

Abstract
The goal of targeted therapy is to match a selective drug with a genetic lesion that predicts for drug sensitivity. In a diverse panel of cancer cell lines, we found that the cells most sensitive to focal adhesion kinase (FAK) inhibition are deficient in the expression of the NF2 tumor suppressor gene product, Merlin. Merlin expression is often lost in malignant pleural mesothelioma (MPM), an asbestos-induced aggressive cancer with limited treatment options. Our data demonstrate that low Merlin expression predicts for increased sensitivity of MPM cells to a FAK inhibitor, VS-4718, in vitro and in tumor xenograft models. Disruption of MPM cell-cell or cell-extracellular matrix (ECM) contacts with blocking antibodies suggests that weak cell-cell adhesions in Merlin-negative MPM cells lead to their greater dependence on cell-ECM-induced FAK signaling. This provides one explanation of why Merlin-negative cells are vulnerable to FAK inhibitor treatment. Furthermore, we validated ALDH as a marker of cancer stem cells (CSCs) in MPM, a cell population thought to mediate tumor relapse after chemotherapy. Whereas pemetrexed and cisplatin, standard-of-care agents for MPM, enrich for CSCs, FAK inhibitor treatment preferentially eliminates these cells. These preclinical results provide the rationale for a clinical trial in MPM patients using a FAK inhibitor as a single agent after first-line chemotherapy. With this design, the FAK inhibitor could potentially induce a more durable clinical response due to reduction of CSCs along with a strong antitumor effect. Furthermore, our data suggest that patients with Merlin-negative tumors may especially benefit from FAK inhibitor treatment.
Introduction
Focal adhesion kinase (FAK) is an important cancer target, because FAK gene amplification and protein overexpression have been demonstrated in a wide range of malignancies (1). FAK is a non-receptor protein tyrosine kinase that integrates signals from integrins and growth factor receptors to regulate cell proliferation, survival, migration, invasion and cancer stem cell (CSC) renewal (1–3). FAK inhibitors have been shown to decrease tumor growth and metastasis in preclinical models, and have shown initial clinical activity in cancer patients (4–6).
Although elevated FAK expression is often observed in human tumors, no specific mutations or translocations have been identified to predict which patient population is most likely to respond to a FAK inhibitor. Successful targeted therapies that pair small molecule inhibitors with specific activated oncogenes include agents targeting BCR-ABL and EML4-ALK translocations, HER2 gene amplification, and activating mutations in EGFR and B-RAF (7). Alternatively, identification of a synthetic lethal relationship between a drug target and loss of a tumor suppressor is exemplified by the efficacy of PARP inhibitors in breast cancer bearing BRCA1 or BRCA2 mutations (7). An analogous therapeutic strategy could greatly facilitate the clinical development of a FAK inhibitor.
The neurofibromatosis type 2 (NF2) tumor suppressor gene encodes the Merlin protein, which has sequence homology with the Ezrin/Radixin/Moesin (ERM) protein family (8). Merlin mediates tumor suppression and cell contact inhibition through reduction of Rac-PAK, mTORC1, Hippo, EGFR and FAK-Src signaling (9). Merlin localizes to cell-cell boundaries where it plays a role in the maturation of adherens junctions (10, 11). Germline mutations in NF2 contribute to development of type 2 neurofibromatosis, which is characterized by growth of meningiomas, ependymomas and schwannomas (12). In addition, NF2 is frequently inactivated in human malignant pleural mesothelioma (MPM), where biallelic inactivation of NF2 occurs in 40–50% of tumors (12, 13). MPM is an aggressive tumor of the pleural lining of the lung and is often associated with prior exposure to asbestos (13). It has been estimated that as many as 43,000 people worldwide die from MPM each year (14). Median overall survival following frontline chemotherapy with pemetrexed and cisplatin is approximately 12 months (15). New therapies are urgently needed to improve the prognosis of patients with MPM.
Cancer stem cells (CSCs) comprise a subpopulation of tumor cells that possess self-renewal capacity, exhibit elevated resistance to chemotherapeutic agents and are often responsible for tumor recurrence (16). CSCs have been identified in many cancer types, including colorectal, breast, ovarian, pancreatic, prostate and head and neck cancers (17). Several studies found cells with CSC properties in MPM (18, 19). Moreover, an elevated CSC population has been demonstrated in a mouse model of aggressive NF2-deficient asbestos-induced mesothelioma (20). FAK plays a role in self-renewal, tumorigenicity and maintenance of mammary CSCs (2). Therefore, therapeutic targeting of FAK may diminish CSCs in a variety of malignancies including MPM.
In the present study, we aimed to identify cancers most sensitive to FAK inhibition and discover biomarkers to identify patients most likely to benefit from a FAK inhibitor treatment. VS-4718, previously known as PND-1186 (21), is a potent and selective FAK inhibitor (Fig. S1). We found that VS-4718 is especially effective against Merlin-negative cell lines of certain cancer types including MPM in vitro and in vivo and have uncovered a mechanism governing sensitivity to the FAK inhibitor. The preferential inhibitory effect of VS-4718 on CSCs, in addition to eradication of non-CSCs, provides a rationale for clinical use of a FAK inhibitor as a single agent after first-line chemotherapy. Altogether, these studies establish the effectiveness of a FAK inhibitor for treatment of Merlin-negative tumors and identify MPM as a promising setting for a Merlin-stratified clinical trial.
Results
Effects of FAK inhibitor on Merlin-positive and Merlin-negative cancer cell lines
Depletion of FAK by shRNA or pharmacological inhibition of FAK protein kinase activity has been demonstrated to reduce cell proliferation in 3-dimensional (3D) culture more effectively than in 2D culture (22). To assess the effects of VS-4718 treatment on cell growth, we modeled the tumor microenvironment using the ‘Matrigel on-top’ (MoT) method (adapted from (22), see Methods). A panel of 47 human cancer cell lines representing diverse tumors, including renal, thyroid, ovarian and breast carcinomas, non-small-cell lung cancer (NSCLC), melanoma and mesothelioma, was tested for sensitivity to VS-4718 in MoT culture. Interestingly, 7 of the 10 most sensitive cell lines (average EC50 = 0.24 μM) lack Merlin protein expression (Figs. 1A, S2). Among the 37 less sensitive cell lines (average EC50 = 1.85 μM), most (92%) expressed high Merlin levels, while only a minority (8%) exhibited loss of Merlin protein expression (Fig. 1A, S2). Hence, loss of Merlin correlated with increased sensitivity to the VS-4718 FAK inhibitor in vitro.
Fig. 1. Cancer cell lines especially sensitive to VS-4718 in vitro and in vivo have low Merlin expression.

