

Abstract
Background
Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are neoplastic disorders of hematopoietic stem cells. DNA methyltransferase inhibitors, 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine), benefit some MDS/AML patients. However, the role of DNA methyltransferase inhibitor-induced DNA hypomethylation in regulation of gene expression in AML is unclear.
Results
We compared the effects of 5-azacytidine on DNA methylation and gene expression using whole-genome single-nucleotide bisulfite-sequencing and RNA-sequencing in OCI-AML3 cells. For data analysis, we used an approach recently developed for discovery of differential patterns of DNA methylation associated with changes in gene expression, that is tailored to single-nucleotide bisulfite-sequencing data (Washington University Interpolated Methylation Signatures). Using this approach, we find that a subset of genes upregulated by 5-azacytidine are characterized by 5-azacytidine-induced signature methylation loss flanking the transcription start site. Many of these genes show increased methylation and decreased expression in OCI-AML3 cells compared to normal hematopoietic stem and progenitor cells. Moreover, these genes are preferentially upregulated by decitabine in human primary AML blasts, and control cell proliferation, death, and development.
Conclusions
Our approach identifies a set of genes whose methylation and silencing in AML is reversed by DNA methyltransferase inhibitors. These genes are good candidates for direct regulation by DNA methyltransferase inhibitors, and their reactivation by DNA methyltransferase inhibitors may contribute to therapeutic activity.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-014-0406-2) contains supplementary material, which is available to authorized users.
Background
Myelodysplastic syndrome (MDS) is a collection of neoplastic disorders of hematopoietic stem cells (HSCs) characterized by inefficient hematopoiesis, peripheral blood cytopenia, morphologic dysplasia, and susceptibility to acute myeloid leukemia (AML). AML is characterized by accumulation of immature myeloid ‘blasts’ in the bone marrow and peripheral blood [1]. Accrual of epigenetic abnormalities likely contributes to development of MDS and AML. For example, promoter DNA hypermethylation and associated silencing of tumor suppressor gene CDKN2b, encoding p15INK4b, has been reported in up to 80% of AML [2]. Accordingly, there has been substantial interest in application of so-called epigenetic therapies to combat MDS and AML, most notably, DNA methylation inhibitors and histone deacetylase inhibitors [3].
In the US, two DNA methyltransferase inhibitors (DNMTi), 5-Azacitidine (AzaC) and 5-aza-2'-deoxycytidine (decitabine), are licenced for therapeutic use in MDS/AML [4]. In the UK, AzaC is approved for use in some adults with MDS, chronic myelomonocytic leukemia or AML. Decitabine is not approved for use in the UK. These drugs act as ‘fraudulent bases’ mimicking cytosine, and once incorporated into DNA in S phase are able to trap DNMTs. Trapped DNMTs are degraded by the proteasome resulting in passive hypomethylation of the DNA during subsequent replication cycles [3].
Initial studies focused on the DNA hypomethylating activity of DNMT inhibitors as being the basis of their therapeutic effects. Approximately 60% of human gene promoters are associated with CpG rich regions termed CpG islands [5]. CpG islands are typically maintained free of DNA methylation and this is permissive for gene expression. However, in many human cancers, a proportion of CpG islands is hypermethylated and this is linked to silencing of some tumor suppressor genes [6]. Hypermethylation of regions of lower CpG density adjacent to islands, termed CpG island shores, are also linked to silencing [7].
Accordingly, it has been proposed that DNMT inhibitors cause hypomethylation of promoter regulatory regions of tumor suppressor genes silenced by DNA methylation, thereby reactivating cell growth arrest and differentiation. For example, treatment of AML cell lines and patient blasts with decitabine induced hypomethylation and reactivation of expression of p15INK4b [8]. However, other studies have failed to confirm a strong correlation between promoter and CpG island hypomethylation and activation of gene expression [9,10]. Indeed, in addition to causing DNA hypomethylation, DNMTi cause damage to DNA, and AzaC is also incorporated into RNA, and these activities might also contribute to their biological effects [3].
To date, studies investigating the relationship between AzaC- and decitabine-induced DNA hypomethylation and gene expression have employed analysis methods that fail to survey methylation across the entire epigenome [8–10]. For example, frequently used Illumina 27 K and 450 K arrays sample only a small number of CpGs per CpG island, and only 27,000 and 450,000 respectively of the approximately 56 million cytosines in CpG context in the genome (that is, approximately 28 million dyad CpGs). To date, no study has compared methylation changes across all CpGs with changes in gene expression. Therefore, we set out to investigate the effects of AzaC in a model AML cell line, using more comprehensive whole genome bisulfite sequencing (WGBS) to map the DNA methylation landscape in AzaC untreated and treated cells, and employing a sophisticated computational approach tailored to whole genome data to unveil relationships between altered methylation and altered gene expression (Washington University Interpolated Methylation Signatures (WIMSi)). By this approach, we identified a set of genes whose is expression is aberrantly repressed by DNA methylation in AML and reversed by DNMTi, perhaps contributing to therapeutic effects of DNMTi.
Results
To investigate the effects of AzaC on DNA methylation and gene expression at the whole genome level, we chose to work with OCI-AML3 cells as a model. AML3 cells are derived from AML (FAB M4) harboring a mutation in nucleophosmin (NPM1) exon 12 and a DNMT3A R882C mutation [11,12]. Approximately, 35% and 22% of primary human AML harbor such mutations in NPM1 and DNMT3a, respectively [13,14]. Since the action of AzaC as a DNA demethylating agent depends on passive demethylation due to downregulation of DNMT1, we first established an AzaC treatment protocol that downregulated DNMT1 but was not so toxic as to acutely arrest DNA synthesis and cell proliferation. We found that treating cells with 0.5 μM AzaC three times at 24-h intervals (0, 24, and 48 h) and harvesting at 96 h after the first treatment resulted in marked downregulation of DNMT1 at 96 h (Figure 1a). However, this dose of AzaC resulted in only a modest decrease in the number of viable cells, compared to untreated controls over the same time course (Figure 1b). Moreover, by this protocol AzaC induced only low levels of DNA damage as measured by γH2AX (Figure 1c), and apoptosis measured by PARP cleavage, caspase 3 activation, and <2n DNA content (Figure 1d and Additional file 1: Figure S1a-c). Most important, by this regimen AzaC did not markedly inhibit cell division, cell cycle distribution, DNA synthesis, and cell proliferation (Figure 1e, f and Additional file 1: Figure S1c, d). Based on these pilot data, we anticipated that treating AML3 cells with 0.5 μM AzaC three times at 24-h intervals (0, 24, and 48 h) and harvesting at 96 h should permit DNA synthesis in the absence of DNMT1, and thus passive genome demethyation.
Figure 1.
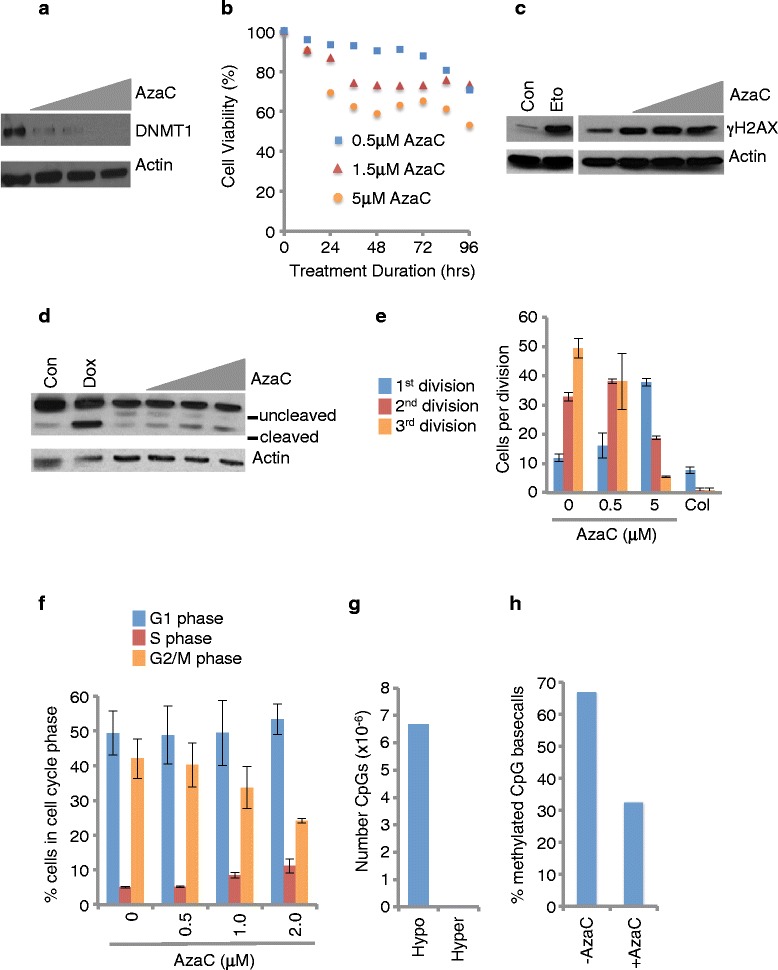
Optimization of AzaC treatment protocol. (a) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC (triangle) at 0, 24, 48 h, harvested at 96 h, and western blotted for DNMT1. (b) AML3 cells were treated with vehicle, 0.5, 1.5, or 5 μM AzaC at 12-h intervals and viability determined by fluorescence assay (Resazurin) at indicated time points. (c) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC (triangle) at 0, 24, 48 h and whole cell lysates western blotted for γH2AX at 72 h. As a positive control, cells were treated with vehicle or 1 μM etoposide (Eto). (d) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC (triangle) at 0, 24, 48 h, harvested at 72 h, and western blotted for uncleaved and cleaved PARP. As a positive control, p53 inducible SAOS2 cells were treated with vehicle or doxycycline (Dox). (e) AML3 cells were treated three times with vehicle, 0.5 or 5 μM AzaC (as indicated by triangle) at 0, 24, 48 h and cumulative cell divisions scored by CFSE at 96 h. Results from three replicate experiments with SD. (f) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC (triangle) at 0, 24, 48 h, pulse labelled with BrdU for 1 h at 96-h time point, and DNA content (7-AAD) and DNA synthesis (BrdU) determined by FACS. (g) AML3 cells were treated three times at 24-h intervals with 0.5 μM AzaC in triplicate and harvested 96 h after the first treatment. Number of CpGs showing decreased (hypo) and increased (hyper) methylation in AzaC treated cells, compared to untreated cells. (h) Overall percent methylated basecalls for all reference CpGs, in cells from (g).
