

Abstract
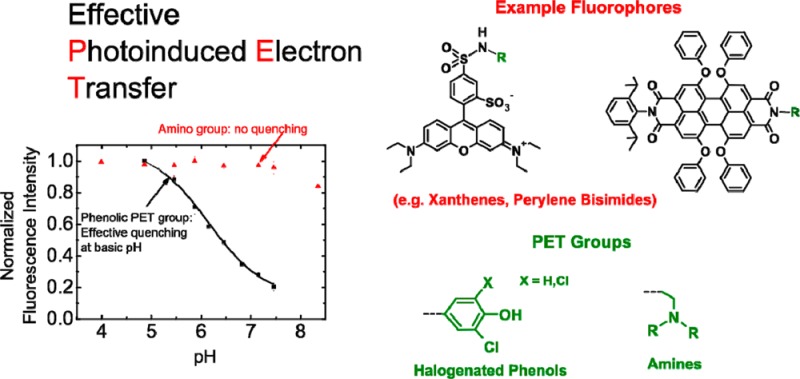
Photoinduced electron transfer (PET), which causes pH-dependent quenching of fluorescent dyes, is more effectively introduced by phenolic groups than by amino groups which have been much more commonly used so far. That is demonstrated by fluorescence measurements involving several classes of fluorophores. Electrochemical measurements show that PET in several amino-modified dyes is thermodynamically favorable, even though it was not experimentally found, underlining the importance of kinetic aspects to the process. Consequently, the attachment of phenolic groups allows for fast and simple preparation of a wide selection of fluorescent pH-probes with tailor-made spectral properties, sensitive ranges, and individual advantages, so that a large number of applications can be realized. Fluorophores carrying phenolic groups may also be used for sensing analytes other than pH or molecular switching and signaling.
Rational dye design is a key concept in molecular recognition, (super)molecular signaling, and optoelectronics. “Modules” capable of a certain function such as interacting with an analyte1 (pH, cations, carbon dioxide, glucose, or complex biomolecules like proteins), molecular switching,2−4 or charge generation5,6 are attached to or integrated into a chromophore. Photoinduced electron transfer (PET)7−9 is a process that can enable all of these processes. A functional group capable of introducing PET (the “PET group”) undergoes a redox process involving the excited state (but not the ground state) of a chromophore. In fluorescent dyes, this results in fluorescence quenching. The process is fast and reversible. The charge separation implied is thus short-lived but can be taken advantage of in photovoltaics.
In this work, we focus on fluorescent dyes suitable for pH-sensing (“probes”). We also aim to provide insights into the PET process in general which may be helpful for the design of dyes and materials applying PET for other purposes. PET-based fluorescent probes and sensors have been presented mostly for recognizing ions10−14 but also for measuring pH.15−19 Those probes and sensors are of great potential for application in diagnostics, life sciences, environmental analytics, or industrial process control.20 In most PET-based probes, fluorescence quenching is caused by an alkylamino group. Upon protonation of the amine or interaction with a cation, PET can be impeded or prevented, which results in fluorescence enhancement. Consequently, there is pH-response affiliated with the protonation of the amine, thus characterized by the corresponding pKA value, with a usual sensitive pH-range of pKA ± 1.5. Phenoxide groups can act in exactly the same way, as was first demonstrated by Gareis et al. by means of a phenol-modified boron-dipyrromethene (BODIPY) dye.21 So far, only a few examples of pH-probes carrying phenolic PET groups have been presented, most of which are BODIPY dyes.22−27 On the other hand, in many fluorescent pH-probes, phenolic groups are fully integrated into the chromophore, for instance in fluoresceins. Those are not true PET probes since the redox properties of the chromophore itself are strongly pH-dependent, even though PET may play a significant role in some.28,29
Here we present, for the first time, a comparison between fluorescent pH-probes carrying phenolic PET groups and those bearing amines on the basis of various fluorophores. It is demonstrated that halogenated phenoxide groups are generally more effective quenchers than amines, and they open the way to alternative strategies for designing fluorescent probes. Because many phenol derivatives are commercially available, and they can be attached to numerous dyes by simple reactions, probes with tailored properties are available in a convenient way. Their spectral properties, sensitive ranges, and stabilities can be selected to meet the variable and demanding requirements of life sciences and diagnostics.
Results and Discussion
Several dye classes with very different spectral and structural properties (Figures 1 and 2 and Table 1) have been selected for this work, including xanthenes (fluoresceins and sulforhodamines), diketopyrrolo[3,4-c]pyrroles (DPPs), and perylene bisimides (PBIs). To introduce PET, both halogenated (o-chloro and o,o-dichloro) phenols and tertiary amines have been attached. Because PET is affected by (de)protonation in both groups, fluorescence pH-calibration has been selected as the primary evaluation method of the PET process. Calibration was carried out in mixtures of ethanol and aqueous buffer [1/1 (v/v)], to provide sufficient solubility for all the investigated dyes which include both water-soluble and highly hydrophobic structures. To further suppress any potential aggregation and other effects perturbing the fluorescence measurement (dimerization, self-quenching), calibration was performed in a highly dilute (from 6 × 10–8 to 2 × 10–6 M) solution. For all dyes, effects of buffer pH on maxima and shape of both absorption and fluorescence spectra were absent or negligible (≤3 nm), emphasizing the absence of major pH-dependent effects other than PET. Only data measured in acidic solution is thus stated in Table 1.
