays open-channel block behavior. The molecular model of fluoxetine in Nav1.5 was in agreement with mutational experiments in which F1760 and Y1767 were found to be the key residues in binding fluoxetine. We concluded that fluoxetine blocks Nav1.5 by binding to the class 1 antiarrhythmic site. The blocking of cardiac Na+ channels should be taken into consideration when prescribing fluoxetine alone or in association with other drugs that may be cardiotoxic or for patients with conduction disorders.
Introduction
Fluoxetine (Prozac) is a selective serotonin reuptake inhibitor (SSRI) (Wong et al., 1995) that is widely prescribed for the treatment of central nervous system–linked cognitive, emotional, and behavioral disorders. Since its discovery in 1974 (Wong et al., 1974), the beneficial psychotropic effects of fluoxetine have led to its being used to treat disorders other than depression, including obsessive compulsive disorders and bulimia nervosa (Wong et al., 1995). The multiple side effects of fluoxetine (Sghendo and Mifsud, 2012) have raised questions about its supposed selective serotonin-mediated effect. Fluoxetine inhibits the serotonin transporter (SERT) in the low nanomolar range (Torres et al., 2003), but its therapeutic effect appears only at much higher plasma and brain concentrations (Muscettola et al., 1978; Bolo et al., 2000). At low micromolar concentrations, fluoxetine also targets other proteins and inhibits several types of ion channels and receptors, including the nicotinic acetylcholine receptor (Hennings et al., 1999; Eisensamer et al., 2003), voltage-gated Ca2+ channels (Deák et al., 2000; Pacher et al., 2000), volume-regulated anion channels (Maertens et al., 2002), neuronal Na+ channels (Lenkey et al., 2006), and human ether-a-go-go-related gene, a cardiac K+ channel (Thomas et al., 2002). The inhibition of the ether-a-go-go-related gene K+ channel by fluoxetine occurs via two different mechanisms: 1) direct channel blockade and 2) disruption of channel protein trafficking (Rajamani et al., 2006). This may explain some of the cardiovascular side effects observed during chronic fluoxetine treatments, including bradycardia and QT interval prolongation (Pacher and Kecskemeti, 2004; Timour et al., 2012). Dysfunctions of Nav1.5, which are responsible for the rapid upstroke of the action potential caused by the rapid entry of Na+ ions into cardiomyocytes, also lead to arrhythmia complications. The prolongation of QT intervals may be due to the improper inactivation of the Nav1.5 as in Romano-Ward syndrome (LQT3), while the reduction of Na+ currents through Nav1.5 may lead to arrhythmias such as Brugada syndrome (Herbert and Chahine, 2006). The major cause of the higher mortality rate in psychiatric patients versus the general population is sudden cardiac death, which mainly results from arrhythmias that occur during treatments with psychotropic drugs. It has been reported that fluoxetine decreases the maximum rate of rise of the depolarization phase (Vmax) of ventricular cell preparations (Pacher et al., 2000; Magyar et al., 2003), but little is known about the direct effect of fluoxetine on the biophysical properties of Nav1.5.
In the present study, we investigated the electrophysical properties of fluoxetine (racemic and enantiomers) and its metabolite norfluoxetine as well as other psychotropic drugs on Nav1.5 stably expressed in HEK-293 cells. We showed that racemic fluoxetine, its metabolite norfluoxetine, and its enantiomers act as potential antagonists of human Nav1.5 unlike the other classes of antidepressants tested.
We also studied the effect of the F1760C and Y1767C mutations of the class I antiarrhythmic binding site on the use-dependent blockade of cardiac Na+ channels by fluoxetine and showed that fluoxetine behaves like a class I antiarrhythmic drug.
Materials and Methods
Cell Culture.
Human embryonic kidney (HEK-293) cells stably expressing human Nav1.5 were used as previously described elsewhere (Huang et al., 2011). In brief, the cells were grown under standard tissue culture conditions (5% CO2, 37°C) in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml of penicillin, and 10 mg/ml of streptomycin (Gibco-BRL/Life Technologies, Burlington, ON, Canada). For experiments with the F1760C, Y1767C, and L409C/A410W mutants, the HEK-293 cells were transiently transfected with the pcDNA3.1 vector containing mutant Nav1.5 cDNA (5 μg) and with the vector after CD8 pIRES/CD8 (5 μg) in 10-cm petri dishes using the calcium phosphate method, as previously described elsewhere (Huang et al., 2011). Transfected cells were briefly preincubated with CD8 antibody-coated beads (Dynabeads M450 CD8-a; Life Technologies, Burlington, ON, Canada) before we recorded the currents. HEK-293 cells expressing the pIRES/CD8 vector were decorated with CD8 beads, which were used to identify cells for recording Na+ currents.
Whole-Cell Patch-Clamp Recordings.
Macroscopic Na+ currents from HEK-293 cells were recorded using the whole-cell patch-clamp technique. Patch-clamp recordings were obtained using low-resistance, fire-polished electrodes (<1 MΩ) made from 8161 Corning borosilicate glass coated with Sylgard (Dow-Corning, Midland, MI) to minimize electrode capacitance. Currents were recorded using an Axopatch 200 amplifier with the pClamp software (Molecular Devices, Sunnyvale, CA). The series resistance was 70–80% compensated. Whole-cell currents were filtered at 5 kHz, digitized at 10 kHz, and stored on a microcomputer equipped with an analog-to-digital converter (Digidata 1300; Molecular Devices). The cells were allowed to stabilize for 5 minutes after the whole-cell configuration was established before we recorded the currents. The experiments were performed at room temperature (22°C). The pipettes were filled with an intracellular solution composed of 35 mM NaCl, 105 mM CsF, 10 mM EGTA, and 10 mM Cs-HEPES. The pH was adjusted to 7.4 with CsOH. The external solution was composed of 150 mM NaCl, 2 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES. The pH was adjusted to 7.4 with NaOH.
The drugs were applied using a constantly running ValveLink8.2 gravity-driven perfusion system (Automate Scientific, Berkeley, CA) equipped with a glass syringe with a 250-µM tip. Different concentrations of the same drug were applied on the same cell. We used silicone-free tubing because we had observed changes in fluoxetine concentrations when silicon tubing was used, most likely because fluoxetine adheres to silicone, which can change the applied concentrations considerably.
