

Abstract
Hepadnaviruses replicate through reverse transcription of an RNA pregenome, resulting in a relaxed circular DNA genome. The first 3 or 4 nucleotides (nt) of minus-strand DNA are synthesized by the use of a bulge in a stem-loop structure near the 5′ end of the pregenome as a template. This primer is then transferred to a complementary UUCA motif, termed an acceptor, within DR1* near the 3′ end of the viral pregenome via 4-nt homology, and it resumes minus-strand DNA synthesis: this process is termed minus-strand transfer or primer translocation. Aside from the sequence identity of the donor and acceptor, little is known about the sequence elements contributing to minus-strand transfer. Here we report a novel cis-acting element, termed the β5 region (28 nt in length), located 20 nt upstream of DR1*, that facilitates minus-strand DNA synthesis. The deletion or inversion of the sequence including the β5 region diminished minus-strand DNA synthesis initiated at DR1*. Furthermore, the insertion of the β5 region into its own position in a mutant in which the sequences including the β5 region were replaced restored minus-strand DNA synthesis at DR1*. We speculate that the β5 region facilitates minus-strand transfer, possibly by bringing the acceptor site in proximity to the donor site via base pairing or by interacting with protein factors involved in this process.
Hepadnaviruses infect the liver tissue of their mammalian and avian hosts, resulting in acute and chronic liver diseases such as hepatitis, cirrhosis, and hepatocellular carcinoma (5). Prototypic members of the family include human hepatitis B virus (HBV), woodchuck hepatitis virus, and duck hepatitis B virus (DHBV). Hepadnaviruses have a DNA genome which replicates through an RNA intermediate via reverse transcription (5, 20). Genome replication of hepadnaviruses, catalyzed by a viral reverse transcriptase, involves the conversion of the single-stranded RNA genome into double-stranded DNA through a complex series of reactions. The strategy of hepadnaviral reverse transcription parallels that of retroviruses in many respects (30). For instance, both of its reverse transcription reactions require multiple template switching events for the completion of viral genome replication. These template switching events are mediated primarily through complementarity between donor sites and acceptor sites present in the terminal redundancy region of the RNA genomes (2, 19, 29).
Despite its fundamental similarity to retroviral reverse transcription, many features of hepadnaviral reverse transcription are distinct. Hepadnaviral reverse transcription occurs inside nucleocapsids after encapsidation of the pregenomic RNA (pgRNA) (5, 20). A stem-loop structure (i.e., ɛ) near the 5′ end of the pgRNA serves not only as an encapsidation signal (6, 10, 11, 21) but also as the initiation site for minus-strand DNA synthesis (19, 23, 29, 32). Consequently, unlike the case for retroviruses, the initiation of minus-strand DNA synthesis is mechanistically coupled to encapsidation of the pgRNA. Furthermore, the viral polymerase (P) acts as both a primer and a reverse transcriptase for the initiation of minus-strand DNA synthesis. Minus-strand DNA synthesis, initiated by protein priming using the ɛ bulge as a template (31), leads to the synthesis of a 3- or 4-nucleotide (nt) oligomer which is covalently linked to P (25). The P-linked oligomer is then transferred to a complementary UUCA motif within direct repeat 1 (DR1*) near the 3′ end of the pgRNA, where minus-strand DNA synthesis resumes (Fig. 1); this process is termed minus-strand transfer or primer translocation (19, 22, 29, 32). Minus-strand DNA elongation and completion are accompanied by degradation of the pgRNA up to the last 18 ribonucleotides, which serve as an RNA primer for plus-strand DNA synthesis (12, 14). However, before the hepadnavirus initiates plus-strand DNA synthesis, an additional template switch occurs. The plus-strand primer translocates to an acceptor site (DR2) near the 5′ end of the minus-strand DNA template and initiates the synthesis of plus-strand DNA (27). For the continuation of plus-strand DNA synthesis, an intramolecular template switch must occur (13). This third template switch event, termed circularization, leads to the formation of the relaxed circular (RC) DNA found in virions. Alternatively, a subset of the plus-strand RNA primer does not transfer to DR2 but initiates the synthesis of plus-strand DNA in situ, resulting in the formation of a duplex linear (DL) DNA genome (27).
FIG. 1.

Model for minus-strand transfer during hepadnaviral reverse transcription. The pgRNA with cis-acting elements relevant to minus-strand transfer is shown. The pgRNA contains direct repeat sequences of 11 nt, designated DR1 and DR2. DR1 is present twice in the pgRNA due to terminal redundancy (R); the one near the 3′ end is designated DR1*. The encapsidation signal (ɛ) near the 5′ terminus, which folds into a stem-loop structure, is shown. The P protein (shaded oval), serving as a primer, synthesizes the first 4 nt of minus-strand DNA by using the UUCA motif within the bulge of the stem-loop structure as a template. The P protein covalently linked to the first 4 nt of minus-strand DNA is transferred to the UUCA motif at DR1*, which is referred to as the acceptor.
The mechanism of hepadnaviral minus-strand transfer is poorly characterized (Fig. 1). Previous mutagenesis studies have shown that minus-strand transfer is mediated primarily by complementarity between the donor and the acceptor, the UUCA motif at DR1* on the pgRNA, or in the case of DHBV, the UUAC motif overlapping DR1* (15, 19, 25). However, the observed complementarity is insufficient to account for the remarkable specificity of transfer to the acceptor site at DR1*, given the presence of numerous UUC motifs in the RNA template (25). Intriguingly, previous studies have shown that in mutants in which the complementarity has been partially disrupted, the P-linked oligomer is still transferred to the mutated DR1* but not to numerous other UUC motifs present in the pgRNA, suggesting that sequence elements surrounding the acceptor site may contribute to the specificity of the acceptor site (15, 19). Nonetheless, the current model for minus-strand transfer does not specify the role of other sequence elements other than the 4-nt acceptor site within the DR1* (15, 19). To gain more insight into the mechanism controlling P-linked oligomer transfer to DR1*, we evaluated the role of sequence elements surrounding DR1*, the acceptor site, in minus-strand DNA synthesis. Here we report the identification of a novel cis-acting sequence, a 28-nt sequence located just upstream of DR1*, that is essential for minus-strand DNA synthesis and possibly facilitates accurate and efficient minus-strand transfer.
MATERIALS AND METHODS
Cell culture.
Huh7 cells were used for transfection (16). Cells were grown in Dulbecco's modified Eagle's medium (GIBCO-BRL) supplemented with 10% fetal bovine serum (GIBCO-BRL) and 10 μg of gentamicin per ml at 37°C in 5% CO2 and were split every third day.
Transfection.
Cells were plated at a confluency of 60% per 100-mm-diameter plate. On the following day, the cells were washed twice with phosphate-buffered saline and fed with fresh medium. After 2 h, the cells were transfected with 20 μg of supercoiled plasmid DNA per 100-mm-diameter plate by the calcium phosphate coprecipitation method, as described previously (24). In brief, 20 μg of DNA was mixed with 250 mM CaCl2 to a final volume of 693 μl. This solution was added drop by drop to 693 μl of 2× HEPES-buffered saline (283 mM NaCl, 23 mM HEPES acid, 1.5 mM Na2HPO4 [pH 7.05]) and was incubated for 30 min at room temperature. The resulting solution, containing a fine white precipitate, was slowly poured over a plate of cells. After 16 h of incubation at 37°C in a 5% CO2 incubator, the culture medium was replaced with fresh medium. The transfected cells were grown until harvest.