A) Average EC50 values for VS-4718 sensitivity in a panel of cancer cell lines. Blue dotted line marks EC50=0.4 μM. N=3 biological replicates. B) Relative growth of MDA-MB-468 and MDA-MB-231 breast cancer cells in response to VS-4718 shown as percent of control. Data are representative of three experiments and are presented as mean ±SEM (N=6). C) Cell lysates from MDA-MB-468 and MDA-MB-231 cell lines, as indicated, analyzed by SDS-PAGE and blotted with anti-Merlin antibody. Actin was used as a loading control. D) Tumor growth inhibition of MDA-MB-231 s.c. xenograft tumors in response to PO BID VS-4718 treatment at 25 mg/kg or 100 mg/kg, as indicated. Data are presented as mean ±SEM (N=10). ****p<0.0001, **p=0.006 (unpaired t-test with Welch’s correction). E) Tumor growth inhibition of MDA-MB-468 s.c. xenograft tumors in response to PO BID VS-4718 treatment at 25 mg/kg or 100 mg/kg, as indicated. Data are presented as mean ± SEM (N=10). **p= 0.0092; non-significant (ns) p=0.056 (unpaired t-test with Welch’s correction).
To test whether enhanced FAK inhibitor sensitivity of Merlin-negative cells occurs in vivo, we assessed the antitumor efficacy of VS-4718 in Merlin-negative MDA-MB-231 and Merlin-positive MDA-MB-468 human triple negative breast cancer xenograft models. In vitro, MDA-MB-231 cells were >60 fold more sensitive to VS-4718 than MDA-MB-468 cells (Fig. 1B, C). In vivo, in the MDA-MB-231 xenograft model, mice dosed orally twice daily with VS-4718 at 25 mg/kg had significantly smaller tumors (p=0.006) than mice receiving vehicle control after 29 days of treatment. Moreover, tumor regression was observed in mice treated with 100 mg/kg VS-4718 (p< 0.0001; Fig. 1D) with corresponding significant reduction of tumor FAK (pY397) autophosphorylation (p=0.03; Fig. S3). In contrast, the Merlin-positive MDA-MB-468 human breast cancer xenograft model showed substantially less antitumor efficacy after 29 days of treatment with VS-4718 at 25 or 100 mg/kg (Fig. 1E). Final average tumor weights in VS-4718 treated groups were reduced only by ~30% relative to the vehicle control group despite significant inhibition of tumor pFAK-Y397 (p=0.0002; Fig. 1E, S3). Hence, tumor growth was suppressed in a dose-dependent manner in the Merlin-negative MDA-MB-231 xenograft model, with less tumor growth inhibition in the Merlin-positive MDA-MB-468 model. Thus, we conclude that Merlin-negative cancer cells are especially sensitive to the FAK inhibitor VS-4718 in vitro and in vivo.
FAK inhibitor sensitivity of Merlin-negative mesotheliomas in vitro
NF2 mutations are common in MPM, with approximately 40–50% of tumors exhibiting biallelic inactivation of NF2 leading to absent or low Merlin expression (12, 13). Accordingly, we examined a panel of mesothelioma cell lines for their FAK inhibitor sensitivity and assessed Merlin expression by Western blotting. Mero-25, MSTO-211H, H28 and H2452 MPM cell lines had high levels of Merlin, while Merlin expression was essentially undetectable in Mero-41, Mero-48a, Mero-83, MM87 and MM129 mesothelioma cell lines (Fig. 2A). Mero-14 expressed a short (~56kDa) isoform 7 variant of Merlin.
Fig. 2. The FAK inhibitor is most efficacious in Merlin-negative mesotheliomas in vitro.

A) Cell lysates from MPM cell lines analyzed by SDS-PAGE, as indicated, and blotted with anti-Merlin antibody. Actin was used as a loading control. B) Relative growth of MPM cells in response to VS-4718 shown as percent of control. Data are representative of three experiments and are presented as mean ± SEM (N=6). C) Cell lysates from MM87/GFP or MM87/NF2-GFP cell lines, as indicated, analyzed by SDS-PAGE and blotted with anti-Merlin antibody. Actin was used as a loading control. D) Relative growth of MM87/GFP or MM87/NF2-GFP cells in response to VS-4718 shown as percent of control. Data are representative of two experiments and are presented as mean ± SEM (N=6). E) Bar graph depicting percent of Ki67-positive cells in Mero-41 and MSTO-211H cell lines treated with siRNA, as indicated. Data are presented as mean ± SD (N=6, 150 cells each).
We assessed the effect of VS-4718 on the proliferation of these MPM cell lines in 3D MoT culture. Merlin-positive MPM cell lines exhibited an average EC50 of ~3.5 μM, while the average EC50 for Merlin-negative MPM cell lines was 0.3 μM (Fig. 2B, S4). In addition to greater potency of VS-4718, all Merlin-negative cell lines demonstrated a greater magnitude of response, exhibiting cell viability curves that approached zero, suggesting a cytotoxic effect of VS-4718 (Fig. 2B). Taken together, these data demonstrated that Merlin-negative mesothelioma cells are more sensitive to VS-4718 than Merlin-positive cells in vitro in 3D culture.
To test for a causal contribution of Merlin expression in regulating the effect of VS-4718 on cell viability, we created isogenic MPM and breast cancer cell lines by ectopically expressing full-length NF2 fused to GFP. Merlin expression was confirmed by Western blot analysis (Fig. 2C). Cell growth inhibition analysis demonstrated decreased sensitivity of MM87 MPM cells and MDA-MB-231 breast cancer cells expressing NF2-GFP to VS-4718, in contrast to cells infected with GFP vector control (Fig. 2D, S5). The VS-4718 EC50 in control MM87/GFP cells was ~300 nM, while in MM87/NF2-GFP cells the EC50 was increased by approximately 3-fold (Fig. 2D). This demonstrated that ectopic expression of full-length Merlin reduced sensitivity to the FAK inhibitor VS-4718, consistent with our hypothesis that FAK inhibitor sensitivity in Merlin-negative mesothelioma and breast cancer cells is enhanced by loss of Merlin.
To determine whether biological depletion of FAK would mimic the effect of VS-4718, we depleted FAK by siRNA in MPM cell lines and assessed cell proliferation in 3D MoT culture. Treatment of MPM cell lines with FAK siRNA resulted in reduction of FAK mRNA levels by 89–93% (Fig. S6). Ki67 staining was reduced approximately 3.5-fold in Merlin-negative Mero-41 cells (Fig. 2E), indicating reduced proliferation in response to FAK depletion. In contrast, reduction in Ki-67 positivity in Merlin-positive MSTO-211H cells in response to FAK siRNA treatment was marginal (Fig. 2E). These data demonstrate that depletion of FAK by siRNA mimics VS-4718 treatment and reduces proliferation especially in Merlin-negative cells.
