

Abstract
Although a number of anti HIV drugs have been approved, there are still problems with toxicity and drug resistance. This demonstrates a need to identify new compounds that can inhibit infection by the common drug resistant HIV-1 strains with minimal toxicity. Here we describe an efficient assay that can be used to rapidly determine the cellular cytotoxicity and efficacy of a compound against WT and mutant viral strains.
The desired target cell line is seeded in a 96-well plate and, after a 24 hr incubation, serially dilutions of the compounds to be tested are added. No further manipulations are necessary for cellular cytotoxicity assays; for anti HIV assays a predetermined amount of either a WT or drug resistant HIV-1 vector that expresses luciferase is added to the cells. Cytotoxicity is measured by using an ATP dependent luminescence assay and the impact of the compounds on infectivity is measured by determining the amount of luciferase in the presence or the absence of the putative inhibitors.
This screening assay takes 4 days to complete and multiple compounds can be screened in parallel. Compounds are screened in triplicate and the data are normalized to the infectivity/ATP levels in absence of target compounds. This technique provides a quick and accurate measurement of the efficacy and toxicity of potential anti HIV compounds.
Keywords: Immunology, Issue 86, HIV, cytotoxicity, infectivity, luciferase, drug resistance, integrase, reverse transcriptase
Introduction
The availability of drugs targeting several essential steps in the HIV-1 viral life cycle has led to combination drug therapy (called highly active anti retroviral therapy, or HAART) that has greatly improved the treatment of HIV-1 infections, and the long term survival of patients. HAART, which typically uses combinations of two nucleoside reverse transcriptase (RT) inhibitors and either a protease inhibitor or a non nucleoside RT inhibitor, has now converted a deadly disease into a life long condition1-5. However, despite the many successes of HAART in sustained suppression of viral replication, it has limitations. HAART does not eradicate HIV, so the patients are not cured and therapy is life long. There are problems with drug toxicity and with the emergence of drug resistant strains. Resistance can arise to all of the approved anti HIV drugs, including the newly approved drugs that target HIV integrase (IN). Drug resistance likely arises because of spontaneous mutations that occur during viral replication (error rate of 3 x 10-5 mutations/base/replication cycle)6. When mutations arise in the genes coding for the targets of anti retroviral drugs, a small subset of these mutations will lead to a reduction in susceptibility of the viral strain to the drugs. To avoid the development of drug resistance, drug concentrations must be maintained at levels that fully suppress HIV replication. Poor adherence to the therapeutic regimen exacerbates the problem, and can lead to the rapid development of resistance7-9. Although drug concentrations can vary in patients due to differing absorption, metabolism, distribution, and excretion levels, high drug concentrations can lead to toxicity10. Since therapy continues for the life of the patient, there are serious safety concerns about the long term toxicity of anti retrovirals. The anti retrovirals commonly used in HAART can have adverse effects and there have been incidences where the side effects have been life threatening11-15. The problems patients encounter with the development of resistance and toxicity underlie the need to develop new drugs that effectively block the replication of the common drug resistant strains of the virus with little or no long term toxicity.
Thus, there is a need for an assay that can screen for compounds that block essential steps in the viral life cycle quickly. Here we describe an efficient assay that can be used to evaluate the cytotoxicity of a compounds and their ability to block the replication of both WT and drug-resistant HIV strains rapidly and efficiently. The assay we use is similar to an assay that was developed to screen for drug resistance in virus isolated from patients16-18.
Although the assay can be used without modification to screen for compounds that can block HIV reverse transcription, we will describe using the assay to evaluate IN inhibitors. IN is an essential viral enzyme that inserts the viral DNA into the cellular genome19. Although a number of promising IN inhibitors are being developed, some of which are currently undergoing clinical trials, only Isentress20,21 (also known as Raltegravir or RAL) and more recently, Elvitegravir (EVG)22 and Dolutegravir (DTG)23 have been approved by the FDA. These compounds are active against both HIV B and non B subtypes in cell culture and in patients24,25. However, treatment with RAL selects for drug-resistant HIV-1 IN mutants, including Y143R, N155H, and G140S/Q148H24,26-31. N155H and G140S/Q148H also reduce the efficacy of EVG, which emphasizes the need to design and develop second generation IN strand transfer inhibitors (INSTIs) that are effective against these resistance mutations.
