

Abstract
Electrostatic interaction-mediated enzymatic-hydrolysis of poly(lactide)-containing nanoscale assemblies is described. At physiological pH, degradable core–shell morphologies with charged shells can readily attract or repel enzymes carrying opposite or similar charges, respectively.
Construction of nanoscale assemblies from enzyme-responsive polymers is of significance for the development of functional, smart materials. For instance, changes in the physicochemical characteristics of polymers in the presence of enzymes can lead to formation,1 destruction2 or morphological transformation2b,3 of assemblies. Enzyme-catalyzed reactions are highly selective and efficient toward specific substrates,4 and have expression patterns that are often associated with diseased sites.3a Therefore, enzymes serve as unique stimuli with great potential in nanomedicinal applications, specifically toward triggered release of therapeutics and disassembly of nanomaterials to facilitate eventual biological clearance in vivo. A noteworthy design strategy that exploits enzyme-substrate interactions in the design of nanomaterials is the synthesis of poly(ethylene glycol) (PEG)-tethered iron oxide nanoparticles that self-assemble by removal of the PEG via cleavage of peptide sequences that are sensitive to matrix metalloproteinases overexpressed in tumor environments.1c,d Additionally, in the design of stimuli-responsive polymeric materials, although several studies have focused on combining the concepts of electrostatic interactions and enzyme-responsiveness, most work primarily discusses either the electrostatic interactions between polymers that lead to adaptations in the presence of enzymes, or the utilization of enzyme-responsive small molecules that can alter the configuration of polymers by responding to enzymes. As one example, Ulijn and coworkers have reported the synthesis of enzyme-reconfigurable polymeric nanostructures that undergo self-assembly through ionic interactions between polymers composed of oppositely-charged surfactant-like molecules, which then reconfigure upon enzymatic hydrolysis of one of the polymeric components.3b In another system, Zhang and coworkers have described the construction of enzyme-responsive superamphiphiles via electrostatic interactions between double-hydrophilic block copolymers and a natural enzyme-responsive molecule.2a
Nanomaterials that are capable of selectively responding and adapting to specific enzymes based on electrostatic interactions at physiological pH, are of interest in a fundamental sense and may be desirable for various biological and/or medical applications. An interesting approach that exploits the electrostatic interactions between enzymes and oppositely-charged polymers has been investigated for the construction of enzyme immobilization materials. For example, positively-charged lysozomes have been formulated into anionic PEG-poly(α,β-aspartic acid) block copolymers5 and anionic catalase has been incorporated into cationic polyethyleneimine–PEG complexes.6 Additionally, Rotello et al. have used electrostatic interactions between polymer-coated cationic gold nanoparticles and anionic lipase to fabricate crosslinked microparticle scaffolds,7 and anionic β-galactosidase for the construction of catalytic micro-capsules.8 Given the tremendous promise in the use of enzymes for the design of nanomaterials,9 we were interested in utilization of electrostatic interactions between enzymes and charged nanoassemblies as accelerating or decelerating mechanisms for disassembly.
In this report, we extend the concept of enzyme-triggered disassembly of block copolymer micelles,2b and explore the role of electrostatic interactions between charged micelles and oppositely-charged enzymes, as an accelerating mechanism for hydrolysis of nanoparticles. Inspired by our previous findings from proteinase K-mediated rapid hydrolysis of poly(lactide) (PLA)-containing shell crosslinked nanoparticles,2b we hypothesized that degradable charged micelles in the presence of oppositely-charged enzymes (e.g. anionic micelles in the presence of cationic enzymes at physiological pH) would result in promoted hydrolysis due to electrostatic attractive forces, while the micelle–enzyme combinations with similar charges would result in relatively slower hydrolysis due to electrostatic repulsion forces between the micelles and enzymes. In this study, we highlight a novel approach in the design of smart nanomaterials with tunable hydrolysis rates, in which the extent of enzyme-responsiveness can be tailored based on electrostatic-interactions between charged micelles and enzymes with varying isoelectric points (pIs).