FAK inhibitor efficacy in Merlin-negative mesotheliomas in vivo
To extend our observation to an in vivo mesothelioma model, we first tested the efficacy of VS-4718 in a Merlin-negative MPM lung tumor model. Merlin-negative MM87 murine mesothelioma cells were injected into the tail vein of mice, allowed to seed in the lungs for 7 days, and then animals were treated orally twice daily with VS-4718 at 75 mg/kg for 2 weeks. By initiating treatment 7 days after tail vein injection, this assay was intended to measure tumor growth in the lungs, rather than assessing elements of the initial metastatic process. At the end of the VS-4718 treatment on day 21 after initial tail vein injection, the tumor burden in the lungs of VS-4718-treated animals averaged 3.5-fold less than in the lungs of vehicle control-treated animals (p<0.0001; Fig. 3A), suggestive of an inhibitory effect of the FAK inhibitor on tumor growth in the lungs.
Fig. 3. Merlin-negative mesothelioma cells are especially sensitive to VS-4718 in vivo.

A) Bar graph depicting average tumor weights from MM87 orthotopic lung tumor model treated with VS-4718, as indicated. Data are presented as mean ± SD (N=10). ****p<0.0001 (Mann-Whitney test). B) Dot plot of tumor volumes in MM87 IP xenograft model treated with VS-4718, as indicated. Black lines represent the mean, whiskers depict 25% and 75% of the distribution, N=5. **p=0.0079 (Mann-Whitney test). C) Relative pFAK-Y397 levels measured by ELISA in tumor samples from (B). Black lines represent the mean, whiskers depict 25% and 75% of the distribution, N=5. *p=0.0159 (Mann-Whitney test). D) Tumor volumes from Merlin-positive human MPM PDX model treated with VS-4718 at 50 mg/kg p.o. BID. Data are presented as mean ± SEM (n=10). Tumor weights on day 42 are not significantly different (ns). p=0.19 (unpaired t-test with Welch’s correction). E) Tumor volumes from a Merlin-negative human MPM PDX model treated with: control (blue), VS-4718 at 50 mg/kg p.o. BID (purple), cisplatin at 4 mg/kg i.p. (once every 7 days) and pemetrexed at 75 mg/kg i.p. (once daily, 5 days on, 2 days off) for 14 days (green), cisplatin at 4 mg/kg i.p. (once every 7 days) and pemetrexed at 75 mg/kg i.p. (once daily, 5 days on, 2 days off) for 14 days followed by VS-4718 at 50 mg/kg p.o. BID started on day 16 (red). Data are presented as mean ±SEM (N=10). **p= 0.0089 (Mann-Whitney test); *p=0.035 (unpaired t-test with Welch’s correction).
We also tested the effect of VS-4718 in an MM87 intraperitoneal orthotopic xenograft model. Animals treated with 25 mg/kg VS-4718 orally twice daily for 2 weeks had small tumors with an average volume of approximately 100 mm3, in contrast to control-treated animals that had large tumors with an average volume of approximately 500 mm3. A measurable tumor was observed in only one animal out of ten treated for 2 weeks with 50 mg/kg or 75 mg/kg of VS-4718 orally twice daily (Fig. 3B). This indicated that the FAK inhibitor was efficacious in a Merlin-negative mesothelioma model in a dose-dependent manner. pFAK-Y397 was significantly inhibited in the tumors treated with VS-4718 (p=0.016; Fig. 3C), establishing a clear correlation between target inhibition and antitumor efficacy. No significant change in tumor burden was observed in Merlin-positive MSTO-211H xenografts treated with VS-4718 orally twice daily at 75 mg/kg for 11 days (Fig. S7), despite ~67% inhibition of tumor FAK-Y397 phosphorylation (p=0.0051; Fig. S7).
As a closer approximation of human cancer, we assessed the effect of VS-4718 in mice bearing human MPM patient-derived xenografts (PDX). Merlin-negative and Merlin-positive PDX models (Fig. S8) were chosen for this experiment, and FAK inhibitor treatment was initiated when tumors reached 200 mm3. FAK autophosphorylation was effectively inhibited in both PDX models treated with 50 mg/kg VS-4718 (Fig. S8). Merlin-positive PDX tumors treated with VS-4718 at 50 mg/kg averaged only 23% smaller than control tumors at the end of the study on day 42 (Fig. 3D). In contrast, in the Merlin-negative PDX study, mice treated orally twice daily with 50 mg/kg VS-4718 exhibited 56% tumor growth inhibition on day 35 as compared to vehicle-treated control animals (p=0.0089; Fig. 3E). Collectively, these observations demonstrate that Merlin-negative mesotheliomas are sensitive to the FAK inhibitor in vivo in mouse xenograft models and in a human PDX model.
Effects of FAK inhibitor treatment on cell proliferation and apoptosis
Reduced cell number in response to VS-4718 could potentially result from reduction of cell proliferation and/or induction of apoptosis. We therefore assessed effects of VS-4718 on caspase3/7 activity as a marker of apoptosis, and Ki67 positivity as an indicator of cell proliferation in mesothelioma cells. We observed 2–3 fold induction of caspase3/7 activity in response to VS-4718 treatment in a panel of human Merlin-negative MPM cell lines, Mero-41, Mero-48a and Mero-83, with only weak caspase3/7 activation in Merlin-positive Mero-25 and H28 cells (Fig. 4A). Ki67 staining was markedly reduced in Merlin-negative Mero-48a and Mero-41 cells treated with VS-4718, whereas little reduction in Ki67 staining was observed in Merlin-positive MSTO-211H and H28 cells (Fig. 4B). These results indicate that the FAK inhibitor reduced both proliferation and viability of Merlin-negative cells.
Fig. 4. VS-4718 induces apoptosis and reduces proliferation in Merlin-negative MPM cells.

A) Relative caspase3/7 activation in response to VS-4718 treatment depicted as a percent of control. Data are representative of three experiments and are presented as mean ± SEM (N=6). B) Bar graph of an average number of Ki67 positive cells treated with DMSO or 1 μM of VS-4718 for 4 days, depicted as percent of control. Data are representative of two experiments and are presented as mean ±SEM (N=6). C) and D) Protein lysates from Mero-41 cells (C) or H28 cells (D) treated with 1 μM VS-4718, as indicated, in MoT culture analyzed on SDS-PAGE and blotted with antibodies, as indicated. Actin was used as a loading control. E) and F) Bar graphs depicting quantification of Western blots in C) (red) and D) (blue) represent a ratio of phosphorylated protein band density to the total protein band density normalized to the control in each case, as indicated.
