

Abstract
Rationale
Related transcriptional enhancer factor-1 (RTEF-1) plays an important role in endothelial cell function by regulating angiogenesis. However, the mechanism underlying the role of RTEF-1 in the endothelium in vivo is not well defined.
Objective
We investigated the biological functions of RTEF-1 by disrupting the gene that encodes it in mice endothelium (RTEF-1-/-).
Methods and Results
RTEF-1-/- mice showed significantly increased blood glucose levels and insulin resistance, accompanied by decreased levels of insulin-like growth factor binding protein-1 (IGFBP-1) mRNA in the endothelium and decreased serum IGFBP-1 levels. Additionally, the RTEF-1-/- phenotype was exacerbated when the mice were fed a high fat diet, correlating with decreased IGFBP-1 levels. In contrast, vascular endothelial-cadherin (VE-Cad)/RTEF-1 overexpressing transgenic mice demonstrated improved glucose clearance and insulin sensitivity in response to a high fat diet. Furthermore, we demonstrated that RTEF-1 up-regulates IGFBP-1 through selectively binding and promoting transcription from the insulin response element (IRE) site. Insulin prevented RTEF-1 expression and significantly inhibited IGFBP-1 transcription in endothelial cells in a dose-dependent fashion.
Conclusion
To our knowledge, this is the first report demonstrating that RTEF-1 stimulates promoter activity through an IRE element and also mediates the effects of insulin on gene expression. These results show that RTEF-1-stimulated IGFBP-1 expression may be central to the mechanism by which RTEF-1 attenuates blood glucose levels. These findings provide the basis for novel insights into the transcriptional regulation of IGFBP-1 and contribute to understanding the role of vascular endothelial cells in metabolism.
Keywords: RTEF-1, IGFBP, vascular endothelium, metabolic syndrome
Introduction
The endothelium is a complex organ with a multitude of properties essential for controlling vascular functions. Dysfunction of the vascular endothelium plays an important role in the pathogenesis of diabetic micro- and macro-angiopathy. Sustained hyperglycemia in metabolic syndrome causes alterations in a large number of transcription factors and mRNA transcripts, ultimately leading to tissue damage. However, transcription factors involved in glucose homeostasis in response to insulin stimulation in the endothelium have not been fully studied. Previous studies from our laboratory have demonstrated that related transcriptional enhancer factor-1 (RTEF-1, also known as TEF-3 or TEAD4) plays an important role in endothelial cells via regulating angiogenesis1,2. More recently, studies have indicated that RTEF-1 drives communication between the endothelium and myocardium3, as well as enhances endothelial-dependent microvascular relaxation4. RTEF-1 is a member of the transcriptional enhancer factor (TEF) family5-7, a multigene family comprised of four vertebrate members that share a highly conserved DNA binding domain capable of binding to M-CAT elements found in the promoters of many genes expressed in cardiac and skeletal muscle5,8,9. In endothelial cells, RTEF-1 is induced by hypoxia as a result of enhanced vascular endothelial growth factor (VEGF) promoter activity2. In addition, transgenic mice overexpressing RTEF-1 specifically in the endothelium show a transcriptional regulation of Hypoxia-inducible factor-1α (HIF-1α), as well as angiogenic ability in the hind limb ischemia model compared with wild-type1. Ablation of the mouse RTEF-1 gene causes defects in the production of trophoblast stem cells, trophectoderm or blastocoel cavities and consequently preimplantation lethality10.
Insulin-like growth factor binding proteins (IGFBPs) are key regulators of insulin-like growth factor (IGF) type 1 and 2 bioavailability at the cellular level, and may also exert IGF-independent effects on target cells. The IGFBP superfamily contains six high affinity binding IGFBPs and nine low affinity binding IGFBP related proteins11-13. Growing evidence has associated large and small vessel diseases with certain IGFBP-1 abnormalities, suggesting that they may be significant factors in the pathophysiology of cardiovascular disease (CVD)14,15. Furthermore, acute steady-state hyperinsulinemia decreases serum IGFBP-1 concentration to values that are 40–70% lower than baseline in normal individuals as well as in diabetic patients, suggesting that insulin is involved in the regulation of serum IGFBP-1 levels16,17. Moreover, low levels of IGFBP-1 are associated with metabolic syndrome and cardiovascular diseases in cross-sectional studies 18,19 and can predict ischemic heart disease mortality20.
In this report, we examined RTEF-1 involvement in the endothelium-related effect of glucose regulation and its potential transcriptional target in vivo by using diet-induced obese transgenic mice, and in vitro using cultured endothelial cells to analyze the related mechanism. We demonstrate that RTEF-1 increases IGFBP-1 gene expression through interacting with its insulin response element. Evidence for a biologic role of RTEF-1 in IGFBP-1 regulation includes decreased expression of IGFBP-1 in endothelial specific RTEF-1 knockout mice (RTEF-1-/-), and in endothelial cells deficient in RTEF-1. Conversely, increased IGFBP-1 expression was observed in overexpressing RTEF-1 transgenic mice and in endothelial cells with forced RTEF-1 overexpression. Additionally, RTEF-1-/- mice exhibited increased blood glucose and insulin sensitivity, which was exacerbated in a high fat diet, correlating with decreasing IGFBP-1 levels. Our findings suggest that RTEF-1 is integral in the endothelial regulation of IGFBP-1 and subsequent blood glucose homeostasis.
Materials and Methods
Generation of RTEF-1 transgenic mice
A conditional knockout (KO) line of RTEF-1 was generated by crossing homozygous TEAD4lox/lox mice (a gift from Dr. Andres Buonanno, NICHHD, NIH) with transgenic mice expressing Cre recombinase under control of the endothelial cell-specific Tie2 promoter/enhancer (a gift from Dr. Anthony Rosenzweig, BIDMC)21. RTEF-1 transgenic mice were generated at the BIDMC Transgenic Core Facility using the vascular endothelial (VE)-cadherin promoter to drive endothelial-specific expression of human RTEF-122. The investigation conforms to the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, 1996) and was approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center. For detailed protocols, please refer to the Online Methods section.
Cell culture and transfection
Human microvascular endothelial cells (HMEC-1, CDC) were cultured in MCDB-131, and human embryonic kidney cells 293 (HEK 293) were cultured in DMEM. Wild-type IGFBP-1 (IGFBP-1FL) or TK.IRS3 or VF-2 mutant promoter luciferase construct, control vector, PXJ40 and/or an increasing amount of RTEF-1 expression vector (generous gifts from Dr. Alexandre Stewart, University of Ottawa, Canada) were transfected into HEK 293 cells, as shown in more detail in the Online Methods section.
Retroviral construction
The RTEF-1 (NM_014059.1) coding sequence was cloned into pBMN–green fluorescence protein (GFP) vector (Obigen, CA) for retrovirus packaging. pBMN-GFP or pBMN-GFP– RTEF-1 was transfected to 293T using polyethylenimine with pVSV-G, pJK3, and pCMVtat. The medium with retrovirus/RTEF-1 or retrovirus/GFP control was collected and filtered before being used to infect HMEC-1 cells.
siRNA transfection
siRNAs targeting human RTEF-1 were synthesized by Genepharma, Inc. (Shanghai, China). Knockdown efficiency of two duplexes of RTEF-1 siRNAs or a nontarget control was determined by transfection into HMEC-1 cells at a final concentration of 50 nM according to the manufacturer's protocol. For details on this protocol please refer to the Online Methods section.