Plasmid construction.
All HBV constructs were derived from a wild-type HBV expression clone, pCMV-HBV/30 (10). All mutants were sequenced to confirm base changes. The nucleotide sequence of the HBV genome was numbered starting from the unique EcoRI site of the HBV ayw subtype, according to the method of Galibert et al. (4); nucleotide position 1 of HBV subtype ayw is the first “A” (underlined) of the EcoRI site (GAATTC). In this numbering system, the 5′ end of the pgRNA is at nt 1820 (18). Most point mutations or substitution and deletion mutations were generated by an overlap extension PCR method (8).
Deletion mutants created by overlap PCR mutagenesis.
For the creation of each deletion mutation, a mutagenic primer (forward mutagenic primer [FMP]) was designed to contain an 18-bp linker spanning each desired deleted region, with 9 bp to each side of the deletion (Table 1). A second mutagenic primer (reverse mutagenic primer [RMP]) was designed to encode an 18-bp linker complementary to the FMP (Table 1). Two PCRs were performed to produce overlapping products: the first was performed with an upstream primer and an RMP, and the second was performed with a downstream primer and an FMP, with the wild type used as a template. A mixture of two purified PCR products which overlapped by 18 bp served as the template for a third PCR, which was performed with the upstream (5′-CATCAGCGCATGCGTGGAAC-3′ [the SphI site is underlined]) and the downstream (5′-TAGAATAGGGCCCTCTAGAA-3′ [the ApaI site is underlined]) primers. This final PCR product was gel purified, digested with SphI and ApaI, and then inserted into the SphI and ApaI sites of the wild-type plasmid. The primers used are described in Table 1.
TABLE 1.
Nucleotide sequences of primers employed for the generation of deletion mutants by overlap extension PCR
| Mutant | FMP sequencea (5′-3′) | RMP sequencea (5′-3′) |
|---|---|---|
| R035 (HBVΔ1607-1804) | TCTGCACGTGCACCAGCACCATGCAAC | TGCTGGTGCACGTGCAGAGGTGAAGCG |
| R811 (HBVΔ1637-1670) | GCCCACCAAGACTCTCAGCAATGTCAA | CTGAGAG TCTTGGTGGGCGTTCACGGT |
| R812 (HBVΔ1670-1740) | CTCTTGGACGTTGGGGGAGGAGATTAG | TCC CCCAACGTCCAAGAGTCCTCTTAT |
| R813 (HBVΔ1780-1807) | TGTACTAGGCCAGCACCATGCAACTTT | TGGTGCT GGCCTAGTACAAAGACCTTT |
| R814 (HBVΔ1637-1740) | GCCCACCAAGTTGGGGGAGGAGATTAG | TCCCCCAACTTGGTGGGCGTTCACGGT |
| R815 (HBVΔ1610-1636) | GCACGTCGCATATTGCCCAAGGTCTTA | GGGCAA TATGCGACGTGCAGAGGTGAA |
| R816 (HBVΔ1741-1779) | CTGGGAGGAAGGCTGTAGGCATAAATT | CTACAGC CTTCCTCCCAGTCTTTAAAC |
| R817 (HBVΔ1637-1740, Δ1780-1807) | GCCCACCAAGTTGGGGGAGATTAG | TCCCCCAACTTGGTGGGCGTTCACGGT |
| R818 (HBVΔ1826-1836) | ATGCGACGTGCGAAGTGCACACGGTCC | GCACTTC GCACGTCGCATGGAGACCAC |
| R819 (HBVΔ1592-1602) | TGCAACTTTCTAATCATCTCTTGTTCA | GATGATTAGAAAGTTGCATGGTGCTGG |
The linker sequences in each primer are underlined; the linker was designed to span each deletion but contains a 9-nt sequence at both sides of the deletion. Thus, each primer contains an 18-nt linker that is complementary to the template.
Substitution or deletion mutants. (i) Plasmid R063 (a helper plasmid).
The helper plasmid R063 was designed to transcribe pgRNA lacking the encapsidation signal (i.e., 5′ ɛ). To facilitate the subcloning process, we first made plasmid R062. Briefly, an insert fragment was made by a PCR using a forward primer (5′-CATGGAATTCATGGACATCGACCCT-3′ [the EcoRI site is underlined]), a reverse primer (5′-CCGCTCGAGCTAACATTGAGATTCCCGAGA-3′ [the XhoI site is underlined]), and a wild-type template. The resulting 551-bp fragment (nt 1903 to 2454) was then digested with EcoRI and XhoI and inserted into the EcoRI (nt 939) and XhoI (nt 972) sites of pcDNA3/amp (Invitrogen). A helper plasmid was constructed by inserting the BspEI-to-ApaI fragment (nt 2331 to 1992) of the wild type into the BspEI (nt 2331) and ApaI (nt 1992) sites of the R062 plasmid.
(ii) Plasmid R029 (pCMV-HBVΔ1804-1884).
To facilitate the subcloning process, we made plasmid R050, in which the EcoRI (nt 3182)-to-ApaI fragment of the wild type was inserted into the multiple cloning site of pBluescriptII KS (+) (Stratagene). A mutant with a deletion of nt 1804 to 1884 was then constructed by inserting the StyI-blunted-EcoRI fragment (nt 3182 to 1804) of the wild-type plasmid into the EcoRI and FspI sites of the R050 plasmid. Subsequently, the HBV fragment with the deletion of nt 1804 to 1884 was transferred into the wild-type plasmid to form R029.
(iii) Plasmid R806.
A unique PmlI site was created at nt 1606 by site-directed mutagenesis using native Pfu DNA polymerase according to the manufacturer's instructions (Stratagene). Briefly, a mutagenic PCR product was made with a forward primer (5′-TTCACCTCTGCACGTGGCATGGAGACCACCGTGAAC-3′ [the PmlI site is underlined]) and a reverse primer (5′-GTTCACGGTGGTCTCCATGCCACGTGCAGAGGTGAA-3′ [the PmlI site is underlined]). The mutation was confirmed by digestion with PmlI.
(iv) Plasmid R808.
A unique HindIII site was created at nt 1808 by overlap PCR mutagenesis as described above, using the primer 5′-TGCGCAAAGCTTCCATGCAACTTTTTCACC-3′ (the HindIII site is underlined), and then fragment 2 (nt 1814 to 1992) was made by a PCR with a forward primer 5′-GCATGGAAGCTTTGCGCAGACCAATTTAT-3′ (the HindIII site is underlined) and a downstream reverse primer.
(v) Plasmid R809.