The peak current amplitudes at different drug concentrations were subtracted from the value obtained with the control solution and were normalized to the control value to obtain the dose-response curves and IC50 values. Each point on the dose-response curves represents the mean of inhibition calculated from all recorded cells at a specific drug concentration. The values were fit to a Hill equation of the following form:
where I is the peak current, a is the maximum inhibition, b is the Hill coefficient, c is the IC50, and x is the concentration of agonist. To obtain activation curves, Na+ conductance (GNa) was calculated from the peak current (INa) using the following equation: GNa = INa/(V − ENa), where V is the test potential and ENa is the reversal potential. Normalized GNa values were plotted against the test potentials. To obtain the inactivation curves, the peak current was normalized to the maximal value and was plotted against the conditioning pulse potential. Steady-state activation and inactivation curves were fit to a Boltzmann equation of the following form:
where G is the conductance, I is the current, V1/2 is the voltage at which the channels are half-maximally activated or inactivated, and kv is the slope factor. To determine the recovery from inactivation, the test pulse peak current (Itest) was normalized to the corresponding prepulse current (Icont). Itest/Icont was plotted against the pulse interval and was fitted to a double or triple exponential function of the following form:
or
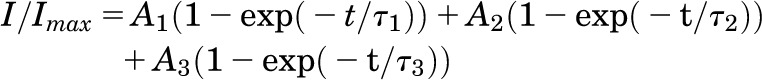 |
where τ1, τ2, and τ3 are the time constants, t is the time and A1, A2, and A3 are the amplitudes of the time constants.
The results were analyzed using a combination of pCLAMP software version 10.2 (Molecular Devices), Microsoft Excel (Microsoft, Redmond, WA), and SigmaPlot version 11.0 (IBM/SPSS, Chicago, IL).
Statistical Analysis.
Results are expressed as mean ± S.E.M. Statistical significance was calculated using Student’s unpaired t test, and P < 0.05 was considered statistically significant. The statistical significance for the IC50 was calculated using R software and the drc package (R Foundation for Statistical Computing, Vienna, Austria).
Drugs.
Racemic fluoxetine, S(+) fluoxetine, R(−) fluoxetine, norfluoxetine, and (+) fenfluramine were obtained from Sigma-Aldrich (St. Louis, MO). Nisoxetine and methylphenidate were obtained from Tocris Bioscience (Bristol, United Kingdom). Stock solutions (5 mM) were prepared in water and were diluted in the external solution before use.
Homology Modeling of Fluoxetine Binding Site in the Nav1.5.
The human cardiac Nav1.5 was modeled in the closed and open states based on the closed NavAb (3RVY.pdb) and open NavMs (3ZJZ.pdb) x-ray structures (Payandeh et al., 2011; Bagnéris et al., 2013). To describe the symmetric positions of residues in four homologous domains in the channel, we used a universal residue-labeling scheme (Zhorov and Tikhonov, 2004). A residue is labeled by its domain number (1–4), segment (i, inner helix S6; o, outer helix S5; p, P-loop), and the relative number from the N end of a transmembrane helix or from the DEKA (i.e., the four amino acids thought to form the selectivity filter of the Na+ channel: aspartate, glutamate, lysine, and alanine) locus positions 1p50, 2p50, and so on. For example, F4i15(1760) designates phenylalanine in the domain IV inner helix, 15 positions downstream from the start of the segment. In some cases, the sequence-based residue number is included in the label in parentheses.
The alignment of bacterial NavAb and NavMs with eukaryotic sodium channels was taken as previously proposed elsewhere (Payandeh et al., 2011; McCusker et al., 2012; Tikhonov and Zhorov, 2012). An insertion downstream from the DEKA locus was proposed (Tikhonov and Zhorov, 2012), but in our models this insertion was not introduced as the ligand was docked in the pore and residues above the DEKA locus would not affect ligand binding. The models contained the pore region (S5, P, and S6) of the human Nav1.5. The closed model also contained the L4-5 linker (the linker between domain 4 and 5) because it is available in the x-ray structure. The extracellular linkers between P-loops and transmembrane helices were truncated to match the length of the x-ray structure templates, which does not affect ligand docking in the inner pore as they are distant. Ionizable residues were modeled as neutral, but the ionizable residues of DEKA locus were modeled as charged. S-fluoxetine was modeled as protonated because its ammonium group has a pKa of ∼10.
All calculations were performed using the ZMM program (ZMM Software, Flamborough, Ontario, Canada). The nonbonded energy was calculated using the AMBER force field (Weiner et al., 1984, 1986) with a cutoff distance of 8 Å. Atomic charges at fluoxetine were calculated with the MOPAC software using the semiempirical method AM1 (Dewar et al., 1985). The hydration energy was calculated by using the implicit-solvent method (Lazaridis and Karplus, 1999). Electrostatic energy was calculated using the environment- and distance-dependent dielectric function without desolvation energy (Garden and Zhorov, 2010). The DEKA locus was loaded with an explicit water molecule, which was initially constrained to the Asp and Lys side chains; subsequently once constraints were removed the water did not move away from the DEKA locus. The Monte Carlo minimization method (Li and Scheraga, 1987) was used to optimize the models. All torsional angles of the protein and ligand were allowed to vary during energy calculations, while bond angles were rigid in the protein and flexible in the ligand. To prevent large deviations of the channel models from the x-ray structure templates during energy minimizations, the α-carbons of the model were constrained to the template using a flat-bottom energy function that allows atoms to deviate penalty-free up to 1 Å, but imposes a penalty of 10 kcal mol−1 Å−1 for larger deviations. All molecular images were created using MVM (ZMM Software). No specific energy terms were used for cation-π interactions, which were accounted for with partial negative charges at the aromatic carbons (Bruhova et al., 2008).
The homology models were first MC-minimized without ligand until the 3000 consecutive energy minimizations did not improve the apparent global minimum found. The optimal binding modes of S-fluoxetine were searched by a two-stage random-docking approach. In the first stage, 60,000 different binding modes of the ligand were randomly generated within a cube with 14-Å edges. This sampling volume covered the entire inner pore including the domain interfaces. Each binding mode was MC-minimized for only five steps to remove steric overlaps with the protein. Energetically favorable conformations within 200 kcal/mol from the apparent global minimum were accumulated and then clustered based on ligand-generalized coordinates. In the second stage, the 500 energetically best conformations found in the first stage were further MC-minimized for 1000 MC-minimization steps. The energetically most favorable ligand-receptor complexes within 4 kcal/mol were collected and analyzed.
Results
Fluoxetine and Its Optical Isomers Block the Nav1.5 Channel.