Figure 1.

Modules used for the design of fluorescent dyes in this work and mechanism of photoinduced electron transfer (PET).
Figure 2.

Synthesis of the fluorescent dyes. Synthetic conditions: (a) CH3SO3H, 145 °C, 1 h; (b) SO2Cl2, RT, 2 h; (c) EDC, NHS, DMF, RT, 1.5 h; R-H, Et(iPr)2N, 4 h; (d) R-H, Et3N, DMF, 16 h, 0–25 °C; (e) ClSO3H, 60 °C, 3 h; R-H; (f) R-NH2, 2,6-diisopropylaniline, 1-methyl-2-pyrrolidone (“NMP”), C2H5COOH, 110 °C, 22 h; (g) PhOH, K2CO3, NMP, 110 °C, 3.5–22 h; and (h) morpholine, NMP, 40 °C, 2 h.
Table 1. Absorption and Fluorescence Maxima and Individual Advantages of the Presented Dyes in Ethanol:Aqueous Buffer Mixture [1:1 (v/v)]; pH 6.4 for 1B and 1C and pH 4.0 for All Other Dyes; Ionic Strength, 10 mMa.
| dye | fluorophore | PET group | λmax abs (ε 10–4)/nm (M–1 cm–1) | λmax fluo/nm | individual advantages |
|---|---|---|---|---|---|
| 1B | FLUO | MCP | 513 (8.58) | 534 | two sensitive ranges |
| 1C | FLUO | amine | 514 (9.07) | 535 | very bright |
| 2A | RHOD | DCP | 564 (9.84) | 589 | very bright |
| 2C | RHOD | amine | 567 (9.13) | 590 | very bright |
| 3A | DPP | DCP | 502 (1.82), 533 (1.84) | 591 | valuable imaging probes30 |
| 3B | DPP | MCP | 504 (1.79), 533 (1.81) | 589 | valuable imaging probes30 |
| 3C | DPP | amine | 505 (1.66), 535 (1.71) | 594 | |
| 5A | PhOPBI | DCP | 447 (1.44), 541 (2.80), 578 (4.24) | 620 | outstanding photostability |
| 5B | PhOPBI | MCP | 452 (1.21), 543 (3.08), 578 (4.66) | 620 | outstanding photostability |
| 5D | PhOPBI | amine | 451 (1.30), 541 (2.77), 578 (4.19) | 620 | outstanding photostability31 |
| 6A | AmPBI | DCP | 461 (1.60), 662 (1.74) | 756 | near infrared emission |
| 6D | AmPBI | amine | 449 (1.77), 658 (1.85) | 761 | near infrared emission32 |
Dye concentrations are reported in Experimental Section.
Quenching caused by PET at basic pH was significantly stronger for the dyes carrying phenolic groups (dyes 1–6 A,B) than for those bearing amines (dyes 1–6 C,D) (Figure 3 and Table 2). Indeed, quenching was almost complete for DPPs 3A,B and PBIs 5A,B and 6A, while it was partial for 5D and absent for 3C and 6D. For xanthene (FLUO, RHOD) dyes, there was partial quenching by phenolic groups (dyes 1B, 2A), but no quenching by amino groups (dyes 1C, 2C) was found. Note that even though PET is often associated with on–off fluorescence quenching,8 partial quenching by PET is not uncommon for some systems in certain environments.33 For ClPBI dyes (4) which are intermediates in the preparation of PhOPBI (5) we previously found strong quenching by amines.31 For 4A,B, we found fluorescent signals undistinguishable from background both at acidic and basic pH. A possible reason is strong quenching by both the phenolic and phenoxide form. In contrast to all other investigated dyes, the probe 1B shows two dynamic ranges; it shows a strong fluorescence increase from pH 3 to pH 6 and moderate fluorescence quenching at basic pH (Figure 3). This behavior is explained by formation of a colorless lactone in acidic environment and is typical for fluoresceins bearing a carboxyl group in the 2′ position of the phenyl ring.34 The behavior of 1B and 1C in the acidic pH range is virtually identical.
Figure 3.