For the generation of a variant encoding two unique restriction sites (the PmlI site at nt 1606 of the R806 mutant and the HindIII site at nt 1808 of the R808 mutant), a fragment containing the HindIII site of the R808 mutant was transferred to R806. Briefly, an insert fragment was made by a PCR using R808 as a template and using a forward primer (5′-CCTCTGCACGTGGCATGGAGACCACCG-3′ [the PmlI site is underlined]) and a reverse primer (5′-TAGAATAGGGCCCTCTAGAA-3′ [the ApaI site is underlined]). The resulting 386-bp fragment (nt 1606 to 1992) was digested with PmlI and ApaI and inserted into the PmlI (nt 1606) and ApaI (nt 1992) sites of R806.
(vi) Plasmid R810.
To invert the fragment encoding the β region, we designed two PCR primers to contain the PmlI site at nt 1808 and the HindIII site at nt 1606. First, an insert fragment was made by a PCR using a forward primer (5′-CCCAAGCTTGGCATGGAGACCACCGTG-3′ [the HindIII site at nt 1606 is underlined]), a reverse primer (5′-CCTCTGCACGTGTTGCGCAGACCAATTTAT-3′ [the PmlI site at nt 1808 is underlined]), and a wild-type template. The resulting fragment (nt 1606 to 1808) was digested with HindIII and PmlI and inserted into the PmlI (nt 1606) and HindIII (nt 1808) sites of R809. The resulting R810 plasmid contains the β region in reverse orientation.
lacZ substitution constructs. (i) Plasmid R825 (pCMV-β/lacZ).
For the creation of plasmid R825, an insert fragment was first made by a PCR using a forward primer (5′-CCTCTGCACGTGATTGAAGCAGAAGCCTGC-3′ [the PmlI site is underlined]), a reverse primer (5′-CCCAAGCTTCAGCGCACGGCGTTAAAG-3′ [the HindIII site is underlined]), and pCH110 (carries lacZ; Pharmacia) as a template. The resulting 199-bp fragment was digested with PmlI and HindIII and then inserted into the PmlI (nt 1606) and HindIII (nt 1808) sites of R809.
(ii) Plasmid R826 (pCMV-β/lacZ/SLIII).
For the creation of plasmid R826, an insert fragment was first made by a PCR using a forward primer (5′-CCGGATATCTTGTACTAGGAGGCTGTA-3′ [the EcoRV site is underlined]), a reverse primer (5′-TAGAATAGGGCCCTCTAGAA-3′ [the ApaI site is underlined]), and a wild-type template. The resulting 222-bp fragment (nt 1770 to 1992) was digested with EcoRV and ApaI and inserted into the EcoRV (nt 1770) and ApaI (nt 1992) sites of the R825 plasmid.
Isolation of viral DNAs.
Viral DNAs from cytoplasmic capsids were isolated from Huh7 cells 3 days after transfection as described by Nassal (17).
Southern blot analysis.
Viral DNAs extracted from the cytoplasmic capsids of transfected Huh7 cells were analyzed by Southern blotting to measure the viral replication intermediates. Typically, one-third of the viral DNA isolated from a single 100-mm-diameter plate was loaded in each lane. The viral DNA was electrophoresed through a 1.3% agarose gel at 50 V for 2 h in 0.5× Tris-acetate-EDTA buffer. The DNAs were then transferred to a nylon membrane (Hybond-XL; Amersham) in 20× SSC buffer (3.0 M NaCl, 0.3 M sodium acetate [pH 7.0]). The membrane was dried and exposed to 250 mJ of 254-nm-wavelength UV light per cm2. The membrane was prehybridized and hybridized with a 32P-labeled full-length HBV DNA probe in a hybridization solution (1% bovine serum albumin, 1 mM EDTA, 0.2 M sodium phosphate [pH 7.2], 7% sodium dodecyl sulfate, and 50 mg of sheared salmon sperm DNA per ml) for 12 h at 65°C. The membrane was then washed with small volume of 2× SSC containing 0.1% sodium dodecyl sulfate for 4 h at 65°C. After being dried, the membrane was autoradiographed on film (Hyperfilm-MP; Amersham) at −70°C. Phosphorimages were quantified by use of a bio-imaging analyzer (BAS-2500; Fujifilm).
Primer extension analysis.
Primer extension analysis was performed with viral DNAs extracted from cytoplasmic core particles of transfected Huh7 cells. End-labeled oligonucleotides M2 (nt 1548 to 1570) and M3 (nt 614 to 633) were used in these reactions. Briefly, a primer extension with the M2 primer was used to determine the 5′ end of minus-strand DNAs from various deletion mutants and to measure the levels of minus-strand DNAs initiated at DR1* (15). The M3 primer was used to determine the 5′ end of minus-strand DNAs of the β-region mutants, such as mutant R035. Primer extension reaction mixtures contained 1× ThermoPol reaction buffer [20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100], a 0.2 mM concentration of each deoxynucleoside triphosphate, 1 U of Vent (Exo−) DNA polymerase (New England Biolabs), 0.6 pmol of 5′-end-labeled oligonucleotide, and approximately 300 pg of viral DNA. The reaction mixtures were incubated in a thermocycler (GeneAmp PCR system 2400; Perkin-Elmer). The thermocycling parameters used for extension with the M2 primer were 20 cycles of 95°C for 40 s, 56°C for 40 s, and 72°C for 40 s. The annealing temperature for the M3 primer was 52°C (the thermocycling parameters were otherwise identical to those employed with the M2 primer). As an internal standard for the analysis with the M2 primer, approximately 1 ng of the wild-type construct digested with AatII (nt 1419) and BglII (nt 1986) was added to the reaction mixtures. Samples were denatured at 95°C for 5 min prior to electrophoresis through a 6% polyacrylamide-8 M urea sequencing gel. Gels were dried and autoradiographed on Hyperfilm-MP (Amersham) at −70°C.
RPA.
RNAs from whole cells and cytoplasmic capsids were extracted from the cells 3 days after transfection as described previously (10). RNase protection assays (RPAs) were performed essentially as previously described (10). Briefly, samples of total RNA (30 μg) or core-associated RNA were hybridized with 105 cpm of a [α-32P]UTP (3,000 Ci/mmol; Amersham)-labeled probe (see below) for 16 h at 42°C. RNase digestion was carried out with a mixture of RNase A and RNase T1 (Ambion) at 37°C for 30 min. The digested products were separated in 6% acrylamide-8 M urea gels. The gels were dried and autoradiographed on Hyperfilm-MP (Amersham) at −70°C. Phosphorimages were quantified by use of a bio-imaging analyzer (BAS-2500; Fujifilm).
The RNA probe used to detect HBV-specific RNAs was generated by the in vitro transcription of plasmid R267, which encodes the sequence from nt 1038 to 1238. Plasmid R267 was constructed by an insertion of a PCR fragment (nt 1038 to 1238) produced with a forward primer (5′-CCGGAATTCTGTGGTTATCCTGCGTTGA-3′ [the EcoRI site is underlined]), a reverse primer (5′-TTGGTCTGCGCACGCGTTCCATCCTTTCTT-3′), and a wild-type template. The resulting 200-bp fragment was digested with EcoRI and SphI and inserted into the EcoRI (nt 3014) and SphI (nt 3056) sites of pcDNA1/amp (Invitrogen). The RNA probe used to detect lacZ-specific RNAs was generated by the in vitro transcription of plasmid R282, which encodes the sequence from nt 401 to 550 of pCH110 (Pharmacia). Plasmid R282 was constructed by insertion of the PCR fragment (nt 401 to 550) created with a forward primer (5′-GCTCTAGACCGATCGCCCTTCCCAAC-3′ [the XbaI site is underlined]), a reverse primer (5′-ATAGGGCCCCGTGCATCTGCCAGTTTG-3′ [the ApaI site is underlined]), and pCH110 plasmid as a template. The resulting 150-bp fragment was digested with XbaI and ApaI and inserted into the XbaI (nt 984) and ApaI (nt 994) sites of pcDNA3/amp.
