

Abstract
Lyssaviruses, which are members of the Rhabdoviridae family, induce apoptosis, which plays an important role in the neuropathogenesis of rabies. However, the mechanisms by which these viruses mediate neuronal apoptosis have not been elucidated. Here we demonstrate that the early induction of apoptosis in a model of lyssavirus-infected neuroblastoma cells involves a TRAIL-dependent pathway requiring the activation of caspase-8 but not of caspase-9 or caspase-10. The activation of caspase-8 results in the activation of caspase-3 and caspase-6, as shown by an increase in the cleavage of the specific caspase substrate in lyssavirus-infected cells. However, neither caspase-1 nor caspase-2 activity was detected during the early phase of infection. Lyssavirus-mediated cell death involves an interaction between TRAIL receptors and TRAIL, as demonstrated by experiments using neutralizing antibodies and soluble decoy TRAIL-R1/R2 receptors. We also demonstrated that the decapsidation and replication of lyssavirus are essential for inducing apoptosis, as supported by UV inactivation, cycloheximide treatment, and the use of bafilomycin A1 to inhibit endosomal acidification. Transfection of cells with the matrix protein induced apoptosis using pathways similar to those described in the context of viral infection. Furthermore, our data suggest that the matrix protein of lyssaviruses plays a major role in the early induction of TRAIL-mediated apoptosis by the release of a soluble, active form of TRAIL. In our model, Fas ligand (CD95L) appears to play a limited role in lyssavirus-mediated neuroblastoma cell death. Similarly, tumor necrosis factor alpha does not appear to play an important role.
Apoptosis is an active physiological process of cellular self-destruction with specific morphological and biochemical cellular changes (65). External apoptotic stimuli and signals generated from within the cell can activate signal transduction pathways involving a family of cysteine proteases (caspases) that play a central role in apoptosis (15, 60). In this context, caspases with death effector domains (caspases 8 and 10) play a crucial role in signal transduction downstream of the death receptors (DRs) (15, 32). The ligation of specific death ligands such as Fas ligand (FasL; CD95L/Apo1 ligand), tumor necrosis factor alpha (TNF-α), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL/Apo2 ligand) to the DRs leads to the recruitment of adaptor proteins such as TRADD and FADD, which trigger the cleavage of procaspase-8 into an active form (3). The caspase cascade ultimately results in the morphological hallmarks of apoptosis, including DNA fragmentation and cell shrinkage (18, 19, 23, 61, 64, 66).
FasL and TNF-α are widely expressed in the nervous system (6). These ligand-DR systems are involved in neuronal cell death (6, 45, 49, 77) during the development of the cerebral cortex (5, 11), tumorigenesis, and adult neurological diseases (28). Similarly, the expression of TRAIL is involved in neuronal cell apoptosis during brain injury and tumorigenesis (2, 17, 26, 56, 73). TRAIL-mediated cell death uses the DRs TRAIL-R1 (DR4) and TRAIL-R2 (DR5), which contain death domains that can initiate the apoptotic signal pathway at their cytoplasmic ends (46, 63, 72, 75).
Lyssaviruses are highly neurotropic (53, 76), and there is much evidence indicating that apoptosis interferes with the transport of the virus within the infected neurons and thereby plays a major role in the neuropathogenesis of rabies (1, 21, 27, 36, 37, 59). Lyssaviruses are enveloped, bullet-shaped viruses of the Rhabdoviridae family. They cause rabies, which is responsible for the death by meningoencephalitis of approximately 50,000 people worldwide each year. The pathogenicity of lyssavirus variants is inversely correlated with apoptosis in vivo, ex vivo, and in vitro (35, 51, 52). However, little is known about the viral mechanism responsible for the induction of apoptotic pathways in neurons.
Taking advantage of the fact that Mokola virus and Lagos bat virus, two members of genotypes 2 and 3 of lyssavirus, respectively, are known to be less pathogenic for mice (4), we explored, in the present study, the roles of DRs and their respective ligands in the induction of apoptosis mediated by lyssavirus infection. Dissection of the signal transduction pathway allowed us to report for the first time, in a model using neuroblastoma cells, the involvement of caspase-8 activation through a TRAIL/TRAIL receptor-dependent interaction. Expression experiments with viral proteins indicated that the matrix (M) protein can induce apoptosis using the same molecular pathways as those described for viral infection, in the absence of any other viral components.
MATERIALS AND METHODS
Infection of cells and virus titrations.
Mouse neuroblastoma cells (N2a), HeLa cells, and BSR cells (a clone of BHK-21) were grown at 37°C under a 5% CO2 atmosphere in Dulbecco's minimal essential medium supplemented with glutamine (1%), gentamicin (50 μg/ml), and 10% heat-inactivated fetal bovine serum. Cells were used when monolayers had reached 80% confluence in 35-mm-diameter dishes. The standard inoculation procedure consisted of washing monolayers once with phosphate-buffered saline (PBS) and overlaying them with serum-free medium containing the virus at a multiplicity of infection (MOI) of 1 or 30. Lyssaviruses belonging to three different genotypes (8) were used to infect cells. Thailand virus, referred to below as THA (isolate 8743THA, genotype 1), was isolated in Thailand from a human bitten by a dog (69). Lagos bat virus, referred to below as LAG (isolate 8619NGA, genotype 2), was isolated from a frugivorous bat in Nigeria (7). Mokola virus, referred to below as MOK (isolate 8720MOK, genotype 3), was isolated from a domestic cat in Zimbabwe (25). MOK and LAG belong to phylogroup II of lyssavirus, which is nonpathogenic when injected intramuscularly (4). After 2 h at 37°C, unadsorbed virus particles were removed by washing cells with PBS. Growth medium was added, and the cells were incubated for various periods at 37°C. Virus suspensions were centrifuged at 200 × g and 4°C for 10 min. The supernatant was collected and stored at −80°C until titration. Titration was performed on BSR cells in 96-well plates as described by Bourhy and Sureau (9).
Cell viability assay.
Cells adherent to culture flasks were collected by treatment with a solution of 0.25% trypsin-1 mM EDTA at 37°C for 2 min and were combined with the floating cells collected from the culture medium. Cell viability was determined by the trypan blue dye exclusion assay, and cells were counted under a microscope by using a hemacytometer chamber.
LDH release and filtration assay.
A Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, Wis.) was used to measure the amount of lactate dehydrogenase (LDH) in supernatant aliquots (300 μl) at different times after infection. Background lysis was measuring by using cells cultured in medium alone. Cytotoxicity is expressed as the percentage of specific LDH release, which was calculated as 100% × (LDH released by infected cells − background release)/(LDH released by uninfected cells − background release). All experiments were carried out in triplicate and conducted at least twice. For the filtration assay, the supernatant was collected at different times postinfection (p.i.) and filtered on a 100-kDa regenerated cellulose membrane (Microcon; Millipore). For Western blotting, filtered supernatants were concentrated on a 10-kDa membrane (Centricon; Millipore).
UV inactivation and drug treatments.