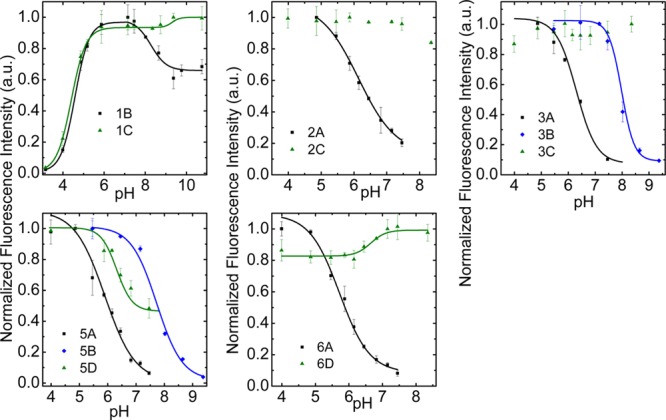
Fluorescence pH calibration curves for the dyes in the ethanol:aqueous buffer mixture [1:1 (v/v), ionic strength of 100 mM] modified with phenolic (1–6 A,B) and amine (1–6 C,D) PET groups. Dye concentrations are stated in Experimental Section.
Table 2. Photophysical and pH-Sensitive Properties of the Dyes in Ethanol: Aqueous Buffer Mixture [1:1 (v/v)], Buffer Concentration: 5 mM, Ionic Strength 50 mM); Acidic Buffer, pH 4.0, Basic Buffer, pH 9.4 (for 1B and 1C, pHs 6.4 and 11.7, respectively)a.
| ΦF (EtOH/H2O 1:1 (V/V)) | ΦF (CH2Cl2) | τ/ns (EtOH/H2O 1:1 (V/V)) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| dye | fluorophore | PET group | acidic | basic | pKA | acidic | basic | acidic | basic |
| 1B | FLUO | MCP | 0.72 | 0.48 | 4.6b; 8.3 | 3.9 | 2.2 | ||
| 1C | FLUO | Amine | 0.79 | 0.85 | 4.4b | 3.9 | 3.9 | ||
| 2A | RHOD | DCP | 0.4 | 0.084 | 6.2 | 2.2 | 2.9; 0.6c | ||
| 2C | RHOD | Amine | 0.37 | 0.35 | -- | 2.0 | 2.2 | ||
| 3A | DPP | DCP | 0.18 | <0.01 | 6.3 | 0.70d | <0.01d· | 5.5 | n.m. |
| 3B | DPP | MCP | 0.26 | <0.01 | 8.0 | 0.66d | <0.01d | 3.4 | n.m. |
| 3C | DPP | Amine | 0.29 | 0.33 | -- | -- | -- | 6.0 | 6.2 |
| 5A | PhOPBI | DCP | 0.16 | <0.01 | 5.9 | 0.99 | 0.025 | 5.4 | n.m. |
| 5B | PhOPBI | MCP | 0.56 | <0.01 | 7.7 | -- | -- | 4.6 | n.m. |
| 5D | PhOPBI | Amine | 0.38 | 0.18 | 6.3 | 0.90 | 0.16 | 5.8 | 2.8 |
| 6A | AmPBI | DCP | 0.052 | <0.01 | 5.8 | 0.17 | 0.038 | 1.1 | n.m. |
| 6D | AmPBI | Amine | 0.043 | 0.051 | (6.7)e | 0.24 | 0.0055 | 1.6 | 1.5 |
In organic solvents, acidic solutions contain trifluoroacetic acid, basic ones ethyldiisopropylamine [0.1% (v/v) each]. Dye concentrations are stated in Experimental Section. ΦF: fluorescence quantum yield; τ: fluorescence decay time; n.m.: not measurable.
Intrinsic pH-sensitivity of the fluorescein derivative, not related to PET.
Biexponential fit was required, with 79% abundance for 2.9 ns.
In tetrahydrofuran, according to30
Slight enhancement of fluorescence at basic pH, not related to PET.
All pH-probes are synthetically easily accessible, FLUO, RHOD, and DPP dyes (1–3) via a single step, PBIs (4–6) via two simple steps. In particular, the xanthene-based probes were prepared from commercially available dyes (Lissamine rhodamine B and 5(6)-carboxy-2′,7′-dichlorofluorescein NHS ester; the latter was synthesized for this work but is also commercially available at a comparatively low cost) under mild conditions (see Figure 2; because the lengthy synthetic section is exclusively based on standard methods, detailed description is included only in the Supporting Information). These and many other dyes, which have been designed to perform bioconjugation, are widely commercially available and easy to handle as “kits”. In this way, pH-probes with selectable spectral properties are easily accessible also to the synthetically inexperienced user. This work clearly emphasizes that phenolic PET groups are better suited for creating a selection of new pH-probes since they can be combined with a significantly larger number of fluorophores to yield PET-based probes. The phenol-based probes presented here cover a wide range (excitable at 400–660 nm, emissive at 500–750 nm, Figure 4) of the visible spectrum. They feature individual advantages so that many applications can be addressed with them. For example, the xanthenes 1B and 2A are very bright and compatible with light sources and filters widely used in fluorescence microscopy. Both feature sensitivity at near-neutral pH, and good photostability is common for rhodamines and dichlorofluoresceins, in contrast to nonchlorinated fluoresceins.351B is an example for a probe featuring two sensitive ranges and can therefore be applied for measuring both near-neutral and acidic pH. Advantages of the other dye classes have been discussed in previous publications, as listed in Table 1.