RESULTS
Experimental strategy.
It has been suggested that sequences near the acceptor site may facilitate minus-strand transfer during hepadnaviral reverse transcription (15). To explore this notion and to investigate the mechanism of minus-strand transfer, we decided to delineate cis-acting elements that are required for efficient minus-strand DNA synthesis. To this end, we generated two deletion mutants near the acceptor site, DR1*, namely mutant R035, lacking 198 nt between DR2 and DR1*, and mutant R029, lacking sequences from nt 1805 to 1884, including DR1* (Fig. 2A). Experiments were carried out by transfecting the human hepatoma cell line Huh7 with a cloned plasmid DNA of HBV that is competent to support viral DNA synthesis (10). To determine whether the sequences deleted from the mutants are essential for HBV genome replication, we transfected each mutant into Huh7 cells, along with pR063, a helper plasmid that provides C (core) and P (polymerase) proteins in trans (Fig. 2A). As expected, analysis of the viral DNA extracted from cytoplasmic core particles after transfection of a wild-type HBV construct revealed three major replication intermediates: RC DNA, DL DNA, and single-stranded DNA (ssDNA) (Fig. 2B, lane 1).
FIG. 2.


A mutant with a deletion of the sequence between DR2 and DR1* is defective in minus-strand DNA synthesis. (A) Map of deletion mutants. The structure of pgRNA is shown at the top. E, EcoRI restriction site (nt 3182 to 3180). The nucleotide sequences that were deleted are depicted by shaded lines along with the deleted nucleotide numbers. A map of the R063 construct, a helper plasmid lacking 5′ ɛ, the encapsidation signal, is shown. (B) Southern blot analysis of the replication-intermediate DNAs extracted from cytoplasmic core particles. Huh7 cells were transfected with each deletion mutant or the wild type (WT), along with a helper plasmid (R063) providing the C and P proteins, and the viral replication intermediates were extracted as described in Materials and Methods. Two restriction fragments, of 3.3 and 2.0 kb, were employed as size markers (SM). The positions of RC DNA, DL DNA, and ssDNA, in order from the top, are indicated by open arrowheads. The fast migrating DL DNA found in the deletion mutants is indicated by closed arrowheads. Only one-half of the DNAs extracted from the wild type were loaded relative to those of the mutants. (C) RPAs were performed to measure the encapsidation efficiency of the deletion mutants. Huh7 cells were cotransfected with each deletion mutant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper and the pCMV-lacZ/30 plasmid as an internal control (10). The protected RNA fragments derived from HBV RNA and the lacZ transcript are indicated by closed and open left-facing arrowheads, respectively. The probes used for the analysis of HBV RNA and the lacZ RNA are indicated by closed and open right-facing arrowheads, respectively. Only one-third of the RNAs extracted from total cells (T) were loaded relative to those of capsids (C). (D) Viral DNA synthesis relative to the wild type after normalization to the amount of encapsidated RNA. Error bars represent the standard deviations from six independent transfections.
Identification of a novel cis-acting element essential for hepadnaviral minus-strand DNA synthesis.
Southern blot analyses of the two deletion mutants described above allowed the identification of a novel cis-acting element that is essential for the efficient synthesis of minus-strand DNA (Fig. 2B; Table 2). First, the R035 mutant, lacking the sequences between DR2 and DR1* (nt 1607 to 1804), displayed a significantly reduced accumulation of DL DNA and a lack of RC DNA, suggesting that the region deleted from the R035 mutant is essential for the efficient synthesis of viral DNAs (Fig. 2B, lane 2). In addition, another DNA species with a higher level of mobility than that of DL DNA was faintly detected. Cleavage by BstEII, but not by SphI, suggested that this fast migrating DNA species was double-stranded DNA representing only a part of the viral genome (i.e., truncated DL DNA) (data not shown; refer to Fig. 3D for the relevant restriction sites). This interpretation was supported by the subsequent characterization of this DNA species by primer extension analysis (see Fig. 3C). Likewise, transfection of the R029 mutant lacking DR1* led to significantly reduced synthesis of the viral replication intermediates (Fig. 2B, lane 3). This finding was consistent with a previous observation for a DHBV mutant lacking DR1* (1). Significantly, a faster migrating DNA species was also detected with the R029 mutant (Fig. 2B, lane 3).
TABLE 2.
Quantitative analysis of viral replication intermediates measured by Southern blottinga
| Virus | % (mean ± SD) of replication intermediate
|
Total | ||
|---|---|---|---|---|
| RC DNA | DL DNAb | SS DNA | ||
| Wild type | 19.67 ± 2.87 | 52.61 ± 1.75 | 27.72 ± 4.35 | 100 |
| R035 | 0.90 ± 0.29 | 6.67 ± 0.15 | 5.10 ± 0.53 | 12.67 |
| R029 | 0.60 ± 0.16 | 2.49 ± 1.17 | 4.83 ± 0.36 | 7.92 |
Data for six independent experiments are shown.
The amount of fast migrating DL DNA was included.
FIG. 3.

A mutant lacking the β region synthesized a significantly reduced amount of minus-strand DNA initiated at DR1*. (A) Strategies for primer extension analysis to determine the 5′ ends of minus-strand DNAs produced by mutants lacking the β region. M2 and M3 primers with nucleotide numbers are depicted on the minus-strand DNA. The oval circle represents the P protein covalently linked to the 5′ end of the minus-strand DNA. (B) Primer extension analysis with the M2 primer. A primer extension analysis was performed to determine the 5′ termini of minus-strand DNAs produced by the deletion mutants. A sequencing ladder generated with the M2 primer flanks the extension reactions. DR1* (wild type) is denoted by a closed arrowhead. The position of the predicted DR1* site for the R035 mutant is denoted by an open arrowhead. Huh7 cells were cotransfected with each deletion mutant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Equal amounts of wild-type and mutant samples were analyzed. (C) Primer extension analysis with the M3 primer. A sequencing ladder generated with the M3 primer flanks the extension reactions. The extension product derived from DNAs initiated at the UUC at nt 813 is denoted by an arrowhead. The extension product labeled DR1* (wild type) was not directly determined by the sequencing ladder but by comparison with the data shown in Fig. 2B. Equal amounts of wild-type and mutant samples were analyzed. (D) Map of pgRNA showing UUC motifs. Vertical bars below the line depict the positions of numerous UUC motifs in the pgRNA. Arrows denote the acceptor sites at DR1* and the UUC at nt 813, respectively. Three restriction sites in the DNA genome are shown: B, BstEII; E, EcoRI; S, SphI.