Accordingly, AML3 cells were treated three times at 24-h intervals with 0.5 μM AzaC in triplicate and harvested 96 h after the first treatment. Genomic DNA was purified from two replicates and subjected to whole genome bisulfite sequencing (in excess of 15× coverage of each replicate), yielding a total of 237Gb of sequence data (Additional file 2: Table S1). In parallel, RNA was purified from three replicates and analyzed by RNA seq of poly (A) RNA. Analysis of the DNA methylation data confirmed that individual replicates of untreated and treated cells were highly concordant (Additional file 2: Tables S2 and S3), with paired Spearman coefficients in the range of 0.79 to 0.94 between like samples (Additional file 2: Table S4). Importantly, in untreated cells there was also a strong correlation in promoter CpG methylation and gene expression between AML3 and primary AML cells (data from TCGA); Spearman correlation coefficient of 0.79 and 0.85 for CpG methylation and gene expression, respectively (Additional file 3: Figure S2). Absolute levels and changes (between untreated and treated) in methylation at non-CpG sites, CHG, and CHH (defined in Material and Methods), were negligible (untreated to treated, 0.44% to 0.40% (CHG) and 0.43% to 0.39% (CHH)) (Additional file 2: Table S5 and S6), compared to the frequency of failed bisulfite conversion of unmethylated C to U (Additional file 2: Table S7). Of 56,328,604 cytosines in a CpG context in the hg18 reference genome, 6,679,526 showed lower methylation (hypomethylated) in AzaC treated cells, compared to untreated cells (FDR corrected P value level of 0.05) (Figure 1g) (Additional file 2: Table S8). One hundred and ninety-two individual CpGs gained DNA methylation (hypermethylated) in AzaC treated cells (FDR corrected P value level of 0.05) (Figure 1g, Additional file 2: Table S8). As expected, analysis revealed an overall decrease in cytosine methylation in AzaC treated cells (Figure 1h), from 66.97% to 32.32% methylcytosine basecalls at reference CpG sites.
As a platform for understanding the effects of AzaC on DNA methylation and gene expression, we first analyzed the relationship between DNA methylation and gene expression in AzaC untreated proliferating AML3 cells. In untreated cells, percent methylation was in the range of 0% to 100% (Figure 2a). However, most individual CpGs were scored as ‘methylated’ (>80% methylated) or ‘partially methylated’, with only 20% scoring as ‘unmethylated’ (<20% methylated) (Figure 2b). When plotted across a linear chromosome, a landscape of highly methylated domains interspersed with partially methylated domains was apparent (Figure 2c). Highly methylated CpGs mapped predominantly to genic regions (including introns) and SINE elements (Additional file 4: Figure S3). Compared to a random distribution across the genome, unmethylated regions were enriched at CpG islands, 5′ UTRs and promoters (Figure 2d). In these general terms, these features of the global methylation landscape of AML3 cells are qualitatively similar to those previously described in cancer cells, differentiated cells, primary tissues and senescent cells [15–20].
Figure 2.
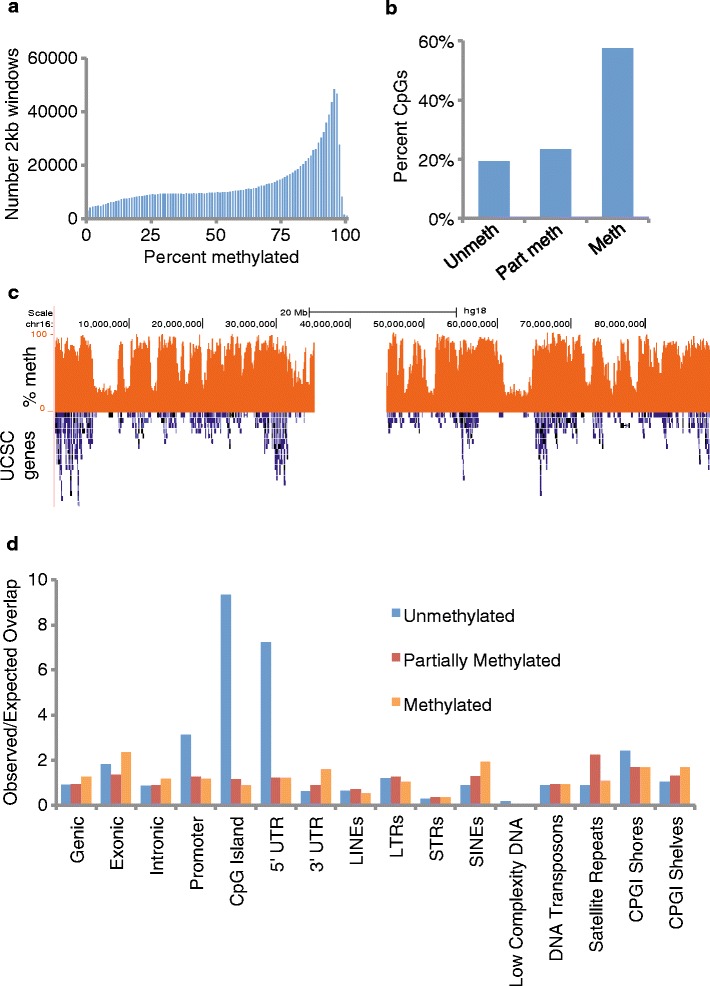
DNA methylation landscape in untreated cells. (a) Histogram of global percentage methylation. The genome is split into non-overlapping 2 kb windows, and the percentage methylation at CpGs calculated for each window. Y-axis indicates the number of windows with a given methylation level. (b) Proportion of individual CpGs that are unmethylated (<20% methylated), partially methylated (20% to 80% methylated) or methylated (>80% methylated). (c) Bisulphite sequencing percentage methylation (orange) and Ensembl genes (blue) over chromosome 16. (d) Ratios of observed to expected overlap (enrichment) of methylated, partially methylated and unmethylated CpGs (defined as in (b)) with specified genomic features.
Next, we plotted average percent methylation across gene bodies, promoters, and up- and downstream regions of composite genes, where transcript start and end sites (TSS and TES) were the outermost start and end sites, as defined in Ensembl Human Genes (version 54) for each gene. Composites were comprised of subgroups of all genes in the expression dataset, grouped according to level of expression in proliferating AzaC untreated cells. Within gene bodies, there was a general trend towards increasing methylation with increasing expression, as reported previously [18,21–25]. This trend was most marked among the more lowly expressed genes (Figure 3a); the highest expressed genes were not the most highly methylated in gene bodies. This bimodal pattern was also apparent when percent methylation along individual genes (including up and downstream regions) was plotted as a heatmap, with genes vertically rank ordered according to level of expression (Figure 3b). At promoters, increasing expression was associated with decreased methylation; interestingly, this trend was most marked in the extent of the unmethylated region downstream of the TSS (Figure 3a). Again, this trend was also apparent in the heatmap analysis (Figure 3b). We reasoned that the correlation between low-level methylation at promoters and high-level gene expression was likely to reflect hypomethylation of CpG islands. Consistent with this idea, the most highly expressed genes also exhibited the highest CpG ratio at promoters (Figure 3c). Of course, in many cancers, a proportion of CpG islands is methylated and this is associated with silencing of those genes [26]. In AML3 cells, about 12% of CpG islands overlapping a TSS showed at least 80% methylation (Figure 3d). As expected, these were expressed at a lower level than genes with unmethylated CpG islands at the TSS (Figure 3e). In sum, as a general trend, the highest expressed genes are those harboring hypomethylated CpG islands at the TSS. A subset of CpG islands is methylated in AML3 cells and this is associated with lower expression.
Figure 3.
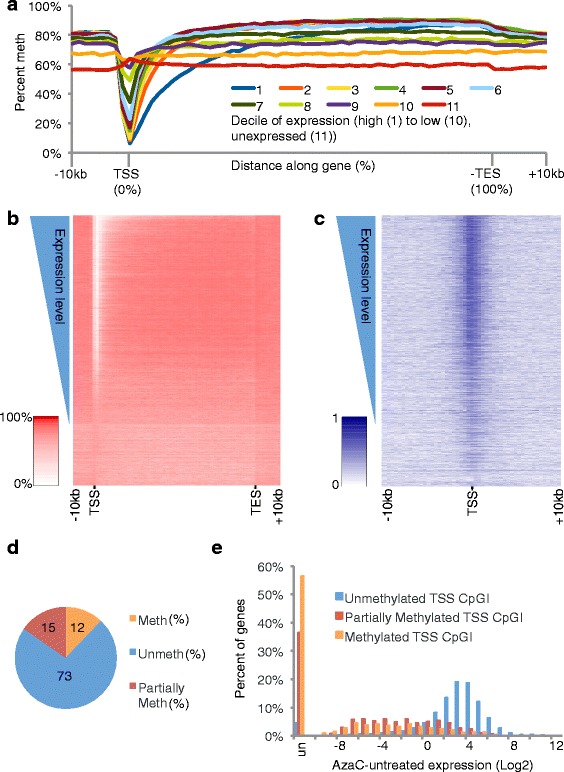
Relationship between DNA methylation and gene expression in untreated cells. (a) A plot of methylation across composite genes, grouped into deciles by level of expression in untreated cells. Along the gene, DNA sequence is split into windows, each representing 2% of the total gene body, with 10 additional 1 kb windows both up- and downstream of the gene. The y-axis shows the mean percentage methylation for each decile and window. (b) Heatmap of percentage methylated CpGs over genes. Genes are split into windows as (a) and rank ordered according to the expression level of the corresponding transcript (y-axis, increasing expression at top). The intensity of the heatmap indicates the percentage methylation from 0% to 100%, white to red. (c) Heatmap of observed-to-expected CpG ratio in a 10 kb window centered on TSS. Genes are rank ordered as (b). The intensity of the heatmap indicates the value of observed-to-expected CpG ratio from 0 to 1, white to blue. (d) Proportion of CpG islands that are methylated (>80% methylation), partially methylated (20% to 80% methylated) and unmethylated (<80% methylated). (e) Histogram of expression at genes with methylated (>80% methylation), partially methylated (20% to 80% methylated) and unmethylated (<80% methylated) CpG island within 10 kb of the TSS.