Protocol
1. Preparation of Master Stocks
Make master stocks of the compounds to be tested in DMSO. Prepare stocks at a standard concentration of 20 mM. Note: any concentration above 10 mM can be used. Compounds that can be used as positive controls to validate this assay include RAL, EVG, and DTG.
Ensure the compounds are dissolved in DMSO by vortexing the solutions multiple times for 15 sec and incubating at RT for 1 hr. Store the 20 mM stock solutions in the dark at -20 °C until use.
2. Preparation of the 96-well Plates for Compound Screening
Choose the cell line to be tested (e.g. HOS or TZM-bl), and seed 100 µl of those cells at a density of 4 x 104 cells/ml (4,000 cells/well) in media (e.g. Dulbecco’s modified Eagle’s or DMEM medium supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/ml) plus streptomycin).
3. Generation of Virus Stocks
Produce VSV-g-pseudotyped HIV by transfecting 293 cells (as shown in Figure 1, step 1)32-34.
On the day prior to transfection, plate 293 cells on 100 mm diameter dishes at a density of 1.5 x 106 cells.
On the day of transfection, transfect 293 cells with 16 µg of wild type or mutant HIV (pNLNgoMIVR-ΔLUC) and 4 µg of VSV (pHCMV-g) using the calcium phosphate method35.
Approximately 6 hr after the calcium phosphate precipitate is added, wash 293 cells twice with phosphate buffered saline (PBS) and incubate with fresh media for 48 hr. [DMEM supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/ml) plus streptomycin (50 µg/ml)].
Harvest the virus containing supernatants by removing the media from the 100 mm diameter dishes, clarify the supernatants by low speed centrifugation at 3,000 rpm for 10 min , filter the supernatants through a 45 µm pore size syringe filter, treat the supernatants with Turbo DNase for 30 min at RT and dilute the supernatants in media for preparation in infection assays. Store the viral supernatants frozen, in aliquots, at -80 °C. Note: the amount of p24 in the supernatant is determined by using a HIV-1 p24 enzyme-linked immunosorbent assay kit. The p24 concentration is used to control the amount of virus in the sample. Approximately 500 ng of virus is added to HOS cells plated on 60 mm diameter dishes at a density of 1.5 x 105 cells/dish on the day prior to infection. After a 48 hr of incubation, the cells are harvested, collected by centrifugation, washed, and resuspended in 100 µl of PBS. Add an equal amount of Luminescence reporter gene assay reagent and measure luciferase activity as described in sections 5.4.1 and 5.4.2. From this, an appropriate dilution of the virus can be made as discussed in step 4.6.
4. Compound Screening in 96-well Plates
Screen each compound in triplicate and average the results.
Note: the effect of each compound on viral replication is corrected by normalizing to the level of replication obtained in the absence of any compound.
Determine the empirical concentration range to be screened. Note: typically, screens with 11 serial dilutions are made by adding the compound to the plate column by column and screens with 7 serial dilutions are made by adding the compound by row. One triplicate set of wells must be reserved for the no compound control. In addition, one column or one row must remain blank to act as the negative/background control. Lastly, whether it is a cellular cytotoxicity or infectivity assay will dictate if virus is added.
Prepare serial dilutions from the 20 mM stock solution. The concentrations are chosen depending on the empirically determined range of concentrations to be tested (as shown in Table 1). Prepare the dilutions in media at 10x the final concentration desired, i.e. if the final concentration is going to be 100 µM, make a 1 mM working stock. Note: compounds with IC50s above 5-10 μM are not usually good candidates for drug development. In the initial assays we test the compounds only against the WT vector. Promising compounds that effectively inhibit the WT vector are then tested against a panel of drug resistant mutants.