We studied the effect of fluoxetine on Nav1.5 stably expressed in HEK-293 cells. Figure 1A shows an example of whole-cell current traces before (control) and after superfusion of 25 and 100 μM racemic fluoxetine. Fluoxetine inhibited Na+ currents, with a maximum blockade occurring at 100 μM. The inhibition was partially reversible. The superfusion of increasing concentrations of fluoxetine (1, 10, 25, 50, 100, and 200 μM) showed that the blockade by fluoxetine was dose dependent. The dose-response curves (Fig. 1B) showed that the sensitivities of the optical isomers were similar, with an IC50 of 39.4 μM for racemic fluoxetine, and 40.0 μM and 46.7 μM for the (+) and (−) isomers, respectively. When the cells were maintained at a holding potential of −90 mV instead of −140 mV, where a proportion of the channels are inactivated, the affinity of racemic fluoxetine for Nav1.5 significantly increased with an IC50 of 4.7 μM. Surprisingly, norfluoxetine, a fluoxetine metabolite, displayed a significantly higher affinity than fluoxetine, with an IC50 of 29.5 μM at holding potential of −140 mV.
Fig. 1.

Tonic block of Nav1.5/WT currents. (A) Superimposed INa recordings obtained before and after perfusion with two concentrations of racemic fluoxetine from a holding potential of −140 mV. The dashed line represents zero current. (B) Dose-response curves of the inhibitory effect of norfluoxetine and different optical isomers of fluoxetine on Nav1.5/WT currents. HEK-293 cells stably expressing Nav1.5/WT were perfused with different concentrations of norfluoxetine, racemic fluoxetine, S(+) fluoxetine, or R(−) fluoxetine. There was no significant difference between the IC50 of racemic fluoxetine (IC50 = 39.4 ± 2.0 μM, n = 3–7) and its two optical isomers (IC50 = 40.0 ± 2.6 μM, n = 6–14 and 46.7 ± 3.1 μM, n = 3–10). However, norfluoxetine had a significantly lower IC50 (29.5 ± 1.0 μM, n = 8–15). The IC50 of fluoxetine was significantly reduced to 4.7 ± 0.5 μM (n = 7–10) when recorded at a holding potential of −90 mV (B, open triangle). (C) Dose-response curves of the inhibitory effect of nisoxetine (n = 3–11), methylphenidate (n = 4–9), and fenfluramine (n = 4–6) on Nav1.5/WT currents recorded at a holding potential of −140 or −90 mV. The IC50 of the three drugs at a holding potential of −90 mV were significantly lower than those recorded at −140 mV. The insets in (B) and (C) show the IC50 for each compound. The values were fitted to a Hill equation. Currents were elicited from a holding potential of −140 mV or −90 mV, and a −30 mV test pulse lasting 50 milliseconds was delivered every 5 seconds. ***P < 0.001.
The effects of three other monoamine transporter (MAT)-targeting drugs were also tested using HEK-293 cells stably expressing Nav1.5. The norepinephrin reuptake inhibitor nisoxetine, the dopamine reuptake inhibitor methylphenidate, and fenfluramine, which like fluoxetine targets SERT, were all less effective in blocking the channels than fluoxetine, with an IC50 of 104.5, 618.7, and 203.5 μM, respectively, at a holding potential of −140 mV (Fig. 1C). The inhibition potency of these three compounds was also increased at a holding potential of −90 mV, with an IC50 of 20.2, 239.5, and 65.5 μM for nisoxetine, methylphenidate, and fenfluramine, respectively (Fig. 1C).
Effect of Fluoxetine on the Steady-State Gating Properties of Nav1.5 Channels.
The availability of Na+ channels after depolarization depends on a number of parameters, including the membrane potential. Fewer channels become available as the membrane potential progressively becomes more depolarized. This is due to the buildup of channels in the inactivated nonconducting state. We studied this phenomenon using a double-pulse protocol: a 500-millisecond conditioning pulse to voltages ranging from −140 mV to 0 mV, and a test pulse to −30 mV. The current measured after the test pulse is an indicator of the fraction of available channels. The normalized currents after the test pulse were plotted against the conditioning voltage (Fig. 2A). Fluoxetine (30 μM) significantly shifted the V1/2 of inactivation of Nav1.5 by 6.7 mV toward more hyperpolarized voltages and resulted in a less steep slope factor (Table 1 and Fig. 2A).
Fig. 2.

Gating properties of Nav1.5/WT treated with fluoxetine. (A) Voltage dependence of steady-state activation and inactivation of Nav1.5. Cells were perfused with external solution as a control (activation, n = 14; inactivation, n = 19) or with 30 µM racemic fluoxetine (activation, n = 18; inactivation, n = 17). Activation curves were elicited with 50-millisecond depolarizing steps from −100 to 80 mV in 10 mV increments. Cells were held at a holding potential of −140 mV. Fluoxetine caused no significant shift in the activation curve. Steady-state inactivation was determined using 4-millisecond test pulses to −30 mV after a 500-millisecond prepulse to potentials ranging from −140 mV to 0 mV (see the inset under the inactivation curves for the protocol). The application of 30 µM fluoxetine induced a significant −6.7 mV shift of the inactivation curve (Table 1). The activation and inactivation curves were fitted to a single Boltzman function (see Materials and Methods). (B) Recovery from inactivation of Nav1.5 in the absence (n = 10) or presence (n = 9) of 30 µM fluoxetine. The cells were depolarized to −30 mV for 40 milliseconds from a holding potential of −140 mV to inactivate all the Na+ channels. Test pulses were then applied to −30 mV for 20 milliseconds to measure current amplitudes, with an interval ranging from 0.1 to 4000 milliseconds. The resulting curves were fitted to a double (control) or a triple (+ fluoxetine) exponential equation, which yielded two or three time constants (τ1, τ2, τ3). The application of 30 µM fluoxetine strongly slowed the recovery from inactivation with the appearance of a third time constant (Table 1).
TABLE 1.
Biophysical properties of Nav1.5 channels
| Property | Nav1.5/WT Control |
Nav1.5/WT Fluoxetine |
||
|---|---|---|---|---|
| Mean ± S.E.M. | n | Mean ± S.E.M. | n | |
| Steady-state activation | ||||
| V1/2, mV | −43.49 ± 1.46 | 14 | −41.52 ± 1.17 | 18 |
| kv | −6.13 ± 0.32 | 14 | 7.28 ± 0.17a | 18 |
| Steady-state inactivation | ||||
| V1/2, mV | −87.34 ± 0.94 | 19 | −94.04 ± 1.64b | 17 |
| kv | 6.37 ± 0.19 | 19 | 7.67 ± 0.29b | 17 |
| Recovery from inactivation | ||||
| τ1 | 1.50 ± 0.1 | 10 | 1.63 ± 0.1 | 6 |
| A1 | 76.3 ± 2.7 | 10 | 35.7 ± 2.1 | 6 |
| τ 2 | 9.13 ± 1.0 | 10 | 14.90 ± 2.6 | 6 |
| A2 | 23.7 ± 0.8 | 10 | 22.0 ± 2.1 | 6 |
| τ 3 | — | — | 1598.23 ± 41.6 | 6 |
| A3 | — | — | 42.3 ± 1.7 | 6 |
A, fraction of the τ components (%); kv, slope factor for activation or inactivation; n, number of cells; τ, time constant; V1/2, midpoint for activation or inactivation.