Quantitative real-time PCR analysis
Total RNA was extracted from the apex of mouse hearts from wild type and transgenic mice, as well as the RTEF-1 o/e and GFP stably expressing HMEC-1 cell lines A total of 2.0μg of RNA from both endothelial cell lines stably transfected with RTEF-1 or isolated from the apex of wildtype (WT), RTEF-1 o/e and RTEF-1 EC-specific KO transgenic mouse hearts were reverse-transcribed. Quantitative real-time PCR (QPCR) amplification was done using SYBR Green Master Mix (Applied Biosystems, CA) according to the manufacturer's protocol, as outlined detail in the Online Methods section.
ELISA and blood glucose analysis
Blood glucose levels were obtained from feed-deprived (overnight), restrained un-anesthetized mice. Blood was obtained via submandibular bleed, and plasma IGFBP-I concentration was measured by ELISA Insulin resistance (IR) was assessed from fasting insulin and glucose levels by the previously validated homeostasis model assessment (HOMA-IR)23. Please refer to the Online Methods section for details.
Metabolic studies
GTT and ITT assays were performed in 5-month-old conscious WT and transgenic mice For GTT glucose was injected intraperitoneally. Blood glucose was measured by tail bleeding at serial time points after glucose injection. For ITT, mice were injected with insulin and blood glucose was measured at the same timepoints for GTT, as shown in detail in the Online Methods section.
Promoter activity and chromatin immunoprecipitation (ChIP)
HEK293 cells were transfected with constructs with or without RTEF-1, indicated in the figure legends. Luciferase activity was determined using the dual luciferase assay system (Promega). Chromatin immunoprecipitation (ChIP) was performed with the ChIP-IT Express Kit (Active Motif, Carlsbad, CA) in accordance with the manufacturer's instructions, with primers shown in detail in the Online Methods section.
Immunoblot analysis
RTEF-1 and GFP-stably transfected HMEC-1 cells were lysed and protein concentrations were determined. Samples were subjected to SDS–PAGE, transferred to nitrocellulose membranes (Whatman, Springfield Mill, UK) and subsequently blocked in TBS–Tween 20 containing 5% non-fat milk for 1h. The membranes were incubated with the indicated primary antibodies: polyclonal anti-IGFBP-1 antibody; polyclonal anti-RTEF-1 antibody, monoclonal anti-vinculin followed by incubation with horseradish peroxidase-conjugated secondary antibodies anti-rabbit or anti-mouse IgG Blots were developed using the chemiluminescence detection system according to the instructions of the manufacturer (Thermo Fisher, PA). Densitometric analysis was done using the NIH software program, Image J. Please see Online Materials section for more details.
Immunofluorescence
Hearts were removed from transgenic and littermate control mice, embedded in O.C.T. compound and frozen at -80°C. Tissues were sectioned, fixed in 4% paraformaldehyde, and stained with antibodies against RTEF- and followed by incubation with goat anti-rabbit FITC and goat anti-mouse TRITC secondary antibodies. Immunofluorescence stained sections were visualized with a DM5000B upright microscope, as shown in the detailed Online Methods section.
Blood pressure measurements
Blood pressure was measured non-invasively on conscious mice using a volume pressure recording tail-cuff system (CODA™ system by Kent Scientific Corporation). Additionally, blood pressure was confirmed using telemetry, as described in detail in the Online Methods section.
NMR determination of body composition
Body composition was measured in both WT and RTEF-1-/- on both a NCD and a HFD using quantitative nuclear magnetic resonance (NMR). Live, conscious, unrestrained mice were placed in small tubes and inserted into a Brucker model mq10 NMR analyzer (Brucker, Canada, Milton ON, Canada). Total fat, lean and water mass were recorded after less than 1 min. Measurements were done in triplicate.
Statistical analysis
Data was obtained from at least three independent cell cultures or animals, as denoted in the figure legends. Data are presented as means ± SEM. The trapezoidal rule was used to determine the area under the curve (AUC). Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as (fasting glucose level × fasting insulin level)/22.4. The level of statistical significance was determined using Student's two-tailed t-test when differences between the means of two populations were considered. Comparison of multiple time points between groups was made using a 1-way or 2-way repeated measures ANOVA.
Results
IGFBP-1 expression is attenuated in RTEF-1-deficient transgenic mice
RTEF-1 is one of the earliest transcription factors expressed during mammalian development and elimination of RTEF-1 expression in mice causes cardiac defects resulting in embryonic lethality10,24. To examine the importance of the RTEF-1 gene in endothelium, we generated a conditional endothelial cell-specific RTEF-1 KO mouse (RTEF-1-/-). To conditionally delete the functional mouse RTEF-1 locus, two loxP sites were inserted into the introns flanking its second exon (Fig. 1A). The targeting vector contained a pGK-neo cassette for positive selection, and a pGK-tk cassette for negative selection against non-homologous recombinants10. Homozygous mice were bred as previously described10 and mated with Tie2-Cre mice that express Cre recombinase specifically in endothelial cells21. RTEF-1-/- mice were genotyped by RT-PCR (Fig. 1B). There were no gross morphological or developmental changes in the RTEF-1-/- transgenic mice and no evidence indicating that strong expression of Cre recombinase-induced abnormalities in wild-type littermate mice. Additionally, to rule out the potential loss of RTEF-1 in hematopoietic cells by using a Tie2-Cre promoter, bone marrow from WT and RTEF-1-/- mice were analyzed for RTEF-1 levels in Supplemental Figure I. No significant differences were found.
Figure 1. Generation of endothelial-specific RTEF-1-/- transgenic mice.

A. Schematic of the conditional knockout (RTEF-1-/-) construct, including the Tie2Cre promoter. The conditional RTEF-1-/- line was generated by crossing homozygous TEAD4lox/lox mice with transgenic mice expressing Cre recombinase under control of the endothelial cell-specific Tie2 promoter/enhancer. B. Mice were screened by PCR to verify germline transmission C. Hearts from both RTEF-1-/- and littermate control mice were isolated and examined for RTEF-1 and IGFBP-1 mRNA levels. Results are presented in-terms of a fold change after normalizing with GAPDH mRNA ± SD (n=3, * p≤0.05; p ** p≤0.01). D. Immunoblot analysis of hearts from both WT and RTEF-1-/- (top panel) showed a significant decrease in both RTEF-1 expression and IGFBP-1 expression. Quantitative densitometry data are shown in the bottom panel, (n=3, * p≤0.05). E. Images of heart sections from WT and RTEF-1-/- mice immunostained for DAPI, CD31, together with RTEF-1. Merged images of the left and middle columns are shown in the right column with DAPI staining, showing that RTEF-1 is not present in the RTEF-1-/- endothelium (representative picture from n = 4).
Originally, DNA microarray data from RTEF-1 overexpressing endothelial cells illustrated that a handful of insulin-regulated genes had significant changes including IGFBP family members (data not shown), which was verified by quantitative PCR (Fig. 1C) and western blot (Fig. 1D). IGFBP-1 levels were significantly decreased in the apex of the RTEF-1-/- hearts compared with littermate controls. We further confirmed that the absence of the RTEF-1 was localized in the endothelium by double immunostaining with antibodies to endothelial marker CD31 and RTEF-1 in the hearts from RTEF-1-/-and littermate controls (Fig. 1E).