Two classes of polymeric core–shell morphologies possessing identical hydrolyzable poly(DL-lactide) (PDLLA) cores, but oppositely-charged shells were synthesized, and their enzymatic-hydrolysis behaviors were investigated in the presence of two electrostatically-complementary model enzymes. The synthetic parameters for the design of micelles included shells composed of poly(acrylic acid) (PAA) with pKa ~ 4.5,10 and poly(acrylamidoethylamine) (PAEA) with pKa ~ 9.1,11 resulting in anionic and cationic surface charges, respectively, at neutral pH. Additionally, because the net charge on a protein at any given pH is determined by its pI,12 the net charge on an enzyme is expected to be positive at pH values below the pI, and negative at pH values above the pI. Consequently, two model enzymes possessing positive and negative charges at physiological pH, proteinase K (PK) with pI ~ 8.9 and porcine liver esterase (PLE) with pI ~ 4.8, were employed in this study. Of the two model enzymes used in this study, PK (MW ~ 29 kDa) is a serine endopeptidase with an active site of Asp-His-Ser,13 and PLE (MW ~ 60 kDa) is a serine hydrolase in which its functionality is attributed to the catalytic triad of Ser-His-Asp/Glu,14 each of which can catalyze hydrolysis of PLA.15
Amphiphilic diblock copolymers, poly(acrylic acid)80-block-poly-(DL-lactide)40 (PAA80-b-PDLLA40) and poly(acrylamidoethylamine)90-block-poly(DL-lactide)40 (PAEA90-b-PDLLA40) were synthesized, by employing sequential ring opening polymerization (ROP) and reversible addition-fragmentation chain transfer (RAFT) polymerization followed by acidolysis (Scheme S1, ESI†), utilizing similar methodologies reported previously.16 The degrees of polymerization and well-defined structures for the polymers were confirmed by a combination of 1H NMR spectroscopy (Fig. S2, ESI†) and gel permeation chromatography (GPC) (Fig. S1, ESI†). The oppositely-charged micelles were prepared by the aqueous supramolecular assembly of the anionic and cationic amphiphilic diblock copolymers (Scheme 1).
Scheme 1.

Schematic illustration of the preparation of anionic and cationic micelles via self assembly of amphiphilic diblock copolymers PAA80-b-PDLLA40 and PAEA90-b-PDLLA40, and their enzymatic hydrolysis by two electrostatically-complementary enzymes. TC = dodecyl trithiocarbonate group.
The average core diameters (Dav) and hydrodynamic diameters (Dh) of the anionic and cationic micelles were analyzed by transmission electron microscopy (TEM) and dynamic light scattering (DLS), respectively. Although synthetic parameters were optimized to maintain similar block copolymer lengths of both amphiphilic diblock copolymers, with the expectation to preserve comparable diameters of their supramolecular assemblies, the overall dimensions of the cationic micelles were larger than those of their anionic analogs. As shown in Fig. 1A, Dav of the anionic micelles in the dry state was 20 ± 3 nm, and the monomodal size distributions observed by DLS were consistent with the TEM measurements with number-averaged hydrodynamic diameters of Dh(n) = 65 ± 18 nm (Fig. S3A, ESI†). For the cationic micelles, Dav was approximately twice as that of the anionic analog (44 ± 7 nm), and monomodal size distributions by DLS showed hydrodynamic diameters of Dh(n) = 101 ± 26 nm (Fig. 1B and Fig. S3B, ESI†). Furthermore, zeta potentials of the micelles prepared from PAA80-b-PDLLA40 were −55 mV and PAEA90-b-PDLLA40 was +26 mV at pH 7.4, confirming the anionic and cationic surface charges of the nanoassemblies.
Fig. 1.
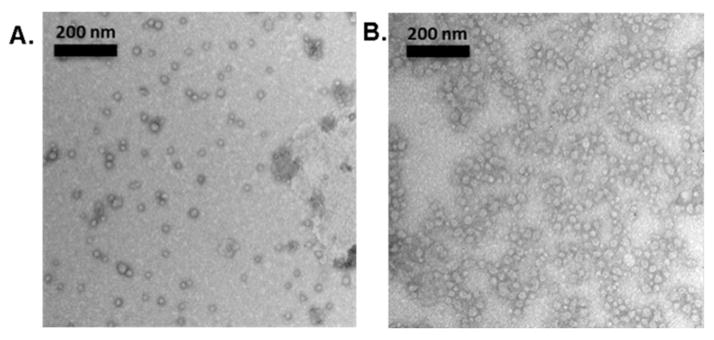
Characterization by TEM showing average core diameters of (A) anionic micelles and (B) cationic micelles.
In order to evaluate and quantify whether accelerated enzymatic core hydrolysis is related to electrostatic interactions between oppositely-charged enzyme–micelle pairs, the core hydrolysis rates of each of the micelles were investigated in the presence of enzymes PK and PLE. In the degradation study, in addition to maintaining similar substrate (PDLLA) concentrations in both anionic and cationic micellar aqueous solutions, the amounts of enzymes being added to each solution were adjusted to maintain similar enzyme activities per substrate concentration (ca. 0.1 U mL−1). Poly(lactide) core hydrolysis of the micellar assemblies was quantitatively determined by monitoring the production of the final degradation product, lactic acid, using a lactate colorimetric assay.