P < 0.01.
P < 0.001.
We also investigated the effect of fluoxetine on the steady-state activation of Nav1.5. The activation curves were derived from I/V curves (see Materials and Methods). The activation curves of Nav1.5 in the absence and presence of 30-μM fluoxetine were plotted against voltage (Fig. 2A). Fluoxetine did not significantly shift the midpoint of steady-state activation, but had a little effect on the slope factor by reducing its steepness.
Fluoxetine Slows the Recovery from Inactivation of Nav1.5 Channels.
A prominent characteristic of many class 1 antiarrhythmics is their ability to slow the recovery from inactivation of drug-modified Na+ channels. We used a two-pulse protocol to investigate the effect of fluoxetine on the recovery from inactivation. We used a 40-millisecond −30 mV conditioning pulse and a 20-millisecond −30 mV test pulse with an interval ranging from 0.1 to 4000 milliseconds to induce recovery from inactivation. The amplitudes of the Na+ currents measured after the test pulse were then normalized to the control currents and were plotted against the duration of the recovery interval. Channels that recovered from inactivation displayed a progressive increase in currents after the increase in the recovery interval (Fig. 2B). The recovery from inactivation of Nav1.5 after fluoxetine treatment was strongly slowed with the appearance of a third time constant. In comparison, the control curve had a τ1 and τ2 of 1.50 and 9.13 milliseconds, respectively, whereas the fluoxetine had a τ1, τ2, and τ3 of 1.63, 14.90, and 1598.23 milliseconds, respectively (Table 1).
Fluoxetine Blocks Nav1.5 Channels in a Use-Dependent Manner.
During depolarization, Na+ channels cycle from the resting to the activated and inactivated states. However, when they are subjected to a train of depolarizing pulses, the number of channels available to open is reduced because they are gradually trapped in the inactivated state. This phenomenon is referred to as use-dependence or “frequency-dependent” current reduction. In the presence of a drug, further decreases in currents are likely due to the accumulation of drug-modified channels. For example, lidocaine, a class 1 antiarrhythmic drug, is known to cause the use-dependent inhibition of Na+ channels.
We tested the effect of rapid pulsing on Nav1.5 by applying a series of 50 short 10-millisecond depolarizing −30 mV pulses. We first characterized the effect of fluoxetine on Nav1.5/WT (wild type), and then on the Nav1.5/F1760C and Nav1.5/Y1767C mutant channels. As shown in Fig. 3A, in the absence of fluoxetine, there was no significant change in the availability of Nav1.5/WT channels when they were pulsed up to 10 Hz. However, in the presence of 30 μM fluoxetine, the availability of Nav1.5/WT channels was dramatically reduced by 44% (P50/P1) when they were pulsed at 2 Hz (Fig. 3, B and C) in comparison with the control without drug. When 5 and 10 Hz pulses were used, 30 μM fluoxetine reduced the currents of the Nav1.5/WT by 58 and 67%, respectively, compared with the control without drug.
Fig. 3.

Frequency-dependent inhibition. (A) Representative whole-cell traces recorded from Nav1.5/WT (±30 µM fluoxetine), Nav1.5/F1760C (+30 µM fluoxetine), and Nav1.5/Y1767C (+ 30 µM fluoxetine) when pulsing at 10 Hz. The dashed line represents zero current. (B) Use-dependent blocks of Nav1.5/WT, Nav1.5/F1760C, and Nav1.5/Y1767C currents in the presence of 30 µM fluoxetine. A 50-pulse train was applied at −30 mV for 10 milliseconds from a holding potential of −140 mV when pulsing at 2 Hz, 5 Hz, and 10 Hz. Peak currents were measured, normalized to the peak amplitude at P1, and plotted against the corresponding pules. (C) Relative currents amplitudes (P50/P1) of the 50th sweep recorded from Nav1.5/WT, Nav1.5/F1760C, and Nav1.5/Y1767C. After the fluoxetine treatment, Nav1.5/WT (n = 8) currents were significantly reduced by 44, 58, and 67% compared with the control when pulsing at 2, 5, and 10 Hz, respectively (***P < 0.001). Fluoxetine significantly reduced Nav1.5/F1760C (n = 11) currents by 15 and 20% when pulsing at 5 and 10 Hz (###P < 0.001), respectively, and Nav1.5/Y1767C (n = 6) currents by 5% when pulsing at 10 Hz (ϕϕP < 0.01) compared with control. There was no significant use-dependent inhibition of Nav1.5/WT (n = 7), Nav1.5/F1760C (n = 5), or Nav1.5/Y1767C (n = 6) currents before the fluoxetine treatment. The controls curves of Nav1.5/WT, Nav1.5/F1760C, and Nav1.5/Y1767C without fluoxetine treatment were removed from the graphics for (B) and (C) for clarity.
To further investigate the role of class 1 antiarrhythmic binding in the current block caused by fluoxetine, we inserted the F1760C or Y1767C mutation into Nav1.5. As shown in Fig. 3, B and C, 30 μM fluoxetine reduced the current by 8, 15, and 20% when Nav1.5/F1760C was pulsed at 2, 5, and 10 Hz, respectively, in comparison with the control without drug. The Y1760C mutation almost completely prevented the use-dependent inhibition of fluoxetine, with a maximal current inhibition of 5% when pulsed at 10 Hz. These results indicated that fluoxetine blocks Nav1.5/WT currents in a use-dependent manner and that the F1760C and Y1767C mutations dramatically reduce the use-dependent inhibition.
Fluoxetine Has a Lower Affinity for Nav1.5/F1760C Mutant Channels.
We studied the effect of the F1760C and Y1767C mutations on the concentration-dependent block of Nav1.5 currents by fluoxetine. Figure 4A shows examples of current traces recorded from Nav1.5/WT and the mutant channels before and after a treatment with 50 μM fluoxetine. As shown in Fig. 4B, the IC50 value of fluoxetine for Nav1.5/Y1767C (50.1 μM) was slightly higher to that of Nav1.5/WT (39.4 μM), but the IC50 value for Nav1.5/F1760C (82.8 μM) was more than twice that of the WT channels.
Fig. 4.