The viruses were inactivated by exposure to a germicidal lamp (wavelength, 254 nm) at a distance of 5 cm for 35 min at 4°C. Inactivation was confirmed by titrating supernatant aliquots for the presence of infectious virus at 48 h p.i. as described above. Confluent cells were treated with bafilomycin A1 (BAF) (10 μM) or cycloheximide (CHX) (10 μg/ml) (Calbiochem) for 1 h before or 2 h after exposure to rabies virus. To examine the involvement of caspases in apoptosis, infected N2a cells were plated out in eight-well plates and incubated with a broad synthetic caspase inhibitor, Z-VAD-fluoromethyl ketone (fmk), and a specific caspase-8 inhibitor, Z-IETD-fmk (Calbiochem).
Caspase-1, -2, and -3 activity assays.
Briefly, 2 × 106 cells were harvested by centrifugation at 450 × g for 5 min and resuspended for 15 min at 4°C in hypotonic cell lysis buffer (25 mM HEPES, 5 mM MgCl2, 5 mM EDTA, 5 mM dithiothreitol, 2 mM phenylmethylsulfonyl fluoride, 10 μg of pepstatin A/ml, and 10 μg of leupeptin/ml). Cell lysates were centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant (cytosolic fraction) was collected. The protein concentration of the cell extract was determined by using the bicinchoninic acid (BCA) protein assay reagent (Pierce). Total proteins (40 to 80 μg) were incubated for 30 min at 37°C in a reaction mixture containing a caspase assay buffer {312 mM HEPES, 31% sucrose, and 0.3% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]}, dimethyl sulfoxide, and 100 mM dithiothreitol (final volume, 98 μl). Two microliters of the appropriate caspase substrate was added, and the mixture was incubated at 37°C for 90 min. Caspase-1 and -3 activities were measured by using specific substrates labeled with Ac-YVAD-7-amino-4-methyl coumarin (Ac-YVAD-AMC) and Ac-DEVD-AMC, respectively, two fluorochromes provided in the CaspAce assay system (Promega). Caspase-2 activity was measured by using a specific substrate labeled with VDVAD-p-nitroanilide (VDVAD-pNA), a chromophore provided in the caspase-2 colorimetric protease assay kit (Chemicon, Temecula, Calif.). The enzyme-catalyzed release of free AMC was quantified by spectrofluorimetry (excitation wavelength, 350 nm; emission wavelength, 460 nm), and the enzyme-catalyzed release of pNA was quantified with a spectrocolorimeter at 405 nm. Activity is expressed as fold change over the activity of the control after correction for baseline activity.
Caspase-8, -9, -10, and -6 activities.
Caspase-8, -10, and -6 activities were detected and quantified at the single-cell level by using the CaspaTag activity kit (Q-Biogene). Briefly, cells grown in duplicate on a glass slide were incubated for 1 h at 37°C in a medium containing a specific FAM-labeled peptide inhibitor (FAM-LETD-fmk, FAM-LEHD-fmk, FAM-AEVD-fmk, or FAM-VEID-fmk, recognizing the activated form of caspase-8, -9, -10, or -6, respectively). Hoechst 33342 was added to the medium for 5 min to label the nuclei. Cells were washed twice with 1× wash buffer and fixed for 30 min at 4°C with 4% paraformaldehyde. Cells were observed under a fluorescence microscope by using a band-pass filter (excitation, 490 nm; emission, 520 nm) for fluorescein isothiocyanate (FITC) analysis and a UV filter (excitation, 365 nm; emission, 480 nm) for Hoechst analysis.
Detection of rabies virus nucleocapsid (NC) antigen, activated caspase-8, and TUNEL signals.
Briefly, cells grown in duplicate on a glass slide were incubated for 1 h at 37°C in a medium containing the caspase-8-specific labeled peptide inhibitor, FAM-LETD-fmk, as described above. Apoptosis was detected and quantified at the single-cell level by immunostaining using the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay according to the manufacturer's instructions (Apoptag Red in situ apoptosis detection kit; Q-Biogene) with minor modifications. Briefly, cells grown on a glass slide were fixed for 30 min at 4°C with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100 or 0.2% saponin. Terminal deoxynucleotidyltransferase (TdT) was added for 1 h at 37°C to incorporate digoxigenin-11-dUTP at the sites of DNA breaks. Cells were then incubated with a rhodamine-conjugated anti-digoxigenin antibody for 45 min at room temperature. Cells were then washed twice with PBS for 5 min each time and treated with FITC-conjugated anti-rabies virus NC antibodies (Bio-Rad) for 1 h at 37°C. The slide was mounted in a mixture of 4′,6′-diamidino-2-phenylindole (DAPI; 1 μg/ml in PBS) or TOPRO-3 (Molecular Probes) and antifade (Vectashield; Vector Laboratories) and was then examined under a fluorescence microscope (Leica) or an inverted laser scanning confocal microscope (Zeiss LMS 410).
Flow cytometry (fluorescence-activated cell sorter [FACS] analysis).
Cells grown in 35-mm-diameter dishes were removed by use of an enzyme-free cell dissociation buffer in a PBS base (Invitrogen). Cells were then suspended in 500 μl of wash buffer (PBS containing 1% fetal calf serum) and incubated for 35 min to block nonspecific binding. Cells were harvested by centrifugation at 900 × g for 3 min and fixed in 400 μl of 4% paraformaldehyde for 15 min at 4°C. Cells were then washed twice with wash buffer, resuspended in 475 μl of primary antibodies (1 μg total of a rat anti-mouse TRAIL monoclonal antibody [clone N2B2] or of a purified rat monoclonal immunoglobulin isotype control antibody [Pharmingen] in PBS containing 1% fetal calf serum), and incubated at 37°C for 1 h. Cells were washed twice with wash buffer and incubated with FITC-labeled anti-rat antibodies at 37°C for 30 min. Cells were then washed, fixed in 2% paraformaldehyde for 5 min at 4°C, and analyzed by flow cytometry. Data were collected (10,000 events) by using a FACScalibur and were analyzed with CellQuest software (both from Becton Dickinson, San Jose, Calif.).
Quantification of TNF-α and neutralizing assays.
Murine TNF-α (mTNF-α) was quantified by enzyme-linked immunosorbent assay (Bender MedSystems). Briefly, the wells of a microtiter plate were coated with monoclonal anti-mTNF-α antibodies, such that any mTNF-α present in samples (supernatant) bound to the antibodies. A biotin-conjugated polyclonal anti-mTNF-α antibody was added, followed by streptavidin-horseradish peroxidase. A colored product was formed following the addition of a substrate that reacts with horseradish peroxidase. The absorbance of the sample was measured by spectrocolorimetry at 450 nm, and the concentration of TNF-α in the solution was determined by use of an mTNF-α standard curve. For the neutralizing assay, cells were infected with the rabies virus and incubated with a mouse anti-mouse FasL neutralizing monoclonal antibody (clone MFL3) (Pharmingen, BD), an anti-mouse TRAIL neutralizing monoclonal antibody (clone N2B2) (Pharmingen, BD), an anti-human TRAIL neutralizing monoclonal antibody (clone RIK-2), an anti-mouse TNF-α neutralizing antibody, or the recombinant human decoy receptor TRAIL-R1-Fc or TRAIL-R2-Fc (Alexis Corporation). Apoptosis assays were performed after 48 and 72 h of incubation.
Western blot analysis.