Remove the 96-well plates from the incubator and add the serial dilutions of the compounds to be tested to the wells in triplicate (as shown in Figure 1, step 2). Note: the volume added to the well should be 1/10 volume of the final concentration. Thus, add 22 µl/well (final volume post virus addition is 220 µl) for infectivity assays. For cytotoxicity assays, add 11 µl/well (final volume is 110 µl/well).
Return the 96-well plates to the incubator. For a cellular cytotoxicity screen, incubate the 96-well plates 48 hr at 37 °C and no further manipulations are needed in Protocol 4: proceed to Protocol 5. For infectivity assays, continue following the instructions in Protocol 4.
Infectivity assays only. Remove the plates from the incubator after a minimum of 3 hr incubation at 37 °C with the compounds to be analyzed. Note: this allows the compound to be taken up by the cells prior to infection with the HIV vector.
Prepare a stock dilution of the virus33,34 (usually approximately 1:3) that will produce a luciferase signal between 0.2-1.5 relative luciferase units (RLUs) in untreated cells. An entire plate will take approximately 10 ml of diluted virus. Add 100 µl of virus to each of the wells, using either an 8 or 12 multichannel pipettor. Do not add virus to the negative/background control wells. Return the plates to the 37 °C incubator for 48 hr. Note: a 1:3 stock dilution of the virus is typical dilution that will produce a luciferase signal between 0.2 -1.5 RLUs based on a p24 assay showing that the viral concentration in the supernatant is approximately 500 ng in 1.0 ml.
5. Preparation and Measurement of Cytotoxicity and Infectivity in 96-well Plates
Aspirate the media from the wells (phenol red in the media can interfere with the luciferase signal). Use a glass pipette with a 200 µl pipette tip attached to the end. Start at the top of the media and slowly work down toward the bottom corner of the well. Do not spend too much time at the bottom of the well, or cells can be removed from the well.
Add 100 µl of PBS supplemented with 0.5 mM MgCl2 to each well. This needs to be done immediately after the media is removed so that the cells do not dry out.
- Only for cytotoxicity assays, add 5 ml of substrate buffer from the Luminescence ATP detection assay to each vial of supplied lyophilized reagent. One vial is sufficient for a 96-well plate.
- Add 50 µl of cell lysis buffer from the Luminescence ATP detection assay to each well. Shake the 96-well plate at 700 rpm at RT for 5 min using a compact thermomixer.
- Add 50 µl of reconstituted Luminescence ATP detection assay reagent to all wells except for the negative control/background wells. Shake at 700 rpm at room temperature for 5 min using a compact thermomixer. Incubate the plates at RT for 20 min to allow time for signal development.
- Read the 96-well plate using the microplate luminometer. Note: open the SoftMax Pro microplate luminometer program. Make sure the luminometer is set to measure luminescence at a sensitivity of 5 readings/well.
- Only for infectivity assays, add 10 ml of substrate buffer from the Luminescence reporter gene assay to each vial of supplied lyophilized reagent. One vial is sufficient for each 96-well plate.
- Add 100 µl of reconstituted Luminescence reporter gene assay reagent to each well. Incubate at room temperature for 20 min to allow time for signal development.
- Read the 96-well plate using a microplate luminometer as performed in section 5.3.3.
6. Determination of CC50 and IC50 Values for Compounds
- Transfer the luciferase data from the microplate luminometer into an excel spreadsheet.
- Average both the luciferase data in triplicate and the background/control signal data. Subtract the average background/control signal from the average triplicate signal for the entire concentration range.
- Normalize the background corrected average signal for the concentration ranges against the activity, whether it is cytotoxicity or infectivity, in the absence of any compound to determine the percent inhibition. Note: percent inhibition is defined as the luciferase activity in the presence of drug divided by the luciferase activity in the absence of drug multiplied by 100.