To rule out the possibility that some of these mutants were defective in pgRNA processing or subsequent encapsidation, we performed RPAs as described in Materials and Methods. As shown in Fig. 2C, the pgRNAs derived from these deletion mutants were as efficiently encapsidated as the wild-type pgRNAs, as indicated by the ratios of core-associated RNAs to total cellular RNAs. A lacZ transcript derived from a previously described cotransfected pCMV-lacZ/30 construct (10) was employed as an internal control to estimate the variability between samples. The RPA results clearly excluded the possibility that the reduced synthesis of the viral replication intermediates by these deletion mutants was caused by a defect in RNA processing or encapsidation (Fig. 2C). A quantitative analysis of the reduced DNA synthesis by these deletion mutants corroborated this conclusion (Fig. 2D; Table 2).
To determine the 5′ ends of the minus-strand DNAs, we performed primer extension analyses with primers M2 and M3 according to the strategy depicted in Fig. 3A. The analysis with the M2 primer enabled us to determine the 5′ end of the DNA positioned at DR1*. As shown in Fig. 3B, the minus-strand DNA initiated at DR1* was detected in the wild type, as anticipated. In contrast, in the case of the R035 mutant, the extended product was barely detectable at the predicted DR1* position, as denoted by an open triangle (Fig. 3B, lane 2); this faint band was more apparent upon a longer exposure time (data not shown). Likewise, in the R029 mutant lacking DR1*, no extended product was detected with the M2 primer (Fig. 3B, lane 3). In other words, in these two deletion mutants, DNAs initiated at other UUC motifs present between DR2 and DR1* were not detected (Fig. 3D), suggesting that aberrant transfer did not occur. These data suggested that the sequence deleted from the R035 mutant is essential not only for efficient minus-strand DNA synthesis but also for the proper selection of the acceptor site during minus-strand DNA synthesis.
A subsequent analysis with the M3 primer allowed us to determine the 5′ ends of the fast migrating DNA species. The fast migrating DNA species shown in Fig. 2B could have been a partial duplex DNA with a single-stranded region around the SphI restriction site, as it was resistant to SphI digestion (data not shown). If this were the case, the partial duplex DNA would likely have had the 5′ end of its minus-strand DNA at DR1*. However, this possibility was excluded by the primer extension analysis described above (Fig. 3B). As shown in Fig. 3C, the analysis with the M3 primer revealed that the UUC motif at nt 813 was used instead as an acceptor for the R035 and R029 mutants (Fig. 3C and D). Therefore, the data are consistent with the interpretation that the fast migrating DNA species of approximately 2.2 kbp found for the two deletion mutants (Fig. 2B) is a partial DL DNA whose minus-strand DNA synthesis initiated at the UUC motif of nt 813. Unexpectedly, this internal acceptor site was used even by the wild type (Fig. 3C, lane 1), indicating that the wild type routinely initiates minus-strand DNA synthesis at the internal UUC site, albeit far less frequently than at DR1*. In retrospect, the fast migrating DNA species that should exist in trace amounts in the wild type appeared to be obscured by the far more abundant DL DNA species (Fig. 2B, lane 1). In addition, it is worth noting that the UUC motif at nt 813 was also detected previously by others as an internal acceptor site when the genome length was increased beyond a threshold level (7). More importantly, in these deletion mutants, aberrant transfer, if any, to UUC motifs other than those at DR1* and nt 813 did not occur, as evidenced by the analysis with the M3 primer (Fig. 3C and D). These data, in conjunction with the observations made with Fig. 2B, led us to conclude that the sequence deleted from the R035 mutant is essential for the efficient synthesis of minus-strand DNA that initiates at DR1*; this novel cis-acting element was termed the β region. Moreover, the observation that the elimination of either the β region or DR1* resulted in an indistinguishable phenotype led us to speculate that the β region might act together with the acceptor site (i.e., DR1*) to facilitate minus-strand DNA synthesis during viral reverse transcription.
The β region fails to exert its function in a reverse orientation.
To further investigate the relevant features of the β region in viral DNA synthesis, we generated a mutant containing the β region in a reverse orientation. To facilitate the construction of the mutant, we introduced two unique restriction sites in sequential order at both ends of this region to form R806, R808, and R809 mutants (Fig. 4A). Subsequently, the mutant containing the inverted β region (R810) was generated as described in Materials and Methods. The phenotypes of these mutants were examined by a Southern blot analysis after transfection as described above. The results shown in Fig. 4B suggest that the three intermediate constructs, R806, R808, and R809, supported the synthesis of wild-type levels of three viral replication intermediates (Fig. 4B, lanes 3 to 5). In contrast, the R810 mutant failed to accumulate a detectable level of viral DNAs (Fig. 4B, lane 6). The data suggested that a novel element carried in this region exerted its functions in a forward orientation, but not in a reverse orientation. Thus, the phenotype of the mutant containing the inverted β region paralleled that of a mutant lacking the β region (i.e., R035) (Fig. 4B, lane 2 versus lane 6). One interpretation of this result is that a secondary structure generated by this β region might play a role during minus-strand transfer. This interpretation raised the intriguing possibility that the β region mediates the juxtaposition of the primer-linked P protein with the acceptor site to facilitate minus-strand transfer. An alternative possibility, that the pgRNAs derived from the R810 mutant were not encapsidated normally into capsid particles, was excluded by an RPA, as shown in Fig. 4C.
FIG. 4.

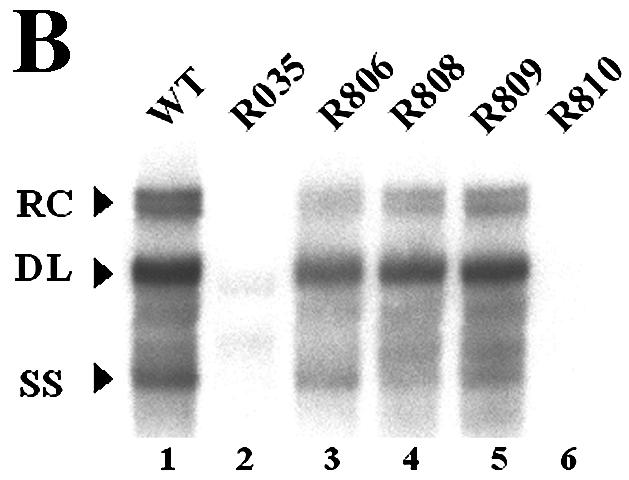

The β region failed to exert its function in reverse orientation. (A) Map of the mutants. Two unique restriction sites, for PmlI (P) and HindIII (H), were introduced to facilitate the inversion of the β region. The R806, R808, and R809 mutants, with either one or both novel restriction sites, were intermediate constructs used to construct the R810 mutant, in which the β region was inserted in the reverse orientation, as indicated by a left-facing arrow. (B) Southern blot analysis performed as described in the legend for Fig. 2B. Huh7 cells were cotransfected with each deletion mutant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Only one-half of the DNAs extracted from the wild type were loaded relative to those of the mutants. (C) RNase protection analysis was performed as described in the legend for Fig. 2C.
Further deletion analysis to delineate the boundary of the β region.