Protein concentrations were determined by using the BCA protein assay reagent (Pierce). Proteins (20 to 50 μg) were subjected to electrophoresis in a sodium dodecyl sulfate (SDS)-12% polyacrylamide gel, blotted onto a Hybond-C pure nitrocellulose membrane (Amersham), and blocked with a solution of PBS-Tween containing 3% nonfat dry milk powder for 1 h. The membranes were then incubated with an anti-mouse caspase-8 polyclonal antibody (clone AB1879) (Chemicon), an anti-human caspase-8 monoclonal antibody (clone 12F5) (Apotech Co.), an anti-mouse TRAIL monoclonal antibody (clone N2B2) (Pharmingen, BD), anti-human TRAIL-R2 antibodies (Calbiochem), or anti-β-actin antibodies (clone AC-74) (Sigma) overnight at 4°C, washed three times with PBS-Tween containing 3% nonfat dry milk powder, and incubated with a biotin-conjugated anti-rat or anti-rabbit antibody followed by streptavidin-conjugated peroxidase. Bound antibodies were detected by the ECL SuperSignal Western blotting detection system (Amersham) using Biomax-MR film (Kodak).
Plasmid construction and DNA transfection.
pM-GFP or pM-(His)6, which encodes the M protein fused to a six-His tag, was generated by PCR amplifying the M genes of the three virus strains and cloning the amplified fragments into pcDNA3.1/CT-GFP or TOPO pcDNA3.1D/V5-His directional, respectively, according to the protocol of Invitrogen, Inc. The sequences and orientations of the M protein genes were checked by sequencing using the BigDye Terminator kit (Applied Biosystems, Inc.). The GenBank accession numbers of the complete coding nucleotide sequences of the M gene of viruses THA, LAG, and MOK are AY540348, AY540349, and AY540347, respectively. An eight-well culture chamber containing coverslips was seeded with 6 × 104 HeLa cells per well 24 h before use. The cells were transfected with 0.5 μg of pM-GFP or pM-(His)6 by using 3 μl of Fugene-6 (Roche Molecular, Inc.).
RESULTS
Lyssavirus-induced apoptosis of neuroblastoma cells is associated with caspase-8 activation.
Apoptosis, as assessed by the TUNEL assay and LDH release, was induced in lyssavirus-infected neuroblastoma cells 48 h after MOK or LAG infection and 96 h after THA infection (Fig. 1A and 2). Chromatin condensation and fragmentation, which are typical features of apoptosis, were visible in MOK-infected cells and to a lesser extent in LAG-infected cells, but not in THA-infected or uninfected neuroblastoma cells at 72 h p.i. (Fig. 2). The effector phase of apoptosis requires the activation of several caspases (15, 55). To determine which caspases are involved in lyssavirus-induced apoptosis, the tetrapeptide fluorogenic and chromogenic substrates DEVD-AFC, YVAD-AFC, and VDVAD-pNA were used to assess caspase-3 (CPP32), caspase-1 (ICE), and caspase-2 activities, respectively (Fig. 1B). Cleavage of these substrates by cytoplasmic cell extracts is indicative of the proteolytic activation of these caspases in response to lyssavirus-induced apoptotic signals. Following infection with MOK or LAG, there was a significant and time-dependent increase in the level of DEVD-AFC cleavage, indicating that caspase-3 was progressively activated. Caspase-3 activity was detected in MOK-infected cells and to a lesser extent in LAG-infected cells at 48 h p.i. This activity was greatly enhanced at 72 h. However, caspase-3 activity was detected in THA-infected cells only at 72 h p.i. (Fig. 1B). Similarly, FAM-VEID-fmk, a specific caspase-6 inhibitor, detected caspase-6 activity at 72 h p.i. in MOK- and LAG-infected cells and to a lesser extent in THA-infected cells (Fig. 1C). No cleavage of YVAD-AFC or VDVAD-pNA was detected at any time after infection with any of the three lyssaviruses, indicating that the levels of caspase-1 and caspase-2 activity in infected neuroblastoma cells remained stable and similar to those in control cells during the first 48 h of infection (Fig. 1B). Thus, the lack of activity of these caspases suggests that they are not involved in the process of initiation of apoptosis in neuroblastoma cells.
FIG.1.

Caspase activation in lyssavirus-infected neuroblastoma cells. Cells were infected with THA, MOK, or LAG at an MOI of 1. After incubation at 37°C for the indicated times, cells were processed. Results are expressed as means of data obtained in three independent experiments. Error bars, standard deviations. Significant effects are indicated by asterisks, pound signs, and plus signs (P < 0.05). (A) Percentages of lyssavirus-infected cells undergoing apoptosis and cell death according to the TUNEL assay and the amount of LDH released. (B) Caspase-1, -2, and -3 activity assays were evaluated by using Ac-DEVD-AMC-, VDVAD-pNA-, and Ac-YVAD-AMC-labeled substrates, respectively. (C) Caspase-6 activity was quantified at the single-cell level by using a medium containing a specific labeled peptide inhibitor (FAM-VEID-fmk) that recognizes the activated form of caspase-6. (D) Caspase-8 activity was quantified at the single-cell level by using a medium containing a specific labeled peptide inhibitor (FAM-LETD-fmk) that recognizes the activated form of caspase-8. LETDase activities were quantified in neuroblastoma cells infected at MOIs of 1 and 30. (E) Caspase-10 activity was quantified at the single-cell level by using a medium containing a specific labeled peptide inhibitor (FAM-AEVD-fmk) that recognizes the activated form of caspase-10. AEVDase activity was quantified in infected neuroblastoma cells (MOI, 30). (F) Western blot analysis of caspase-8 revealed the p54 fragment of the precursor form of caspase-8 (Pro casp-8). β-Actin was included as a loading control. NEG, negative control. (G) Caspase-9 activity was quantified at the single-cell level by using a medium containing a specific labeled peptide inhibitor (FAM-LEHD-fmk) that recognizes the activated form of caspase-9. LEHDase activities were quantified in neuroblastoma cells infected at MOIs of 1 and 30. (H) Cells were incubated in the presence or absence of a cell-permeant, irreversible pan-caspase inhibitor, Z-VAD-fmk, and a specific caspase-8 inhibitor, Z-IETD-fmk. CONTR, control.
FIG. 2.

Apoptosis and detection of caspase-8 activity in lyssavirus-infected neuroblastoma cells. Cells were either mock infected (A, E, and I) or infected with THA (B, F, and J), MOK (C, G, and K), or LAG (D, H, and L) (MOI, 1). (A through D) After 72 h at 37°C, cells were processed for TUNEL staining (red) and viral NC antigen staining (green) and were analyzed with a laser scanning confocal microscope (magnification, ×600). (E through H) Active caspase-8 was detected 48 h p.i. by using the carboxyfluorescein-labeled caspase inhibitor FAM-LETD-fmk (green). (I through L) Cells were counterstained with Hoechst 33342 (blue) and analyzed by fluorescence microscopy (magnification, ×1,000). (M) Matrix protein fused to His6 was visualized by use of an anti-His6 antibody conjugated with FITC (green). (N) Cells were transfected with the matrix-His6 fusion protein (M) and treated for the TUNEL assay (red). (O) Cells were counterstained with TOPRO-3 (blue) and analyzed by confocal microscopy (magnification, ×600).