The AKT family of kinases regulates cell survival and proliferation (23). To better understand the inhibitory effect of VS-4718 on cell survival, we measured AKT activation in cells grown in 3D MoT culture. After 1 hr of VS-4718 treatment, FAK activation (pFAK-Y397) was inhibited to a similar extent in Merlin-negative Mero-41 and Mero-48a cells as in Merlin-positive H28 and MSTO-211H cells (Fig. 4C– F; S9) and this effect was sustained for at least 48 hrs. In contrast, after 1 hr of VS-4718 treatment we observed reduced AKT activation (pAKT-S473) in Merlin-negative Mero-41 and Mero-48a cells sustained for 48 hrs, whereas no inhibition of AKT activation was detected in Merlin-positive H28 or MSTO-211H cells (Fig. 4C– F; S9). VS-4718 had no effect on ERK1/2 activation in both Merlin-negative and Merlin-positive cells. These observations suggest that in the absence of Merlin, inhibition of AKT activation may mediate the pro-apoptotic effect of the FAK inhibitor on MPM cells. Taken together, these data indicate that Merlin-negative MPM cells are dependent on FAK signaling for both cell survival and proliferation in a 3D environment.
Potential mechanism of FAK inhibitor sensitivity in cells lacking Merlin expression
Merlin has been shown to play an important role in the establishment of stable adherens junctions in epithelial and Schwann cells in vitro and in vivo (10, 11). We hypothesized that survival and proliferation of tumor cells may depend on a balance between cell-cell contacts supported by Merlin and cell-ECM adhesion supported by integrin and FAK signaling. To confirm Merlin’s role in cell-cell adhesion, we examined whether Merlin expression status would affect the morphology of cell colonies in MoT culture. Indeed, Merlin-positive MSTO-211H cells exhibited round mass colony morphology in 3D, suggestive of robust cell-cell adhesions (24), while Merlin-negative H2052 cells formed invasive stellate-like colonies (Fig. 5A). Merlin-negative parental MDA-MB-231 cells formed stellate, poorly consolidated colonies in MoT culture, as expected (24), which changed into round, smooth-surfaced morphology upon forced expression of NF2 (Fig. 5A), consistent with the known role of Merlin in stabilizing cell-cell junctions.
Fig. 5. FAK inhibitor sensitivity of Merlin-negative cells is caused by increased cell-ECM engagement and β1 integrin activation.

A) Representative phase-contrast images of cells grown in 3D MoT culture, as indicated. Scale bar = 400 μm. B) pFAK-Y397 levels measured by ELISA in MDA-MB-231/GFP and MDA-MB-231/NF2-GFP cells and represented as percent of control. Data are representative of two experiments and are presented as mean ± SEM (N=6). C) Relative growth of MSTO-211H cells treated with control IgG or N-cadherin blocking antibody in response to VS-4718, depicted as percent of control. Data are representative of two experiments and are presented as mean ±SEM (N=6). D) Relative growth of Mero-41 and Mero-25 cells, as indicated, treated with control IgG or β1 integrin blocking antibody, depicted as percent of control. Data are representative of two experiments and are presented as mean ±SEM (N=6). E) Relative growth of Mero-41 cells treated with control IgG or β1 integrin blocking antibody in response to VS-4718, depicted as percent of control. Data are representative of three experiments and are presented as mean ± SEM (N=6). F) Relative growth of Mero-25 cells treated with control IgG or β1 integrin blocking antibody in response to VS-4718, depicted as percent of control. Data are representative of three experiments and are presented as mean ± SEM (N=6).
The destabilization of adherens junctions in Merlin-negative cells may result in an upregulation of integrin-FAK signaling and increased levels of pFAK as cells compensate for the loss of trophic signals normally derived from cell-cell contacts. An inhibitory effect of Merlin expression on FAK activation has been previously demonstrated in mesothelioma and glial cells (25, 26). To confirm this observation, we performed pFAK-Y397 immunofluorescence analysis in Mero-41 and Mero-48a Merlin-negative cells ectopically expressing NF2 fused to GFP. pFAK-Y397 signal was clearly reduced at focal adhesions in cells with detectable Merlin expression (Fig. S10). To test FAK activation in a 3D environment, we performed an ELISA to detect the activated form of FAK (pFAK-Y397) in Merlin-negative MDA-MB-231 parental cells and in cells engineered to express NF2. We found that activated FAK levels were decreased by approximately 70% upon expression of NF2 (Fig. 5B), suggesting that ectopic Merlin expression diminished FAK activation.
We hypothesized that Merlin-positive cells forced to lose cell-cell interactions might become more dependent on cell-ECM interactions and thus on FAK signaling. To test this directly, we assessed FAK inhibitor sensitivity in 3D culture of Merlin-positive MSTO-211H and H28 cells, both of which express N-cadherin (Fig. S11), in the presence of an N-cadherin blocking antibody employed to disrupt cell-cell adhesions (27). The N-cadherin blocking antibody reduced β–catenin recruitment to cell-cell junctions indicative of defective cell-cell adhesions (Fig. S11). While Merlin-positive MSTO-211H cells were only modestly sensitive to VS-4718, exhibiting an EC50 of 11 μM, the N-cadherin blocking antibody increased the potency of VS-4718 by approximately 60-fold resulting in an EC50 of 180 nM (Fig. 5C). Similarly, the N-cadherin antibody increased the sensitivity of Merlin-positive H28 cells to VS-4718 (Fig. S11). These results support the concept that blocking cell-cell contacts in a Merlin-positive context renders cells more dependent on cell-ECM stimulated FAK signaling.
We further hypothesized that reducing cell-ECM interactions through disruption of β1 integrin activity in Merlin-negative cells would supplant FAK inhibition, decreasing sensitivity to the FAK inhibitor. Therefore, we assessed whether a β1 integrin blocking antibody would inhibit FAK activation and reduce proliferation of MPM cells. In Merlin-negative Mero-41 and Mero-83 cells, β1 integrin blockade indeed reduced FAK activation in contrast to Merlin-positive H28 and Mero-25 cells (Fig. S12). Proliferation of Merlin-negative Mero-41 cells was dramatically reduced (90%) by treatment with the β1 integrin blocking antibody (Fig. 5D), thus mimicking the effect of the FAK inhibitor. Moreover, blocking of β1 integrin activity rendered Merlin-negative Mero-41 cells relatively insensitive to further reduction of proliferation by VS-4718, with an EC50 of 12 μM compared to an EC50 of 0.31 μM in the control IgG treated cells (Fig. 5E). In contrast, in Merlin-positive Mero-25 cells, the β1 integrin-blocking antibody had no effect on proliferation (Fig. 5D) or response to VS-4718 (Fig. 5F), suggesting lack of dependence of Merlin-positive cells on β1 integrin/FAK signaling. Altogether, we conclude that a balance between cell-cell adhesion and β1 integrin–mediated cell-ECM interaction regulates sensitivity to FAK inhibitor in MPM cells.