To determine the boundary of the β region, we made seven smaller deletion mutants (Fig. 5A). As shown in Fig. 5B, for most mutants, transfection resulted in a slightly reduced accumulation of viral DNA compared to the wild type. In contrast, a rather dramatic reduction of viral DNA synthesis was observed in cells transfected with mutants R813 and R817, from which the β5 region was commonly deleted (Fig. 5B, lanes 6 and 8, and E; Table 3). In addition, although the β2 and β3 regions were commonly deleted from both the R814 and R817 mutants, only the R817 mutant, which lacks the β5 region, led to a greatly reduced accumulation of minus-strand DNA (Fig. 5B, compare lanes 7 and 8), suggesting that the β5 region is the most critical region for the efficient synthesis of minus-strand DNA.
FIG. 5.

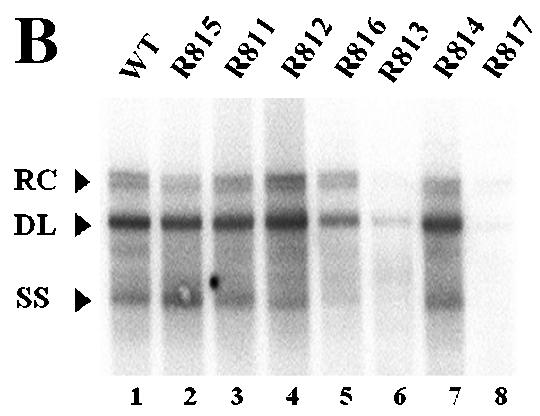



Determination of the boundary of the β region by deletion analysis. (A) Map of seven deletion mutants and pgRNA. The nucleotide sequences deleted from each mutant are depicted by shaded boxes. The mutant names, along with the β-region nucleotide numbers deleted from each construct, are presented to the left. The PRE is indicated by a hatched box, along with two minimal essential elements, HSLα and HSLβ1 (26). (B) Southern blot analysis performed as described in the legend for Fig. 2B. Huh7 cells were cotransfected with each deletion mutant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Only one-half of the DNAs extracted from the wild type were loaded relative to those of the mutants. (C) The β5 region is the most critical sequence for its function. A primer extension analysis was performed with the M2 primer to determine the 5′ termini of minus-strand DNAs synthesized by each mutant. A sequencing ladder generated with the M2 primer flanks the extension reactions. The acceptor site at DR1* utilized by the wild type is indicated. Faster migrating extension products detected for each deletion mutant correspond to the minus-strand DNA initiated at DR1* of each mutant. Huh7 cells were cotransfected with each deletion mutant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Equal amounts of wild-type and mutant samples were analyzed. (D) None of the β-region deletion mutants were defective in RNA processing and encapsidation. An RPA analysis was performed as described in the legend for Fig. 2C to examine the efficiency of RNA processing and encapsidation. (E) Quantitative analysis of the total DNA synthesized normalized to the amount of encapsidated RNA for each mutant. Error bars represent standard deviations from four independent experiments.
TABLE 3.
Quantitative analysis of viral replication intermediates measured by Southern blottinga
| Virus | % (mean ± SD) of replication intermediate
|
Total | ||
|---|---|---|---|---|
| RC DNA | DL DNAb | SS DNA | ||
| Wild type | 18.10 ± 0.21 | 49.94 ± 0.66 | 31.96 ± 0.98 | 100 |
| R815 | 4.80 ± 0.49 | 23.71 ± 1.97 | 23.16 ± 1.59 | 51.67 |
| R811 | 8.10 ± 2.49 | 31.10 ± 5.14 | 18.41 ± 2.76 | 57.61 |
| R812 | 8.93 ± 3.38 | 34.17 ± 7.35 | 13.49 ± 1.36 | 56.69 |
| R816 | 9.04 ± 1.23 | 22.32 ± 6.25 | 14.31 ± 4.59 | 45.67 |
| R813 | 0.36 ± 0.18 | 3.75 ± 0.95 | 4.73 ± 0.60 | 8.84 |
| R814 | 8.56 ± 1.48 | 37.78 ± 2.57 | 23.30 ± 2.30 | 69.64 |
| R817 | 1.19 ± 0.49 | 7.14 ± 4.3 | 5.03 ± 2.38 | 13.36 |
Data for four independent experiments are shown.
The amount of fast migrating DL DNA was included.
A primer extension analysis was performed next to measure the level of minus-strand DNA initiated at DR1* relative to that of the wild type. The analysis was carried out with the M2 primer, as depicted in Fig. 3A. As shown in Fig. 5C, the sizes of the extension products derived from these mutants varied, but each of them was predictably smaller than those of the wild type according to the size of each deletion, consistent with the idea that the 5′ end of the minus-strand DNA synthesized by each of the seven deletion mutants corresponds to the DR1* site. More importantly, the minus-strand DNA initiated at DR1* was detected at a level comparable to that in the wild type for most of the β-region deletion mutants, whereas far less minus-strand DNA initiated at DR1* was detected in the R813 (Fig. 5C, lane 6) and R817 mutants (data not shown). These data further confirmed that the β5 region deleted from these two mutants represents the most critical element for the efficient synthesis of minus-strand DNA. Intriguingly, despite the deletions (27 to 67 nt), none of these mutants used nearby UUC motifs that would have been detected in this analysis (for instance, UUCA at nt 1712 or UUCA at nt 1595 within DR2) as acceptors for minus-strand DNA synthesis (Fig. 3D). The observation that only the UUC motif at DR1* was selected as an acceptor is consistent with the results of a previous study which showed that minus-strand transfer in DHBV prefers a specific location (15).
The β-region mutants are not defective in RNA processing.
Alternatively, though unlikely, it was formally possible that some of these mutants were defective in the pgRNA processing or subsequent encapsidation step. In this regard, it is worth noting that the posttranscriptional regulatory element (PRE) was not impaired in any of these deletion mutants (3, 9); specifically, neither of the two minimal elements (i.e., HSLα and HSLβ1) essential for full PRE function was disrupted (26) (Fig. 5A). Nonetheless, to rule out this alternative possibility, we performed RPAs. As shown in Fig. 5D, the encapsidation efficiencies of pgRNAs from these mutants were similar to that of the wild type. The data clearly demonstrated that none of these deletion mutants was defective in RNA processing or encapsidation, corroborating the hypothesis that they were defective in minus-strand DNA synthesis.
Ectopic insertion of the β5 region restores minus-strand transfer.
We next sought to determine whether the β5 region could function in the absence of surrounding β-region sequences. To this end, we first constructed a variant in which a heterologous sequence of the same size (i.e., the lacZ sequence) substituted for the β region (Fig. 6A, R825). Subsequently, the sequence encoding the β5 region was inserted back into its own position (Fig. 6A, R826). To address whether the ectopically inserted β5 region in a heterologous context could support minus-strand DNA synthesis, we transfected each variant and analyzed the viral DNAs as described above. As shown in Fig. 6B, transfection of the R825 variant, which lacked the β5 region, led to a significantly reduced accumulation of viral DNAs. In contrast, the insertion of the β5 region did indeed significantly restore minus-strand DNA synthesis (Fig. 6B). In addition, primer extension analysis with the M2 primer indicated that the insertion of the β5 region restored the minus-strand DNA initiated at DR1* to a level comparable to that of the wild type and confirmed that the acceptor site at DR1* was primarily used for the synthesis of minus-strand DNA by the R826 variant (Fig. 6C). An RPA excluded the possibility that the pgRNA transcribed from the R825 variant was defective in encapsidation (Fig. 6D). These results clearly indicated that the β5 region exerted its function in the absence of the rest of the β-region sequences.