Figure 4.
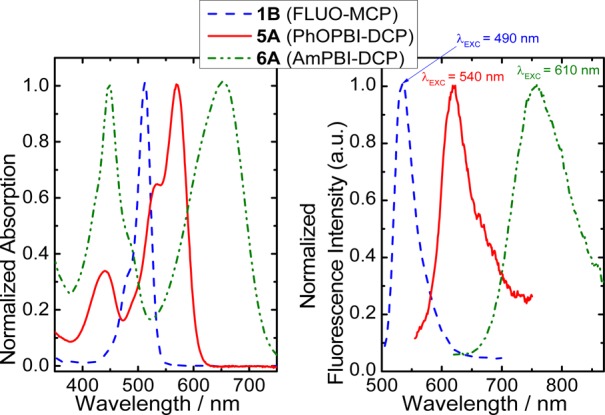
Absorption (left) and emission (right) spectra of the dyes presented in this work covering the entire visible range and additionally including near-infrared emission (AmPBI dyes). Example spectra for RHOD, DPP, and ClPBI dyes (2–4 A–D) are not shown for the sake of clarity.
Since many halogenated phenols with different substitution patterns are commercially available, it is possible to select the pKA value of the resulting pH-probe and thus its sensitive range. For instance, the di- and monochlorinated phenolic groups presented here are useful for slightly acidic and neutral to slightly basic values, respectively. The similar pKA values for fluorophores featuring very different structure and hydrophilicity (Table 2) underline that the sensitive range can be selected beforehand to match the pH range of interest. In fact, the pKa values for the 2,6-dichlorophenol-subsituted dyes (A series) are in the range of 5.8–6.3, whereas the pKa values for the 2-monochlorophenol-substituted dyes (B series) vary from 7.7 to 8.3. These values are slightly lower than for the corresponding free 2,6-dichlorophenol (pKa = 6.8)36 and 2-chlorophenol (pKa = 8.5),37 which can be explained by an electron-withdrawing character of the carboxamnido (1B), sulfonamido (2A, 3A, 3B), and imide (5A, 5B, 6A) groups located in the para position with respect to the hydroxyl group.
Phenolic PET groups can be associated with pH-probes bearing neutral/anionic charge at acidic/basic pH, while amino groups rather come with cationic/neutral charge. This implies different interactions with the environment (e.g., biomolecules present in biological applications), underlining that phenolic groups may provide a reasonable alternative, even in combination with fluorophores which are effectively quenched by amines. It should be considered that the cell-penetration properties of the dyes are significantly affected by their charge and therefore some of the anionic probes may not be suitable for intracellular measurements. However, other strategies can be used to ensure cell-penetration (e.g., by incorporating the dyes inside silica or polymeric nanoparticles).38 For example, efficient passive cell loading was previously reported for cationic oxygen-sensitive nanoparticles39 and applicability of such materials for intracellular pH sensing is currently being investigated in our group.
Probes carrying phenolic PET groups are not limited to pH-sensing but are also potentially useful for measuring carbon dioxide and ions. The sensitivity in most optical carbon dioxide probes is affiliated with the protonation of a phenoxide group40,41 and PET-based indicators (mostly based on amines, so far) have been widely applied for optical sensing of metal cations.10,12,42
Because PET is a quenching process, we expected that fluorophores subject to PET may feature shorter fluorescence decay times in their basic forms than in their acidic forms, which are not affected by PET. While decay times are not expected to be pH-dependent in the case of complete quenching since the decay time of the basic form is not measurable, we anticipated that a shorter decay time should be detectable for dyes undergoing partial quenching. This is in agreement with our experimental findings for 1B and 5D, indicating that PET is a quenching process that diminishes both fluorescence intensity and decay time. For 2A, a biexponential decay curve was found in basic solution and a diminishment in decay time could not be verified, indicating that in this case there may be more than just two species contributing to the decay times. Still, pH-probes undergoing partial quenching by PET are potentially very useful for quantitative pH measurement and imaging as they enable decay time readout and do not require further referencing.
The free enthalpy of the PET process, ΔGQ,PET, is given by Weller’s equation:7
| 1 |
Eox,rec is the first oxidation potential of the PET group (receptor), Ered,flu the first reduction potential of fluorophore, ΔEexc,flu the singlet excitation energy of the fluorophore, and EIP the ion pairing energy. Consequently, PET will be observed if the PET group is easily oxidized and the fluorophore is easily reduced and absorbs at short wavelengths.9,16,43 PET is also more favorable in polar solvents.44 Kinetic aspects can play a significant role, in particular, PET becomes less effective with increasing distance between fluorophore and the PET group and an adverse nature and orientation of molecular orbitals may inhibit the process.45−47 Nevertheless, these thermodynamic considerations generally enable a valuable estimation whether a PET process will take place and are suitable for identifying systems in which PET is impossible.