Preferential effect of FAK inhibitor on mesothelioma CSCs in vitro and in vivo
FAK has been shown to play a role in self-renewal and tumor-initiating capability of cancer stem cells (CSCs) in a mouse model of breast cancer (2). Therefore, we were interested to assess the role of CSC depletion in the anti-tumor efficacy of the FAK inhibitor in MPM cells. Cancer stem cells (CSCs) are defined as a subpopulation of cancer cells that 1) can self-renew, 2) has tumor initiating capability, and 3) can differentiate into heterogeneous cell types within tumors (17). An Aldefluor assay, which measures aldehyde dehydrogenase (ALDH) activity, has been used in various cancer types to define and isolate CSCs (17). To validate Aldefluor as a CSC marker in MPM, we assessed self-renewal, tumor-initiating capability and differentiation within tumors.
The MM87 MPM cell line has on average 3.8%±1.4% Aldefluor+ cells. Self-renewal potential of Aldefluor+ MM87 MPM cells was assessed in a tumorsphere assay (17). Increased tumorsphere-forming efficiency of Aldefluor+ MM87 cells was evident in primary and secondary tumorsphere assays. MM87 cells isolated from primary tumorspheres generated 85.6±6.5 secondary spheres while Aldefluor− cells only generated 8.6±5.3 spheres (Fig. S13) suggesting increased self-renewal capability of Aldefluor+ MPM cells. To assess tumor-initiating potential of Aldefluor+ MPM cells, Aldefluor+ or Aldefluor− MM87 cells were sorted and injected subcutaneously into NOD/SCID mice at various cell numbers per injection from 100,000 down to 50 cells (limiting dilution) and tumor burden was evaluated 3 weeks later. Large tumors of ~1000 mm3 were formed in mice injected with as few as 50 Aldefluor+ cells within 3 weeks, whereas Aldefluor− cells typically required injection of 1000 cells to initiate tumors (Fig. 6A, B). Overall, Aldefluor+ cells had ~35-fold higher tumor-initiating potential than Aldefluor− cells (p<0.0001; Fig. 6B). To assess the ability of Aldefluor+ MPM cells to differentiate within tumors, we analyzed Aldefluor+ and Aldefluor− cell content in MPM tumors formed from MM87 Aldefluor+ cells. After three weeks, tumors from Aldefluor+ cell injection retained only ~1% Aldefluor+ cells, whereas the bulk of the tumor consisted of Aldefluor− cells (Fig. S13), suggesting that Aldefluor+ cells differentiated into Aldefluor− cells during tumor growth. Taken together, these data validate Aldefluor as a tumor-initiating cell/CSC marker in MPM.
Fig. 6. FAK inhibitor VS-4718 has a preferential effect on CSCs in MPM.

A) Representative photographs of mice injected with Aldefluor+ MM87 cells (A+) on one side and Aldefluor− MM87 cells (A−) on the other, as indicated. B) Table summarizing tumor initiating frequency (TIF) of MM87 A+ and A− cells (p<0.0001). Total number of injected cells is indicated in the column ‘Cell#’. Total number of tumors/total number of mice is indicated in columns ‘A+’ and ‘A−’. C) Bar graph showing the number of Aldefluor+ H2052 cells in response to drug treatment, as indicated, depicted as a percent of control. Compounds were used at the following concentrations: pemetrexed 1 nM, cisplatin 2 nM, gemcitabine 1 nM, vinorelbine 1 nM, VS-4718 300 nM. Data are representative of two experiments and are presented as mean ±SEM (N=6). D) Bar graph showing the number of Aldefluor+ H2052 cells, depicted as a percent of control, in response to pemetrexed alone or pemetrexed in combination with VS-4718, as indicated. Data are representative of two experiments and are presented as mean ±SEM (n=6). E) Representative immunofluorescence images of MM87 intraperitoneal xenograft tumors treated with VS-4718 and stained with anti-ALDH1 antibody (green) and DAPI (blue). Scale bars = 20 μm. F) Percent of ALDH1-positive cells in MM87 xenograft tumors from (E) depicted relative to control. Data are presented as mean ± SEM (N=100).
We next determined whether the FAK inhibitor VS-4718 has an effect on tumor-initiating cells in MPM. Changes in the Aldefluor+ cell subpopulation in Merlin-negative H2052 mesothelioma cells were assessed in response to VS-4718 or treatment with the standard-of-care cytotoxic agents pemetrexed, cisplatin, vinorelbine or gemcitabine. VS-4718 treatment reduced the percentage of Aldefluor+ cells in direct contrast with the standard cytotoxic agents, which enriched the Aldefluor+ cell subpopulation (Fig. 6C). Similar effects of VS-4718 on Aldefluor+ CSCs were observed in the Mero-83 and Mero-48a MPM cell lines (Fig. S14). Moreover, when used in combination, VS-4718 blocked the enrichment of Aldefluor+ cells by pemetrexed (Fig. 6D), suggesting that FAK activity is necessary for survival and/or proliferation of mesothelioma tumor-initiating CSCs in vitro.
ALDH1 expression, like Aldefluor activity has been widely used as a CSC marker (17). Accordingly, we attempted to extend the above observations to an in vivo model of MPM. ALDH1 immunofluorescence analysis demonstrated that control MPM tumors in an MM87 intraperitoneal xenograft model had dispersed ALDH1-positive cells. In contrast, tumors from animals treated with VS-4718 at 50 mg/kg PO BID showed a marked reduction of ALDH1-positive cells (Fig. 6E, F). Collectively, these data demonstrate that the FAK inhibitor decreased tumor-initiating CSC content in Merlin-negative MPM in vitro and in vivo.
Standard front-line therapy for patients with mesothelioma typically consists of pemetrexed in combination with a platinum agent (15). Because pemetrexed and platinum increase the proportion of CSCs in MPM cells (Fig. 6C), we reasoned that use of a FAK inhibitor as a single agent after front line chemotherapy to suppress CSCs and tumor recurrence might effectively extend survival. To model this preclinically, we used a Merlin-negative MPM PDX model (Fig. 3E). Mice bearing Merlin-negative PDX tumors were first treated with cisplatin and pemetrexed for 14 days, followed by vehicle control or VS-4718 treatment for an additional 32 days to model maintenance treatment. Combination treatment with cisplatin and pemetrexed resulted in complete tumor growth inhibition on day 14, however, tumors regrew after cessation of treatment, reaching 1000 mm3 by day 46 (Fig. 3E). In contrast, treatment with VS-4718 as a single agent starting on day 16, after cessation of the cisplatin/pemetrexed regimen, substantially delayed tumor regrowth resulting in 77% tumor growth inhibition by day 46 as compared to the cisplatin/pemetrexed arm (p=0.035; Fig. 3E). These results demonstrate that a FAK inhibitor can indeed extend the duration of tumor growth inhibition, providing a more durable response when used as a single agent after cisplatin/pemetrexed treatment in a PDX model of mesothelioma.