Interestingly, PK possessing a net positive charge at pH 7.4, showed preferential hydrolysis of the anionic micelles with the degradation of ca. 35% of the PDLLA core into lactic acid, as opposed to only ca. 15% degradation from the cationic analog, within 24 h (Fig. 2A). To confirm whether the differences in hydrolysis between the two oppositely-charged micelles were a result of electrostatic interactions between the enzyme and the charged micelles, and not necessarily due to the differences in hydrodynamic diameters of the micelles, an analogous experiment was carried out in the presence of PLE (net negative charge at physiological pH) in the place of PK. In contrast to the PK-catalyzed anionic micelles, in the presence of PLE, preferential hydrolysis was observed for the cationic micelles with the degradation of ca. 15% as opposed to negligible core hydrolysis from the anionic analog, within 24 h (Fig. 2B), confirming our hypothesis. As evident by the degradation profiles, enzymatic-hydrolysis of the micelles by both PK and PLE was faster compared to their uncatalyzed analogs that reached only <3% hydrolytic-degradation within 24 h. Moreover, when the two enzymes are compared, the slower hydrolysis observed for the PLE-catalyzed nanoassemblies is supported by previous studies that have shown relatively low enzymatic activity of PLE toward bulk PLA as opposed to PK,15b,17 and may also be attributed to the large size of PLE that exists in a native trimeric complex of ca. 180 kDa (ca. 60 kDa monomer)14b that perhaps hinders its ability to migrate through the polymeric shell of the nanoassemblies.
Fig. 2.
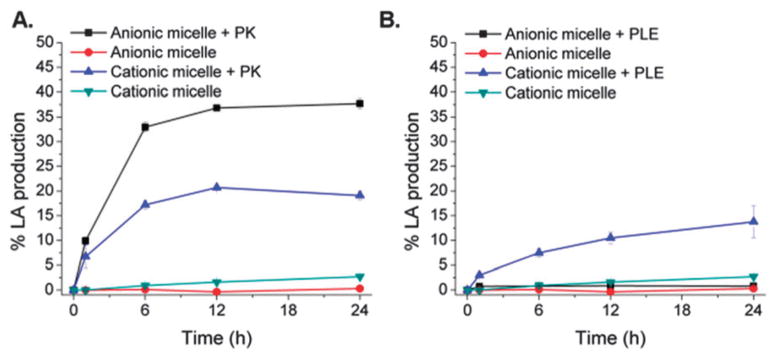
Degradation profiles of anionic and cationic micelles in 0.1 M tris-HCl buffer (pH 7.4) at 37 °C within 24 h, in the presence and absence of (A) PK and (B) PLE. Most of the error bars are smaller than the symbols used in the plot.
To assess the dimensional changes upon enzyme-catalyzed core hydrolysis, the micellar solutions were observed under TEM after treatment with each of the enzymes. In our previous work, micellar disassembly was observed under TEM at >50% core hydrolysis in the presence of PK.2b Similar to these findings, partial disassembly was observed for the enzymatically-hydrolyzed micelles (Fig. S4, ESI†). These dimensional changes may be due to micellar reorganization events, including polymer chain exchange and micelle aggregation, as the hydrophobic chain segment lengths decrease over time and hydrophilic surface chains are released from the micelle assemblies.
In summary, we have demonstrated that preferential core hydrolysis of anionic micelles proceeds in the presence of a positively-charged model enzyme (PK), and of cationic micelles in the presence of a negatively-charged model enzyme (PLE), compared to their oppositely-charged micellar analogs. Furthermore, the overall rates of hydrolysis by PK were greater than those of PLE. While these experimental findings support the electrostatic complementarity of enzyme catalysis,18 they also provide valuable insight toward understanding fundamental mechanisms involved in enzymatic-substrate interactions, and may be of potential significance toward designing charge-mediated enzyme-responsive nanomaterials that are capable of undergoing environmentally-triggered therapeutic release, disassembly or morphological alterations under selective enzyme conditions.
Supplementary Material
Acknowledgments
Financial support from the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C), the National Institutes of Health (NIH GM084266), and the Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001 is gratefully acknowledged.