We therefore measured the redox potentials of fluorophores and PET groups to determine ΔGQ,PET (Table 3, measurement curves shown in the Supporting Information). Model compounds for fluorophore and PET group were used in some cases, as listed in Table 3 and discussed in the following.
Table 3. Redox Properties and PET Driving Forces Using Model Compounds. ΔEexc,flu (Singlet Excitation Energy) Was Calculated from the Fluorescence Spectra as Stated in More Detail in the Text. Redox Potentials of the Model Compounds ΔUox,rec and ΔUred,flu Were Determined from Cyclic Voltammograms (Displayed in the Electronic Supplementary Information, ESI) Measured in Ethanol/Water Containing K2CO3 (5% w/w), except for DPP-MonoSA and 5A,B,D Which Were Measured in Tetrahydrofuran/Water 9:1 (V/V) Containing K2CO3 (0.2% w/w). (i) Denotes Irreversible Reaction.
| dye | ΔEexc,flu (eV) | model fluorophore | ΔUred,flu (V) | model for PET group | ΔUox,rec (V) | ΔGQ,PET (eV) |
|---|---|---|---|---|---|---|
| 1B | 2.37 | 2,7-dichlorofluorescein | –1.16 | 5B | 0.64 (i) | –0.67 |
| 1C | 2.37 | 2,7-dichlorofluorescein | –1.16 | triethylamine | 0.82 | –0.49 |
| 2A | 2.15 | sulforhodamine B | –0.99 | 5A | 0.71 (i) | –0.55 |
| 2C | 2.15 | sulforhodamine B | –0.99 | triethylamine | 0.82 | –0.44 |
| 3A | 2.21 | DPP-MonoSAa | –0.79 (i) | 5A | 0.71 (i) | –0.81 |
| 3B | 2.21 | DPP-MonoSAa | –0.79 (i) | 5B | 0.64 (i) | –0.87 |
| 3C | 2.21 | DPP-MonoSAa | –0.79 (i) | triethylamine | 0.82 | –0.69 |
| 4A | 2.29 | Cl-DAPBIb | –0.21 | 5A | 0.71 (i) | –1.47 |
| 4B | 2.29 | Cl-DAPBIb | –0.21 | 5B | 0.64 (i) | –1.54 |
| 4D | 2.29 | Cl-DAPBIb | –0.21 | triethylamine | 0.82 | –1.36 |
| 5A | 2.07 | 5A | –0.55 | 5A | 0.71 (i) | –0.91 |
| 5B | 2.07 | 5B | –0.55 | 5B | 0.64 (i) | –0.98 |
| 5D | 2.07 | 5D | –0.55 | triethylamine | 0.82 | –0.81 |
| 6A | 1.75 | MOP3Cl-DAPBIc | –0.37 | 5A | 0.71 (i) | –0.77 |
| 6D | 1.75 | MOP3Cl-DAPBIc | –0.37 | triethylamine | 0.82 | –0.66 |
1,4-Diketo-3-({4-[N-(2-ethylhexyl)amino]sulfonyl}phenyl)-6-phenylpyrrolo[3,4-c]pyrrole.
1,6,7,12-Tetrachloro-N,N′-di(2,6-diisopropylphenyl)perylene-3,4:9,10-tertracarboxylic bisimide.
1-(4-Morpholinyl)-6,7,12-trichloro-N,N′-di(2,6-diisopropylphenyl)perylene-3,4:9,10-tertracarboxylic bisimide.
In accordance with the method used by DeSilva and co-workers,16 model compounds available at low cost and with presumably very similar photo- and electrochemical properties were measured for most fluorophores. Singlet excitation energies were calculated as intersection wavelengths between fluorescence excitation and emission spectra as outlined by Ford et al.48 For the ion pairing energy, the value of 0.1 can be assumed.16,49 The redox potentials of the PET groups may be estimated measuring those of amines and chlorophenoxide or p-aminochlorophenoxide ions. However, since in most of the obtained indicators, the PET groups are located closely to electron-withdrawing groups (imide, sulfonamide, and carboxamide), we decided to use 5A,B,D as model compounds. All measurement curves are shown in the Supporting Information. The oxidation potential found for the basic form of 5A (0.708 V) is indeed higher than the one of 2,6-dichlorophenol (0.56 V, irreversible oxidation) and significantly higher than the one of 4-amino-2,6-dichlorophenol (−0.09 V, reversible oxidation which may originate from amine rather than phenoxide oxidation). Using 5A,B, in which the phenoxide groups are substituted by strongly electron-withdrawing imide groups, as model compounds should prevent an overestimation of the thermodynamic driving force for PET since the phenoxide groups in 1–4 A,B and 6A are not expected to feature significantly lower electron densities. In 5D, the oxidation potential cannot be reliably determined due to background oxidation, which complicates readout at >0.8 V. The value for triethylamine (0.82 V) was used instead. The true oxidation potentials of the indicators may be higher due to the presence of β-nitrogen atoms carrying electron-withdrawing substituents. Estimation with β-substituted model compounds (tetramethyldiaminoethane and dimethylaminoethanol), showed oxidation at only slightly lower voltage (0.85 and 0.87 V, respectively, irreversible; the values were obtained considering a blank sample to account for background oxidation, as shown in the Supporting Information). Even in case calculated ΔGQ,PET values are slightly too negative, positive values seem very unrealistic (note that amine oxidation potentials as high as 1.2–1.6 V would be required to make PET thermodynamically unfavorable for all of 1–6 C,D).