Fluorescence microscopy using a specific fluorogenic peptide inhibitor (FAM-LETD-fmk) showed that caspase-8 was activated in a virus-, dose-, and time-dependent manner (Fig. 1D and 2). The amount of caspase-8 mRNA did not change between 4 and 48 h p.i. (data not shown). At 48 h p.i., the percentages of activated caspase-8 rose from 14 and 30% at an MOI of 1 to 17 and 43% at an MOI of 30 in MOK- and LAG-infected neuroblastoma cells, respectively. In THA-infected cells, caspase-8 activity increased only at 96 h p.i. (Fig. 1D). A similar experiment was performed using a caspase-10-specific fluorogenic peptide inhibitor. Virtually no caspase-10 activity was detected except at 72 h p.i. in the case of LAG-infected cells (Fig. 1E). Western blot analysis revealed that the amount of p54, the precursor form of caspase-8, decreased at 72 h p.i. for MOK- and LAG-infected cells but not for THA-infected cells relative to the amount in the control (Fig. 1F). These results suggest that procaspase-8 is processed into the active form at 72 h p.i. for both LAG and MOK but not for THA, confirming previous results obtained by fluorescence microscopy. The role of the caspase-9 pathway in the induction of apoptosis was investigated by using a caspase-9-specific fluorogenic peptide inhibitor (Fig. 1G). It appears not to be critical. Indeed, in the case of MOK, the activation of caspase-9 (72 h p.i.) followed that of caspase-8 (48 h p.i.) and the induction of apoptosis (48 h p.i.). In the case of LAG, the activation of caspase-9 did not exceed 4% of the cells 68 h after infection at an MOI of 1.
Caspase inhibitor prevents lyssavirus-induced apoptosis in neuroblastoma cells.
We next investigated the effect of a broad caspase inhibitor, zVAD-fmk, on apoptosis induction in lyssavirus-infected host cells. Addition of 50 μM zVAD-fmk prevented lyssavirus-infected neuroblastoma cells from undergoing apoptosis, as assessed by the TUNEL assay after 72 h of culture (Fig. 1H). These results suggest that the activation of caspases is a central process in lyssavirus-induced cell death. Treatment of infected cells with IETD-fmk, a specific and irreversible inhibitor of caspase-8, also prevented lyssavirus-induced apoptosis in a dose-dependent manner (Fig. 1H). IETD-fmk (50 μM) decreased the levels of virus-induced apoptosis by 34 and 42%, respectively, for MOK- and LAG-infected cells, whereas 150 μM IETD-fmk almost completely abolished apoptosis in both cases (Fig. 1H). These results indicate that caspase-8 is the main caspase involved in lyssavirus-induced apoptosis. Although the caspase inhibitors prevented apoptosis, they did not change viral production as judged by viral titration. The titers differed by less than 0.53, 0.23, and 0.18 log unit for MOK, LAG, and THA, respectively. Moreover, real-time PCR in the absence and presence of zVAD-fmk indicated that the ratios of viral genomic RNA were not significantly different for the three viruses in terms of real-time PCR sensitivity (data not shown).
Lyssavirus-induced apoptosis of neuroblastoma cells is mediated by TRAIL.
The observation that caspase-8 is activated at an early stage in infected neuroblastoma cells prompted us to explore whether extrinsic pathways (DRs) are involved following lyssavirus infection. To assess the roles of DRs and their ligands, we used antibodies directed against CD95L, TNF-α, and TRAIL molecules as well as decoy TRAIL-R1/R2 receptors that block the interaction between TRAIL and the DRs. In the first set of experiments, cells were treated with different concentrations of a neutralizing anti-TRAIL antibody, decoy TRAIL-R1/R2 receptors, an anti-CD95L antibody, or an anti-TNF-α antibody. The percentage of apoptotic cells was evaluated by the TUNEL assay 72 h p.i. (Fig. 3A). The amount of soluble TNF-α detected in the supernatant did not increase during lyssavirus infection and remained below 4 pg/ml (data not shown). The fact that high concentrations of the neutralizing TNF-α antibody did not have a preventive effect confirmed that TNF-α does not mediate apoptosis (Fig. 3A). Treatment of MOK- and LAG-infected cells with an anti-TRAIL antibody or decoy TRAIL-R1/R2 receptors significantly inhibited lyssavirus-induced apoptosis in a dose-dependent manner (Fig. 3A) without enhancing viral infection (data not shown). Moreover, the anti-TRAIL antibody not only prevented cell death but also decreased the level of caspase-8 activation (Fig. 3B). The neutralizing anti-CD95L antibody also prevented MOK- and LAG-mediated cell death, although to a lesser extent than the neutralizing anti-TRAIL antibody, even at saturating concentrations (eight times the lowest concentration). To determine whether lyssaviruses sensitize cells to TRAIL-induced apoptosis by altering the amount of TRAIL, we assessed TRAIL expression in neuroblastoma cells at 24 to 48 h p.i. We analyzed cell surface and cytosolic TRAIL expression in mock- and lyssavirus-infected neuroblastoma cells by flow cytometry (Fig. 3C). Our data showed that TRAIL is essentially a membrane-bound protein (data not shown) and that the level of TRAIL cell surface expression was unchanged following infection with lyssaviruses (Fig. 3C). These results suggest that differences in the sensitivity of TRAIL are the major changes involved in lyssavirus-mediated cell death.
FIG. 3.

TRAIL is necessary but not sufficient to activate caspase-8 and to induce apoptosis in lyssavirus-infected neuroblastoma cells. Neuroblastoma cells were pretreated with anti-TRAIL antibodies (2.75 to 5.5 μg/ml; 1× to 2×), soluble TRAIL receptors (22 μg/ml), anti-FasL antibodies (11 to 22 μg/ml; 4× to 8×), or anti-TNF-α antibodies (22 μg/ml; 8×) for 1 h before virus infection (MOI, 1). They were maintained in media containing anti-ligands for 72 h. Results are means of data obtained in two independent experiments. Error bars, standard deviations.Significant effects are indicated by asterisks and pound signs (P < 0.05). (A and B) The percentage of apoptotic cells was evaluated by the TUNEL assay (A) and caspase-8 activity (B). (C) FACS analysis of cytosolic and cell surface TRAIL expression using a purified rat anti-mouse TRAIL monoclonal antibody or a purified rat monoclonal immunoglobulin isotype control followed by an FITC-conjugated anti-rat antibody. Data were collected (10,000 events) by using a Becton Dickinson FACScalibur and were analyzed with CellQuest software. NT, not treated; CONT, control.
Lyssavirus-induced apoptosis is mediated by an extrinsic death signal in a time- and virus-dependent manner through caspase-8 activation.