FIG. 6.

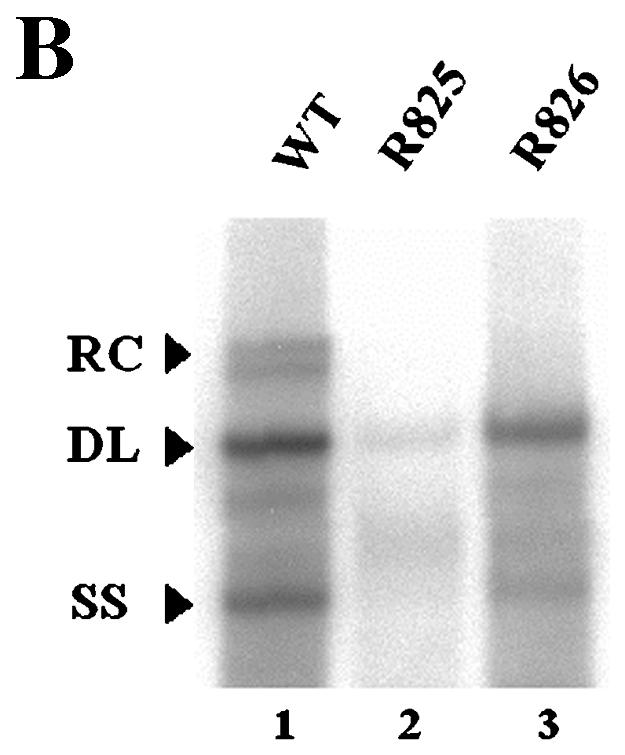
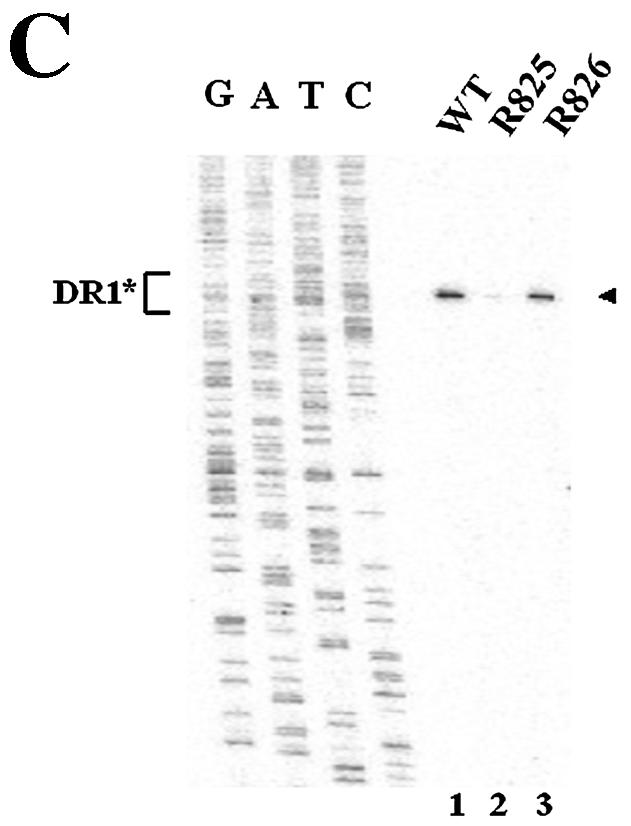

Ectopic insertion of the β5 region is sufficient to restore the initiation of minus-strand DNA synthesis at DR1*. (A) Map of two variants with the heterologous lacZ sequence in place of the β region. In R825, the heterologous lacZ sequence substituted for the β region. The nucleotide sequence encoding the β5 region was inserted back into its own position in R826. (B) Southern blot analysis performed as described in the legend for Fig. 2B. Huh7 cells were cotransfected with each variant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Only one-half of the DNAs extracted from the wild type were loaded relative to those of the variants. (C) Primer extension analysis with the M2 primer to determine the 5′ termini of minus-strand DNAs produced by the variants. A sequencing ladder generated with the M2 primer flanks the extension reactions. The acceptor site at DR1* utilized by the wild type and R826 is indicated by an arrowhead. Huh7 cells were cotransfected with each variant or the wild type, as indicated above each lane, along with the R063 plasmid as a helper. Equal amounts of wild-type and variant samples were analyzed. (D) An RPA analysis was performed as described in the legend for Fig. 2C to examine the efficiency of RNA processing and encapsidation.
DISCUSSION
In this study, we identified a novel cis-acting element, termed the β region, that is essential not only for the efficient synthesis of minus-strand DNA but also for the proper selection of the acceptor site for minus-strand transfer during hepadnaviral reverse transcription. Moreover, our data indicated that the β5 region within the β region, which is 28 nt in length, is the most critical sequence for its function. We speculate that the β5 region, positioned just 20 nt upstream of DR1*, directs efficient minus-strand transfer from the donor site at the 5′ end to the acceptor site near the 3′ end of the pgRNA. The necessity of a cis-acting element besides the donor and acceptor sites underscores the complexity of the mechanisms involved in minus-strand transfer in hepadnaviral reverse transcription.
How does the β5 region work during minus-strand DNA synthesis? We reasoned that the reduced synthesis of minus-strand DNA initiated at DR1* could be ascribed to one of the following: (i) a defect in protein priming at 5′ ɛ, (ii) a defect in minus-strand transfer to the acceptor, and (iii) a failure in elongation from DR1* after the transfer. We favor the second possibility for the following reason, although the other two possibilities are not formally excluded. In fact, sequence complementarity between the donor and acceptor sites is crucial for template switching in both retroviruses and hepadnaviruses (2, 19, 29). In particular, in retroviruses, a minimum homology length of 12 nt is required for accurate and efficient minus-strand transfer (2). In contrast, in hepadnaviruses, the extent of homology between the donor and the acceptor is limited to 4 nt, which is presumably insufficient to provide the binding energy (thermodynamically) that is necessary for accurate and efficient minus-strand transfer. In this regard, it seems reasonable that a cis-acting element besides the donor and acceptor sites contributes to minus-strand transfer. Therefore, we speculate that the β5 region identified in this report provides the binding energy necessary to facilitate template switching.