Consequently, for all indicators, PET has been estimated to be thermodynamically favorable, even for the amines which experimentally showed no PET (1–3C, 6D). Hence, even though the smaller thermodynamic driving force for PET may play a role in the much lower efficiency of the PET process observed with amines, it does not seem convincing as the sole explanation. Kinetic aspects probably play an additional role, and indeed, we had previously found highly effective PET both for rhodamines50 and 1-aminoperylene bisimides32 carrying amino groups in different positions than those investigated in this work. Thus, we present here another example for how the PET process can be influenced by structural parameters. Effective PET requires certain proximity between the HOMO of the donor and the LUMO of the acceptor, which may be easier to realize with phenoxide systems. Note that amino groups usually require a minimum spacer length of two atoms if attached by the easy-to-use techniques presented in this work. A single-carbon spacer would result in aminal structures which are unstable.
Experimental Section
Materials and Methods
1,6,7,12-Tetrachloroperylene-3,4:9,10-tetracarboxylic bisanhydride was purchased from Beijing Wenhaiyang Industry and Traiding Co. Ltd. (http://china.zhaoteng.com), 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole (Irgazin Scarlet) from Kremer Pigmente (http://kremer-pigmente.de/en). The preparation of Cl-DAPBI and DPP-MonoSA is published elsewhere.31,51 MOP3Cl-DAPBI was prepared according to a procedure described previously.32 1-Methyl-2-pyrrolidone was from ABCR (www.abcr.de), and chlorophenols were from TCI Europe (www.tcichemicals.com). Deuterated solvents were obtained from Eurisotop (www.eurisotop.com), silica gel from Acros (www.fishersci.com), and solvents used for column chromatography from VWR (www.vwr.at). Inorganic salts were supplied by Carl Roth (www.roth.de). All other chemicals were from Sigma-Aldrich (www.sigmaaldrich.com).
Absorption measurements were performed on a Cary 50 UV–vis spectrophotometer from Varian (www.varianinc.com). Dye concentration was 2 μM for 1 and 2; 5 μM for 4 and 5; and 10 μM for 3 and 6. Fluorescence spectra were recorded on a Fluorolog3 spectrofluorimeter (www.horiba.com). Fluorescence decay times were determined using a time-correlated single photon counting technique on a Fluorolog3 spectrometer equipped with a NanoLED 455 nm laser diode. Decay curves were subjected to monoexponential fit using DAS6 supplied by the manufacturer. Dye concentration was 0.05 μM for 1, 2, and 5; 0.3 μM for 3; and 2 μM for 6.
Relative fluorescence quantum yields were determined using rhodamine 101, rhodamine B, and Nile Red as standards (quantum yields 0.96, 0.65,52 and 0.2753 in ethanol, respectively). NMR spectra were recorded on a 300 MHz instrument (Bruker) in CDCl3 with TMS as a standard. MALDI-TOF mass spectra were recorded on a Micromass TofSpec 2E. The spectra were taken in reflectron mode at an accelerating voltage of +20 kV.
The pH of the buffer solutions (phosphate, acetate, and TRIS) was controlled by a digital pH meter (Seven Easy, Mettler Toledo, www.mt.com) calibrated at 25 °C with standard buffers of pH 7.0 and 4.0 (WTW GmbH & Co. KG, www.wtw.com). The buffers were adjusted to constant ionic strength using sodium chloride as the background electrolyte.
Electrochemical measurements were performed using a VMP3 electrochemical workstation (Biologic) and a multinecked, airtight glass cell. The measurements were carried out at room temperature. A 1 mm diameter glassy carbon disk (BAS Inc.) was employed as the working electrodes. Prior to use, the working electrode was polished with a 0.05 μm alumina slurry in water and rinsed with copious amounts of water and ethanol followed by drying. A platinum wire served as the counter electrode. Measurements were performed using a Ag/AgCl reference electrode (BAS Inc.). Electrolytes were saturated with Ar prior to the measurements. E1/2 was determined from the peak potentials for quasi-reversible processes. For irreversible processes, the E1/2 was estimated from the half peak potential Ep/2 via E1/2 = Ep/2 – 1.09 RT/nF.54
Calibration
The following sigmoidal function was used for sensor calibration:
| 2 |
where I is fluorescence intensity and Amax and Amin, and are numerical coefficients.