Tonic block of Nav1.5/F1760C and Nav1.5/Y1767C by fluoxetine. (A) Representative whole-cell traces recorded from Nav1.5/WT (left), Nav1.5/F1760C (middle), and Nav1.5/Y1767C (right) channels before and after the application of 50 μM fluoxetine. The dashed line represents zero current. (B) Dose-response curves of the inhibitory effect of racemic fluoxetine on Nav1.5/WT, Nav1.5/F1760C, and Nav1.5/Y1767C. The IC50 values of Nav1.5/F1760C (83 μM) and Nav1.5/Y1767C (50 μM) were significantly higher than Nav1.5/WT value (39 µM). **P < 0.01; ***P < 0.001. The different concentrations of drugs were applied using a perfusion system. Currents were elicited from a holding potential of −140 mV with a 50-millisecond test pulse at −30 mV delivered every 5 seconds. The inset in (B) shows the IC50 for each compound. Normalized current (INa) values were fitted to a Hill equation.
Fluoxetine Acts as an Open-Channel Blocker.
To investigate the role of inactivation in the blockade of Nav1.5 by fluoxetine in greater detail, we used Nav1.5/L409C/A410W mutant transiently expressed in HEK-293 cells. These channels exhibit a significant reduction in fast inactivation in HEK-293 (Wang et al., 2013). A large persistent current was detected in the absence of fluoxetine (Fig. 5A). We applied different concentrations of fluoxetine and determined the IC50 at the peak current and at the end of the test pulse (90–100 milliseconds). The block at the end of the pulse represents the affinity of the fluoxetine for open channels. As shown in Fig. 5B, the IC50 (3.5 μM) at the end of the pulse was slightly lower than the IC50 at the peak current (9.6 μM), suggesting that fluoxetine is an open-channel blocker.
Fig. 5.

Open-channel block of Nav1.5 by fluoxetine. (A) Superimposed INa recordings obtained after the application of different concentrations of fluoxetine on Nav1.5/L409C/A410W-expressing cells. The dashed line represents zero current. (B) Dose-response curves of the inhibitory effect of fluoxetine on Nav1.5/L409C/A410W at the peak current (blue circle) and 90–100 milliseconds after the beginning of the pulse (green square). The IC50 value at the end of the pulse (3.5 μM) was significantly lower than the IC50 value at the peak current (9.6 μM) (***P < 0.001). Currents were elicited from a holding potential of −140 mV with a 50-millisecond test pulse at 0 mV delivered every 5 seconds. Normalized current (INa) values were fitted to a Hill equation. Dotted gray boxes represent the peak current (left box) and the 90- to 100-millisecond (right box) areas used to construct the dose-response curves.
Molecular Modeling of Fluoxetine in the Nav1.5.
To discover the molecular details of the fluoxetine binding site, we have homology modeled the pore domain of Nav1.5 in the closed and open states based on the x-ray structures of bacterial Na channels, NavAb (Arcobacter butzleri sodium channel) (Payandeh et al., 2011) and NavMs (Magnetococcus sp. sodium channel) (Bagnéris et al., 2013), respectively (see Protein Data Bank file in the Data Supplement). A random sampling approach was used to search for the energetically most favorable binding modes of fluoxetine in the Nav1.5. We seeded 60,000 random orientations of fluoxetine inside the channel within a volume to cover the entire pore cavity and inner helix interfaces (Fig. 6A, B). After two rounds of Monte Carlo energy minimizations, the energetically best fluoxetine complexes bound inside the inner pore. Fluoxetine adopts two distinct binding modes: a horizontal and a vertical binding mode (Fig. 6, C–F). These two binding modes were energetically favorable in both the closed and the open-channel pore.
Fig. 6.

Searching for the binding site of fluoxetine in the closed and open Nav1.5. The P-loops and S6 helices of domains I, II, III, and IV are colored blue, orange, green, and violet, respectively. The outer helices and the L4–5 linker are shown as gray strands. The side chains of residues in the DEKA locus, Q1p49(372), S4p49(1712), F4i15(1760), and Y4i22(1767), are shown as sticks with yellow carbons. The water molecule at the DEKA locus is rod-shaped. (A and B) The side and extracellular views of the randomly generated starting points of fluoxetine in the closed Nav1.5. Fluoxetine is presented in wire-frame with gray carbons. For clarity, only 6000 of the 60,000 starting points are shown. (C–F) The side views of the lowest energy vertical (C and E) and horizontal (D and F) binding modes of fluoxetine in the closed (C and D) and open (E and F) channel. Fluoxetine is shown in thick sticks with gray carbons. The side chain of F3p49(1236) is shown in (D) and (F). For clarity, the outer helices are not shown in (C–F) (see the Protein Data Bank file in the Data Supplement).
Fluoxetine resembles a three-pointed star with a chiral center in the middle linking three arms comprising an ammonium group, a benzene ring, and a trifluoromethyl benzene ring. In both of the two binding modes of fluoxetine, its ammonium group localizes to the channel’s central axis under the DEKA locus, near the focus of the P-helices (Fig. 1, C–F). Just one position upstream of the DEKA locus, a ring of QGFS residues in position p49 (see description of relative number scheme in Materials and Methods) favorably interacts with fluoxetine because their side chains face downward into the pore. Particularly, Q1p49(372) and S4p49(1712) form favorable electrostatic contacts with fluoxetine’s nitrogen, each contributing 4–9% to the ligand-receptor energy. The ammonium group of fluoxetine was also attracted by the two negatively charged residues of the DEKA locus, which outweighed the repulsion from the Lys of the DEKA locus. Further, the backbone carbonyl groups of residues two to three positions upstream of the DEKA locus (positions p47 and p48) also stabilize fluoxetine.
The horizontal and vertical binding modes are distinguished by the two benzene arms of fluoxetine. In the vertical binding mode (Fig. 6, C and E), one benzene arm is parallel, and the other arm is perpendicular to the pore axis. In this mode, one benzene ring π-stacks with Y4i22(1767) and the other interacts with F4i15(1760). Y4i22 and F4i15 were found to be the two most significant residues in binding fluoxetine; each contributes 16–33% to the ligand-receptor energy. In the horizontal binding mode (Fig. 6, D and F), the two benzene arms point away ∼45–90° from the pore axis. The ligand leans against the P-loop and protrudes between the III–IV domain inner helices. Here, one of the benzene rings π-stacks with F4i15, while the other arm extends toward F3p49(1236) and IIIS6. In the horizontal binding mode, F4i15 has the strongest interaction with fluoxetine, contributing 23–33% to the ligand-receptor energy, while other residues contributed <10%.