Importantly, we observed that the phenotype of the β5 region deletion mutants (e.g., R035) was indistinguishable from that of the acceptor site deletion mutant (i.e., R029) (Fig. 2 and 3). This finding led us to speculate that these two elements might work, temporally or physically, in a concerted manner. If this is true, then how does the β5 region exert its function to facilitate minus-strand transfer? It could be suggested that the 5′ and 3′ termini of the pgRNA are juxtaposed within the capsid during minus-strand transfer, with the β5 region mediating such a juxtaposition. Intriguingly, an RNA structure analysis of the β5 region by use of the Zuker program (Institute for Biomedical Computing, Washington University, St. Louis, Mo. [http//www.bioinfo.rpi.edu/applications/mfold/old/rna/form1.cgi]) revealed a small stem-loop structure (M.-K. Shin and W.-S. Ryu, unpublished observation). On the other hand, Tang and McLachlan recently reported the identification of a cis-acting element, termed φ, that is essential for minus-strand DNA synthesis (28). It turned out that the φ sequence is a subset of the β5 region reported here (Fig. 5A). Tang and MaLachlan proposed a novel closed-loop structure of pgRNA mediated by significant base pairing between 5′ ɛ and the φ sequence (28). Importantly, the proposed structure that brings the donor and the acceptor into proximity appears to be conserved among hepadnaviruses (28). Thus, it is of considerable interest to determine experimentally whether such a global closed-loop structure of the β5 region is relevant for its function. Alternatively, but not mutually exclusively, it is possible that the β5 region is recognized by a viral or cellular protein that facilitates minus-strand transfer.
Template switching is the hallmark of the reverse transcription reaction catalyzed by the viral reverse transcriptases. In retroviruses, homology between two repeat (R) regions mediates minus-strand transfer during reverse transcription (30). The R regions of different retroviruses vary greatly in size, from 15 nt in mouse mammary tumor virus to 247 nt in human T-cell leukemia virus type 2. On the other hand, in hepadnaviruses, it is conceivable that due to the limited sequence homology (4 nt), sequences besides the donor and acceptor sites might well contribute to minus-strand transfer. Despite mechanistic similarities between retroviral and hepadnaviral genome replication, significant differences regarding the sequence requirements for minus-strand transfer are evident.
An important question that remains to be addressed regarding hepadnaviral reverse transcription is how the viral reverse transcriptase recognizes these multiple cis-acting elements (i.e., the donor, acceptor, and β5 region) to facilitate minus-strand transfer and whether host factors are involved in this process. The identification of the cis-acting element as a mediator of hepadnaviral minus-strand transfer provides important clues toward a complete molecular description of the template switching event, which is a pivotal process shared among retrovirus-like elements.
Acknowledgments
This work was supported by a National Research Laboratory Grant from the Ministry of Science and Technology of the Korean Government.
REFERENCES
- 1.Condreay, L. D., T. T. Wu, C. E. Aldrich, M. A. Delaney, J. Summers, C. Seeger, and W. S. Mason. 1992. Replication of DHBV genomes with mutations at the sites of initiation of minus- and plus-strand DNA synthesis. Virology 188:208-216. [DOI] [PubMed] [Google Scholar]
- 2.Dang, Q., and W. S. Hu. 2001. Effects of homology length in the repeat region on minus-strand DNA transfer and retroviral replication. J. Virol. 75:809-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donello, J. E., A. A. Beeche, G. J. Smith, III, G. R. Lucero, and T. J. Hope. 1996. The hepatitis B virus posttranscriptional regulatory element is composed of two subelements. J. Virol. 70:4345-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galibert, F., E. Mandart, F. Fitoussi, P. Tiollais, and P. Charnay. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646-650. [DOI] [PubMed] [Google Scholar]
- 5.Ganem, D., and R. Schneider. 2001. Hepadnaviridae: the viruses and their replication. Lippincott-Raven Publishers, Philadelphia, Pa.
- 6.Hirsch, R. C., D. D. Loeb, J. R. Pollack, and D. Ganem. 1991. cis-Acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 65:3309-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho, T. C., K. S. Jeng, C. P. Hu, and C. Chang. 2000. Effects of genomic length on translocation of hepatitis B virus polymerase-linked oligomer. J. Virol. 74:9010-9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horton, R. M., S. N. Ho, J. K. Pullen, H. D. Hunt, Z. Cai, and L. R. Pease. 1993. Gene splicing by overlap extension. Methods Enzymol. 217:270-279. [DOI] [PubMed] [Google Scholar]
- 9.Huang, Z. M., and T. S. Yen. 1995. Role of the hepatitis B virus posttranscriptional regulatory element in export of intronless transcripts. Mol. Cell. Biol. 15:3864-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong, J. K., G. S. Yoon, and W. S. Ryu. 2000. Evidence that the 5′-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J. Virol. 74:5502-5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junker-Niepmann, M., R. Bartenschlager, and H. Schaller. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lien, J. M., C. E. Aldrich, and W. S. Mason. 1986. Evidence that a capped oligoribonucleotide is the primer for duck hepatitis B virus plus-strand DNA synthesis. J. Virol. 57:229-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeb, D. D., K. J. Gulya, and R. Tian. 1997. Sequence identity of the terminal redundancies on the minus-strand DNA template is necessary but not sufficient for the template switch during hepadnavirus plus-strand DNA synthesis. J. Virol. 71:152-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loeb, D. D., R. C. Hirsch, and D. Ganem. 1991. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: implications for other reverse transcribing elements. EMBO J. 10:3533-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeb, D. D., and R. Tian. 1995. Transfer of the minus strand of DNA during hepadnavirus replication is not invariable but prefers a specific location. J. Virol. 69:6886-6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 17.Nassal, M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66:4107-4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nassal, M., M. Junker-Niepmann, and H. Schaller. 1990. Translational inactivation of RNA function: discrimination against a subset of genomic transcripts during HBV nucleocapsid assembly. Cell 63:1357-1363. [DOI] [PubMed] [Google Scholar]
- 19.Nassal, M., and A. Rieger. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nassal, M., and H. Schaller. 1996. Hepatitis B virus replication—an update. J. Vir. Hepat. 3:217-226. [DOI] [PubMed] [Google Scholar]
- 21.Pollack, J. R., and D. Ganem. 1993. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 67:3254-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollack, J. R., and D. Ganem. 1994. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 68:5579-5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rieger, A., and M. Nassal. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryu, W. S., M. Bayer, and J. Taylor. 1992. Assembly of hepatitis delta virus particles. J. Virol. 66:2310-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seeger, C., and J. Maragos. 1991. Identification of a signal necessary for initiation of reverse transcription of the hepadnavirus genome. J. Virol. 65:5190-5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith, G. J., III, J. E. Donello, R. Luck, G. Steger, and T. J. Hope. 1998. The hepatitis B virus post-transcriptional regulatory element contains two conserved RNA stem-loops which are required for function. Nucleic Acids Res. 26:4818-4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staprans, S., D. D. Loeb, and D. Ganem. 1991. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J. Virol. 65:1255-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang, H., and A. McLachlan. 2002. A pregenomic RNA sequence adjacent to DR1 and complementary to epsilon influences hepatitis B virus replication efficiency. Virology 303:199-210. [DOI] [PubMed] [Google Scholar]
- 29.Tavis, J. E., S. Perri, and D. Ganem. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telesnisky, A., and S. P. Goff. 1997. Reverse transcriptase and the generation of retroviral DNA, p. 121-160. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Press, Cold Spring Harbor, N.Y. [PubMed]
- 31.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 32.Wang, G. H., and C. Seeger. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]