Syntheses
All dyes presented in this work were obtained by established procedures, as summarized in Figure 2. A detailed description is available in the Supporting Information.
Conclusions
In conclusion, we have demonstrated that employing phenolic groups, pH-probes derived from a wide range of fluorophores can be created. They feature selectable sensitive pH ranges, spectral properties, and individual advantages that can be chosen to meet the requirements of many particular applications. In particular, phenolic groups allow higher flexibility than the much more commonly used amines as they can quench many fluorophores due to photoinduced electron transfer (PET) at basic pH, which are not quenched by amines. Unlike amines, they do not introduce positive charge into the molecule. The probes are accessible via simple reactions similar to those common in bioconjugation chemistry, which makes the concept applicable for a wide research field, without requiring a profound expertise in synthetic organic chemistry. Fluorophores carrying phenolic PET groups are not limited to pH sensing but can be applied for the numerous other application possibilities affiliated with PET, such as ion sensing, molecular switching, and signaling or organic electronics.
Acknowledgments
Financial support from the Austrian Science Fund FWF (project P 21192-N17) and European Commission (7th Framework Programme, Project SenseOcean, N 614141) is gratefully acknowledged.
Supporting Information Available
Detailed synthetic procedures, electrochemical measurement curves (cyclic voltammograms), NMR, and MALDI-TOF spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Valeur B.Molecular Fluorescence: Principles and Applications; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Pischel U. Angew. Chem., Int. Ed. 2007, 46, 4026. [DOI] [PubMed] [Google Scholar]
- de Silva A. P.; Uchiyama S. Nat. Nanotechnol. 2007, 2, 399. [DOI] [PubMed] [Google Scholar]
- Credi A. Angew. Chem., Int. Ed. 2007, 46, 5472. [DOI] [PubMed] [Google Scholar]
- Kaake L. G.; Jasieniak J. J.; Bakus R. C.; Welch G. C.; Moses D.; Bazan G. C.; Heeger A. J. J. Am. Chem. Soc. 2012, 134, 19828. [DOI] [PubMed] [Google Scholar]
- Grätzel M. Acc. Chem. Res. 2009, 42, 1788. [DOI] [PubMed] [Google Scholar]
- Weller A. Pure Appl. Chem. 1968, 16, 115. [Google Scholar]
- de Silva A. P.; Vance T. P.; West M. E. S.; Wright G. D. Org. Biomol. Chem. 2008, 6, 2468. [DOI] [PubMed] [Google Scholar]
- Bissell R. A.; de Silva A. P.; Gunaratne H. Q. N.; Lynch P. L. M.; Maguire G. E. M.; Sandanayake K. R. A. S. Chem. Soc. Rev. 1992, 21, 187. [Google Scholar]
- De Silva A. P.; Gunaratne H. Q. N.; Gunnlaugsson T.; Huxley A. J. M.; McCoy C. P.; Rademacher J. T.; Rice T. E. Chem. Rev. 1997, 97, 1515. [DOI] [PubMed] [Google Scholar]
- Jeong Y.; Yoon J. Inorg. Chim. Acta 2012, 381, 2. [Google Scholar]
- Kubo K.; Mori A. J. Mater. Chem. 2005, 15, 2902. [Google Scholar]
- Bozdemir O. A.; Guliyev R.; Buyukcakir O.; Selcuk S.; Kolemen S.; Gulseren G.; Nalbantoglu T.; Boyaci H.; Akkaya E. U. J. Am. Chem. Soc. 2010, 132, 8029. [DOI] [PubMed] [Google Scholar]
- Mbatia H. W.; Burdette S. C. Biochemistry (Mosc.) 2012, 51, 7212. [DOI] [PubMed] [Google Scholar]
- Han J.; Burgess K. Chem. Rev. 2010, 110, 2709. [DOI] [PubMed] [Google Scholar]
- Daffy L. M.; de Silva A. P.; Gunaratne H. Q. N.; Huber C.; Lynch P. L. M.; Werner T.; Wolfbeis O. S. Chem.–Eur. J. 1998, 4, 1810. [Google Scholar]
- Saleh N.; Alsoud Y.; Nau W. Spectrochim. Acta, Part A 2008, 71, 818. [DOI] [PubMed] [Google Scholar]
- Koide Y.; Urano Y.; Hanaoka K.; Terai T.; Nagano T. ACS Chem. Biol. 2011, 6, 600. [DOI] [PubMed] [Google Scholar]
- Cox R. P.; Higginbotham H. F.; Graystone B. A.; Sandanayake S.; Langford S. J.; Bell T. D. M. Chem. Phys. Lett. 2012, 521, 59. [DOI] [PubMed] [Google Scholar]
- Wencel D.; Abel T.; McDonagh C. Anal. Chem. 2014, 86, 15. [DOI] [PubMed] [Google Scholar]
- Gareis T.; Huber C.; Wolfbeis O. S.; Daub J.. Chem. Commun. 1997, 1717.