We next investigated whether a direct or an indirect mechanism is involved in this process of cell death. The supernatant was collected at different times following infection (MOI, 1), filtered through a 100-kDa membrane to remove viral particles, and transferred to fresh neuroblastoma cells. The percentage of apoptosis of target cells was recorded by a TUNEL assay after 72 h of incubation (Fig. 4A). Our results showed that filtered supernatants from MOK-, LAG-, or THA-infected cells induced a paracrine mode of apoptosis on fresh neuroblastoma cells, compared with supernatants from uninfected cells. The apoptogenic effect of the supernatant was not due to the presence of infectious virus particles or viral NCs, because they are too large to cross the membrane of the filtration unit. This result was confirmed by immunofluorescence using a rabies virus NC-conjugated antibody. No viral inclusions were observed in the target cells (data not shown). We next determined the capacity of the supernatants, collected when apoptosis was maximal and filtered, to activate caspase-8 (i.e., supernatants collected at 24, 48, or 72 h p.i. for MOK-, LAG-, or THA-infected cells, respectively). In all cases, supernatant-mediated apoptosis correlated with a 7 to 10% increase in caspase-8 activity at 48 h after incubation with filtered supernatants (Fig. 4B). Because TRAIL is the main effector involved in lyssavirus-induced apoptosis, we next assessed whether the observed paracrine cell death also involves a TRAIL-dependent pathway. Neuroblastoma cells were incubated with the apoptogenic, filtered supernatants (see above) in the presence or absence of a neutralizing anti-TRAIL antibody or a neutralizing anti-CD95L antibody. After 72 h of incubation, the anti-TRAIL antibody significantly inhibited apoptosis and caspase-8 activity, suggesting the presence of an active soluble form of TRAIL (Fig. 4B and C). However, even saturating concentrations of anti-CD95L had no significant effect on the percentage of apoptosis in target cells.
FIG. 4.

Induction in trans of apoptosis of lyssavirus-infected neuroblastoma cells is mediated by TRAIL through a synergistic released factor(s). Neuroblastoma cells were either mock infected (NEG. CONT.) or infected with THA, MOK, or LAG (MOI, 1). (A) At various times p.i., supernatants were filtered through a 100-kDa membrane and transferred to fresh, noninfected neuroblastoma cells. Graph shows the percentages of apoptotic nuclei in neuroblastoma cells following treatment with filtered supernatants taken from infected or mock-infected neuroblastoma cells. Times of supernatant harvest are given along the x axis. Apoptosis was assayed 72 h after supernatant transfer to noninfected cells. Results are means of data obtained in two independent experiments. Error bars, standard deviations. (B and C) Supernatants corresponding to maximum levels of apoptosis (with times of harvest given in parentheses) were incubated with an anti-mouse TRAIL neutralizing antibody (5.5 μg/ml; 2×). Caspase-8 activity (B) and TUNEL (C) assays were performed after 72 h of incubation with supernatants. (D) The release of the soluble form of TRAIL (sTRAIL) was analyzed by Western blotting of filter-concentrated supernatants at various times p.i. Protein concentrations of all samples were standardized by the BCA protein assay. Supernatants (40 μg) were subjected to SDS-polyacrylamide gel electrophoresis, blotted onto nitrocellulose membranes, and probed with anti-mouse TRAIL antibodies. (E) The amount of TRAIL-R2 receptor was analyzed by Western blotting using the solubilized cell membrane at 72 h p.i. Concentrated proteins (40 μg) were subjected to SDS-polyacrylamide gel electrophoresis, blotted, and probed with anti-human TRAIL-R2 antibodies.
Lyssavirus infection of neuroblastoma cells does not modify the expression of soluble TRAIL or the expression of TRAIL-R2 on the cell surface.
We used Western blotting to detect soluble TRAIL released into apoptogenic supernatants. This procedure revealed the presence of the monomeric p45 form of soluble TRAIL in supernatants of both infected and mock-infected neuroblastoma cells. However, the amounts of this monomeric form of soluble TRAIL were equivalent in infected and uninfected cells, whatever virus was used (Fig. 4D). The amount of TRAIL-R2 at the cell surface 72 h p.i. was also assessed by Western blotting. Similarly, no differences in the expression of this receptor were observed between infected and uninfected cells (Fig. 4E). These observations suggest that, although TRAIL is essential for lyssavirus-induced apoptosis in neuronal cells, this requirement is not due to a specific modulation of the release of the monomeric form in the extracellular compartment or to the overexpression of at least one of its receptors on the neuronal cell surface.
Induction of the caspase cascade is dependent on viral decapsidation and replication.
To determine the step in lyssavirus replication that induces apoptosis, we investigated whether virus adsorption or replication was required for apoptosis. In contrast to infectious viruses, UV-inactivated viruses did not induce significant neuroblastoma cell death within 72 h of infection; no cell death was observed by the trypan blue dye exclusion assay (Fig. 5A) or the TUNEL assay (Fig. 5B), and no caspase-3 activity was detected (Fig. 5C). BAF is a vacuolar H+-ATPase inhibitor that prevents endosomal acidification and inhibits the acid-induced conformational change and fusion of the virus envelope and cell membrane (10). Pretreatment of neuroblastoma cells with BAF protected them from LAG- and to a lesser extent from MOK-induced cell death (Fig. 5). Moreover, CHX increased the viability of infected cells and reduced the number of dead cells detected by the TUNEL assay, suggesting that induction of cell death requires viral or cellular protein synthesis (Fig. 5A and B). Altogether, these results indicate that the induction of apoptosis requires the decapsidation and replication of lyssaviruses, since either UV inactivation, inhibition of endosomal acidification during viral adsorption, or inhibition of protein synthesis during viral infection prevented lyssavirus-mediated cell death. Furthermore, recombinant TRAIL added to the medium of neuroblastoma cells incubated with UV-inactivated or live lyssavirus did not increase the percentage of TUNEL-positive cells (data not shown), confirming that the virus infection is responsible for the production of an active form of TRAIL.
FIG. 5.