Discussion
Targeted therapies tailored to cancers with specific genetic lesions have emerged as effective anti-cancer treatments. In the absence of a specific activating mutation or translocation of the drug target, another promising approach is to establish a ‘synthetic lethal’ relationship wherein pharmacological inhibition of one gene product is especially efficacious in the background of loss of function of a second gene product (7). Our studies reveal that a potent and selective FAK inhibitor, VS-4718, is efficacious in several cancer types, and that Merlin deficiency is a potential predictive marker for enhanced response to the FAK inhibitor in MPM. Our work also demonstrates that the FAK inhibitor targets CSCs in MPM, in contrast to cytotoxic agents that enrich the CSC population. Altogether, our data suggest a ‘synthetic lethal’ relationship between Merlin loss and FAK inhibitor sensitivity and provide an attractive strategy for clinical testing of FAK inhibitors in patients with MPM.
NF2 expression and function can be lost in tumors by a variety of mechanisms, including mutation, chromosome loss and focal deletions (13). NF2 is mutated in approximately 40–50% of MPMs, with lower frequency of mutations in several other cancer types (12, 13). Additional promising indications for clinical use of FAK inhibitors may include other tumors with high prevalence of NF2 loss, such as schwannomas, meningiomas and ependymomas resulting from neurofibromatosis type 2 syndrome (89% NF2 mutations; (28)) and high-grade malignant meningioma (29). Additional studies are needed to unequivocally extend the ‘synthetic lethal’ relationship between Merlin loss and FAK inhibitor sensitivity to other tumor types beyond the MPM tumors studied here.
A few reports have suggested a potential role of Merlin in regulating FAK activity in MPM and glial cells (25, 26). FAK has been demonstrated to play a role in proliferation, migration and survival of NF2−/− schwannoma cells through activation of PI3K/AKT and ERK signaling pathways (9). Ectopic expression of Merlin in NF2−/− MPM cells inhibited invasion, decreased FAK Tyr397 phosphorylation and impaired FAK interaction with its binding partners Src and p85 (26). However, an effect of Merlin expression on the anti-proliferative and pro-apoptotic activities of FAK inhibitors has not previously been investigated.
Merlin has been shown to play a role in the establishment of stable adherens junctions in several epithelial cell types (11, 30). Our data support a model in which the well-established cell-cell junctions in Merlin-positive MPM cells provide survival and proliferation signals independent of FAK signaling, explaining their reduced sensitivity to inhibition of FAK kinase activity (Fig. 7). In contrast, Merlin-negative cells with weak cell-cell contacts depend more on cell-ECM induced FAK activation for proliferation and resistance to apoptosis and are therefore more sensitive to FAK kinase inhibition (Fig. 7). Accordingly, it may be possible to extend this observation to other cancers with weak cell-cell adhesions. For example, others have suggested that another FAK inhibitor, BI 853520, may be most effective in treatment of tumors with low expression of E-cadherin, another essential mediator of adherens junctions (31). The enhanced sensitivity of CAL-62, COV318 and CAOV-4 ovarian cancer cell lines to VS-4718 (Fig. 1A) despite wild-type Merlin expression supports the notion that additional molecular markers and mechanisms predictive of FAK inhibitor sensitivity remain to be elucidated.
Fig. 7. FAK inhibitor sensitivity depends on the balance between cell-ECM and cell-cell interactions.
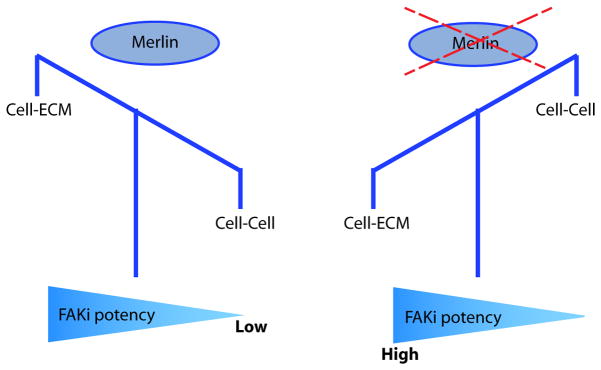
In Merlin-positive cells, stable cell-cell adhesions decrease dependence on FAK signaling for survival and proliferation, thus causing low sensitivity to FAK inhibition. In Merlin-negative cells, strong dependence on cell-ECM interactions is mediated through β1 integrin/FAK signaling important for cell survival and proliferation, and leads to greater sensitivity to FAK inhibition. Genetic or pharmacological manipulations of this balance influence FAK inhibitor sensitivity accordingly.
Our data also demonstrate that a FAK inhibitor preferentially targets tumor-initiating CSCs in MPM. CSCs are thought to play an integral role in metastasis and recurrence following chemotherapy (17). Although several putative CSC markers have been proposed in MPM (18, 19), they have not been rigorously validated. ALDH activity has been used to define CSCs in numerous solid tumor types (17). We validated ALDH activity (Aldefluor) as a marker of CSCs in MPM through use of tumor initiation, tumorsphere self-renewal and in vivo differentiation assays. In murine and human mesothelioma samples, VS-4718 decreased ALDH-positive cells in vitro and in vivo, establishing the FAK inhibitor as an anti-CSC agent in mesothelioma. Recent evidence in the literature implies a potential role for FAK in CSC proliferation, differentiation, motility and invasion (32). Interestingly, distinct roles for kinase-dependent and independent activities of FAK have been suggested in regulation of mammary stem cell biology (33). Our data clearly demonstrate the importance of FAK kinase activity for the maintenance of CSCs in mesothelioma.
Frontline cytotoxic therapy with pemetrexed and cisplatin is the standard of care for treatment of MPM (15). Our data demonstrate that treatment of mesothelioma cells with these agents increases the proportion of CSCs, which may account for the tumor recurrence that invariably follows initial response. Treatment with a FAK inhibitor after completion of first line therapy might extend survival through MPM tumor growth inhibition coupled with the preferential suppression of CSCs. The striking tumor growth delay observed with FAK inhibitor administration after combination treatment with cisplatin and pemetrexed in an MPM patient-derived xenograft model provides preclinical proof of concept for this strategy. Recent data from a Phase I clinical trial demonstrated increased progression free survival (PFS) of MPM patients treated with a FAK inhibitor (GSK2256098). The disease stabilization was especially pronounced in Merlin-low mesothelioma patients (34). Altogether, the data presented here provide strong rationale for a clinical trial of a FAK inhibitor administered to MPM patients after first line therapy with stratification based on Merlin protein expression (ClinicalTrials.gov NCT01870609).