The effect of RTEF-1 ablation on diet-induced obesity
We next examined the metabolic effect of RTEF-1 deletion on mice fed a normal chow diet (NCD) versus a high fat diet (HFD). Five-week-old male RTEF-1-/- and WT mice were fed either NCD or HFD (42% kcal from fat) for 15 weeks. The HFD is a well-established model for obesity-induced insulin resistance25. Although the HFD increased growth rate and final body weight compared with the mice on the NCD, RTEF-1 did not significantly alter body weight with either diet (Table 1), nor did it change the percentage of skeletal muscle or adipose tissue in WT and RTEF-1-/- mice (Supplemental Figure IIA and B). Obesity is typically associated with decreased insulin sensitivity and elevated circulating concentrations of glucose and insulin. Initially, blood glucose levels were screened in RTEF-1-/- mice and WT mice, and, as shown in Fig. 2A, fasting blood glucose levels were modestly increased in the RTEF-1-/- compared with WT mice on the NCD. Despite having a similar body weight and fat pad mass, overnight fasting blood glucose (Fig. 2A) and 6 hour fasting plasma insulin concentrations (Fig. 2B) were increased in obese RTEF-1-/- mice compared with littermate controls on the HFD, suggesting an impaired insulin sensitivity. Serum insulin levels were increased approximately 2-fold (Fig. 2B) in RTEF-1-/- versus littermate controls fed both the NCD and HFD. Furthermore, obese RTEF-1-/- mice demonstrated a decrease in IGFBP-1 levels by approximately 4-fold compared with their littermate controls (Fig. 2C). Additionally, serum IGFBP-1 levels were decreased in the WT HFD cohort compared with the previous WT NCD (Fig. 2C), suggesting that obesity decreases serum IGFBP-1 levels.
Table 1. Systolic BP and cardiac function of the RTEF-/- compared with the control littermates.
| WT | RTEF-/- | p value | WT p value | RTEF-/- p value | ||
|---|---|---|---|---|---|---|
| NCD vs HFD | NCD vs HFD | |||||
| Body weight (g) | NCD | 28.4 ± 3.1 | 30 ± 1.1 | 0.23 | ||
| HFD | 37.2 ± 3.8 | 38.45 ± 2.6 | 0.43 | 0.0008 | 0.006 | |
| Body composition | ||||||
| Lean mass | NCD | 68.2 ± 5.2 | 69.72 ± 6 | 0.62 | ||
| HFD | 55.7 ± 5.4 | 49.81 ± 5.8 | 0.65 | 0.001 | 0.002 | |
| Fat mass | NCD | 12.2 ± 5.7 | 11.72 ± 7.1 | 0.41 | ||
| HFD | 29.9 ± 8.9 | 36.87 ± 6.6 | 0.52 | 0.0007 | 0.0002 | |
| Heart Rate (bpm) | NCD | 674.9 ± 55.7 | 704.8 ± 38.2 | 0.08 | ||
| HFD | 711.9 ± 32.6 | 736.7 ± 25.8* | 0.03 | 0.12 | 0.0007 | |
| Systolic BP (mmHg) | NCD | 99.7 ± 3 | 109.1 ± 7 | 0.07 | ||
| HFD | 106.5 ± 5 | 111 ± 6 | 0.32 | 0.04 | 0.88 | |
| % FS | NCD | 51.0 ± 3.6 | 50 ± 1.4 | 0.57 | ||
| HFD | 45.8 ± 2.9 | 37.5 ± 6.2** | 0.01 | 0.02 | 0.001 | |
| LVDd | NCD | 2.61 ± 0.1 | 3.21 ± 0.2** | 0.01 | ||
| HFD | 3.5 ± 0.3 | 3.53 ± 0.2 | 0.11 | 0.89 | 0.07 | |
| LVDs | NCD | 1.27 ± 0.04 | 1.58 ± 0.06* | 0.04 | ||
| HFD | 2.2 ± 0.03 | 1.64 ± 0.1** | 0.0008 | 0.06 | 0.08 | |
| IVSd + LVPWd | NCD | 1.48 ± 0.06 | 1.44 ± 0.1 | 0.56 | ||
| Total Wall Thickness | HFD | 1.3 ± 0.1 | 1.15 ± 0.2 | 0.74 | 0.04 | 0.04 |
| IVSs | NCD | 1.6 ± 0.03 | 1.37 ± 0.08* | 0.03 | ||
| HFD | 1.3 ± 0.04 | 1.1 ± 0.1** | 0.01 | 0.00002 | 0.02 | |
| LVPWS | NCD | 1.3 ± 0.1 | 1.2 ± 0.1 | 0.6 | ||
| HFD | 1.0 ± 0.05 | 0.83 ± 0.09** | 0.01 | 0.23 | 0.54 |
Figure 2. RTEF-1-/- transgenic mice have lower IGFBP-1 levels and increased HOMA-IR.

A. Blood glucose levels were obtained from food-deprived (overnight), restrained, unanesthetized mice. Blood was obtained via submandibular bleed, and glucose was quantified using a CVS TRUEtrack glucose monitor at 8 weeks of age on WT and RTEF-1-/- mice fed a normal chow or high fat diet. Blood glucose was significantly higher in the transgenic mice that lacked RTEF-1 in the endothelium in both the NCD and HFD (*p=0.022; n = 18 mice). B. Serum insulin concentration was measured by ELISA with a maxiumum absorbance of 450 nm. The limit of sensitivity is at or above 0.121ng/ml. C. Serum IGFBP-I concentration was measured by ELISA. IGFBP-1 concentration was measured with a maximum absorbance of 450 nm. The limit of sensitivity is at or above 31.2 pg/ml. (n = 3; * p=0.035). The IGFBP-1 levels inversely correlated with blood glucose levels in these mice.
Insulin resistance in obese RTEF-1-/- mice
We evaluated insulin sensitivity and glucose homeostasis in RTEF-1-/- and littermate controls fed either a normal chow or high fat diet. As shown in Fig 3A and B, the ability to regulate blood glucose levels in response to an intraperitoneal glucose (IPGTT) (1 g/kg body weight) or insulin (IPITT) (0.75unit/kg) challenge was not significantly impaired in the transgenic mice versus WT mice maintained on the NCD. In contrast, glucose tolerance was significantly impaired in the obese RTEF-1-/- mice compared to the littermate controls (Fig. 3C). Following intraperitoneal administration of insulin (0.75 unit/kg), glucose excursion from blood was slightly increased in RTEF-1-/- mice compared with the wild-type mice maintained on the HFD (Fig. 3D). However, RTEF-1-/- mice fed a high fat diet were significantly insulin resistant compared with littermate control mice. Additionally, the area under the curve (AUC) was calculated and is significantly different in the transgenic RTEF-1-/- mice fed a HFD, further indicating their insulin resistance (Supplemental Figure IIIA and B). Insulin resistance was also assessed via HOMA-IR and was significantly increased in the normal chow and obese RTEF-1-/- mice compared with littermate controls, indicating that the ability of circulating insulin to regulate fasting blood glucose levels was impaired (Supplemental Figure 3A and B).
Figure 3. Obese RTEF-1-/- transgenic mice have decreased glucose tolerance and increased insulin resistance.