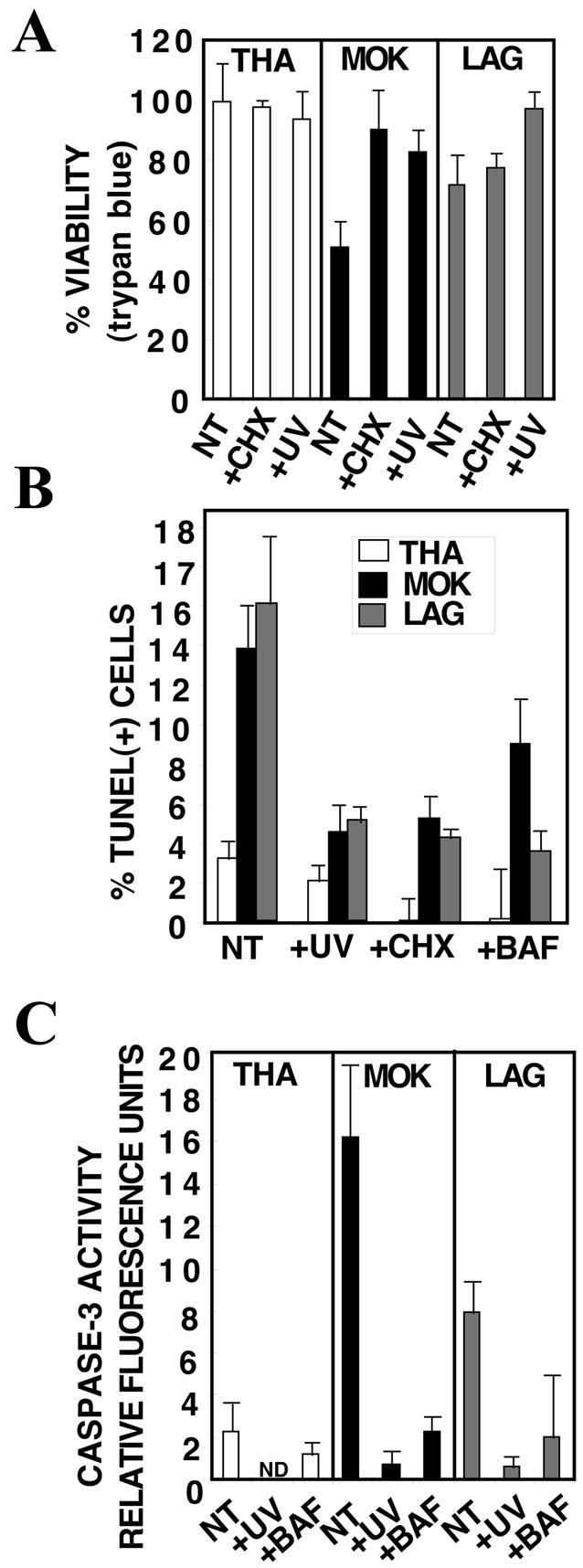
Induction of cell death is dependent on viral decapsidation and replication. At 72 h p.i., the medium was removed, and cells were collected and counted under a microscope by using a hemacytometer chamber (A) or were treated for a TUNEL assay (B). Proteins were extracted and assayed for caspase-3 activity (C). Results are means of data obtained in three independent experiments. Error bars, standard deviations. (A) Virus inactivation by UV or pretreatment of neuroblastoma cells with CHX (10 μg/ml) protected neuroblastoma cells against MOK and to a lesser extent against LAG, as revealed by an increase in cell viability quantified by the trypan blue exclusion assay. (B and C) Pretreatment of neuroblastoma cells with BAF (10 μM), CHX (10 μg/ml), or UV-inactivated virus completely or partially protected them against LAG- or MOK-induced apoptosis, as evidenced by a decrease in the percentage of TUNEL-positive cells (B) and in caspase-3 activity (C). NT, not treated.
Matrix protein alone induces apoptosis by a TRAIL-dependent mechanism involving the caspase-8 pathway.
Infected HeLa cells exhibited the same hallmarks of virus-induced apoptosis as neuroblastoma cells: TUNEL-positive staining and caspase-8 activation (Fig. 6A and B). We focused on the matrix protein, because matrix proteins from members of the Vesiculovirus genus, a related genus also belonging to the Rhabdoviridae family, induce apoptosis in a number of cell types (43, 57, 58, 71). Several plasmids were constructed to express lyssavirus matrix proteins and phosphoproteins (as a control) of MOK, LAG, or THA fused to either His6 or green fluorescent protein (GFP). Surprisingly, we observed the same hierarchy of apoptotic activities in HeLa cells transfected with lyssavirus M proteins as were observed in the context of the viral infections (Fig. 6C). Apoptosis was induced within 24 h of transfection with the M protein from LAG or MOK and within 48 h of transfection with the M protein from THA. These apoptotic activities were M protein specific, as evidenced by the facts that no apoptosis was observed after transfection with plasmids expressing lyssavirus phosphoproteins (data not shown) and no difference was observed after transfection with the M protein fused with His6 or with GFP (data not shown). Similarly, the gradient of caspase-8 induction (greater with MOK or LAG than with THA) was confirmed in HeLa cells 24 h after transfection with M proteins (Fig. 6D). Western blot analysis revealed that the amount of p54, the precursor form of caspase-8, decreased at 48 h p.i. for MOK- and LAG-infected cells but not for THA-infected cells relative to the amount in control cells (Fig. 6E). Finally, the role of TRAIL as an essential paracrine relay in the induction of apoptosis was confirmed by the fact that the number of TUNEL-positive cells was reduced by more than 49% at 48 h after transfection with the M protein and incubation with an anti-TRAIL antibody (Fig. 6F).
FIG. 6.

Matrix protein alone induces apoptosis by a TRAIL-dependent mechanism involving the caspase-8 pathway. (A) HeLa cells were infected with THA, MOK, or LAG (MOI, 15) and processed for the TUNEL assay. (B) Proteins (20 μg) were subjected to SDS-polyacrylamide gel electrophoresis, blotted, and probed with anti-human caspase-8. β-Actin was included as a loading control. (C through F) HeLa cells were either mock transfected (NTF) or transfected with pGFP (positive control), pM-GFP, or pM-(His)6. Cells were then processed for a TUNEL assay at the indicated time points (C), a caspase-8 activity assay at 24 h posttransfection (D), and Western blotting with anti-human caspase-8 (E). Cells were also processed for a neutralizing assay using an anti-human TRAIL neutralizing antibody at 48 h posttransfection (4.4 μg/ml) (F). Results are means of data obtained in two independent experiments. Error bars, standard deviations.
DISCUSSION
Virus-induced cell injury plays an important role in the pathogenesis of viral infections of the nervous system. Therefore, it is crucial to define the steps of the apoptotic signal transduction cascade that are triggered by viruses. In this study, we investigated the mechanism by which lyssaviruses induce the apoptosis of neuroblastoma cells. We found that lyssavirus infection of neuronal cells leads to cell death and that this process involves a TRAIL-dependent pathway requiring the activation of caspase-8 but not of caspase-9 and caspase-10. When infected cells were incubated with a broad synthetic caspase inhibitor (z-VAD-fmk) or a specific inhibitor of caspase-8 (IETD-fmk), apoptosis was prevented, indicating that caspase-8 is the main caspase involved in the cytopathic effect of rabies virus. Activation of caspase-8 results in activation of caspase-3 and caspase-6, as shown by an increase in the cleavage of the specific caspase substrate in lyssavirus-infected cells. However, neither caspase-1 nor caspase-2 activity was detected during the early phase of infection. Lyssavirus-mediated cell death involves an interaction between TRAIL receptors and TRAIL, as demonstrated by experiments using neutralizing antibodies and soluble decoy TRAIL-R1/R2 receptors and is not due to a direct interaction between DR and an external viral protein (i.e., glycoprotein) (22, 59). We also demonstrated that the decapsidation and replication of lyssavirus are essential for inducing apoptosis, as supported by UV inactivation, CHX treatment, and the use of BAF to inhibit endosomal acidification. Interestingly, transfection of cells with the matrix protein induced apoptosis using pathways similar to those described in the context of viral infection. TRAIL also appears to be necessary for the activation cascade leading to the induction of apoptosis in M protein-transfected cells. In our model, CD95L appears to play a limited role in lyssavirus-mediated neuroblastoma cell death. Similarly, TNF-α does not appear to play an important role.