The closed NavAb-based and open NavMs-based models exhibit similar channel geometry, except for the intracellular half of the inner helices of the open state that bend to widen the pore. In both the closed and open channel models, residues in position i15 (which includes F4i15) and position i22 (which includes Y4i22) are pore facing. Thus, a vertical and a horizontal binding mode of fluoxetine were found in both the closed and open pore. However, in terms of energy, the horizontal binding mode was favored in the closed state, because the fluoxetine experienced ligand strain in the narrower closed pore. On the other hand, the fluoxetine preferred to adopt the vertical binding mode in the open state as it formed better ligand-receptor contacts with F4i15 and Y4i22. In summary, the molecular model of fluoxetine in Nav1.5 was in agreement with mutational experiments, suggesting that F1760 and Y1767 are the two key residues for its binding.
Discussion
In the present study, we characterized the effects of fluoxetine, a widely used antidepressant drug, on Nav1.5, the cardiac voltage-gated Na+ channel.
Our results showed that racemic fluoxetine and its optical isomers are equally effective blockers of Nav1.5 when current were recorded at a holding potential of −140 mV. Similar results have been reported for cardiac voltage-gated Ca2+ channels in canine ventricular cardiomyocytes, where both fluoxetine enantiomers have a similar IC50 (Magyar et al., 2003). We also conducted dose-response curves experiments for racemic fluoxetine in HEK-293 at a holding potential of −90 mV, which is near the resting potential of cardiomyocytes. These experiments showed that the IC50 of fluoxetine is eight times lower at a holding potential of −90 mV compared with −140 mV, going from 39.4 μM to 4.7 μM. In a manner that is hard to explain, these data are in contradiction with those published by Rajamani et al. (2006), who reported that fluoxetine does not inhibit Na+ currents in HEK-293 cells expressing Nav1.5. However, our IC50 of 4.7 μM is very similar with that published by Harmer et al. (2011), who reported an IC50 of 4.9 μM using IonWorks assays from hNav1.5-expressing Chinese hamster ovary (CHO) cells maintained at a holding potential of −90 mV. These results suggest that the holding potential of the cell is very important to the affinity of fluoxetine for the channel, as it has been also shown in rat hippocampi neurons (Lenkey et al., 2006), suggesting that the fluoxetine binds with higher affinity to inactivated than to resting channels.
In the nervous system, fluoxetine primary targets SERT, which, together with dopamine transporter and norepinephrine transporter, make up the three major MAT classes. To investigate the effect of other MAT-targeting drugs, we investigated the effect of nisoxetine (norepinephrine transporter-targeting drug) (Tejani-Butt, 1992), methylphenidate (dopamine transporter-targeting drug) (Han and Gu, 2006), and fenfluramine (SERT-targeting drug) (Cosgrove et al., 2010) on Nav1.5 currents. Our results showed that the affinity of these drugs for Nav1.5 is dependent on the holding potential. The IC50 of nisoxetine, methylphenidate, and fenfluramine are respectively 5, 2.5, and 3 times lower at a holding potential of −90 mV compared with −140 mV. Similar to fluoxetine, the decrease of IC50 at a more depolarized potential suggests a higher affinity of these three compounds to inactivated than to resting channels. Furthermore, these compounds also exhibited a use-dependent inhibition, especially nisoxetine with a significant current reduction of 26, 36, and 38% when pulsing at 2, 5, and 10 Hz, respectively (data not shown). However, these three compounds are still less potent than fluoxetine at inhibiting Nav1.5.
Our study was designed to investigate the biophysical mechanism of the Nav1.5 block by fluoxetine as well as the possible proarrhythmic properties of this drug. A major finding of our work was that fluoxetine shifts the steady-state inactivation curve by 6.7 mV toward more hyperpolarized values, indicating that it binds to the inactivated state of Nav1.5, as is the case with neuronal Na+ channels (Lenkey et al., 2006). In addition to a tonic block, fluoxetine decreased Nav1.5 currents in a use-dependent manner when pulsing at 2, 5, and 10 Hz. The affinity of fluoxetine for Nav1.5 thus appears to be modulated by the state of the channel, which rapidly switches between the open and inactivated configurations, leading to the progressive accumulation of inactivated Nav1.5. Use-dependence occurs because drug-modified channels slowly recover only at hyperpolarized voltages. Class 1 antiarrhythmic drugs and local anesthetics have a similar effect (Chahine et al., 1992). We thus determined whether fluoxetine could inhibit Na+ currents by mutating residues in the class 1 antiarrhythmic drug binding site.
Amino acids situated near the cytoplasmic ends of the membrane-spanning S6 α-helixes of all four homologous domains (DIS6-DIVS6) form the cytoplasmic entrance of the pore and contribute to the binding sites of both the native inactivation gate and class 1 antiarrhythmic drugs. We previously reported that two highly conserved residues of the DIVS6 segment (F1760, Y1767) contribute directly to the local anesthetic binding site of cardiac Na+ channels (O’Leary and Chahine, 2002). We showed that both mutations (F1760C and Y1767C) markedly reduced the frequency-dependent effect, with the Y1767C mutation having the greatest effect. However, in a tonic block, the F1760C increased the IC50 of fluoxetine more significantly than the Y1767C. These results showed that these residues of DIVS6 are an integral part of the binding site of fluoxetine, as is the case with many class 1 antiarrhythmic drugs. Our data also suggest that F1760 appears to be more involved in binding fluoxetine when the channel is in the resting state, whereas Y1767 appears to be key for fluoxetine binding when the channel is in the open/inactivated sate.
Molecular modeling of fluoxetine in Nav1.5 was in agreement with mutational experiments, in which F4i15(1760) and Y4i22(1767) were found to be the key residues in binding fluoxetine. However, the models predicted that the ligand is able to assume two energetically favorable binding modes. The vertical binding mode was favored in the open state model, and the horizontal mode in the closed state model. This could suggest that open channel block involves both F4i15(1760) and Y4i22(1767) as visualized in the vertical binding mode. With the same assumption, the horizontal binding mode could represent a resting channel block with F4i15(1760) as the essential residue. Fluoxetine share similarities with local anesthetics. Both are drugs sensitive to mutations at F4i15(1760) and Y4i22(1767). Structurally, fluoxetine resembles most classic local anesthetics in approximate size and by possessing an ammonium group and a benzene ring. Fluoxetine adopts similar binding modes in the closed channel homology model as QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], cocaine, and tetracaine (Bruhova et al., 2008; Tikhonov and Zhorov, 2012). Because fluoxetine can protrude between the III–IV inner helix interface while in the horizontal binding mode, it could suggest that fluoxetine may enter or exit through the III–IV domain interface pathway from the extracellular side of the membrane as it has been demonstrated with local anesthetics (Qu et al., 1995; Sunami et al., 2001). Experiments with fluoxetine with a quaternarized ammonium could reveal whether the ligand can block from the extracellular side.