A. Glucose tolerance in WT and RTEF-1-/-; glucose was measured before, and 30, 60, and 120 min after IP glucose injection at 15 weeks of age. B. Insulin tolerance in WT and RTEF-1-/- was analyzed; glucose was measured before, and 30, 60, and 120 min after IP insulin injection. Data are presented as mean ± S.D. (n = 7–9 animals per group). *, p≤0.05 and **, p≤ 0.01 for RTEF-1-/- vs. WT. C. Glucose tolerance in WT and RTEF-1-/-; mice was measured before, and 30, 60, and 120 min after IP glucose injection. D. Insulin tolerance in WT and RTEF-1-/- mice at 15 weeks of HFD; glucose was measured before, and 30, 60, and 120 min after IP insulin injection. Data are presented as mean ± S.D. (n = 7–9 animals per group). *, p≤0.05 and **, p≤ 0.01 for RTEF-1-/- vs. WT.
The effect of RTEF-1 ablation on cardiac function
To assess the effect of endothelium-specific ablation of RTEF-1 and obesity on the heart, cardiac parameters of RTEF-1-/- mice were assessed (Table 1). No significant difference in heart rate or systolic blood pressure was detected between the groups. Additionally, telemetry was used to confirm the blood pressure measurements taken in the RTEF-1-/- and control mice by a VPR tail-cuff system (Supplementary Figure IV). Although no difference in blood pressure was detected, echocardiography revealed a significant decline of heart function in both control and RTEF-1-/- mice fed a high fat diet (Table 1; 45.8 ± 2.9 vs. 37.5 ± 6%).
RTEF-1 induced IGFBP-1 expression in endothelial cells
To confirm that IGFBP-1 might be a potential target gene of RTEF-1 and investigate the mechanism involved, RTEF-1 was overexpressed in human microvascular endothelial cells (HMEC-1) by retroviral infection. RTEF-1 and IGFBP mRNA were examined by QPCR (Fig. 4A). IGFBP-1 mRNA expression, as well as secreted IGFBP-1 (Fig. 4B) were markedly increased in RTEF-1 o/e cells compared with control cells. To verify that RTEF-1 is necessary for increasing IGFBP-1 levels, siRNAs were used to knockdown RTEF-1 expression. RTEF-1 levels were significantly decreased in HUVEC cells, resulting in an almost 70% reduction in IGFBP-1 levels (Fig. 4C). When HMEC-1 cells were treated with insulin, IGFBP-1 mRNA levels decreased (Fig. 4D). Increasing insulin concentrations also dose-dependently decreased RTEF-1 mRNA levels (Fig. 4E). Furthermore, when cells were treated with RTEF-1 siRNA, insulin was unable to significantly regulate IGFBP-1 levels compared with control (Fig. 4F).
Figure 4. RTEF-1 is required for IGFBP-1 gene expression in endothelial cells.

A. Quantitative real time PCR (QPCR) analysis of IGFBP-1 mRNA levels in HMEC-1 cells o/e RTEF-1. IGFBP-1 levels were increased at least 5 fold compared with control cells. B. Immunoblot analysis of IGFBP-1 levels in the concentrated conditioned media from RTEF-1 o/e HMEC-1 cells. C. QPCR analysis of RTEF-1 and IGFBP-1 levels in HMEC-1 cells treated with either 1 of 2 targeting siRNAs to RTEF-1 or nontarget control siRNAs. D. QPCR analysis of RTEF-1 and IGFBP-1 levels in HMEC-1 cells treated with or without insulin (ng/ml). E. QPCR analysis of RTEF-1 levels in HUVEC cells treated with increasing insulin concentrations. F. QPCR analysis of RTEF-1 levels in HMEC-1 cells treated with RTEF-1 siRNA or nontarget control siRNAs with or without 100 ng/ml insulin. These results are from three independent experiments (mean ± S.D.). * p ≤0.05; ** p≤ 0.01.
IGFBP-1 is a potential target gene of RTEF-1
To determine whether IGFBP-1 was regulated by RTEF-1 on a transcriptional level via the IGFBP-1 promoter, we performed luciferase reporter assays. The IGFBP-1 promoter exhibited an RTEF-1 dose-dependent increase in activity, exhibiting a maximum of a 3.7 ± 0.4-fold increase (Fig. 5B). Many of the important cis-regulatory elements are located within 500 bp of the transcription start site in the IGFBP-1 promoter (Fig. 5A). The hepatocyte nuclear factor 1 (HNF1) binding region, cAMP response element (CRE), insulin response element (IRE), gluococorticoid response element (GRE) and TATA element are highly conserved among the human, rat, baboon and mouse IGFBP-1 promoters26. Previously, it has been shown that RTEF-1 regulates its target genes through the M-CAT element, and can also transcriptionally regulate VEGF through its Sp1 site2. Analysis of the IGFBP-1 promoter revealed that there were no typical M-CAT sites or Sp1 sites on the IGFBP-1 promoter, so we examined whether this effect of RTEF-1 is mediated through its insulin response element (IRE) (Fig 5A). Additionally, IGFBP-1 has been previously reported to be regulated by other transcription factors, such as FOXO1, through its IRE27,28. The IGFBP-1 promoter contains an IRE including 2 insulin response sites (IRSs) located approximately 100 bp 5′ to the RNA cap site, IRSA (CAAAACAA) and IRSB (TTATTTTG), and each is sufficient to mediate the negative effects of insulin on promoter activity. To determine if RTEF-1 possibly regulates IGFBP-1 via its IRS, an array of IRSs (TK.IRS3) were introduced immediately upstream from the thymidine kinase promoter27. The TK.IRS3 was sufficient to confer effects of RTEF-1 on promoter activity (Fig. 5C and D), as evidenced by the 4.9 ± 0.03-fold increase in the TK.IRS3 luciferase reporter construct. This was confirmed by the inability of RTEF-1 to increase promoter activity in the IGFBP-1 VF2 mutant, which lacked the IRE sequence (Fig. 5B and E). Additionally, Fig. 5D demonstrates that the IRE mutation disrupted the ability of RTEF-1 to stimulate IGFBP-1 promoter activity in a dose-dependent manner, whereas the IRS sequence was sufficient to confer effects of RTEF-1 on IGFBP-1 promoter activity in Fig. 5C.
Figure 5. RTEF-1 up-regulates IGFBP-1 transcription through its IRE site.