When percent methylation across each linear chromosome was compared between AzaC-untreated and treated cells, decreased methylation was apparent along the length of the chromosome (Figure 4a). Regardless of initial methylation level, the mean level of methylation after AzaC was typically about 50% of the untreated level (Figure 4b), a uniform relative decrease in methylation (Figure 4c). Strikingly, a histogram of relative change of DNA methylation in 2 kb windows conformed to a normal distribution, confirming that across the vast majority of windows there was no difference in tendency to hypomethylation in the presence of AzaC (Figure 4d). Most specific sequence features of the genome underwent a comparable approximate 50% loss of methylation (Figure 4e). Some regions, notably CpG islands, 5′ UTR and gene promoters, underwent a smaller decrease in methylation, likely because their starting methylation was already close to zero (Figure 2d). Of particular note, CpG islands that were heavily methylated in untreated cells underwent substantial hypomethylation after AzaC treatment (Figure 4f). Despite extensive DNA hypomethylation across the whole genome, by RNA-seq analysis, a relatively small proportion of genes significantly altered their expression (Figure 4g and Additional file 2: Table S9). Of 36,119 annotated genes (Ensembl Human Genes Version 54) 792 were significantly upregulated and 426 were downregulated (BH-fdr <0.05) (Additional file 5: Dataset 1, ‘AML3_AzaC_SigDiff_1147’).
Figure 4.
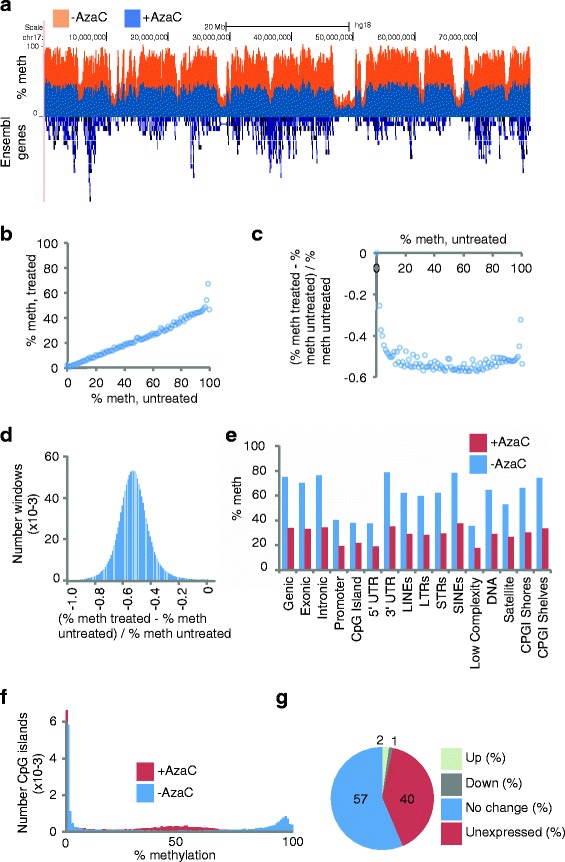
AzaC induces global DNA hypomethylation, but select changes in gene expression. (a) Representative bisulphite sequencing data for AzaC-untreated (orange) and treated (blue), and Ensembl genes (blue (below)) over chromosome 16. Percent methylation on y-axis. (b, c) Relationship between global average untreated methylation and treated methylation. Reference CpGs are placed into integer bins (in the range of 0 to 100) corresponding to AzaC-untreated% methylation. The mean AzaC-treated% methylation (b) and mean relative difference (c) are calculated for the CpGs in each bin, and plotted. (d) Distribution of methylation differences approximates a normal distribution. The genome is split into non-overlapping 2 kb windows, and the relative difference in methylation at CpGs calculated for each window. (e) Methylation changes within genomic features. Mean percentage methylation over specified genomic features, for AzaC-untreated (blue) and AzaC-treated (red) cells. (f) Methylation plotted for untreated and treated CpG islands. Histogram of percentage CpG methylation at CpG islands for AzaC untreated (blue) and AzaC treated (red) cells. (g) Classification of gene expression changes after AzaC treatment. Proportion of genes that are up- and downregulated (BH-fdr <0.05), unchanging (BH-fdr >0.05) and unexpressed in either sample (FPKM = 0). Up, 792; down, 426; no change, 20,385; unexpressed, 14,516. See Additional file 5: Dataset 1.
Since AzaC induced substantial loss of methylation across the whole genome, but only a small proportion of genes altered their expression, we next set out to identify the parameters that determine altered gene expression. First, we considered the hypothesis that activation of gene expression is tightly linked to loss of methylation at a promoter CpG island. Consistent with this idea, some activated genes, for example, DAZL, did undergo CpG island hypomethylation after AzaC treatment (Figure 5a). However, genome-wide analyses did not support a strong link between CpG island hypomethylation and activation of gene expression. Considering the top-most upregulated genes, some are not linked to promoter CpG islands whereas others are linked to CpG islands that are only weakly methylated in untreated cells (Additional file 5: Dataset 2). Moreover, there was a very small and insignificant overlap between those genes harboring a methylated CpG island at the TSS in untreated cells and genes upregulated by AzaC (P = 0.4195) (Figure 5b).
Figure 5.
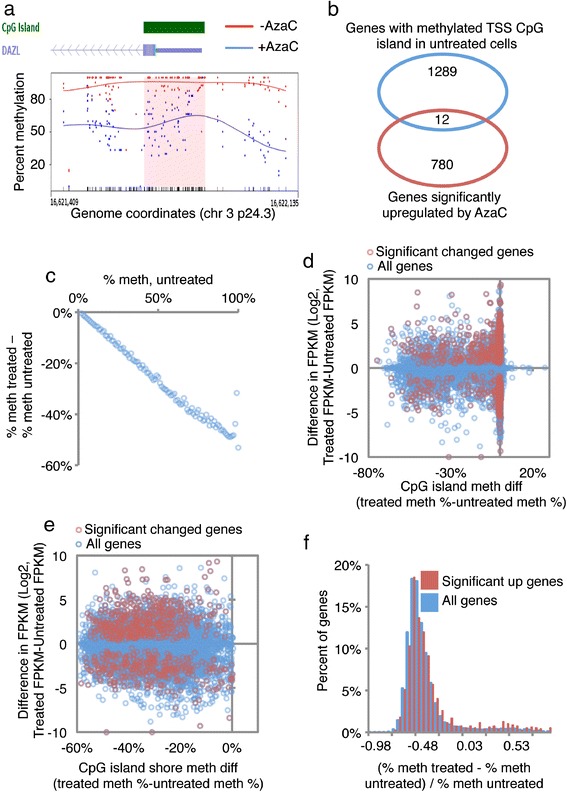
AzaC-induced changes in gene expression are not linked to gross hypomethylation of CpG islands. (a) Smoothed percentage methylation plot of the DAZL promoter region, showing individual CpGs (dots) and smoothed methylation (lines) for untreated (red) and treated (blue). (b) Overlap between genes with a methylated CpG island overlapping TSS (mCpG > 80%) and upregulated genes (BH-fdr <0.05). (c) Reference CpGs are placed into integer bins (in the range of 0 to 100) corresponding to AzaC untreated% methylation. The mean difference in methylation is calculated for the CpGs in each bin, and plotted. (d) Scatter plot of difference in expression versus difference in CpG island methylation at genes with a CpG island within 10 kb of the TSS, showing all genes (blue) and significantly regulated genes (red). (e) Scatter plot of difference in expression versus difference in CpG island shore methylation at genes with a CpG island within 10 kb of the TSS, showing all genes (blue) and significantly regulated genes (red). (f) Histogram of relative methylation differences for upregulated and all genes. The relative difference in the promoter (+/- 2 kb TSS) methylation for significant upregulated (red) and all genes (blue) is calculated and the distribution is plotted.
Although AzaC induced a relatively uniform approximately two-fold decrease in methylation across the whole genome, the absolute difference in percent methylation varied quite widely, depending on the level of methylation in untreated cells (Figure 5c). However, a dot plot of absolute difference in percent methylation at CpG islands versus difference in gene expression between untreated and treated cells failed to show a correlation between altered methylation and altered expression (Figure 5d and Additional file 6: Figure S4a). Similarly, there was not a strong link between absolute difference in percent methylation at CpG island shores and altered expression (Figure 5e and Additional file 6: Figure S4b). This is the case whether changes in expression are assessed by fold change (Additional file 6: Figure S4a, b) or absolute change in expression (Figure 5d, e). Finally, genes whose expression increased significantly after AzaC did not show a greater decrease in promoter methylation than all other genes (Figure 5f). Together, these results do not support the hypothesis that CpG island and/or shore hypomethylation is primarily responsible for activation of gene expression by AzaC.