- Use Kaleidagraph software program to obtain CC50 and IC50 values
- Transfer both the empirically determined concentration range and percent inhibition of luciferase activity into Kaleidagraph.
- Plot the data with the concentration range on the x axis and the percent inhibition of luciferase activity on the y axis.
Representative Results
If the assay (Figure 1, steps 1 and 2) was successfully performed, then the luciferase values should resemble the data presented in Table 2. Scan across the concentration range; a potentially potent compound will reveal increasing luciferase activity from left to right, and the control should have the highest luciferase activity. If the luciferase activity does not exceed 0.1 Relative Luciferase Units (RLUs) across the concentration range, this usually indicates that the compound killed the cells. If the luciferase data is greater than or equal to 2.0 RLUs across all the serial dilutions, then the compounds were not able to inhibit HIV-1 infections at the tested concentrations.
Plotting the concentration of the compounds versus the percent inhibition of luciferase activity in Kaleidagraph (Table 3, part A), after performing linear regression analysis, will produce results similar to those shown in Table 3, part B.
 Figure 1. Preparation of HIV-1 Viral Stocks and the Setup of Cellular Cytotoxicity and Single-round Infectivity Assays. In step 1, 293T cells are transfected with pNL4.3ΔEnv.LUC and VSV-G and incubated for 48 hr to produce virus34. The virus is harvested and stored (frozen at -80 °C in aliquots) until it is used in the infectivity assays. For step 2, HOS cells are seeded in a 96-well plate and incubated for 24 hr. The cells are then preincubated with serial dilutions of the compounds to be tested for 3 hr and then infected with virus (either WT or drug resistant). After a 48 hr incubation, luciferase activity is measured.
Figure 1. Preparation of HIV-1 Viral Stocks and the Setup of Cellular Cytotoxicity and Single-round Infectivity Assays. In step 1, 293T cells are transfected with pNL4.3ΔEnv.LUC and VSV-G and incubated for 48 hr to produce virus34. The virus is harvested and stored (frozen at -80 °C in aliquots) until it is used in the infectivity assays. For step 2, HOS cells are seeded in a 96-well plate and incubated for 24 hr. The cells are then preincubated with serial dilutions of the compounds to be tested for 3 hr and then infected with virus (either WT or drug resistant). After a 48 hr incubation, luciferase activity is measured.
 Table 1.Drug Screening Serial Dilution Prototype. A more stringent screening involves 11 serial dilutions which typically start at 10 µM and end at 0.0005 µM. The serial dilutions are prepared 10x; there are 100 µl of cells and 100 µl of virus in each well). At this volume and following these calculations, the serial dilutions will be enough for 3 rows of an entire 96-well plate. The cytotoxicity assays are prepared similarly; however the 11 serial dilutions start at 250 μM and end at 0.05 μM. Only 11 μl of the dilutions are added to the wells in the plate that contain 100 μl of cells. Please click here to view a larger version of this figure.
Table 1.Drug Screening Serial Dilution Prototype. A more stringent screening involves 11 serial dilutions which typically start at 10 µM and end at 0.0005 µM. The serial dilutions are prepared 10x; there are 100 µl of cells and 100 µl of virus in each well). At this volume and following these calculations, the serial dilutions will be enough for 3 rows of an entire 96-well plate. The cytotoxicity assays are prepared similarly; however the 11 serial dilutions start at 250 μM and end at 0.05 μM. Only 11 μl of the dilutions are added to the wells in the plate that contain 100 μl of cells. Please click here to view a larger version of this figure.