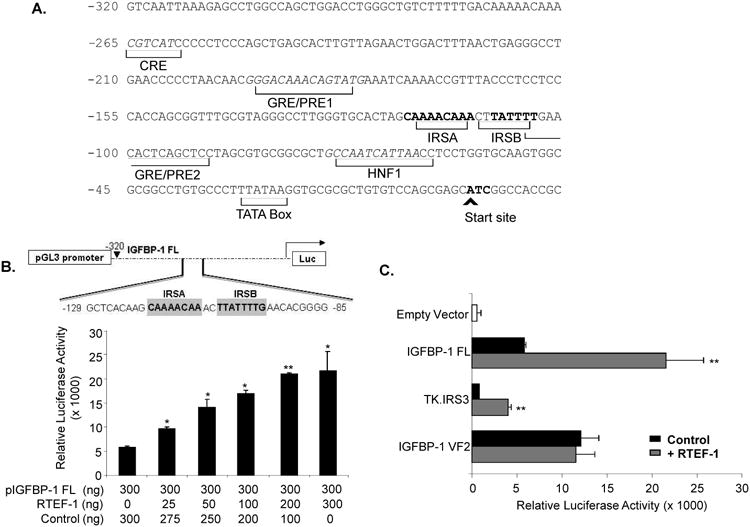
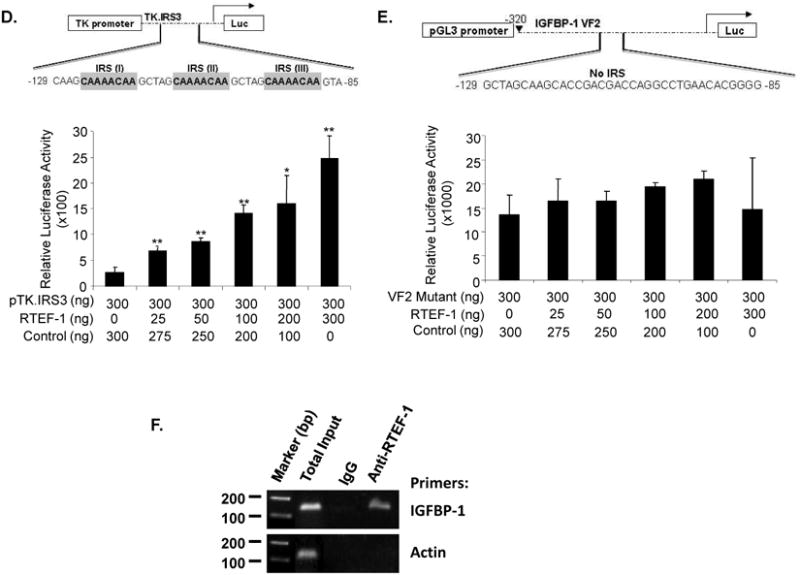
A. IGFBP-1 full length (FL) promoter sequences designating binding elements. B. Schematic of IGFBP-1 promoter. IGFBP-1 full length promoter was transiently co-transfected with increasing concentrations of RTEF-1, and luciferase activity was examined. C. Transient transfections using a set of IGFBP-1 promoter luciferase constructs (300ng) and control vector (600ng, black bar) or equal amount of RTEF-1 cDNA (grey bar). D. The truncated IRS-3 repeat promoter was transiently co-transfected with increasing concentrations of RTEF-1, and luciferase activity was examined. E. The mutant VF-2 promoter was transiently cotransfected with increasing concentrations of RTEF-1, and luciferase activity was examined. F. ChIP assays were performed by immunoprecipitating chromatin from HMEC-1 cells with control IgG or an anti-RTEF-1 antibody and performing RT-PCR with primers to the IRE site on the IGFBP-1 promoter or another control to the actin promoter. The results are from three independent experiments (mean ± S.D.). *, p ≤0.05; p≤ 0.01
To further investigate the IRE as the key element in RTEF-1 regulating IGFBP-1 transcription, a chromatin immunoprecipitation (ChIP) assay was performed (Fig 5F). Primers were designed to flank the IRE sequence of IGFBP-1 and the ChIP assay demonstrated that RTEF-1 specifically bound to the IGFBP-1 IRE promoter region. Taken together, these results imply that RTEF-1, much like the forkhead transcription factors, exerts effects on IGFBP-1 promoter activity through its IRE.
Overexpression of RTEF-1 in the endothelium improved glucose tolerance in obese mice
Some of the physiological consequences of diet-induced obesity are insulin resistance, glucose intolerance, and the eventual development of overt diabetes. We evaluated whether mice overexpressing RTEF-1 showed any changes in glucose tolerance after prolonged exposure to a HFD. Interestingly, Figure 6A demonstrates that blood glucose levels were increased in the high-fat fed WT mice compared with the VE-Cad/RTEF-1 mice. In correlation with the previous data in the RTEF-1-/- mice, serum IGFBP-1 levels in both WT and VE-Cad/RTEF-1 levels were decreased in response to the high fat fed diet, however, VE-Cad/RTEF-1 mice have significantly higher levels compared to WT (Fig. 6B).
Figure 6. Obese VE-Cad/RTEF-1 o/e mice show restored glucose tolerance and circulating IGFBP-1.

A. Blood glucose levels were obtained from food-deprived (overnight), restrained, unanesthetized mice. Blood was obtained via submandibular bleed, and glucose was quantified using a CVS TRUEtrack glucose monitor on WT and VE-Cad/RTEF-1 mice fed a high fat diet. Blood glucose was significantly lower in the transgenic mice that overexpressed RTEF-1 in the endothelium (* = p≤0.05; n = 18 mice). B. Serum IGFBP-I concentration was measured by ELISA. IGFBP-1 concentration was measured with a maximum absorbance of 450 nm. The limit of sensitivity is at or above 31.2 pg/ml. (n = 3; * = p≤0.05). The IGFBP-1 levels inversely correlated with the blood glucose levels in these mice. C. Serum insulin concentration was measured by ELISA with a maxiumum absorbance of 450 nm. The limit of sensitivity is at or above 0.121 ng/ml. E. Glucose tolerance in WT and VE-Cad/RTEF-1; glucose was measured before, and, 30, 60, and 120 min after IP glucose injection. F. Insulin tolerance in WT and VECad/RTEF-1 mice at 15 weeks of HFD; glucose was measured before, and, 30, 60, and 120 min after IP insulin injection. Data are presented as mean ± S.D. (n = 7–9 animals per group). *, p≤0.05 and **, p≤ 0.01 for VE-Cad/RTEF-1 vs. WT.
To determine if IGFBP-1 levels were affected in the VE-Cad/RTEF-1 mice, we analyzed IGFBP-1 levels in these mice. Fig. 6C demonstrates that VE-Cad/RTEF-1 mice had significantly higher IGFBP-1 serum levels in comparison with littermate controls, despite the decrease resulting from the HFD. In addition, both the AUC and HOMA-IR were significantly increased in the WT mice compared with the VE-Cad/RTEF-1 transgenic mice, indicating that the ability of circulating insulin to regulate fasting blood glucose levels is not significantly impaired in VE-Cad/RTEF-1 mice (Supplemental Figure IIIC). When IPGTT experiments were performed after 24 weeks of a high-fat diet, VE-Cad/RTEF-1 mice displayed a markedly improved glucose tolerance and insulin sensitivity compared with the WT mice fed a similar high-fat diet, suggesting protection from the HFD-induced insulin resistance.
Discussion
Our present studies revealed an important role of RTEF-1 in the regulation of IGFBP-1 and potential glucose homeostasis. First, we demonstrated that IGFBP-1 is a target gene of RTEF-1, and that RTEF-1 can increase IGFBP-1 levels in the endothelium both in vitro and in vivo. Second, we identified an IRE element in the IGFBP-1 promoter that RTEF-1 directly binds to. Third, ablation of RTEF-1 from the endothelium significantly decreased circulating IGFBP-1 serum levels, and increased glucose intolerance, insulin resistance and decreased fractional shortening. Finally, IGFBP-1 expression regulated by RTEF-1 overexpression resulted in improved glucose tolerance and insulin sensitivity.