Cleavage of FasL or TNF from the cell surface requires the action of a zinc-dependent metalloprotease, whereas the generation of soluble TRAIL involves the action of one or more cysteine proteases (42, 47, 48, 67). We found that the apoptogenic supernatants of lyssavirus-infected neuroblastoma cells contained a soluble form of TRAIL. However, cytometry and Western blotting revealed no differences in the amounts of soluble TRAIL and of cell surface TRAIL-R2 between infected and uninfected cells. Thus, TRAIL sensitization does not depend on the amount of its soluble monomeric form present or on expression of the TRAIL-R2 DR. Our data further indicated that DRs are not modified: the addition of exogenous TRAIL during infection did not increase the percentage of apoptosis in neuroblastoma cells. Other viruses may also cause sensitization of the DRs. For example, TRAIL and TRAIL receptors are modified following infection with cytomegalovirus or Theiler's murine encephalomyelitis virus (38, 62). In human immunodeficiency virus and reovirus infection, T cells are abnormally sensitive to TRAIL- and CD95L-mediated apoptosis (13, 14, 20, 21, 39-47, 54, 74). In the case of lyssaviruses, the modification of the transduction signal most probably occurred through a modification of TRAIL, either stabilization of the oligomerization or posttranslational modification, mediated by the matrix protein. Such a stabilization phenomenon has already been described for other death ligands (33, 34, 70).
Our data suggest for the first time that lyssavirus M proteins are involved in the induction of apoptosis in neuronal cells. This indicates that the viral induction of apoptosis is probably multigenic and not only related to the viral glycoprotein (22, 59). M is a small (202-amino-acid), multifunctional protein (25 kDa) that is responsible for the assembly and budding of virus particles (29, 30, 50) as well as for the regulation of the balance of virus transcription and replication (24). M also induces cell death in the related vesiculoviruses and fish rhabdoviruses (12, 16, 43, 44). Taken together, these results suggest that apoptosis induced by the M protein is probably a common outcome of rhabdovirus infection. The M protein of vesiculoviruses deregulates nucleocytoplasmic transport and thereby cellular gene expression by targeting the nucleoporin Nup98 (31, 57, 58, 71). Understanding the mechanisms involved in the activation of TRAIL by the M protein is one of the keys to understanding the neuronal cell apoptosis induced by lyssaviruses. We are currently exploring the differential viral regulation involved in cell death signaling and the precise role of the viral M protein in the sensitization of neuronal cells to the TRAIL cell death pathway.
The notion that lyssavirus-infected neuronal cells die via a paracrine, TRAIL-dependent pathway is new. It raises the possibility that uninfected neuronal cells in vivo may die through a TRAIL-dependent paracrine mechanism that in turn prevents viral spread. Thus, this process would benefit the host by limiting viral dissemination. However, some viruses have evolved a number of strategies both to inhibit and to activate apoptosis (68). The sequential induction of TRAIL-mediated apoptosis and particularly the delayed induction observed in the case of THA are intriguing. Whether this delay favors the dissemination of THA into the central nervous system, leading to a more severe pathogenesis than that of MOK or LAG (4), remains to be explored. Comparison of the M protein sequences demonstrated 42 differences between THA and LAG, 42 differences between THA and MOK, and 27 differences between LAG and MOK. This sequence comparison (available on request) does not provide sufficient information to map the domain(s) of the M protein involved in the induction of TRAIL-dependent apoptosis. The delineation of these domains should provide interesting information on how the M protein sequence can regulate the temporal induction of TRAIL-dependent apoptosis.
Acknowledgments
R.K. was supported by a predoctoral scholarship from the regional council of Guadeloupe, France.
We thank S. Susin for helpful discussions and suggestions. We are grateful to Laurent Audry and Rolande Rotsen for expert technical assistance. We thank Patricia Davis for critical comments.
REFERENCES
- 1.Adle-Biassette, H., H. Bourhy, M. Gisselbrecht, F. Chretien, L. Wingertsmann, M. Baudrimont, Y. Rotivel, B. Godeau, and F. Gray. 1996. Rabies encephalitis in a patient with AIDS: a clinicopathological study. Acta Neuropathol. 92:415-420. [DOI] [PubMed] [Google Scholar]
- 2.Ashkenazi, A., and V. M. Dixit. 1999. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 11:255-260. [DOI] [PubMed] [Google Scholar]
- 3.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 4.Badrane, H., C. Bahloul, P. Perrin, and N. Tordo. 2001. Evidence of two Lyssavirus phylogroups with distinct pathogenicity and immunogenicity. J. Virol. 75:3268-3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker, V., G. Middleton, F. Davey, and A. M. Davies. 2001. TNF-α contributes to the death of NGF-dependent neurons during development. Nat. Neurosci. 4:1194-1198. [DOI] [PubMed] [Google Scholar]
- 6.Becher, B., S. D. D'Souza, A. B. Troutt, and J. P. Antel. 1998. Fas expression on human fetal astrocytes without susceptibility to Fas-mediated cytotoxicity. Neuroscience 84:627-634. [DOI] [PubMed] [Google Scholar]
- 7.Boulger, L. R., and J. S. Portefield. 1958. Isolation of a virus from Nigerian fruit bats. Trans. R. Soc. Trop. Med. Hyg. 52:421-424. [DOI] [PubMed] [Google Scholar]
- 8.Bourhy, H., B. Kissi, and N. Tordo. 1993. Molecular diversity of the Lyssavirus genus. Virology 194:70-81. [DOI] [PubMed] [Google Scholar]
- 9.Bourhy, H., and P. Sureau. 1991. Laboratory methods for rabies diagnosis. Institut Pasteur, Paris, France.
- 10.Bowman, E. M., A. Siebers, and K. Altendorf. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells and plant cells. Proc. Natl. Acad. Sci. USA 85:7972-7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheema, Z. F., S. B. Wade, M. Sta, K. Walsh, F. Sohrabji, and R. C. Miranda. 1999. Fas/Apo (apoptosis)-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-κB. J. Neurosci. 19:1754-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiou, P. P., C. H. Kim, P. Ormonde, and J. A. Leong. 2000. Infectious hematopoietic necrosis virus matrix protein inhibits host-directed gene expression and induces morphological changes of apoptosis in cell cultures. J. Virol. 74:7619-7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke, P., S. M. Meintzer, S. Gibson, C. Widmann, T. P. Garrington, G. L. Johnson, and K. L. Tyler. 2000. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 74:8135-8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke, P., S. M. Meintzer, A. C. Spalding, G. L. Johnson, and K. L. Tyler. 2001. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 20:6910-6919. [DOI] [PubMed] [Google Scholar]
- 15.Cohen, G. M. 1997. Caspases: the executioners of apoptosis. Biochem. J. 326:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desforges, M., G. Despars, S. Berard, M. Gosselin, M. O. McKenzie, D. S. Lyles, P. J. Talbot, and L. Poliquin. 2002. Matrix protein mutations contribute to inefficient induction of apoptosis leading to persistent infection of human neural cells by vesicular stomatitis virus. Virology 295:63-73. [DOI] [PubMed] [Google Scholar]
- 17.Dorr, J., S. Waiczies, U. Wendling, B. Seeger, and F. Zipp. 2002. Induction of TRAIL-mediated glioma cell death by human T cells. J. Neuroimmunol. 122:117-124. [DOI] [PubMed] [Google Scholar]
- 18.Enari, M., H. Hug, and S. Nagata. 1995. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature 375:78-81. [DOI] [PubMed] [Google Scholar]
- 19.Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43-50. [DOI] [PubMed] [Google Scholar]
- 20.Estaquier, J., T. Idsiorek, W. Zou, D. Emilie, C. M. Farber, J. M. Bourez, and J. C. Ameisen. 1995. T helper type 1/T helper type 2 cytokines and T cell death: preventive effect of interleukin 12 on activation-induced and CD95 (FAS/APO-1)-mediated apoptosis of CD4+ T cells from human immunodeficiency virus-infected persons. J. Exp. Med. 182:1759-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estaquier, J., M. Tanaka, T. Suda, S. Nagata, P. Golstein, and J. C. Ameisen. 1996. Fas-mediated apoptosis of CD4+ and CD8+ T cells from human immunodeficiency virus-infected persons: differential in vitro preventive effect of cytokines and protease antagonists. Blood 87:4959-4966. [PubMed] [Google Scholar]
- 22.Faber, M., R. Pulmanausahakul, S. S. Hodawadekar, S. Spitsin, J. P. McGettigan, M. J. Schnell, and B. Dietzschold. 2002. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response. J. Virol. 76:3374-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandes-Alnemri, T., R. C. Armstrong, J. Krebs, S. M. Srinivasula, L. Wang, F. Bullrich, L. C. Fritz, J. A. Trapani, K. J. Tomaselli, G. Litwack, and E. S. Alnemri. 1996. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA 93:7464-7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finke, S., R. Mueller-Waldeck, and K. K. Conzelmann. 2003. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 84:1613-1621. [DOI] [PubMed] [Google Scholar]
- 25.Foggin, C. M. 1985. Mokola virus infection in domestic cats in Zimbabwe, p. 65-70. In E. Kuwert, C. Mérieux, H. Koprowski, and K. Bögel (ed.), Rabies in the tropics. Springer-Verlag, Berlin, Germany.