The previous analyses employed a relatively simple quantitative analysis of total DNA methylation over defined regions, CpG islands, or shores. Next, we adopted a more sophisticated approach for discovering differential patterns of methylation associated with changes in gene expression. In this recently described approach (Washington University Interpolated Methylation Signatures (WIMSi) [27]) differential methylation between AzaC-untreated and treated cells for each gene was first represented as an interpolated curve, or signature. Genes were then clustered by the shape-based similarity of their methylation signatures (Figure 6a). Clusters of genes where at least 85% of the genes changed expression in the same direction and the distribution of expression changes was different than the background distribution were then identified. The resultant clusters of genes all have similar methylation signatures and show concordant expression changes. Out of 1,147 genes significantly up- or downregulated by AzaC (753 upregulated and 394 downregulated (Additional file 5: Dataset 1)), this approach identified 246 upregulated genes (32.7% of all upregulated genes) that underwent a similar pattern of decreased methylation on AzaC treatment (Figure 6a, b and Additional file 5: Dataset 3, ‘AML3_AzaC_WIMSi_246’). Specifically, these genes are characterized by a loss of DNA methylation on AzaC treatment greater than 0.5 to 1 kb 5′ and 3′ from the TSS, but minimal change close to the TSS itself (Figure 6b, c). This signature reflects the fact that in untreated AML3 cells, these genes are primarily methylated 5′ and 3′ to the TSS, but devoid of methylation at the TSS (Figure 6d).
Figure 6.
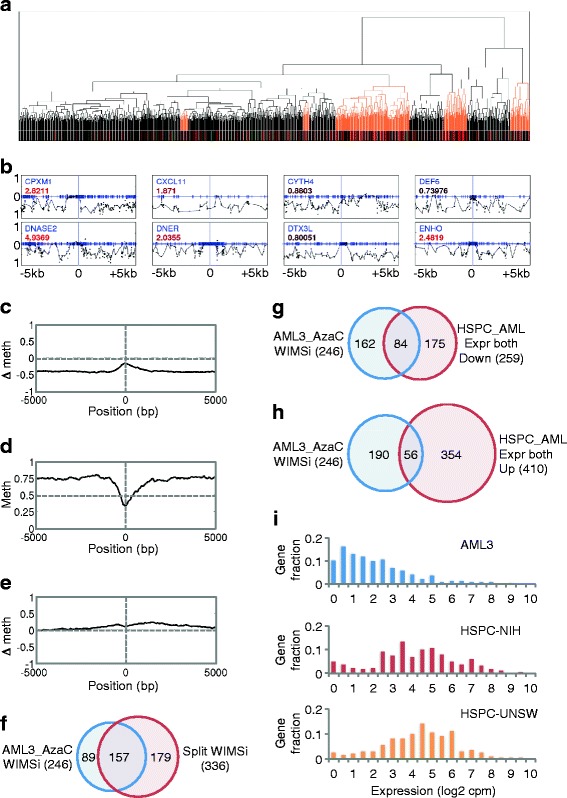
AzaC reverses a specific signature of aberrant promoter DNA methylation and associated gene silencing in AML. (a) Clusters of upregulated genes with similar differential methylation signatures [27]. Orange highlight regions, 75% upregulated. (b) Differential methylation up and downstream of TSS (0 bp). Y-axis, differential methylation, -1 to 1, complete hypomethylation and hypermethylation, respectively. Fold change (log2) gene expression is indicated (>0 = upregulated by AzaC) (Black to red, extent of upregulation). (c) Average differential methylation in AML3 -/+AzaC, over 246 genes identified by the gene list tool. (d) Average fraction methylation (Y-axis) around TSS of 246 genes in AzaC-untreated AML3. (e) Average plot of differential methylation, HSPC, and AML3 cells, over 336 genes identified by the gene list tool. (f) Overlap between 246 genes differentially methylated between AzaC-treated and untreated AML3 ((AML3_AzaC_WIMSi_246), Additional file 5: Dataset 3) and 336 genes differentially methylated between HSPC and AML3 ((Split WIMSi_336), Additional file 5: Dataset 4) (2.18-fold enrichment over random (P < 1 × 10-37), Additional file 5: Dataset 5)). (g) Overlap between 246 genes differentially methylated between AzaC-treated and untreated AML3 (AML3_AzaC_WIMSi_246) and genes downregulated in expression in AML3 cells compared to both HSPC data sets (NIH and UNSW) and significantly regulated by AzaC in AML3) (Additional file 5: Dataset 6, HSPC_AML Expr both Down_259). Eighty-four gene overlap represents a 1.5-fold enrichment over random (P < 3 × 10-6) (Additional file 5: Dataset 7). (h) Overlap between 246 genes differentially methylated between AzaC-treated and untreated AML3 (AML3_AzaC_WIMSi_246) and genes upregulated in expression in AML3 cells compared to both HSPC data sets (NIH and UNSW) and significantly regulated by AzaC in AML3 (HSPC_AML Expr both Up_410) (Additional file 5: Dataset 8). Fifty-six gene overlap represents a 1.57-fold depletion over random (P <1 × 10-6) (Additional file 5: Dataset 9). (i) Fraction of 246 genes differentially methylated between AzaC-treated and untreated AML3 (AML3_AzaC_WIMSi_246) with indicated gene expression in AML3 and HSPC cells (NIH or UNSW datasets).
To further interrogate the significance of this gene set, we repeated the same differential methylation/expression analysis, but compared the same 1,147 genes to the difference in methylation between AML3 cells and normal human hematopoietic stem and progenitor cells (HSPC) previously reported by Hannon and co-workers [28]. This directly tested whether there are methylation changes from HSPC to AML3 cells that correlate with gene expression changes in AML3 cells upon treatment with 5-AzaC. This analysis identified 336 genes that exhibited a similar methylation difference between AML3 and HSPC (Figure 6e and Additional file 5: Dataset 4, ‘Split WIMSi_336’). Specifically, these show more methylation in AML3 compared to HSPC, most marked 5′ and 3′ to the TSS (Figure 6e). Remarkably, all of these genes were upregulated on AzaC treatment of AML3 (Additional file 5: Dataset 4). Of the 246 genes showing decreased methylation on AzaC treatment of AML3 and the 336 genes showing increased methylation between HSPC and AML3, 157 were in common, a significant (P <1 × 10-37) 2.18-fold enrichment over random (Figure 6f and Additional file 5: Dataset 5, ‘Overlap_157’). Thus, genes in the AML3_AzaC_WIMSi_246 gene set tend to show increased promoter methylation in AML3 compared to HSPC.
Accordingly, we reasoned that genes in the AML3_AzaC_WIMSi_246 gene set might tend to be silenced in AML3 compared to HSPC. To test this, we identified all those genes whose expression is downregulated in AML3 compared to HSPC, based on two previously published HSPC gene expression datasets [29,30], and whose expression is significantly regulated by AzaC treatment of AML3 (Additional file 5: Dataset 6, ‘HSPC_AML_Expr both Down_259’). Conversely, we identified all those genes whose expression is upregulated in AML3 compared to HSPC and whose expression is significantly regulated by AzaC treatment of AML3 (Additional file 5: Dataset 8, ‘HSPC_AML_Expr both Up_410’). We then assessed overlap between the AML3_AzaC_WIMSi_246 gene set and these two gene sets. A substantial proportion of the 246 gene set were silenced in AML3 cells compared to normal HSPC (Additional file 5: Dataset 7, ‘Overlap_84’, 84 gene overlap represents a 1.5-fold enrichment over random (P < 3 × 10-6)), but a much smaller proportion was activated in AML3 cells compared to HSPC (Additional file 5: Dataset 9, ‘Overlap_56’, 56 gene overlap represents a 1.57-fold depletion over random (P <1 × 10-6)). Consistent with this, the AML3_AzaC_WIMSi_246 genes were expressed at relatively lower levels in AML3 than in HSPC (Figure 6i). In sum, the AML3_AzaC_WIMSi_246 gene set tends to be silenced in AML3 compared to HSPC.
Analysis of public TCGA primary AML data to compare CpG island methylation and expression of the AML3_AzaC_WIMSi_246 genes according to mutation status of known epigenetic regulators, NPM1 and DNMT3A (any somatic mutation in NPM1 or DNMT3A, and irrespective of normal or abnormal karyotype), revealed very high correlation coefficients between WT/WT AML and the other three genotypes (DNMT3A/NPM1 mutant/WT, WT/mutant, and mutant/mutant (somatic mutation status determined from TCGA)), for both gene expression and promoter CpG methylation (Additional file 7: Figure S5). Thus, across a broad spectrum of AML, NPM1, and/or DNMT3A mutation status does not greatly affect promoter methylation and expression of the 246 genes. However, previous studies have shown that normal karyotype DNMT3A R882 mutant AML exhibit focal CpG hypomethylation at sites throughout the genome [31], whereas normal karyotype IDH1 R132 or IDH2 R140 mutant AML exhibit global hypermethylation [32]. In line with this, we observed a trend towards relative hypomethylation of the 246 genes in normal karyotype AML harboring DNMT3A R882 point mutations, and hypermethylation in normal karyotype AML harboring IDH1 R132 or IDH2 R140 mutations (Additional file 8: Figure S6a). In the case of DNMT3A R882 and IDH2 R140 mutations, the methylation differences compared to normal karyotype AML wild type at these positions were P <0.05 (Additional file 8: Figure S6b). Most notably, in the case of DNMT3A R882 mutant AML, the 246 genes exhibited a significantly greater loss of methylation than all genes (or three randomly selected groups of 246 control genes), whereas in IDH1 R132 and IDH2 R140 mutant AML the 246 genes showed significantly greater gain of methylation than all and control genes (Figure 7a, b, and c). Despite these differences in expression, there was no significant difference in the level of gene expression between any of the wild-type and mutant genotypes (data not shown).
Figure 7.