As a member of the transcriptional enhancer factor (TEF) family, RTEF-1 binds to the M-CAT regulatory element in promoters of muscular genes to direct gene expression5. However, the abilities of RTEF-1 in non-muscle cells have not been fully investigated. We have previously reported that RTEF-1 is involved in hypoxia-induced angiogenesis through its target genes including HIF-1α1 and VEGF3 in endothelial cells, and that RTEF-1 increases endothelial-specific coronary microvascular relaxation55, indicating an importance in further understanding the role of RTEF-1 in endothelium. Endothelium-specific RTEF-1-deficient transgenic mice provide an efficient model to dissect the function of RTEF-1 in endothelium. Interestingly, RTEF-1-/- transgenic mice fed a normal chow diet were normoglycemic in the fasting state, appeared to be glucocompetent when assessed by an intraperitoneal glucose tolerance test, and showed only slightly impaired insulin sensitivity. In contrast, when these mice were fed a high fat diet, both the transgenic mice and littermate controls demonstrated impaired glucose tolerance, hyperinsulinemia, and impaired insulin sensitivity. However, the RTEF-1-/- mice showed significantly increased hyperglycemia, impaired glucoregulation and HOMA-IR scores, the main features of insulin-resistant states. These findings revealed a novel ability of RTEF-1, as a transcription factor, to be involved in glucose regulation via IGFBP-1.
The present study indicates that IGFBP-1 is a potential target gene of RTEF-1 and plays a key role in RTEF-1-regulation of glucose homeostasis. Previous reports indicated that lower concentrations of IGFBP-1 were found in subjects with diabetes and macrovascular disease29,30. Current findings substantiate the conclusion that IGFBP-1 is negatively associated with insulin resistance31-35. This indicates IGFBP-1 may simply be a marker of metabolic improvements and reduced insulinemia; yet more current research suggests that IGFBP-1 exerts direct metabolic effects. For example, IGFBP-1 binds integrin receptors which leads to cross-talk between integrin receptors and insulin signaling pathways that may lead to increased insulin sensitivity36,37. In human longitudinal studies, IGFBP-1 concentrations were lower in obese children with metabolic syndrome as compared to obese children without35. Additionally, mean IGFBP-1 levels were significantly lower in human subjects with abdominal obesity (p<.001), elevated fasting glucose (p<.001), hypertension (p<.001), low high-density lipoprotein cholesterol (p<.001), and metabolic syndrome (p<.001) compared to control subjects without these metabolic abnormalities32. In rodent models, overexpression of IGFBP-1 resulted in dysregulation of the insulin/IGFBP axis and is a model of reduced sensitivity of IGFBP-1 to insulin regulation38,39. However, more recent studies in mice that backcrossed human-IGFBP-1 to a C57BL/6 background demonstrated that IGFBP-1 had a protective effect on susceptibility to glucose intolerance and insulin resistance when challenged with nutritional obesity, which correlates with other recent data pooled from rodent studies on IGFBP-1 in glucose regulation40.
In order to clarify more precise information regarding RTEF-1's role in the regulation of IGFBP-1 and the physiological effect, we used an endothelial cell-specific conditional knockout (Tie2-Cre/loxp) and an overexpressing (VE-Cad) transgenic model. Tie2 is not only expressed in the endothelium, but also in early hematopoietic progenitors, which differentiate into hematopoietic, lymphoid, and endothelial cell lineages21,41. While no significant differences were found in RTEF-1-/- mice in hematopoietic cells (Supplemental Figure I), it is possible that this could be due to the low expression of RTEF-1 in the bone marrow, and deletion of RTEF-1 in hematopoietic and lymphoid cells may contribute to the phenotype observed in our study.
Differential distributions of IGFBP-1 have been found in ECs; IGFBP-1 expression in the endothelium is influenced by various conditions, growth factors and molecules15,42-47. Recent data demonstrates novel actions of IGFBP-1 with rescue of endothelial function in a diabetic model of insulin-resistance via increased eNOS production and preserved insulin-sensitivity43. IGFBP-1 levels were attenuated in human retinal endothelial cells (HRECs) of diabetic origin compared with non-diabetic47. In the present study, we have demonstrated a novel pathway in which RTEF-1 regulates IGFBP-1 expression in endothelial cells. Endothelium-specific RTEF-1 transgenic mice provide an efficient model to dissect the function of RTEF-1 in endothelium. This study has demonstrated that RTEF-1 regulates IGFBP-1 promoter activity through its IRS in endothelial cells.
Insulin influences gene expression in multiple tissues and can suppress expression of a number of genes that contain a conserved IRS, (CAAAA(C/T)AA), including IGFBP-1. One of the main branching pathways activated by insulin is the IRS pathway, termed the “metabolic signal”, which leads to activation of kinases dependent upon phosphoinositide 3-kinase (PI3-K). Based on growing evidence that impaired signaling through PI3-K contributes to obesity-induced insulin resistance in peripheral tissues, the hypothesis that obesity similarly impairs IGFBP-1 signaling through PI3K provides a plausible mechanism to explain this phenomenon48. Regulation of IGFBP-1 and modulation of the IRS/PI3-K pathway are highly dynamic and remains to be further examined.
These findings represent a novel mechanism by which RTEF-1 regulates IGFBP-1 in the endothelium and suggest a protective role of RTEF-1 in the pathogenesis of diabetes and subsequent vasculopathy.
Supplementary Material
Novelty and Significance.
What Is Known?
Related transcriptional enhancer factor-1 (RTEF-1) plays an important role in cardiac and endothelial cell function.
IGFBPs (Insulin-like growth factor binding proteins) are key regulators of insulin-like growth factor (IGF) at the cellular level.
Low levels of IGFBP-1 are associated with metabolic syndrome and cardiovascular diseases.
What New Information Does This Article Contribute?
RTEF-1 increases IGFBP-1 gene expression by interacting with its insulin response element.
RTEF-1 deficiency in endothelial cells exhibited increased blood glucose and insulin sensitivity in vivo.
The increased blood glucose and insulin sensitivity shown in RTEF-1 deficiency in vivo was exacerbated in a high fat diet, correlating with decreasing IGFBP-1 levels.
The transcriptional enhancer factor family regulates multiple genes expressed in both cardiac myocytes and endothelial cells. In the present study, we identified that RTEF-1 directly regulates IGFBP-1, and determined a novel interaction site between RTEF-1 and the IGFBP-1 promoter. Through regulating IGFBP-1, RTEF-1's signaling cascade impacts glucose levels and insulin resistance both in vitro and in vivo. These findings represent a novel mechanism by which RTEF-1 regulates IGFBP-1 in the endothelium and suggest a protective role of RTEF-1 in the pathogenesis of diabetes and subsequent vasculopathy.
Acknowledgments
Dr. Andres Buanonno (NIH, NCIH) for TEAD4 loxp mice; Dr. Anthony Rosenzweig (BIDMC, Harvard) for the Tie2-Cre mice and Drs. Glenn Rowe and Pablo Quintero for helpful scientific discussions.
Sources of Funding: This work was supported by NIH grant HLR01082837 (Li) and T32 training grant (Messmer-Blust).
Non-Standard Abbreviations
- RTEF-1
Related transcriptional enhancer factor-1
- IGFBP
Insulin-like growth factor binding protein
- VE-Cad
Vascular endothelial-Cadherin
- M-CAT
Muscle-CAT
- VEGF
Vascular endothelial growth factor
- HIF-1α
Hypoxia-inducible factor-1α
- IGF
Insulin-like growth factor
- TEF
Transcriptional enhancer factor
Footnotes
Disclosures: None.