- Baruah M.; Qin W.; Basarić N.; De Borggraeve W. M.; Boens N. J. Org. Chem. 2005, 70, 4152. [DOI] [PubMed] [Google Scholar]
- Baki C. N.; Akkaya E. U. J. Org. Chem. 2001, 66, 1512. [DOI] [PubMed] [Google Scholar]
- Boens N.; Leen V.; Dehaen W. Chem. Soc. Rev. 2012, 41, 1130. [DOI] [PubMed] [Google Scholar]
- Qin W.; Baruah M.; De Borggraeve W. M.; Boens N. J. Photochem. Photobiol. Chem. 2006, 183, 190. [Google Scholar]
- Zhang X.-F. J. Fluoresc. 2011, 21, 1559. [DOI] [PubMed] [Google Scholar]
- Sun K. M.; McLaughlin C. K.; Lantero D. R.; Manderville R. A. J. Am. Chem. Soc. 2007, 129, 1894. [DOI] [PubMed] [Google Scholar]
- Murtagh J.; Frimannsson D. O.; O’Shea D. F. Org. Lett. 2009, 11, 5386. [DOI] [PubMed] [Google Scholar]
- Isgor Y. G.; Akkaya E. U. Tetrahedron Lett. 1997, 38, 7417. [Google Scholar]
- Aigner D.; Ungerböck B.; Mayr T.; Saf R.; Klimant I.; Borisov S. M. J. Mater. Chem. C 2013, 1, 5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner D.; Borisov S. M.; Klimant I. Anal. Bioanal. Chem. 2011, 400, 2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner D.; Borisov S. M.; Petritsch P.; Klimant I. Chem. Commun. 2013, 49, 2139. [DOI] [PubMed] [Google Scholar]
- Lin H.-J.; Herman P.; Kang J. S.; Lakowicz J. R. Anal. Biochem. 2001, 294, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov S. M.; Herrod D. L.; Klimant I. Sens. Actuators, B 2009, 139, 52. [Google Scholar]
- Weidgans B.; Krause C.; Kliamnt I.; Wolfbeis O. S. Analyst 2005, 129, 645. [DOI] [PubMed] [Google Scholar]
- Xie T. M.; Dyrssen D. Anal. Chim. Acta 1984, 160, 21. [Google Scholar]
- Liptak M. D.; Gross K. C.; Seybold P. G.; Feldgus S.; Shields G. C. J. Am. Chem. Soc. 2002, 124, 6421. [DOI] [PubMed] [Google Scholar]
- Han J.; Burgess K. Chem. Rev. 2010, 110, 2709. [DOI] [PubMed] [Google Scholar]
- Fercher A.; Borisov S. M.; Zhdanov A. V.; Klimant I.; Papkovsky D. B. ACS Nano 2011, 5, 5499. [DOI] [PubMed] [Google Scholar]
- Wolfbeis O. S.; Weis L. J.; Leiner M. J. P.; Ziegler W. E. Anal. Chem. 1988, 60, 2028. [Google Scholar]
- Mills A.; Lepre A.; Wild L. Sens. Actuators, B 1997, 39, 419. [Google Scholar]
- Walkup G. K.; Burdette S. C.; Lippard S. J.; Tsien R. Y. J. Am. Chem. Soc. 2000, 122, 5644. [DOI] [PubMed] [Google Scholar]
- de Silva A. P.; Gunnlaugsson T.; Rice T. E. Analyst 1996, 121, 1759. [Google Scholar]
- Bissell R. A.; de Silva A. P.; Thilak W.; Fernando M. L.; Patuwathavithana S. T.; Shantha T. K.; Samarasinghe D. Tetrahedron Lett. 1991, 32, 425. [Google Scholar]
- de Silva A. P.; Gunaratne H. Q. N.; Habib-Jiwan J.-L.; McCoy C. P.; Rice T. E.; Soumillion J.-P. Angew. Chem., Int. Ed. Engl. 1995, 34, 1728. [Google Scholar]
- Closs G. L.; Miller J. R. Science 1988, 240, 440. [DOI] [PubMed] [Google Scholar]
- Lewis F. D.; Burch E. L. J. Am. Chem. Soc. 1994, 116, 1159. [Google Scholar]
- Ford W. E.; Kamat P. V. J. Phys. Chem. 1987, 91, 6373. [Google Scholar]
- Graboski Z. R.; Dobkowski J. Pure Appl. Chem. 1983, 55, 245. [Google Scholar]
- Aigner D.; Borisov S. M.; Orriach Fernández F. J.; Fernández Sánchez J. F.; Saf R.; Klimant I. Talanta 2012, 99, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutting S.; Borisov S. M.; Klimant I. Anal. Chem. 2013, 85, 3271. [DOI] [PubMed] [Google Scholar]
- Kubin R. F.; Fletcher A. N. J. Lumin. 1982, 27, 455. [Google Scholar]
- Sens R.; Drexhage K. H. J. Lumin. 1981, 24–25Part 2709. [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.