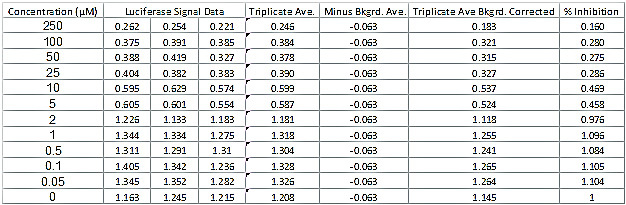 Table 2.Luciferase Signal data Readout and Determination of Percent Inhibition of Luciferase Activity. The data table shows a typical set of luciferase data for a successful compound. The table also shows the additional calculations needed to determine the percent inhibition of luciferase activity and the CC50 and IC50 values. Please click here to view a larger version of this figure.
Table 2.Luciferase Signal data Readout and Determination of Percent Inhibition of Luciferase Activity. The data table shows a typical set of luciferase data for a successful compound. The table also shows the additional calculations needed to determine the percent inhibition of luciferase activity and the CC50 and IC50 values. Please click here to view a larger version of this figure.
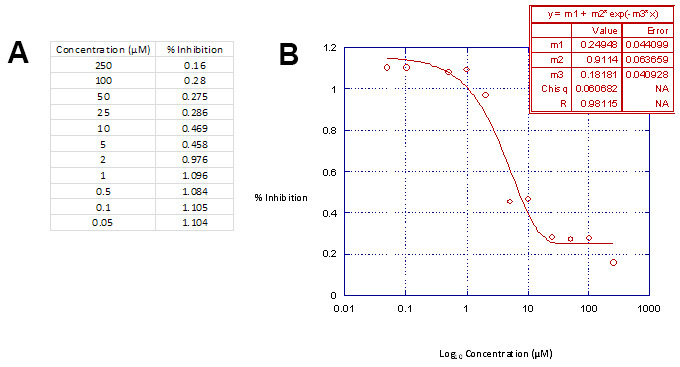 Table 3. Linear Regression Analysis Data Table. Part A. Graphing the concentration range used versus the percent inhibition of luciferase activity in Kaleidagraph will produce the appropriate inhibition curves. Part B, The inhibition curves are defined by the 3 parametric sigmoidal function and fit to the data by linear regression analyses18. This data table is then used in conjunction with Microsoft Excel to calculate the drug concentrations required to inhibit virus integration and cellular cytotoxicity by 50%, e.g. IC50 and CC50. Please click here to view a larger version of this figure.
Table 3. Linear Regression Analysis Data Table. Part A. Graphing the concentration range used versus the percent inhibition of luciferase activity in Kaleidagraph will produce the appropriate inhibition curves. Part B, The inhibition curves are defined by the 3 parametric sigmoidal function and fit to the data by linear regression analyses18. This data table is then used in conjunction with Microsoft Excel to calculate the drug concentrations required to inhibit virus integration and cellular cytotoxicity by 50%, e.g. IC50 and CC50. Please click here to view a larger version of this figure.
Discussion
We describe a quick, efficient, and reproducible assay that can be used to screen compounds for cytotoxicity and for their ability to inhibit the replication of both WT and drug resistant HIV-1. The ability to quickly identify compounds and test their efficacy and cytotoxicity is crucial in the development of new and improved drugs against HIV-1. Once lead compounds are identified, analogues of the lead compound can be produced and tested using the same assay. The assay is relatively simple. There are both positive and negative controls that allow the user to diagnose the most common problems (toxic compounds, problems with the vector stock). The use of a triplicate set of wells with no added compound shows that viral infection has occurred. The fact that cytotoxicity is measured in an independent assay avoids misinterpreting a reduction in luciferase caused by cytotoxicity as a specific effect on viral replication.
The critical steps in the protocol are preparing the plates so that the cells are uniformly distributed in the wells, that the wells have the proper concentrations of the compounds to be tested, adding the same amount of virus to each of the wells, and measuring the luciferase activity to determine the CC50 and IC50 values.
The assay is safe, quantitative, and reproducible. The assay is safe because the vector is replication defective. The assay is quantitative and reproducible because it is based on a single round vector that expresses luciferase, which can be assayed accurately and conveniently. In a multi round virus replication assay, the measured IC50 depends on the number of viral life cycles; this is a particular problem when the assays involve both WT and drug resistant viruses that may have significantly different replication capacities.