In June 2012, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.35 days
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jin Y, Wu J, Song X, Song Q, Cully BL, Messmer-Blust A, Xu M, Foo SY, Rosenzweig A, Li J. RTEF-1, an upstream gene of HIF-1{alpha}, accelerates recovery from Ischemia. J Biol Chem. doi: 10.1074/jbc.M111.237024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shie JL, Wu G, Wu J, Liu FF, Laham RJ, Oettgen P, Li J. RTEF-1, a novel transcriptional stimulator of vascular endothelial growth factor in hypoxic endothelial cells. J Biol Chem. 2004;279:25010–6. doi: 10.1074/jbc.M403103200. [DOI] [PubMed] [Google Scholar]
- 3.Xu M, Jin Y, Song Q, Wu J, Philbrick MJ, Cully BL, An X, Guo L, Gao F, Li J. The Endothelial-Dependent Effect of RTEF-1 in Pressure Overload Cardiac Hypertrophy - Role of VEGF-B. Cardiovasc Res. doi: 10.1093/cvr/cvq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messmer-Blust AF, Zhang C, Shie JL, Song Q, He P, Lubenec I, Liu Y, Sellke F, Li J. Related transcriptional enhancer factor 1 increases endothelial-dependent microvascular relaxation and proliferation. J Vasc Res. 49:249–59. doi: 10.1159/000335180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azakie A, Larkin SB, Farrance IK, Grenningloh G, Ordahl CP. DTEF-1, a novel member of the transcription enhancer factor-1 (TEF-1) multigene family. J Biol Chem. 1996;271:8260–5. doi: 10.1074/jbc.271.14.8260. [DOI] [PubMed] [Google Scholar]
- 6.Gan Q, Yoshida T, Li J, Owens GK. Smooth muscle cells and myofibroblasts use distinct transcriptional mechanisms for smooth muscle alpha-actin expression. Circ Res. 2007;101:883–92. doi: 10.1161/CIRCRESAHA.107.154831. [DOI] [PubMed] [Google Scholar]
- 7.Maeda T, Gupta MP, Stewart AF. TEF-1 and MEF2 transcription factors interact to regulate muscle-specific promoters. Biochem Biophys Res Commun. 2002;294:791–7. doi: 10.1016/S0006-291X(02)00556-9. [DOI] [PubMed] [Google Scholar]
- 8.Anbanandam A, Albarado DC, Nguyen CT, Halder G, Gao X, Veeraraghavan S. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc Natl Acad Sci U S A. 2006;103:17225–30. doi: 10.1073/pnas.0607171103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrance IK, Mar JH, Ordahl CP. M-CAT binding factor is related to the SV40 enhancer binding factor, TEF-1. J Biol Chem. 1992;267:17234–40. [PubMed] [Google Scholar]
- 10.Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, Buonanno A. Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development. 2007;134:3827–36. doi: 10.1242/dev.010223. [DOI] [PubMed] [Google Scholar]
- 11.Rodgers BD, Roalson EH, Thompson C. Phylogenetic analysis of the insulin-like growth factor binding protein (IGFBP) and IGFBP-related protein gene families. Gen Comp Endocrinol. 2008;155:201–7. doi: 10.1016/j.ygcen.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruan W, Lai M. Insulin-like growth factor binding protein: a possible marker for the metabolic syndrome? Acta Diabetol. 47:5–14. doi: 10.1007/s00592-009-0142-3. [DOI] [PubMed] [Google Scholar]
- 13.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–54. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 14.Bayes-Genis A, Conover CA, Schwartz RS. The insulin-like growth factor axis: A review of atherosclerosis and restenosis. Circ Res. 2000;86:125–30. doi: 10.1161/01.res.86.2.125. [DOI] [PubMed] [Google Scholar]
- 15.Delafontaine P, Ku L, Anwar A, Hayzer DJ. Insulin-like growth factor 1 binding protein 3 synthesis by aortic endothelial cells is a function of cell density. Biochem Biophys Res Commun. 1996;222:478–82. doi: 10.1006/bbrc.1996.0769. [DOI] [PubMed] [Google Scholar]
- 16.Holly JM, Biddlecombe RA, Dunger DB, Edge JA, Amiel SA, Howell R, Chard T, Rees LH, Wass JA. Circadian variation of GH-independent IGF-binding protein in diabetes mellitus and its relationship to insulin. A new role for insulin? Clin Endocrinol (Oxf) 1988;29:667–75. doi: 10.1111/j.1365-2265.1988.tb03715.x. [DOI] [PubMed] [Google Scholar]
- 17.Snyder DK, Clemmons DR. Insulin-dependent regulation of insulin-like growth factor-binding protein-1. J Clin Endocrinol Metab. 1990;71:1632–6. doi: 10.1210/jcem-71-6-1632. [DOI] [PubMed] [Google Scholar]
- 18.Heald AH, Cruickshank JK, Riste LK, Cade JE, Anderson S, Greenhalgh A, Sampayo J, Taylor W, Fraser W, White A, Gibson JM. Close relation of fasting insulin-like growth factor binding protein-1 (IGFBP-1) with glucose tolerance and cardiovascular risk in two populations. Diabetologia. 2001;44:333–9. doi: 10.1007/s001250051623. [DOI] [PubMed] [Google Scholar]
- 19.Rajpathak SN, McGinn AP, Strickler HD, Rohan TE, Pollak M, Cappola AR, Kuller L, Xue X, Newman AB, Strotmeyer ES, Psaty BM, Kaplan RC. Insulin-like growth factor-(IGF)-axis, inflammation, and glucose intolerance among older adults. Growth Horm IGF Res. 2008;18:166–73. doi: 10.1016/j.ghir.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laughlin GA, Barrett-Connor E, Criqui MH, Kritz-Silverstein D. The prospective association of serum insulin-like growth factor I (IGF-I) and IGF-binding protein-1 levels with all cause and cardiovascular disease mortality in older adults: the Rancho Bernardo Study. J Clin Endocrinol Metab. 2004;89:114–20. doi: 10.1210/jc.2003-030967. [DOI] [PubMed] [Google Scholar]
- 21.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 22.Gory S, Vernet M, Laurent M, Dejana E, Dalmon J, Huber P. The vascular endothelial-cadherin promoter directs endothelial-specific expression in transgenic mice. Blood. 1999;93:184–92. [PubMed] [Google Scholar]
- 23.Bonora E, Targher G, Alberiche M, Bonadonna RC, Saggiani F, Zenere MB, Monauni T, Muggeo M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care. 2000;23:57–63. doi: 10.2337/diacare.23.1.57. [DOI] [PubMed] [Google Scholar]
- 24.Chen Z, Friedrich GA, Soriano P. Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev. 1994;8:2293–301. doi: 10.1101/gad.8.19.2293. [DOI] [PubMed] [Google Scholar]
- 25.Lovejoy JC. The influence of dietary fat on insulin resistance. Curr Diab Rep. 2002;2:435–40. doi: 10.1007/s11892-002-0098-y. [DOI] [PubMed] [Google Scholar]
- 26.Kim JJ, Taylor HS, Akbas GE, Foucher I, Trembleau A, Jaffe RC, Fazleabas AT, Unterman TG. Regulation of insulin-like growth factor binding protein-1 promoter activity by FKHR and HOXA10 in primate endometrial cells. Biol Reprod. 2003;68:24–30. doi: 10.1095/biolreprod.102.009316. [DOI] [PubMed] [Google Scholar]