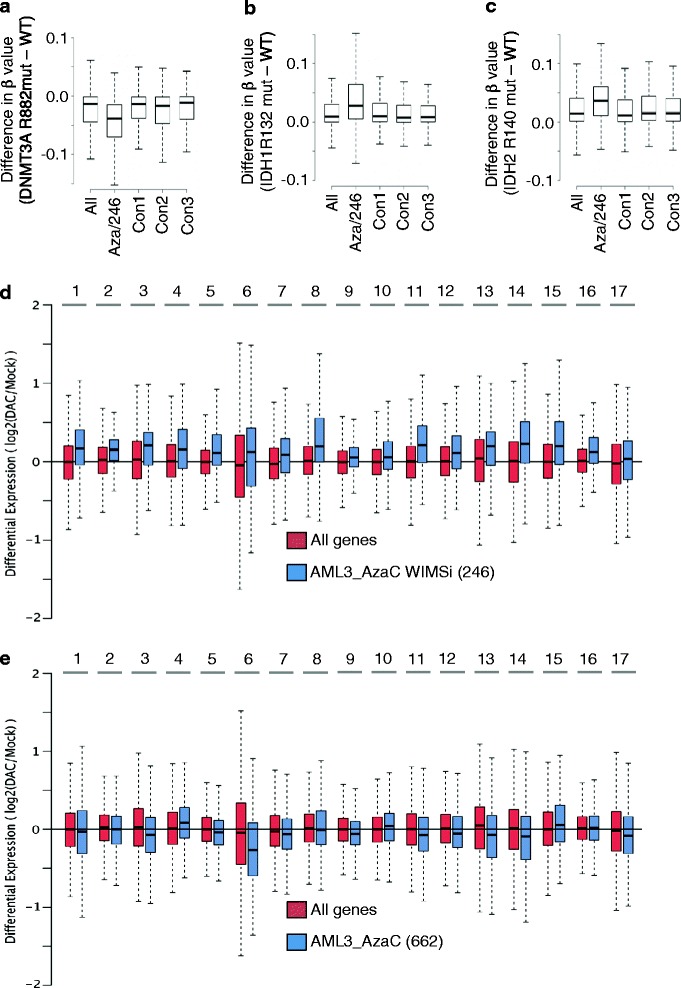
Genes upregulated by AzaC in AML3 are preferentially impacted by mutations that affect DNA methylation and upregulated by Decitabine in patient primary AML blasts. (a) Difference between mean promoter methylation of normal karyotype primary AML WT or mutant at DNMT3A R882 for the indicated groups of genes (all, AML3_AzaC_WIMSi_246 (Aza/246) or three groups of 246 randomly selected genes). P value (Wilcoxon test) of AML3_AzaC_WIMSi_246 versus all other groups <0.001. (b) As (a), but the difference between IDH1 R132 and WT. P value of AML3_AzaC_WIMSi_246 versus all other groups <0.001. (c) As (a), but the difference between IDH2 R140 and WT. P value of AML3_AzaC_WIMSi_246 versus all other groups <0.001. (d) Log2 fold change in expression of AML3_AzaC_WIMSi_246 genes (blue) and all genes (red) in patient primary blasts after treatment with decitabine. Data from [9]. Sixteen out of 17 patients showed a P value <0.001 (Fisher’s Exact). See Additional file 5: Dataset 10 for P value of difference between AML3_AzaC WIMSi (246) genes and all genes. (e) As (a), but 662 genes out of 1,147 genes regulated by AzaC in AML3 (Additional file 5: Dataset 1) that are most divergent from signature identified by WIMSi depicted in Figure 6c (AML3_AzaC_662). See Additional file 5: Dataset 10 for P value of difference between AML3_AzaC WIMSi (246) genes and AML3_AzaC_662 genes.
While analysis of transcription factor binding sites (from [33]) in gene promoters did not reveal any transcription factor binding sites to be substantially enriched in a large proportion of the AML3_AzaC_WIMSi_246 genes (data not shown), assessment of gene ontology using Ingenuity pathway analysis showed that the aforementioned 246 are enriched in genes potentially linked to therapeutic activity of AzaC, including cell movement, cell death and survival, and cell growth and proliferation (Table 1). To further assess the potential therapeutic relevance of this AzaC-responsive 246 gene set, we analyzed their expression levels in patient-derived primary AML blasts treated in vitro with the related DNMTi, decitabine, using a dataset previously published by Ley and co-workers [9]. Of the 246 AzaC-responsive genes, 230 were represented in the Ley and coworkers dataset [9], and these genes were consistently more upregulated by decitabine than all genes, across a group of 17 patients (Figure 7d and Additional file 5: Dataset 10). In contrast, the 662 genes (in the original set of all genes regulated by AzaC in AML3, AML3_AzaC_SigDiff_1147) for which WIMSi did not find evidence of a correlation between methylation and expression (‘AML3_AzaC_662’) were not more upregulated by decitabine than all genes (Figure 7e and Additional file 5: Dataset 10). Thus, using WIMSi’s methylation signature approach, we have identified a subset of genes regulated by AzaC treatment of AML3 that are more likely than other AzaC-regulated genes to be upregulated by decitabine in primary patient blasts.
Table 1.
Results of IPA analysis of 246 genes identified by WIMSi in AzaC-treated AML3 cells (AML3_AzaC WIMSi (246))
| Molecular and cellular function | Number genes | P value |
|---|---|---|
| Cell death and survival | 102 | <0.004 |
| Cellular movement | 68 | <0.004 |
| Cellular function and maintenance | 80 | <0.004 |
| Cellular growth and proliferation | 106 | <0.004 |
| Cell-to-cell signaling and interaction | 63 | <0.004 |
The five top-scoring ‘molecular and cellular functions’ are shown.
Discussion
To date, most WGBS analyses of human cancers have been performed on solid tumors [15–17]. DNA methylation analyses in AML have tended to employ less comprehensive methods, such as Illumina arrays, reduced representation bisulfite sequencing (RRBS) and methylated DNA immunoprecipitation (MeDIP)-seq. While these studies obviously have their own important strengths, such as throughput of multiple primary samples from patients [9,34–36], no previous study has performed WGBS on primary AML blasts or cell lines. Nor have previous studies examined the effects of DNMTi on DNA methylation and gene expression, employing such comprehensive methods as WGBS and RNA-seq. This is important because DNMTi are used in the clinic, yet the relationship between their effects on DNA methylation and gene expression is unclear.
Accordingly, we report here the first WGBS analysis of DNA methylation in an AML cell line. We also report the effects of AzaC treatment on DNA methylation and gene expression. Based on simple quantitative analyses of methylation at promoters, CpG islands, and shores, there was no significant correlation between loss of DNA methylation and change in gene expression. However, a more sophisticated search algorithm identified a subset of upregulated genes with a signature loss of methylation flanking the TSS. Remarkably, many of these same genes gained methylation in AML3 cells compared to normal hematopoietic stem and progenitor cells and this was typically accompanied by their downregulation in AML3 cells. These genes have functions in cell movement, cell death and survival, and cell growth and proliferation and are preferentially upregulated on decitabine treatment of patient-derived primary AML blasts. Hence, these genes are candidates for genes whose expression is aberrantly repressed by DNA methylation in AML and reversed by DNMTi treatment.
Globally, the DNA methylation landscape of proliferating AML cells, without AzaC treatment, is reminiscent of other solid tumor epigenomes analyzed by WGBS [15–17]; large regions of near complete DNA methylation are interspersed with regions of partial methylation and much more focal regions that are largely depleted of DNA methylation. As in normal genomes, regions lacking DNA methylation are predominantly at promoters containing CpG islands [18]. However, as is typical of cancer genomes, a proportion of CpG islands is methylated [26]; in AML3 cells, about 12% of CpG islands overlapping gene TSS are methylated. Consistent with the link between CpG island hypermethylation and gene silencing [26], genes with methylated CpG islands tend to be expressed at a lower level than genes with unmethylated CpG islands. As shown previously in other cell types, at gene bodies there is general trend towards increasing gene body methylation with increasing expression, although this relationship breaks down at the most highly expressed genes [18,21,22,24,25].
Treatment of AML3 cells with AzaC resulted in a near-uniform approxiamte 50% decrease in methylation across the whole genome. Only promoters, CpG islands and 5′ UTRs underwent a slightly more modest decrease, presumably because many of these regions are unmethylated or barely methylated even prior to AzaC treatment. In contrast, previous reports have suggested that AzaC and decitabine cause preferential loss of DNA methylation at some regions of the genome [9,34,37]. While differences between cell lines, primary blasts, and DNMTi treatment protocols might account for some differences, the 15× genome-wide coverage achieved in our study unambiguously reveals a uniform decrease across the genome in this study. Of course, a 50% decrease in methylation across the whole genome results in a greater absolute loss of methylation at highly methylated regions, compared to lowly methylated regions. In fact, from this perspective our data appear consistent with those of Ley and coworkers [9]. In sum, at least in AML3 cells, there is no locus-specific preferential loss or retention of DNA methylation.
In contrast to the uniform loss of methylation across the genome, AzaC caused highly targeted gene-specific changes in gene expression. Specifically, 792 genes were significantly upregulated, and 426 downregulated. Since about 12% of genes with a CpG island overlapping the TSS harbor a methylated CpG island in AzaC-untreated cells, and since methylation is associated with decreased expression in these cells, we initially asked whether upregulation of gene expression was associated with CpG island hypomethylation. However, based on simple quantitative analyses of methylation at promoters, CpG islands, and shores, there was no significant correlation between loss of DNA methylation and change in gene expression. Previous studies, for example employing Illumina 450 K arrays or Sequenom technology targeted to selected genes, similarly failed to observe a strong link in this regard [9,10,38–43]. Conceivably, failure to observe widespread upregulation of hypomethylated genes in in vitro studies depends, in part, on lack of appropriate in vivo signals and environmental factors. Obviously, this issue can only be addressed in humans in the context of clinical studies.