There have been several enzymatic assays reported previously that can be used to screen for IN inhibitors. Assays that involve real time PCR technology to measure integrated DNA require purified recombinant proteins(s), and are, in general, both more labor intensive and expensive36,37. Although it is possible to use enzymatic assays to measure the impact of compounds on the other viral enzymes (RT and protease), each enzyme requires its own assay system. The one round vector assay, as described, can be used, without modification, to screen for RT inhibitors. A similar assay can be used to screen for protease inhibitors; however, in a protease inhibitor assay, the compounds must be added to the cells used to produce the vectors. A related assay, using different cells and vectors, can also be used to screen for envelope (env) and HIV entry fusion inhibitors. Lastly, the assay can be used, on a larger scale, with automated robotic dispensers. Thus, the assay can be used to screen large libraries of compounds against WT and mutant HIV. However, the fact the assay can detect an inhibitor of HIV replication that acts at different stages of the viral life cycle points to a limitation in interpreting the data. By itself the assay does not define which step in the life cycle is blocked by a compound. If this question arises, it can be resolved by using time of addition assays38, and by testing the compound against purified recombinant viral proteins.
Unfortunately, despite the success of anti HIV drugs, there are still problems with both resistance and toxicity. In the absence of an effective anti HIV vaccine, there is a need not only to develop new therapeutic drugs that will be effective against the existing drug resistant mutants, but also to develop prophylactic drugs that can reduce the spread of the virus. If the prophylactic use of anti HIV drugs incudes the treatment of uninfected people, this approach will place a special burden on developing drugs that are have little or no long term toxicity.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This research was supported by the Intramural Research Program of the NCI.
References
- Mouton Y, et al. Impact of protease inhibitors on AIDS-defining events and hospitalizations in 10 French AIDS reference centres. Federation National des Centres de Lutte contre le SIDA. AIDS. 1997. pp. 101–105. [DOI] [PubMed]
- Hammer SM, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less AIDS Clinical Trials Group 320 Study Team. New Eng. J. Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- Hogg RS, et al. Improved survival among HIV-infected patients after initiation of triple-drug antiretroviral regimens. Can. Med. Assoc. 1999;160:659–665. [PMC free article] [PubMed] [Google Scholar]
- Egger M, et al. Impact of new antiretroviral combination therapies in HIV infected patients in Switzerland: prospective multicentre study. Swiss HIV Cohort Study. BMJ. 1997;315:1194–1199. doi: 10.1136/bmj.315.7117.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick RM, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New Eng. J. Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Simoni JM, Amico KR, Pearson CR, Malow R. Strategies for promoting adherence to antiretroviral therapy: a review of the literature. Curr. Infect. Dis. Rep. 2008;10:515–521. doi: 10.1007/s11908-008-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni JM, Amico KR, Smith L, Nelson K. Antiretroviral adherence interventions: translating research findings to the real world clinic. Curr. HIV/AIDS Rep. 2010;7:44–51. doi: 10.1007/s11904-009-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376:49–62. doi: 10.1016/S0140-6736(10)60676-9. [DOI] [PubMed] [Google Scholar]
- Acosta EP, et al. Novel method to assess antiretroviral target trough concentrations using in vitro susceptibility data. Antimicr. 2012;56:5938–5945. doi: 10.1128/AAC.00691-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockstroh JK, et al. Long-term treatment with raltegravir or efavirenz combined with tenofovir/emtricitabine for treatment-naive human immunodeficiency virus-1-infected patients: 156-week results from STARTMRK. Clin. Infect. Dis. 2011;53:807–816. doi: 10.1093/cid/cir510. [DOI] [PubMed] [Google Scholar]