- 27.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 28.Foucher I, Volovitch M, Frain M, Kim JJ, Souberbielle JC, Gan L, Unterman TG, Prochiantz A, Trembleau A. Hoxa5 overexpression correlates with IGFBP1 upregulation and postnatal dwarfism: evidence for an interaction between Hoxa5 and Forkhead box transcription factors. Development. 2002;129:4065–74. doi: 10.1242/dev.129.17.4065. [DOI] [PubMed] [Google Scholar]
- 29.Heald AH, Siddals KW, Fraser W, Taylor W, Kaushal K, Morris J, Young RJ, White A, Gibson JM. Low circulating levels of insulin-like growth factor binding protein-1 (IGFBP-1) are closely associated with the presence of macrovascular disease and hypertension in type 2 diabetes. Diabetes. 2002;51:2629–36. doi: 10.2337/diabetes.51.8.2629. [DOI] [PubMed] [Google Scholar]
- 30.Leinonen ES, Salonen JT, Salonen RM, Koistinen RA, Leinonen PJ, Sarna SS, Taskinen MR. Reduced IGFBP-1 is associated with thickening of the carotid wall in type 2 diabetes. Diabetes Care. 2002;25:1807–12. doi: 10.2337/diacare.25.10.1807. [DOI] [PubMed] [Google Scholar]
- 31.Alderete TL, Byrd-Williams CE, Toledo-Corral CM, Conti DV, Weigensberg MJ, Goran MI. Relationships between IGF-1 and IGFBP-1 and adiposity in obese African-American and Latino adolescents. Obesity (Silver Spring) 2010;19:933–8. doi: 10.1038/oby.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gokulakrishnan K, Velmurugan K, Ganesan S, Mohan V. Circulating levels of insulin-like growth factor binding protein-1 in relation to insulin resistance, type 2 diabetes mellitus, and metabolic syndrome (Chennai Urban Rural Epidemiology Study 118) Metabolism. 2012;61:43–6. doi: 10.1016/j.metabol.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 33.Lewitt MS, Hilding A, Ostenson CG, Efendic S, Brismar K, Hall K. Insulin-like growth factor-binding protein-1 in the prediction and development of type 2 diabetes in middle-aged Swedish men. Diabetologia. 2008;51:1135–45. doi: 10.1007/s00125-008-1016-x. [DOI] [PubMed] [Google Scholar]
- 34.Petersson U, Ostgren CJ, Brudin L, Brismar K, Nilsson PM. Low levels of insulin-like growth-factor-binding protein-1 (IGFBP-1) are prospectively associated with the incidence of type 2 diabetes and impaired glucose tolerance (IGT): the Soderakra Cardiovascular Risk Factor Study. Diabetes Metab. 2009;35:198–205. doi: 10.1016/j.diabet.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Reinehr T, Kleber M, Toschke AM, Woelfle J, Roth CL. Longitudinal association between IGFBP-1 levels and parameters of the metabolic syndrome in obese children before and after weight loss. Int J Pediatr Obes. 2011;6:236–43. doi: 10.3109/17477166.2010.544739. [DOI] [PubMed] [Google Scholar]
- 36.Guilherme A, Torres K, Czech MP. Cross-talk between insulin receptor and integrin alpha5 beta1 signaling pathways. J Biol Chem. 1998;273:22899–903. doi: 10.1074/jbc.273.36.22899. [DOI] [PubMed] [Google Scholar]
- 37.Jones JI, Gockerman A, Busby WH, Jr, Wright G, Clemmons DR. Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the alpha 5 beta 1 integrin by means of its Arg-Gly-Asp sequence. Proc Natl Acad Sci U S A. 1993;90:10553–7. doi: 10.1073/pnas.90.22.10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crossey PA, Jones JS, Miell JP. Dysregulation of the insulin/IGF binding protein-1 axis in transgenic mice is associated with hyperinsulinemia and glucose intolerance. Diabetes. 2000;49:457–65. doi: 10.2337/diabetes.49.3.457. [DOI] [PubMed] [Google Scholar]
- 39.Wolf E, Schneider MR, Zhou R, Fisch TM, Herbach N, Dahlhoff M, Wanke R, Hoeflich A. Functional consequences of IGFBP excess-lessons from transgenic mice. Pediatr Nephrol. 2005;20:269–78. doi: 10.1007/s00467-004-1657-z. [DOI] [PubMed] [Google Scholar]
- 40.Wheatcroft SB, Kearney MT. IGF-dependent and IGF-independent actions of IGF-binding protein-1 and -2: implications for metabolic homeostasis. Trends Endocrinol Metab. 2009;20:153–62. doi: 10.1016/j.tem.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 41.Schlaeger TM, Qin Y, Fujiwara Y, Magram J, Sato TN. Vascular endothelial cell lineage-specific promoter in transgenic mice. Development. 1995;121:1089–98. doi: 10.1242/dev.121.4.1089. [DOI] [PubMed] [Google Scholar]
- 42.Bastian SE, Walton PE, Belford DA. Paracellular transport of insulin-like growth factor-I (IGF-I) across human umbilical vein endothelial cell monolayers. J Cell Physiol. 1997;170:290–8. doi: 10.1002/(SICI)1097-4652(199703)170:3<290::AID-JCP10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 43.Giannini S, Cresci B, Manuelli C, Fujita-Yamaguchi Y, Romagnani P, Mohan S, Rotella CM. Insulin-like growth factor binding protein production in bovine retinal endothelial cells. Metabolism. 1997;46:1367–79. doi: 10.1016/s0026-0495(97)90134-7. [DOI] [PubMed] [Google Scholar]
- 44.Moser DR, Lowe WL, Jr, Dake BL, Booth BA, Boes M, Clemmons DR, Bar RS. Endothelial cells express insulin-like growth factor-binding proteins 2 to 6. Mol Endocrinol. 1992;6:1805–14. doi: 10.1210/mend.6.11.1282670. [DOI] [PubMed] [Google Scholar]
- 45.Tucci M, Nygard K, Tanswell BV, Farber HW, Hill DJ, Han VK. Modulation of insulin-like growth factor (IGF) and IGF binding protein biosynthesis by hypoxia in cultured vascular endothelial cells. J Endocrinol. 1998;157:13–24. doi: 10.1677/joe.0.1570013. [DOI] [PubMed] [Google Scholar]
- 46.Worthmann K, Peters I, Kumpers P, Saleem M, Becker JU, Agustian PA, Achenbach J, Haller H, Schiffer M. Urinary excretion of IGFBP-1 and -3 correlates with disease activity and differentiates focal segmental glomerulosclerosis and minimal change disease. Growth Factors. 28:129–38. doi: 10.3109/08977190903512594. [DOI] [PubMed] [Google Scholar]
- 47.Shaw LC, Grant MB. Insulin like growth factor-1 and insulin-like growth factor binding proteins: their possible roles in both maintaining normal retinal vascular function and in promoting retinal pathology. Rev Endocr Metab Disord. 2004;5:199–207. doi: 10.1023/B:REMD.0000032408.18015.b1. [DOI] [PubMed] [Google Scholar]
- 48.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–6. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.