Regardless, a major advantage of WGBS data lies in the ability to perform unbiased searches for patterns of methylation (at the single nucleotide level) that correlate with expression [20,27]. Indeed, a more sophisticated search algorithm, WIMSi [27], identified a subset of 246 upregulated genes with a shared signature loss of methylation flanking the TSS. Increased expression of these genes after AzaC, associated with a common methylation loss signature, tentatively suggests that these genes might be directly regulated by DNA methylation. In further support of this idea, many of these same genes gained methylation in AML3 cells compared to normal hematopoietic stem and progenitor cells and this was typically accompanied by their downregulation. Conceivably, these are genes whose is expression is aberrantly repressed by DNA methylation in AML and reversed by AzaC treatment of AML; the remainder of the genes regulated by AzaC in AML3 might be regulated as a secondary consequence of these candidate primary targets, or might be regulated via effects of AzaC on RNA or DNA damage. Consistent with these 246 genes being directly regulated by DNA methylation, these genes were also significantly and preferentially upregulated in decitabine-treated human primary AML blasts, compared to all genes and even genes regulated by AzaC in AML3 but lacking the loss of methylation signature characteristic of the 246 genes. Since AzaC and decitabine share the ability to induce DNA hypomethylation, but differ in some other respects such as AzaC’s preferential incorporation into RNA, this points to the 246 genes being regulated by DNA methylation. Underscoring the power of the WGBS and WIMSi analysis approach, the original authors of this decitabine study reported a limited correlation between change in DNA methylation and expression [9], likely because the array-based approach had insufficient coverage of promoter CpGs. The 246 AzaC-regulated genes are involved in processes the can reasonably drive anti-neoplastic activity, cell death, cell movement, and cell proliferation, supporting the view that reversal of silencing of these genes by DNMTi contributes to therapeutic activity. If so, methylation and/or expression status of these genes might have utility as biomarkers to predict and/or monitor patient response to DNMTi.
Conclusions
In sum, our WGBS and WIMSi data analysis approach has identified a set of genes whose methylation and silencing in AML is reversed by DNMTi. Known genetic determinants of altered genome methylation in AML, namely DNMT3A, IDH1, and IDH2 mutations, also preferentially impact methylation of this group of 246 genes in normal karyotype primary AML. These genes are good candidates for direct regulation by DNMTi, and their reactivation by DNMTi may contribute to therapeutic activity. Consequently, regulation of these genes by DNMTi might serve as a biomarker to monitor on-target activity of DNMTi in patients, and perhaps to predict therapeutic response. This study also demonstrates the ability of WIMSi to reveal relationships between DNA methylation and gene expression, based on single-nucleotide bisulfite-sequencing and RNA-seq data.
Materials and methods
Antibodies
Antibodies to the following targets were used in this study: actin (A1978, Sigma); DNMT1 (AB19905, Abcam); γH2AX (05-636, Millipore), PARP (9542P, Cell Signaling), and 5′-BrdU (347580, Becton Dickinson).
Cell culture, AzaC treatment, and cell viability assays
OCI-AML3 cells were obtained from DSMZ [44] and authenticated using Applied Biosystems AMPF/STR identifier kit (short tandem repeat multiplex assay). Cells were cultured in suspension in RPMI media supplemented with FBS 20%, Penicillin 5%, and L-Glutamine 5%, incubated at 37°C in humidified conditions and 5% CO2. To passage, cells were counted by hemocytometer and either centrifuged to a pellet and resuspended in fresh media or resuspended in 50% fresh media at a concentration of 0.5 × 106/mL every 2 to 3 days. For long-term storage cells were centrifuged to a pellet and resuspended in 1 mL freezing media (70% RPMI/20% FBS/10% DMSO) in batches of between 4 and 10 × 106/mL and stored in cryovials at -80°C.
AzaC was dissolved from lyophilised powder into culture-sterile DMSO to produce a 20 mM stock solution and stored as 15 μL aliquots at -80°C. For each new experiment a fresh aliquot of AzaC was diluted in RPMI/20% FBS to produce a 2 mM working stock which was diluted directly onto cells in culture.
Viable cells were counted using vital dye (trypan blue) to assess membrane integrity. All counts were performed in duplicate or triplicate. Where indicated, cell viability was also assessed using the indicator dye Rezasaurin to measure metabolic capacity of the cells, according to the manufacturer’s instructions (CellTiter-Blue, Promega).
Western blotting
Cell lysates were prepared by resuspending cells in freshly-boiled 1x Laemmli SDS sample buffer. Protein quantitation was performed by Bradford assay (Bio-Rad). Western blotting was performed as described previously [45].
Activated caspase assays
NucView 488 Caspase 3 substrate was used to detect caspase 3 activity as a reflection of apoptosis, according to manufacturer’s instructions (Biotium).
Cell cycle analysis
DNA content was measured by FACS in fixed, permeabilized 7-AAD-stained cells, as described previously [46]. Cell cycle distribution was modelled from DNA content using FlowJo [47]. Two color 7-AAD and 5-BrdU cell cycle analysis was performed as described previously [48].
CFSE assays
CFSE staining was used to track cumulative cell divisions, as described previously [49].
Bisulfite sequencing
Bisulfite sequencing of duplicate samples of genomic DNA from untreated and AzaC-treated AML3 cells was performed by BGI Tech.
DNA methylation data analysis/statistics
Processing and alignment of Bisulfite sequencing reads
Sequence reads are transformed in silico to fully bisulfite converted forward (C- > T) and reverse (G- > A) reads. The converted sequences are aligned against a converted human reference genome (hg18) in each combination: (1) forward (C- > T) reads aligned to forward (C- > T) genome, (2) reverse (G- > A) reads aligned to reverse (G- > A) genome, (3) forward (C- > T) reads aligned to reverse (G- > A) genome, (4) reverse (G- > A) reads aligned to forward (C- > T) genome. During the library preparation process genomic fragments representing alignments (3) and (4) are generated in the PCR step however they are not sequenced and only fragments corresponding to alignments (1) and (2) are read. As a result only uniquely matching alignments from (1) and (2) are retained. Alignment was performed using bismark [50] (version 0.5.1), based on the Bowtie [51] aligner (version 0.12.7). Unaligned reads resulting from the initial alignments from these libraries were trimmed 15 bp from the 5′ end in order to remove the adapter sequences, as some libraries contained these sequences and realigned.
For each aligned sequence tag, the original unconverted sequence is compared against the original unconverted reference genome and the methylation status is inferred. Sequences aligned from (1) and (2) give information on cytosines on the forward and reverse strands, respectively.
To remove PCR bias a deduplication step removes potential duplicate reads, where both ends of the fragment align to the same genomic positions on the same strand, only one of the reads is retained. To control for potential incomplete bisulfite treatment any reads with more than three methylated cytosines in non-CpG contexts are discarded. Additional file 2: Table S1 details the sequence yields at each stage of this process. Additional file 2: Tables S2, S5, and S6 detail the number of methylated and unmethylated bases sequenced within CpG, CHG, and CHH contexts (H = A, C, or T). Additionally, reads are mapped against the unmethylated lambda genome which was added to bisulfite sequencing reactions, giving the number of methylated bases allowing combined error rate resulting from sequencing errors and incomplete bisulfite conversion (Additional file 2: Table S7) to be determined.
Identification of methylated cytosines
Processed reads are aggregated on a per CpG basis (number of bases read supporting methylated/unmethylated status). At each reference cytosine the binomial distribution was used to identify whether a subset of the genomes within the sample were methylated at this location. Methylcytosines were identified while keeping the number of false positives below 1%. The probability of sequencing an observed number of cytosines given the identified error rates from the lambda alignment was determined using the binomial distribution. At each reference cytosine the number of trials in the binomial test was the read depth and the number of successes in the test was equal to the number of cytosines sequenced at that base. The probability was then corrected using the BH-FDR function and the list of CpG sites was thresholded at the 0.01 FDR level. See Additional file 2: Table S3.
A two-tailed Fisher’s exact test was used to identify CpGs that were differentially methylated. Only CpG determined using the binomial distribution in at least one sample, with at least three reads in at least one condition and with at least one read in the other condition, were considered for testing. P values were corrected using the BH-FDR function to control false positives at a rate of 5%. At this stage replicates were pooled for subsequent analysis.
Percentage CpG methylation in genomic windows
The percentage CpG methylation for a given window was calculated as the total number of methylated cytosines sequenced (at CpG sites for that window), divided by the total number of cytosines sequenced (at CpG sites for that window), multiplied by 100.
Difference and relative difference in CpG methylation
Difference in CpG methylation was defined as the difference between the treated and untreated percentage methylation. Relative difference in CpG methylation was defined as the difference in CpG methylation divided by the untreated percentage methylation.
Global methylation
The whole genome was split into non-overlapping 2 kb windows. For each window the start position was the nearest upstream reference CpG site to the end of the previous window. The percentage CpG methylation (AzaC untreated and treated), and relative difference in CpG methylation was determined for each window as previously described. Windows with no reads at all CpG sites, in either dataset were omitted.
Genomic features
The genomic features genic, exonic, 5′ UTR, and 3′ UTR were downloaded from Biomart (Ensembl genes version 54), and CpG Islands, SINEs, LINEs, STRs, LTRs, low complexity DNA, DNA transposons, and satellite repeats were downloaded from UCSC (hg18). Promoter regions were defined as TSS +/- 2 kb, and intronic regions as genic and non-exonic (overlapping exons were merged). CpG island shores were defined as the 2 kb flanking the CpG island, and CpG island shelves as the 2 kb flanking the shores [7].
CpG ratio
The observed-to-expected CpG Ratio was calculated as: (CpGs in window * window length)/(Number of Gs in Window * Number of Cs in window) [42].
Determination of observed-to-expected overlaps
Overlaps were computed on a per base pair basis between two datasets (A and B). For every region within A the number of base pairs that were occupied by a region within B was computed. A permutation test was performed in order to determine the background genomic average expected overlap. One thousand sets of regions with properties (length distribution and chromosome distribution) equal to set B were generated. Randomly generated regions of B were prevented from being generated within unsequenced regions of the genome (as defined by UCSC mapping and sequencing track - gap). The overlap of A and B was repeated for each randomly generated set of B to determine the average expected random overlap. P values were estimated empirically from the observed overlaps of the randomly generated sets.
Methylation difference plots
Data bins were created for the integer values 0 to 100 and initialized as empty lists. For each CpG site within the pooled data the methylation percentage for AzaC untreated and treated samples was calculated, considering only CpGs with minimum coverage of 10 reads in at least one sample and three reads in both samples. Methylation percentage of the AzaC untreated sample was truncated to an integer value and used to select an appropriate data bin into which the treated sample methylation percentage was appended and the average of each bin was computed giving the corresponding final methylation percentage in treated cells. Difference in methylation was defined as the difference between final and starting average methylation percentages, and relative difference in methylation was defined as the difference in methylation divided by the average starting methylation percentages.