The blockade of Nav1.5 by fluoxetine should be taken into consideration when prescribing this drug. Blocking the cardiac Na+ channel may cause an intracardiac conduction delay, which may in turn cause a prolongation of the QRS complex on the electrocardiogram (Delk et al., 2007). Given the association between QRS prolongation and mortality, and the potential for drug-induced arrhythmia, caution is required when prescribing fluoxetine (Thanacoody and Thomas, 2005; Delk et al., 2007), especially given that inhibiting the Nav1.5 by as little as 10% may cause a prolongation of the QRS complex in humans (Cordes et al., 2009).
However, a question remains as to how to transpose the significance of the IC50 value of fluoxetine to a pathophysiological setting. The IC50/fCmax ratio, where fCmax represents the unbound (free) plasma concentration in a clinical setting, of a drug that evokes a QRS or a change in QT interval has been proposed as a tool for determining whether a drug can be safely prescribed (Redfern et al., 2003; Harmer et al., 2011). An IC50/fCmax ratio above 30 to 100 has been shown to ensure a suitable degree of safety in terms of drug-induced QRS complex prolongation. The fCmax for fluoxetine is 93 nM (Harmer et al., 2011). Thereby, when we mimic the membrane potential of cardiomyocytes in patch-clamp studies by imposing a holding potential of −90 mV to HEK-293 cells, the IC50/fCmax ratio is 50. This is within the 30 to 100 margin, and it should act as a safety flag for a possible cardiotoxicity.
Furthermore, in the case of fluoxetine, fCmax may not be a good indicator of actual plasma concentrations of total Nav1.5 blockers in vivo because norfluoxetine, an active metabolite of fluoxetine, has a higher affinity for Nav1.5 than fluoxetine itself. Given that norfluoxetine has a half-life of more than a week compared with 70 hours for a single dose of fluoxetine (Rambourg Schepens and Dawling, 1998), there is a possibility of a long-lasting additive effect on cardiac Na+ channels. In fact, in the calculation of the IC50/fCmax ratio, we should take into consideration the unbound (free) plasma concentration of norfluoxetine. Despite the lack of information about the fCmax after a single dose, it is known that the plasma concentration of total (unbound and bound) fluoxetine and norfluoxetine at steady state are very similar after chronic treatment (91–302 ng/ml and 72–258 ng/ml, respectively) (FDA, 2009). This suggests that the IC50/fCmax ratio after fluoxetine treatment is probably underestimated.
In conclusion, caution should be taken when prescribing fluoxetine at same time as other Na+ channel inhibitors such as class 1 antiarrhythmic drugs, especially class 1A and 1C drugs. In addition, fluoxetine should be prescribed with extreme care for patients suffering from ventricular conduction disorders or liver disease. Indeed, as the liver is the primary site of fluoxetine metabolism, the impairment of liver functions as a result of hepetatis or cirrhosis could affect the elimination half-life of fluoxetine and norfluoxetine (Schenker et al., 1988).
Supplementary Material
Abbreviations
- DEKA, the four amino acids forming the putative selectivity filter of the Na+ channel
aspartate (D), glutamate (E), lysine (K), and alanine (A)
- fCmax
unbound (free) plasma concentration in a clinical setting
- HEK-293
human embryonic kidney 293 cells
- MAT
monoamine transporter
- QX-314
N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide
- SERT
serotonin transporter
- WT
wild type
Authorship Contributions
Participated in research design: Poulin, Chahine, Beaulieu, Timour, Theriault.
Conducted experiments: Poulin, Chahine, Beaulieu, Bruhova.
Wrote or contributed to the writing of the manuscript: Poulin, Chahine, Beaulieu, Timour, Frassati, Bruhova.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research [CIHR, MOP-111072], the Heart and Stroke Foundation of Quebec (HSFQ), a research fellowship from the Canadian Institute of Health Research (to I.B.), the Natural Sciences and Engineering Research council of Canada [NSRC:RGPIN 19483302013], and by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS-064969] through Anthony Auerbach (to I.B.). Computations were made possible by the facilities of the Center for Computational Research University at Buffalo (CCR: www.ccr.buffalo.edu).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Bagnéris C, Decaen PG, Hall BA, Naylor CE, Clapham DE, Kay CWM, Wallace BA. (2013) Role of the C-terminal domain in the structure and function of tetrameric sodium channels. Nat Commun 4:2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolo NR, Hodé Y, Nédélec JF, Lainé E, Wagner G, Macher JP. (2000) Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 23:428–438 [DOI] [PubMed] [Google Scholar]
- Bruhova I, Tikhonov DB, Zhorov BS. (2008) Access and binding of local anesthetics in the closed sodium channel. Mol Pharmacol 74:1033–1045 [DOI] [PubMed] [Google Scholar]
- Chahine M, Chen LQ, Barchi RL, Kallen RG, Horn R. (1992) Lidocaine block of human heart sodium channels expressed in Xenopus oocytes. J Mol Cell Cardiol 24:1231–1236 [DOI] [PubMed] [Google Scholar]
- Cordes J, Li C, Dugas J, Austin-LaFrance R, Lightbown I, Engwall M, Sutton M, Steidl-Nichols J. (2009) Translation between in vitro inhibition of the cardiac Nav1.5 channel and pre-clinical and clinical QRS widening (Abstract). J Pharmacol Toxicol Methods 60:221 [Google Scholar]
- Cosgrove KP, Staley JK, Baldwin RM, Bois F, Plisson C, Al-Tikriti MS, Seibyl JP, Goodman MM, Tamagnan GD. (2010) SPECT imaging with the serotonin transporter radiotracer [123I]p ZIENT in nonhuman primate brain. Nucl Med Biol 37:587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Lasztóczi B, Pacher P, Petheö GL, Valéria Kecskeméti, Spät A. (2000) Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology 39:1029–1036 [DOI] [PubMed] [Google Scholar]
- Delk C, Holstege CP, Brady WJ. (2007) Electrocardiographic abnormalities associated with poisoning. Am J Emerg Med 25:672–687 [DOI] [PubMed] [Google Scholar]
- Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. (1985) Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J Am Chem Soc 107:3902–3909 DOI: 10.1021/ja00299a024. [Google Scholar]
- Eisensamer B, Rammes G, Gimpl G, Shapa M, Ferrari U, Hapfelmeier G, Bondy B, Parsons C, Gilling K, Zieglgänsberger W, et al. (2003) Antidepressants are functional antagonists at the serotonin type 3 (5-HT3) receptor. Mol Psychiatry 8:994–1007 [DOI] [PubMed] [Google Scholar]