- Fernandez-Montero JV, Eugenia E, Barreiro P, Labarga P, Soriano V. Antiretroviral drug-related toxicities - clinical spectrum, prevention, and management. Exp. Opin. Drug Safety. 2013. [DOI] [PubMed]
- Lunzen J, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect Dis. 2012;12:111–118. doi: 10.1016/S1473-3099(11)70290-0. [DOI] [PubMed] [Google Scholar]
- Sax PE, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379:2439–2448. doi: 10.1016/S0140-6736(12)60917-9. [DOI] [PubMed] [Google Scholar]
- Sax PE, et al. Abacavir/lamivudine versus tenofovir DF/emtricitabine as part of combination regimens for initial treatment of HIV: final results. Infect. Dis. 2011;204:1191–1201. doi: 10.1093/infdis/jir505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellam P, Larder BA. Recombinant virus assay: a rapid, phenotypic assay for assessment of drug susceptibility of human immunodeficiency virus type 1 isolates. Antimicr. Agents Chemother. 1994;38:23–30. doi: 10.1128/aac.38.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertogs K, et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicr. Agents Chemother. 1998;42:269–276. doi: 10.1128/aac.42.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos CJ, et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicr. Agents Chemother. 2000;44:920–928. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- Nguyen BY, et al. Raltegravir: the first HIV-1 integrase strand transfer inhibitor in the HIV armamentarium. Ann. N.Y. Acad. Sci. 2011;1222:83–89. doi: 10.1111/j.1749-6632.2011.05972.x. [DOI] [PubMed] [Google Scholar]
- Wills T, Vega V. Elvitegravir: a once-daily inhibitor of HIV-1 integrase. Exp. Opin. Invest. Drugs. 2012;21:395–401. doi: 10.1517/13543784.2012.658914. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicr. Agents Chemother. 2011;55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DA, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. Eng. J. Med. 2008;359:355–365. doi: 10.1056/NEJMoa0708978. [DOI] [PubMed] [Google Scholar]
- Briz V, et al. Raltegravir and etravirine are active against HIV type 1 group O. AIDS Res.Human Retroviruses. 2009;25:225–227. doi: 10.1089/aid.2008.0222. [DOI] [PubMed] [Google Scholar]
- Fransen S, et al. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 2009;83:11440–11446. doi: 10.1128/JVI.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goethals O, et al. Primary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profiles. Virology. 2010;402:338–346. doi: 10.1016/j.virol.2010.03.034. [DOI] [PubMed] [Google Scholar]
- Goethals O, et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008;82:10366–10374. doi: 10.1128/JVI.00470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canducci F, et al. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS. 2009;23:455–460. doi: 10.1097/QAD.0b013e328323da60. [DOI] [PubMed] [Google Scholar]
- Ceccherini-Silberstein F, et al. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009;11:17–29. [PubMed] [Google Scholar]
- Charpentier C, et al. Drug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapy. HIV Med. 2008;9:765–770. doi: 10.1111/j.1468-1293.2008.00628.x. [DOI] [PubMed] [Google Scholar]
- Julias JG, et al. Effects of mutations in the G tract of the human immunodeficiency virus type 1 polypurine tract on virus replication and RNase H cleavage. J. Virol. 2004;78:13315–13324. doi: 10.1128/JVI.78.23.13315-13324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol. Pharmacol. 2011;80:565–572. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp SD, et al. A novel polymorphism at codon 333 of human immunodeficiency virus type 1 reverse transcriptase can facilitate dual resistance to zidovudine and L-2',3'-dideoxy-3'-thiacytidine. J. Virol. 1998;72:5093–5098. doi: 10.1128/jvi.72.6.5093-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- Brussel A, et al. Longitudinal monitoring of 2-long terminal repeat circles in peripheral blood mononuclear cells from patients with chronic HIV-1 infection. AIDS. 2003;17:645–652. doi: 10.1097/00002030-200303280-00001. [DOI] [PubMed] [Google Scholar]
- Daelemans D, Pauwels R, De Clercq E, Pannecouque C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011;6:925–933. doi: 10.1038/nprot.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]