Smoothed methylation plots
The pooled whole genome methylation data were processed using the BSmooth algorithm from the bsseq (v0.8.0) package within Bioconductor as described in [52]. A modified version of the bsseq plotSmoothData function was created to plot the smoothed data with individual CpGs shown (addPoints = True) but while suppressing the vertical ablines.
Composite plot generation
To generate composite gene profiles the Ensembl gene annotation (version 54) was used to identify the outermost transcript start and end sites (TSS and TES) for each of the Ensembl genes in the expression dataset. The area between these sites, for each gene, was classified as the gene body and split into 50 windows of equal size (each corresponding to 2% of the total gene body). Ten additional 1 kb windows were prepended (appended) to the start (end) of the gene to provide genomic context. The AzaC untreated percentage methylated CpGs for each window was determined.
Genes by decile plot
Expressed Genes (FPKM > 0) were ranked by expression and placed into appropriate ranked decile bins. For each composite plot window, the percentage methylation was averaged for all genes in each decile.
Gene body heatmap
Heatmaps were generated where the y-axis corresponds to Ensembl genes ordered by expression value in AzaC untreated RNA-seq dataset and the x-axis to percentage CpG methylation in the composite plot windows described previously and their position along genes. Genes for which greater than 50% of windows contained no reference CpGs were omitted.
TSS centered heatmaps
The area around each TSS was split into 200 bp windows spanning 5 kb upstream and downstream of the TSS. The percentage CpG methylation and observed-to-expected CpG ratio for each window was determined. Heatmaps were generated where the y-axis was as the gene body heatmap, and the x-axis to the described windows and their position relative to the TSS. Genes for which greater than 50% of windows contained no reference CpGs were omitted.
RNA sequencing
Samples were prepared for RNA sequencing (RNA-seq) according to the Illumina manufacturer’s instructions. Samples were sequenced using the Illumina GAIIX sequencer. For analysis, RNA-seq paired-end reads are aligned to the human genome (hg18) using a splicing-aware aligner (tophat). Reference splice junctions are provided by a reference transcriptome (Ensembl build 54), and novel splicing junctions are determined by detecting reads that span exons that are not in the reference annotation. Aligned reads are processed to assemble transcript isoforms, and abundance is estimated using the maximum likelihood estimate function (cuffdiff) from which differential expression and splicing can be derived. See Additional file 2: Table S9. RNA-seq gene expression data from HSPC cells [29,30], was processed using edgeR and an FDR cutoff of 0.1.
Comparison of gene expression and DNA methylation data
For direct comparisons of gene expression to DNA methylation in Figure 5 and Additional file 6: Figure S4, the full list of 1,218 significantly changed genes (including 71 small RNAs) was used.
Comparison of AML and AML3 methylation
The Cancer Genome Atlas (TCGA) Illumina Human Methylation 450 k Array data for AML was downloaded from the TCGA data portal [53]. The hg18 genomic coordinates for each CpG probe was identified using the Illumina Human Methylation 450 k Array annotation file obtained from GEO [54]. To allow reasonable comparison, the TCGA methylation data were filtered to remove all CpGs where there were fewer than 10 reads in our pooled AML3 BS-Seq dataset. Next, for each gene (NCBI gene annotation 36.1): the mean beta value across all samples, for all CpGs within 2 kb of the TSS, was calculated. Finally the mean beta values were plotted against the mean% CpG methylation at the equivalent CpGs in the pooled AML3 data. To generate smoothed scatter plots the R (v3.0.2) function smoothscatter was used, using the transformation function x^1. Spearman correlations were calculated using the R function cor.test method = ‘spearman’.
Comparison of AML and AML3 expression
The Cancer Genome Atlas (TCGA) RNA-seq V2 data for AML was downloaded from the TCGA data portal [53]. For each gene, the mean read count across all TCGA samples was calculated. For each gene, the mean read count across all AML3 RNA-seq samples was calculated using HTSeq [55]. Finally expression values (log2) for AML were plotted against the equivalent values (log2) in AML3, using R (v3.0.2) as above.
Comparison of TCGA AML subtypes data preparation
The Cancer Genome Atlas (TCGA) Patient and Mutation data for AML were downloaded from the TCGA data portal [53]. Mutation subtypes (number of samples) were identified as: WT/WT (120 genes), WT/mut (23 genes), mut/WT (26 genes), and mut/mut (28 genes) for DNMT3A and NPM1, respectively. Where WT corresponds to no somatic mutations identified, and mut corresponds to at least one somatic mutation identified in the sample. Additional mutation subtypes (number of samples) were identified as: NK DNMT3A R882+ (20 samples), NK DNMT3A R882- (82 samples), NK IDH1 R132+ (10 samples), NK IDH1 R132- (92 samples), NK IDH2 R140+ (12 samples), and IDH2 R140- (90 samples). Where NK indicates normal karyotype and +/- indicates the presence/absence of the specific mutation in the sample.
Using the TCGA illumina human methylation 450 k array data for AML
For each gene (NCBI gene annotation 37), the mean beta value across all samples of each mutation subtype, for all CpGs within 2 kb of the TSS, was calculated.
Using the RNA-seq V2 data for AML
For each gene, the mean read count across all samples of each mutation subtype was calculated.
Comparison of TCGA AML subtypes, scatter, and boxplots
To generate the TCGA AML mutation subtype scatter plots, the expression and methylation data described above were filtered to include only the 246 upregulated genes identified by WIMSi between AML3 and AML3 + AzaC. Scatter and boxplots were then generated comparing each mutation subset to its appropriate control, for both methylation and expression. For expression a log2 scale was used. Pearson Correlation Coefficients were calculated using Libre Office (v4.0.2.2). Wilcoxon tests were performed using the R (v3.0.2) function wilcox.test. To generate control random gene lists, the python (v2.7.4) library random.py was used.
Identification of altered methylation associated with altered gene expression by WIMSi
This was performed as described in [27]. For this approach, methylation signatures were created for each gene by interpolating the differential methylation scores over a fixed window relative to the gene’s transcription start site (TSS). The geneset was significant changed genes (Additional file 5: Dataset 1, not including 71 small RNAs). A curve similarity metric, Dynamic Time Warping, was used to cluster genes together based on the shape of the differential methylation data in the window. We then identified clusters of genes with similar patterns that have coordinated differential expression. To generate a gene list, we executed this procedure over many 5 kb windows around the TSS and selectively combine the results.
Duplicates from AzaC treated and untreated were averaged and cross-referenced with Ensembl to determine the TSS. Genes with no sites having a differential methylation level of at least 0.2 within the window were removed. Genes for which methylation could not be interpolated due to a low number of sites in the region were removed. Signatures were clustered for 23 5 kb windows overlapping every 500 bp, covering the area from 8 kb upstream of the TSS to 8 kb downstream. Clusters that were significantly up- or downregulated compared to the overall set of expression values were then identified (Kolmogorov-Smirnov test; FDR <0.05 using Benajmini-Hochberg). The Fréchet distance was computed using a scaling factor between the x-axis (bp) and y-axis (differential methylation score) of 2,500 bp to 1 unit. A minimum cluster size of 10 and a minimum cluster purity of 0.85 were used. Gaussian smoothing (σ = 50) was applied to the signatures before clustering. A gene was included in the final list if at least two of its replicates were present in a significant cluster for at least three of the 5 kb windows.
Analysis of decitabine-treated AML cells
Processed expression values were downloaded from GEO (GSE40442). Expression values were averaged across probes for each Refseq gene. For each gene differential expression was computed as log2(decitabine treated expression/Mock treated expression). Differential expression values for each sample from the 246 AzaC responsive genes (AML3_AzaC_WIMSi_246) were compared to all genes using Fisher’s Exact test. To identify genes likely not regulated by methylation, we ran the WIMSi gene list tool and found 662 genes (out of 1,147 genes differentially expressed in AzaC treated AML3 cells) that were not flagged as significant in any window.
Data access
RNA seq data from human CD34+ HSPC (UNSW) GEO accession, GSM1097887 [30].
RNA seq data from human CD34+ HSPC (NIH) GEO accession, GSM651554 [29].
DNA methylation data from human HSPCs GEO accession, GSE31971 [28].
Microarray data from DAC treated AML cells, GEO accession GSE40442 [9].
TCGA data were from the TCGA data portal ([53]).
New datasets in this MS:
RNA-seq supplementary data: [56].
BS-seq supplementary data: [57].
Acknowledgments
We thank Dr. Alice Baudot for p53 inducible SAOS2 cells. Whole genome bisulfite sequencing was performed by BGI Tech. Work in the lab of PDA is supported by CRUK program grant A10250 (renewal A16566). Work in the lab of JRE is supported by grants NIH 4-R00-CA127360-02, NIH 1R21LM011199-01, and DOD BC101296P1. MC was supported by the Scottish Funding Council SSCF scheme (SCD/04).
Additional files
Optimization of AzaC treatment protocol. (a) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC at 0, 24, 48 h and assayed by FACS at 96 h for activated caspase 3. As a positive control, cells were treated with vehicle or 1 μM etoposide (Eto). (b) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC at 0, 24, 48 h and DNA content measured by FACS at 72 h in AAD-stained cells. (c) Cell cycle distribution, including <2n DNA content (Sub G1), was determined from (b). (d) AML3 cells were treated three times with vehicle, 0.5, 1, or 2 μM AzaC at 0, 24, 48 h and proliferation of viable cells measured by trypan blue exclusion assay.
Sequence yield. Table S2. CpG methylation. Table S3. Methylated CpG sites. Table S4. Spearman correlation coefficients. Table S5. CHG methylation. Table S6. CHH methylation. Table S7. Error rates. Table S8. Individual CpG differences. Table S9. RNA sequencing.