- Garden DP, Zhorov BS. (2010) Docking flexible ligands in proteins with a solvent exposure- and distance-dependent dielectric function. J Comput Aided Mol Des 24:91–105 [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH. (2006) Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer AR, Valentin JP, Pollard CE. (2011) On the relationship between block of the cardiac Na+ channel and drug-induced prolongation of the QRS complex. Br J Pharmacol 164:260–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennings ECP, Kiss JP, De Oliveira K, Toth PT, Vizi ES. (1999) Nicotinic acetylcholine receptor antagonistic activity of monoamine uptake blockers in rat hippocampal slices. J Neurochem 73:1043–1050 [DOI] [PubMed] [Google Scholar]
- Herbert E, Chahine M. (2006) Clinical aspects and physiopathology of Brugada syndrome: review of current concepts. Can J Physiol Pharmacol 84:795–802 [DOI] [PubMed] [Google Scholar]
- Huang H, Priori SG, Napolitano C, O’Leary ME, Chahine M. (2011) Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am J Physiol Heart Circ Physiol 300:H288–H299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T, Karplus M. (1999) Effective energy function for proteins in solution. Proteins 35:133–152 [DOI] [PubMed] [Google Scholar]
- Lenkey N, Karoly R, Kiss JP, Szasz BK, Vizi ES, Mike A. (2006) The mechanism of activity-dependent sodium channel inhibition by the antidepressants fluoxetine and desipramine. Mol Pharmacol 70:2052–2063 [DOI] [PubMed] [Google Scholar]
- Li Z, Scheraga HA. (1987) Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci USA 84:6611–6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens C, Droogmans G, Verbesselt R, Nilius B. (2002) Block of volume-regulated anion channels by selective serotonin reuptake inhibitors. Naunyn Schmiedebergs Arch Pharmacol 366:158–165 [DOI] [PubMed] [Google Scholar]
- Magyar J, Rusznák Z, Harasztosi C, Körtvély A, Pacher P, Bányász T, Pankucsi C, Kovács L, Szûcs G, Nánási PP, et al. (2003) Differential effects of fluoxetine enantiomers in mammalian neural and cardiac tissues. Int J Mol Med 11:535–542 [PubMed] [Google Scholar]
- McCusker EC, Bagnéris C, Naylor CE, Cole AR, D’Avanzo N, Nichols CG, Wallace BA. (2012) Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat Commun 3:1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscettola G, Goodwin FK, Potter WZ, Claeys MM, Markey SP, Markey SP. (1978) Imipramine and desipramine in plasma and spinal fluid: relationship to clinical response and serotonin metabolism. Arch Gen Psychiatry 35:621–625 [DOI] [PubMed] [Google Scholar]
- O’Leary ME, Chahine M. (2002) Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels. J Physiol 541:701–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Kecskemeti V. (2004) Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des 10:2463–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Magyar J, Szigligeti P, Bányász T, Pankucsi C, Korom Z, Ungvári Z, Kecskeméti V, Nánási PP. (2000) Electrophysiological effects of fluoxetine in mammalian cardiac tissues. Naunyn Schmiedebergs Arch Pharmacol 361:67–73 [DOI] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, Catterall WA. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475:353–358 Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. (1995) Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci USA 92:11839–11843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, Delisle BP, Anson BD, Makielski JC, et al. (2006) Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol 149:481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambourg Schepens MO and Dawling S (1998) Fluoxetine. International Programme on Chemical Safety (IPCS) INCHEM, http://www.inchem.org/documents/pims/pharm/pim651.htm
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PKS, Strang I, Sullivan AT, Wallis R, et al. (2003) Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58:32–45 [DOI] [PubMed] [Google Scholar]
- Schenker S, Bergstrom RF, Wolen RL, Lemberger L. (1988) Fluoxetine disposition and elimination in cirrhosis. Clin Pharmacol Ther 44:353–359 [DOI] [PubMed] [Google Scholar]
- Sghendo L, Mifsud J. (2012) Understanding the molecular pharmacology of the serotonergic system: using fluoxetine as a model. J Pharm Pharmacol 64:317–325 [DOI] [PubMed] [Google Scholar]
- Sunami A, Glaaser IW, Fozzard HA. (2001) Structural and gating changes of the sodium channel induced by mutation of a residue in the upper third of IVS6, creating an external access path for local anesthetics. Mol Pharmacol 59:684–691 [DOI] [PubMed] [Google Scholar]
- Tejani-Butt SM. (1992) [3H]nisoxetine: a radioligand for quantitation of norepinephrine uptake sites by autoradiography or by homogenate binding. J Pharmacol Exp Ther 260:427–436 [PubMed] [Google Scholar]
- Thanacoody HKR, Thomas SHL. (2005) Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev 24:205–214 [DOI] [PubMed] [Google Scholar]
- Thomas D, Gut B, Wendt-Nordahl G, Kiehn J. (2002) The antidepressant drug fluoxetine is an inhibitor of human ether-a-go-go-related gene (HERG) potassium channels. J Pharmacol Exp Ther 300:543–548 [DOI] [PubMed] [Google Scholar]
- Tikhonov DB, Zhorov BS. (2012) Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol 82:97–104 [DOI] [PubMed] [Google Scholar]
- Timour Q, Frassati D, Descotes J, Chevalier P, Christé G, Chahine M. (2012) Sudden death of cardiac origin and psychotropic drugs. Front Pharmacol 3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. (2003) Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci 4:13–25 [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration (FDA) (2009) Prozac NDA 18-936/S-064 final labeling, March 31, 2009. http://www.accessdata.fda.gov/drugsatfda_docs/label/2003/018936s064lbl.pdf
- Wang GK, Russell G, Wang SY. (2013) Persistent human cardiac Na (+) currents in stably transfected mammalian cells: Robust expression and distinct open-channel selectivity among Class 1 antiarrhythmics. Channels (Austin) 7:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. (1984) A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 106:765–784 DOI: 10.1021/ja00315a051. [Google Scholar]
- Weiner SJ, Kollman PA, Nguyen DT, Case DA. (1986) An all atom force field for simulations of proteins and nucleic acids. J Comput Chem 7:230–252 DOI: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Engleman EA. (1995) Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci 57:411–441 [DOI] [PubMed] [Google Scholar]
- Wong DT, Horng JS, Bymaster FP, Hauser KL, Molloy BB. (1974) A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine. Life Sci 15:471–479 [DOI] [PubMed] [Google Scholar]
- Zhorov BS, Tikhonov DB. (2004) Potassium, sodium, calcium and glutamate-gated channels: pore architecture and ligand action. J Neurochem 88:782–799 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.