Footnotes
Electronic supplementary information (ESI) available: Synthesis and characterization; GPC traces, NMR. TGA, DSC, DLS and TEM data. See DOI: 10.1039/c3cc46013d
Notes and references
- 1.(a) Amir RJ, Zhong S, Pochan DJ, Hawker CJ. J Am Chem Soc. 2009;131:13949. doi: 10.1021/ja9060917. [DOI] [PubMed] [Google Scholar]; (b) Caponi PF, Qiu XP, Vilela F, Winnik FM, Ulijn RV. Polym Chem. 2011;2:306. [Google Scholar]; (c) Harris TJ, von Maltzahn G, Derfus AM, Ruoslahti E, Bhatia SN. Angew Chem, Int Ed. 2006;45:3161. doi: 10.1002/anie.200600259. [DOI] [PubMed] [Google Scholar]; (d) von Maltzahn G, Harris TJ, Park JH, Min DH, Schmidt AJ, Sailor MJ, Bhatia SN. J Am Chem Soc. 2007;129:6064. doi: 10.1021/ja070461l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Wang C, Chen Q, Wang Z, Zhang X. Angew Chem, Int Ed. 2010;49:8612. doi: 10.1002/anie.201004253. [DOI] [PubMed] [Google Scholar]; (b) Samarajeewa S, Shrestha R, Li Y, Wooley KL. J Am Chem Soc. 2012;134:1235. doi: 10.1021/ja2095602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Ku TH, Chien MP, Thompson MP, Sinkovits RS, Olson NH, Baker TS, Gianneschi NC. J Am Chem Soc. 2011;133:8392. doi: 10.1021/ja2004736. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Caponi PF, Winnik FM, Ulijn RV. Soft Matter. 2012;8:5127. [Google Scholar]; (c) Chien MP, Thompson MP, Lin EC, Gianneschi NC. Chem Sci. 2012;3:2690. doi: 10.1039/C2SC20165H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu J, Zhang G, Liu S. Chem Soc Rev. 2012;41:5933. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Harada A, Kataoka K. J Am Chem Soc. 2003;125:15306. doi: 10.1021/ja038572h. [DOI] [PubMed] [Google Scholar]; (b) Harada A, Kataoka K. J Controlled Release. 2001;72:85. doi: 10.1016/s0168-3659(01)00264-4. [DOI] [PubMed] [Google Scholar]
- 6.Batrakova EV, Li S, Reynolds AD, Mosley RL, Bronich TK, Kabanov AV, Gendelman HE. Bioconjugate Chem. 2007;18:1498. doi: 10.1021/bc700184b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong Y, Duncan B, Park MH, Kim C, Rotello VM. Chem Commun. 2011;47:12077. doi: 10.1039/c1cc14448k. [DOI] [PubMed] [Google Scholar]
- 8.Samanta B, Yang XC, Ofir Y, Park MH, Patra D, Agasti SS, Miranda OR, Mo ZH, Rotello VM. Angew Chem, Int Ed. 2009;48:5341. doi: 10.1002/anie.200901590. [DOI] [PubMed] [Google Scholar]
- 9.(a) van Eldijk MB, Wang JCY, Minten IJ, Li CL, Zlotnick A, Nolte RJM, Cornelissen JJLM, van Hest JCM. J Am Chem Soc. 2012;134:18506. doi: 10.1021/ja308132z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma YJ, Nolte RJM, Cornelissen JJLM. Adv Drug Delivery Rev. 2012;64:811. doi: 10.1016/j.addr.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Gebhardt JE, Fuerstenau DW. Colloids Surf. 1983;7:221. [Google Scholar]
- 11.Zou J, Zhang SY, Shrestha R, Seetho K, Donley CL, Wooley KL. Polym Chem. 2012;3:3146. doi: 10.1039/C2PY20324C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanford C. Adv Protein Chem. 1963;17:69. [Google Scholar]
- 13.Betzel C, Pal GP, Saenger W. Eur J Biochem. 1988;178:155. doi: 10.1111/j.1432-1033.1988.tb14440.x. [DOI] [PubMed] [Google Scholar]
- 14.(a) Junge W, Heymann E. Eur J Biochem. 1979;95:519. doi: 10.1111/j.1432-1033.1979.tb12992.x. [DOI] [PubMed] [Google Scholar]; (b) Heymann E, Junge W. Eur J Biochem. 1979;95:509. doi: 10.1111/j.1432-1033.1979.tb12991.x. [DOI] [PubMed] [Google Scholar]
- 15.(a) Lee SH, Song WS. Appl Biochem Biotechnol. 2011;164:89. doi: 10.1007/s12010-010-9117-7. [DOI] [PubMed] [Google Scholar]; (b) Reeve MS, Mccarthy SP, Downey MJ, Gross RA. Macromolecules. 1994;27:825. [Google Scholar]
- 16.(a) Li A, Luehmann HP, Sun G, Samarajeewa S, Zou J, Zhang S, Zhang F, Welch MJ, Liu Y, Wooley KL. ACS Nano. 2012;6:8970. doi: 10.1021/nn303030t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Samarajeewa S, Ibricevic A, Gunsten SP, Shrestha R, Elsabahy M, Brody SL, Wooley KL. Biomacromolecules. 2013;14:1018. doi: 10.1021/bm3018774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuzaki H, Yoshida M, Asano M, Kumakura M. Eur Polym J. 1989;25:1019. [Google Scholar]
- 18.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:501. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.