Materials and Methods
Study design
Mouse models of mesothelioma, human MPM patient derived xenografts, MSTO-211H, MDA-MB-468 and MDA-MB-231 s.c. xenograft experiments are described in detail in the Supplementary Methods. Briefly, endpoints of each study included assessment of the anti-tumor activity of compounds based on tumor growth inhibition (TGI) at the end of the study and quantification of pFAK-Y397 in tumor samples. For s.c. models, animals were randomized into treatment or control groups when tumors reached approximately 125–250 mm3 and dosing was initiated. All of the studies were powered to have an 80% (1−β) chance of detecting a significant difference in tumor burden at the end of the study specifying α = 0.05 (two-tailed testing). Control cohorts had tight tumor growth curves, allowing to be adequately powered with experimental recipient cohorts of average size n = 5–10. Original data for animal studies are provided in Table S1. For mouse models of mesothelioma, all animal experiments were performed in accordance with the regulations of Fox Chase Cancer Center’s Institutional Animal Care and Use Committee.
Cell culture and compound treatment
Cell lines used in this study and culture conditions are described in the Supplementary Methods. Cells were treated with VS-4718, pemetrexed, cisplatin, vinorelbine or gemcitabine for 4 days. VS-4718, pemetrexed, vinorelbine or gemcitabine were added at 15 μl/well in a 96-well plate as 10X DMSO solution (n=6 per concentration point). Cisplatin was diluted from 10X NaCl stock solution.
Plasmids, virus production and infection of target cells
The pMXs-NF2-IRES-GFP construct was generated by inserting NF2 cDNA (Open Biosystems, MHS1011-202832561) into pMXs-IRES-GFP retroviral vector (Cell Biolabs). Retroviral packaging, infection, and fluorescence-activated cell sorting (FACS) were performed as previously described (22).
FAK siRNA
A mix of four individual PTK2 siRNAs (ON-TARGETplus PTK2 siRNA; Dharmacon, Thermo Scientific) was used for depletion of FAK in mesothelioma cell lines using DharmaFECT reagent as described in the ‘siRNA Transfection with DharmaFECT Protocol’. siGENOME Non-Targeting siRNA #1was used as a negative control.
Statistics
Shapiro-Wilk normality test was used to determine whether the data was normally distributed. Unpaired t-test with Welch’s correction or a one-way ANOVA was used to determine statistical significance of the results for normally-distributed datasets. Mann Whitney test or Kruskal-Wallis test was used to determine significance of non-normally distributed data. α = 0.05 (two-tailed testing) was used for all statistical measurements. Statistical analysis was performed using Prism 6 software.
Aldefluor assay, limiting dilution assay, immunofluorescence and immunohistochemistry procedures, Matrigel-on-top, western blotting and antibodies, blocking antibody experiments and mouse models are described in the Supplementary Methods.
Supplementary Material
Table S1. Original data from animal in vivo studies.
Fig. S1. Chemical structure of VS-4718.
Fig. S2. Western blot analysis of lysates from cancer cell lines in Fig. 1A.
Fig. S3. pFAK-Y397 levels in MDA-MB-231 and MDA-MB-468 xenograft tumor samples.
Fig. S4. Average EC50 values for Fig. 2B.
Fig. S5. Relative growth of MDA-MB-231/GFP and MDA-MB-231/NF2-GFP cells in response to VS-4718.
Fig. S6. qPCR analysis of FAK cDNA in MSTO-211H and Mero-41 cells.
Fig. S7. Tumor growth inhibition and pFAK-Y397 expression analysis in MSTO-211H xenograft model.
Fig. S8. Merlin and pFAK-Y397 expression in human mesothelioma PDX models.
Fig. S9. pAKT and pFAK activation in Mero-48a and MSTO-211H cell lines.
Fig. S10. pFAK-Y397 immunofluorescence analysis in Mero-41 and Mero-48a cells expressing NF2-GFP.
Fig. S11. N-cadherin blocking antibody treatment in H28 and MSTO-211H cells.
Fig. S12. Bar graph of pFAK-Y397 levels in H28, Mero-25, Mero-83 and Mero-41 cells.
Fig. S13. Tumorsphere self-renewal and in vivo differentiation analysis of Aldefluor+ MM87 cells.
Fig. S14. Effect of VS-4718 treatment on Aldefluor+ CSCs in Mero-83 and Mero-48a MPM cells.
Acknowledgments
We thank Prof. Robert A. Weinberg and the Verastem team for helpful discussions and critical reading of the manuscript. Human PDX xenograft studies were conducted by Champions (Baltimore, MD). Animal husbandry and in vivo procedures for MPM tumor-initiation studies were conducted by ViviSource Laboratories. S.c. xenograft studies were conducted by TGen Drug Development (TD2).
Funding
This work was funded by Verastem, Inc. A.I.M. was also funded by the National Institutes of Health (R01CA113733) and an MGH Research Scholars Award. J.R.T., C.M.V., Y.K. were also supported by NCI grant CA148805.
Footnotes
Author contributions
I.M.S., J.A.P., J.R.T., M.P., V.N.K., Q.X. designed the experiments. I.M.S., V.N.K, C.M.V., J.E.R., Y.K., Q.W. performed the experiments and analyzed the data. D.T.W and M.P. assisted with human and PDX data analysis and interpretation. A.I.M. provided input on data interpretation and on the manuscript. I.M.S., C.M., Q.X., J.R.T., J.A.P. supervised the research. I.M.S. and J.A.P. wrote the manuscript.
Competing interests
I.S., V.K., C.V., J.R., D.W., M.P., Q.X., J.P. are employees and stockholders of Verastem Inc. Q.W. is a stockholder of Verastem Inc. Verastem has issued and pending patents for VS-4718. Y.K., C.M., A.M., J.T. do not have competing financial interests.
Materials and data availability
An MTA approved by Verastem is required to share VS-4718 compound.