Comparison of promoter DNA methylation and gene expression in AML3 cells and primary AML cells. (a) Smoothed scatter plot of mean promoter (TSS -/+2 kb) methylation of all genes (NCBI36.1) in primary AML (based on β-values from TCGA) versus promoter (TSS -/+2 kb) methylation of all genes in AML3. (b) Smoothed scatter plot of mean expression (log2 read counts) of all genes in primary AML (from TCGA) versus expression of all genes in AML3.
Genome distribution of DNA methylation. Number of methylated, partially methylated, and unmethylated CpGs (defined as in Figure 2 (b)) overlapping specified genomic features.
Datasets 1 to 10.
Relationship of gene expression to methylation. (a) Scatter plot of fold change in expression versus difference in CpG island methylation at genes with a CpG island within 10 kb of the TSS, showing all genes (blue) and significant regulated genes (red). (b) Scatter plot of fold change in expression versus difference in CpG island shore methylation at genes with a CpG island within 10 kb of the TSS, showing all genes (blue) and significant regulated genes (red).
Comparison of expression and promoter methylation of AML3_AzaC WIMSi (246) genes across different primary AML genotypes. (a) Scatter plots of gene expression in primary AML cells (DNMT3A WT and NPM1 WT) versus primary AML of indicated genotypes (DNMT3A/NPM1 status as indicated). Pearson correlation coefficient (PCC) is indicated. (b) Scatter plots of promoter (β-value, TSS -/+2 kb) methylation in primary AML cells (DNMT3A WT and NPM1 WT) versus primary AML of indicated genotypes (DNMT3A/NPM1 status as indicated). Pearson correlation coefficient (PCC) is indicated. Mutation status from TCGA.
Comparison of promoter methylation of AML3_AzaC WIMSi (246) genes across different primary AML normal karyotype genotypes. (a) Scatter plots of mean promoter methylation (mean β-value, TSS -/+2 kb) of all AML3_AzaC WIMSi (246) genes in all normal karyotype primary AML (DNMT3A, IDH1, IDH2 WT, as indicated) versus normal karyotype primary AML of indicated mutant genotypes (DNMT3A R882, IDH1 R132, IDH2 R140, as indicated). Pearson correlation coefficient (PCC) is indicated. Mutation status from TCGA. (b) Box plots of mean promoter methylation (mean β-value, TSS -/+2 kb) of all normal karyotype AML for each AML3_AzaC WIMSi (246) gene, comparing AML WT and mutant at indicated position. P value (Wilcoxon test) WT versus mutant = 0.0097, 0.0600, 0.0236 for DNMT3A R882, IDH1 R132, and IDH2 R140, respectively.
Footnotes
Competing interests
The authors declare that they have no competing interests
Authors’ contributions
KL performed experiments, provided critical intellectual input, and wrote the manuscript. JC performed data analysis and provided critical intellectual input. NDV performed data analysis and provided critical intellectual input. TM performed data analysis and provided critical intellectual input. NAP and WC assisted with experiments MC provided critical intellectual input. JRE performed data analysis and provided critical intellectual input. PDA conceived the study, provided critical intellectual input, and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Kirstin Lund, Email: kirstinlund@nhs.net.
John J Cole, Email: johnjcole@hotmail.co.uk.
Nathan D VanderKraats, Email: ndvk1980@yahoo.com.
Tony McBryan, Email: tony@mcbryan.co.uk.
Nikolay A Pchelintsev, Email: n.a.pchelintsev@gmail.com.
William Clark, Email: w.clark@beatson.gla.ac.uk.
Mhairi Copland, Email: Mhairi.Copland@glasgow.ac.uk.
John R Edwards, Email: JEDWARDS@DOM.wustl.edu.
Peter D Adams, Email: p.adams@beatson.gla.ac.uk.
References
- 1.Weinberg OK, Seetharam M, Ren L, Seo K, Ma L, Merker JD, Gotlib J, Zehnder JL, Arber DA. Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood. 2009;113:1906–1908. doi: 10.1182/blood-2008-10-182782. [DOI] [PubMed] [Google Scholar]
- 2.Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 1997;57:837–841. [PubMed] [Google Scholar]
- 3.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kihslinger JE, Godley LA. The use of hypomethylating agents in the treatment of hematologic malignancies. Leuk Lymphoma. 2007;48:1676–1695. doi: 10.1080/10428190701493910. [DOI] [PubMed] [Google Scholar]
- 5.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul TA, Bies J, Small D, Wolff L. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood. 2010;115:3098–3108. doi: 10.1182/blood-2009-07-233858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N, Erdmann-Gilmore P, Lichti CF, Meyer MR, Townsend RR, Wilson RK, Mardis ER, Ley TJ. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. 2013;121:1633–1643. doi: 10.1182/blood-2012-09-459313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claus R, Pfeifer D, Almstedt M, Zucknick M, Hackanson B, Plass C, Lübbert M. Decitabine induces very early in vivo DNA methylation changes in blasts from patients with acute myeloid leukemia. Leuk Res. 2013;37:190–196. doi: 10.1016/j.leukres.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Quentmeier H, Martelli MP, Dirks WG, Bolli N, Liso A, Macleod RA, Nicoletti I, Mannucci R, Pucciarini A, Bigerna B, Martelli MF, Mecucci C, Drexler HG, Falini B. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760–1767. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 12.Tiacci E, Spanhol-Rosseto A, Martelli MP, Pasqualucci L, Quentmeier H, Grossmann V, Drexler HG, Falini B. The NPM1 wild-type OCI-AML2 and the NPM1-mutated OCI-AML3 cell lines carry DNMT3A mutations. Leukemia. 2012;26:554–557. doi: 10.1038/leu.2011.238. [DOI] [PubMed] [Google Scholar]
- 13.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O'Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF, GIMEMA Acute Leukemia Working Party Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 15.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Stevenson BJ, Ecker JR, Bafna V, Strausberg RL, Simpson AJ, Ren B. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, Van Den Berg D, Laird PW. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44:40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, Gingrich JA, Haghighi F, Nutter R, Bestor TH. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, Adams PD. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15:1495–1506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci U S A. 2009;106:671–678. doi: 10.1073/pnas.0812399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 23.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 24.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sproul D, Meehan RR. Genomic insights into cancer-associated aberrant CpG island hypermethylation. Brief Funct Genomics. 2013;12:174–190. doi: 10.1093/bfgp/els063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanderkraats ND, Hiken JF, Decker KF, Edwards JR. Discovering high-resolution patterns of differential DNA methylation that correlate with gene expression changes. Nucleic Acids Res. 2013;41:6816–6827. doi: 10.1093/nar/gkt482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, Smith AD, Hannon GJ. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beck D, Thoms JA, Perera D, Schütte J, Unnikrishnan A, Knezevic K, Kinston SJ, Wilson NK, O'Brien TA, Göttgens B, Wong JW, Pimanda JE. Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood. 2013;122:e12–e22. doi: 10.1182/blood-2013-03-490425. [DOI] [PubMed] [Google Scholar]
- 31.Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–454. doi: 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 34.Yan P, Frankhouser D, Murphy M, Tam HH, Rodriguez B, Curfman J, Trimarchi M, Geyer S, Wu YZ, Whitman SP, Metzeler K, Walker A, Klisovic R, Jacob S, Grever MR, Byrd JC, Bloomfield CD, Garzon R, Blum W, Caligiuri MA, Bundschuh R, Marcucci G. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood. 2012;120:2466–2474. doi: 10.1182/blood-2012-05-429175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, Allis CD, Roeder RG, Valk PJ, Löwenberg B, Delwel R, Fernandez HF, Paietta E, Tallman MS, Schroth GP, Mason CE, Melnick A, Figueroa ME. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saied MH, Marzec J, Khalid S, Smith P, Down TA, Rakyan VK, Molloy G, Raghavan M, Debernardi S, Young BD. Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS One. 2012;7:e33213. doi: 10.1371/journal.pone.0033213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagemann S, Heil O, Lyko F, Brueckner B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS One. 2011;6:e17388. doi: 10.1371/journal.pone.0017388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, Plass C, Niemeyer CM, Lübbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23:1019–1028. doi: 10.1038/leu.2008.397. [DOI] [PubMed] [Google Scholar]
- 39.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, Brock MV, Stearns V, Feller-Kopman D, Yarmus LB, Lin YC, Welm AL, Issa JP, Minn I, Matsui W, Jang YY, Sharkis SJ, Baylin SB, Zahnow CA. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mossman D, Kim KT, Scott RJ. Demethylation by 5-aza-2′-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists. BMC Cancer. 2010;10:366. doi: 10.1186/1471-2407-10-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmelz K, Sattler N, Wagner M, Lubbert M, Dorken B, Tamm I. Induction of gene expression by 5-Aza-2′-deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia. 2005;19:103–111. doi: 10.1038/sj.leu.2403552. [DOI] [PubMed] [Google Scholar]
- 42.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 43.Qiu X, Hother C, Ralfkiær UM, Søgaard A, Lu Q, Workman CT, Liang G, Jones PA, Grønbæk K. Equitoxic doses of 5-azacytidine and 5-aza-2′deoxycytidine induce diverse immediate and overlapping heritable changes in the transcriptome. PLoS One. 2010;5:e12994. doi: 10.1371/journal.pone.0012994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures. ,[http://www.dsmz.de/home.html]
- 45.Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 46.Crissman HA, Steinkamp JA. Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. J Cell Biol. 1973;59:766–771. doi: 10.1083/jcb.59.3.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flowjo., [http://www.flowjo.com]
- 48.Dolbeare F, Gratzner H, Pallavicini MG, Gray JW. Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad Sci U S A. 1983;80:5573–5577. doi: 10.1073/pnas.80.18.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 50.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.TCGA public data., [https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm?]
- 54.GEO public data., [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL13534]
- 55.Anders S, Pyl PT, Huber W: HTSeq — A Python framework to work with high-throughput sequencing data.; 2014. bioRxiv preprint. [DOI] [PMC free article] [PubMed]
- 56.GEO public data., [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=khcfkyoonngfbof&acc=GSE55123]
- 57.GEO public data., ᅟ:ᅟ [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=erczewmadpsnrep&acc=GSE55124]