References
- 1.McLean GW, et al. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nature reviews Cancer. 2005 Jul;5:505. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 2.Luo M, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer research. 2009 Jan 15;69:466. doi: 10.1158/0008-5472.CAN-08-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nature reviews Molecular cell biology. 2005 Jan;6:56. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 4.Infante JR, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012 May 1;30:1527. doi: 10.1200/JCO.2011.38.9346. [DOI] [PubMed] [Google Scholar]
- 5.Roberts WG, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer research. 2008 Mar 15;68:1935. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 6.Jones SF, Bendell JC, Chen EX, Bedard P, Cleary JM, Pandya S, Pierce KJ, Houk B, Hosea N, Zandi KS, Roberts WG, Shreeve SM, Siu LL. Phase I study of PF-04554878, a second-generation focal adhesion kinase (FAK) inhibitor, in patients with advanced solid tumors. Journal of clinical oncology; 2011; ASCO Annual Meeting Abstracts; 2011. p. abstract 3002. [Google Scholar]
- 7.Polyak K, Garber J. Targeting the missing links for cancer therapy. Nature medicine. 2011 Mar;17:283. doi: 10.1038/nm0311-283. [DOI] [PubMed] [Google Scholar]
- 8.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nature reviews Molecular cell biology. 2002 Aug;3:586. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 9.Li W, Cooper J, Karajannis MA, Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO reports. 2012 Mar;13:204. doi: 10.1038/embor.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaiz C, Utermark T, Parkinson DB, Poetsch A, Hanemann CO. Impaired intercellular adhesion and immature adherens junctions in merlin-deficient human primary schwannoma cells. Glia. 2008 Apr;56:506. doi: 10.1002/glia.20629. [DOI] [PubMed] [Google Scholar]
- 11.Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes & development. 2003 May 1;17:1090. doi: 10.1101/gad.1054603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianchi AB, et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proceedings of the National Academy of Sciences of the United States of America. 1995 Nov 21;92:10854. doi: 10.1073/pnas.92.24.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng JQ, et al. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes, chromosomes & cancer. 1999 Mar;24:238. [PubMed] [Google Scholar]
- 14.Driscoll T, et al. The global burden of disease due to occupational carcinogens. American journal of industrial medicine. 2005 Dec;48:419. doi: 10.1002/ajim.20209. [DOI] [PubMed] [Google Scholar]
- 15.Vogelzang NJ, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003 Jul 15;21:2636. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 16.Podberezin M, Wen J, Chang CC. Cancer stem cells: a review of potential clinical applications. Archives of pathology & laboratory medicine. 2013 Aug;137:1111. doi: 10.5858/arpa.2012-0494-RA. [DOI] [PubMed] [Google Scholar]
- 17.D’Angelo RC, Wicha MS. Stem cells in normal development and cancer. Progress in molecular biology and translational science. 2010;95:113. doi: 10.1016/B978-0-12-385071-3.00006-X. [DOI] [PubMed] [Google Scholar]
- 18.Ghani FI, et al. Identification of cancer stem cell markers in human malignant mesothelioma cells. Biochemical and biophysical research communications. 2011 Jan 14;404:735. doi: 10.1016/j.bbrc.2010.12.054. [DOI] [PubMed] [Google Scholar]
- 19.Yamazaki H, et al. Characterization of cancer stem cell properties of CD24 and CD26-positive human malignant mesothelioma cells. Biochemical and biophysical research communications. 2012 Mar 16;419:529. doi: 10.1016/j.bbrc.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 20.Menges CW, et al. Tumor Suppressor Alterations Cooperate to Drive Aggressive Mesotheliomas with Enriched Cancer Stem Cells via a p53-miR-34a-c-Met Axis. Cancer research. 2014 Feb 15;74:1261. doi: 10.1158/0008-5472.CAN-13-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanjoni I, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer biology & therapy. 2010 May 15;9:764. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer discovery. 2012 Aug;2:706. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breuleux M, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Molecular cancer therapeutics. 2009 Apr;8:742. doi: 10.1158/1535-7163.MCT-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Molecular oncology. 2007 Jun;1:84. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houshmandi SS, Emnett RJ, Giovannini M, Gutmann DH. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Molecular and cellular biology. 2009 Mar;29:1472. doi: 10.1128/MCB.01392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulikakos PI, et al. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene. 2006 Sep 28;25:5960. doi: 10.1038/sj.onc.1209587. [DOI] [PubMed] [Google Scholar]
- 27.Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer research. 2001 May 1;61:3819. [PubMed] [Google Scholar]
- 28.Ahronowitz I, et al. Mutational spectrum of the NF2 gene: a meta-analysis of 12 years of research and diagnostic laboratory findings. Human mutation. 2007 Jan;28:1. doi: 10.1002/humu.20393. [DOI] [PubMed] [Google Scholar]
- 29.Goutagny S, et al. Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clinical cancer research. 2010 Aug 15;16:4155. doi: 10.1158/1078-0432.CCR-10-0891. [DOI] [PubMed] [Google Scholar]
- 30.Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Developmental cell. 2010 Nov 16;19:727. doi: 10.1016/j.devcel.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirt UA, Braunger J, Schleicher M, Weyer-Czernilofsky U, Garin-Chesa P, Bister B, Stadtmueller H, Sapountzis I, Kraut N, Adolf GR. BI 853520, a potent and highly selective inhibitor of protein tyrosine kinase 2 (focal adhesion kinase), shows efficacy in multiple xenograft models of human cancer. Mol Can Ther. 2011;10(Supplement 1):Abstract A249. [Google Scholar]
- 32.Guan JL. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB life. 2010 Apr;62:268. doi: 10.1002/iub.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo M, et al. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer research. 2013 Sep 1;73:5591. doi: 10.1158/0008-5472.CAN-13-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soria JC, et al. Loss of the tumor suppressor merlin as a potential predictive biomarker of clinical activity for the oral, focal adhesion kinase (FAK) inhibitor GSK2256098 in patients with recurrent mesothelioma. EORTC meeting. 2012 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Original data from animal in vivo studies.
Fig. S1. Chemical structure of VS-4718.
Fig. S2. Western blot analysis of lysates from cancer cell lines in Fig. 1A.
Fig. S3. pFAK-Y397 levels in MDA-MB-231 and MDA-MB-468 xenograft tumor samples.
Fig. S4. Average EC50 values for Fig. 2B.
Fig. S5. Relative growth of MDA-MB-231/GFP and MDA-MB-231/NF2-GFP cells in response to VS-4718.
Fig. S6. qPCR analysis of FAK cDNA in MSTO-211H and Mero-41 cells.
Fig. S7. Tumor growth inhibition and pFAK-Y397 expression analysis in MSTO-211H xenograft model.
Fig. S8. Merlin and pFAK-Y397 expression in human mesothelioma PDX models.
Fig. S9. pAKT and pFAK activation in Mero-48a and MSTO-211H cell lines.
Fig. S10. pFAK-Y397 immunofluorescence analysis in Mero-41 and Mero-48a cells expressing NF2-GFP.
Fig. S11. N-cadherin blocking antibody treatment in H28 and MSTO-211H cells.
Fig. S12. Bar graph of pFAK-Y397 levels in H28, Mero-25, Mero-83 and Mero-41 cells.
Fig. S13. Tumorsphere self-renewal and in vivo differentiation analysis of Aldefluor+ MM87 cells.
Fig. S14. Effect of VS-4718 treatment on Aldefluor+ CSCs in Mero-83 and Mero-48a MPM cells.