- 26.Frank, S., U. Kohler, G. Schackert, and H. K. Schackert. 1999. Expression of TRAIL and its receptors in human brain tumors. Biochem. Biophys. Res. Commun. 257:454-459. [DOI] [PubMed] [Google Scholar]
- 27.Galelli, A., L. Baloul, and M. Lafon. 2000. Abortive rabies virus central nervous infection is controlled by T lymphocyte local recruitment and induction of apoptosis. J. Neurovirol. 6:359-372. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann, A., A. Mouatt-Prigent, B. A. Faucheux, Y. Agid, and E. C. Hirsch. 2002. FADD: a link between TNF family receptors and caspases in Parkinson's disease. Neurology 58:308-310. [DOI] [PubMed] [Google Scholar]
- 29.Harty, R. N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Her, L. S., E. Lund, and J. E. Dahlberg. 1997. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 276:1845-1848. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann, K., P. Bucher, and J. Tschopp. 1997. The CARD domain: a new apoptotic signaling motif. Trends Biochem. Sci. 22:155-159. [DOI] [PubMed] [Google Scholar]
- 33.Holler, N., T. Kataoka, J. L. Bodmer, P. Romero, J. Romero, D. Deperthes, J. Engel, J. Tschopp, and P. Schneider. 2000. Development of improved soluble inhibitors of FasL and CD40L based on oligomerized receptors. J. Immunol. Methods 237:159-173. [DOI] [PubMed] [Google Scholar]
- 34.Holler, N., A. Tardivel, M. Kovacsovics-Bankowski, S. Hertig, O. Gaide, F. Martinon, A. Tinel, D. Deperthes, S. Calderara, T. Schulthess, J. Engel, P. Schneider, and J. Tschopp. 2003. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 23:1428-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito, N., M. Takayama, K. Yamada, M. Sugiyama, and N. Minamoto. 2001. Rescue of rabies virus from cloned cDNA and identification of the pathogenicity-related gene: glycoprotein gene is associated with virulence for adult mice. J. Virol. 75:9121-9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson, A. C., and J. P. Rossiter. 1997. Apoptosis plays an important role in experimental rabies virus infection. J. Virol. 71:5603-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson, A. C., and H. Park. 1998. Apoptotic cell death in experimental rabies in suckling mice. Acta Neuropathol. 95:159-164. [DOI] [PubMed] [Google Scholar]
- 38.Jelachich, M. L., and H. L. Lipton. 2001. Theiler's murine encephalomyelitis virus induces apoptosis in gamma interferon-activated M1 differentiated myelomonocytic cells through a mechanism involving tumor necrosis factor alpha (TNF-α) and TNF-α-related apoptosis-inducing ligand. J. Virol. 75:5930-5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeremias, I., I. Herr, T. Boehler, and K. M. Debatin. 1998. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur. J. Immunol. 28:143-152. [DOI] [PubMed] [Google Scholar]
- 40.Jeremias, I., and K. M. Debatin. 1998. TRAIL induces apoptosis and activation of NF-κB. Eur. Cytokine Netw. 9:687-688. [PubMed] [Google Scholar]
- 41.Jeremias, I., C. Kuppat, B. Baumann, I. Herr., T. Wirth, and K. M. Debatin. 1998. Inhibition of nuclear factor κB activation attenuates apoptosis resistance in lymphoid cells. Blood 91:4624-4631. [PubMed] [Google Scholar]
- 42.Kayagaki, N., A. Kawasaki, T. Ebata, H. Ohmoto, S. Ikeda, S. Inoue, K. Yoshino, K. Okumura, and H. Yagita. 1995. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 182:1777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopecky, S. A., and D. S. Lyles. 2003. The cell-rounding activity of the vesicular stomatitis virus matrix protein is due to the induction of cell death. J. Virol. 77:4658-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le-Niculescu, H., E. Bonfoco, Y. Kasuya, F.-X. Claret, D. R. Green, and M. Karin. 1999. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 19:751-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macfarlane, M., M. Ahmad, S. M. Srinivasula, T. Fernandes-Alnemri, G. M. Cohen, and E. S. Alnemri. 1997. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 272:25417-25420. [DOI] [PubMed] [Google Scholar]
- 47.Mariani, S. M., and P. H. Krammer. 1998. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 28:973-982. [DOI] [PubMed] [Google Scholar]
- 48.Mariani, S. M., and P. H. Krammer. 1998. Surface expression of TRAIL/Apo-2 ligand in activated mouse T and B cells. Eur. J. Immunol. 28:1492-1498. [DOI] [PubMed] [Google Scholar]
- 49.Martin-Villalba, A., I. Herr, I. Jeremias, M. Hahne, R. Brandt, J. Vogel, J. Schenkel, T. Herdegen, and K. M. Debatin. 1999. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 19:3809-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mebatsion, T., F. Weiland, and K. K. Conzelmann. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morimoto, K., D. C. Hooper, S. Spitsin, H. Koprowski, and B. Dietzschold. 1999. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J. Virol. 73:510-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morimoto, K., H. D. Foley, J. P. McGettigan, M. J. Schnell, and B. Dietzschold. 2000. Reinvestigation of the role of the rabies virus glycoprotein in viral pathogenesis using a reverse genetics approach. J. Neurovirol. 6:373-381. [DOI] [PubMed] [Google Scholar]
- 53.Murphy, F. A., S. P. Bauer, A. K. Harrison, and W. C. Winn. 1973. Comparative pathogenesis of rabies and rabies-like viruses. Viral infection and transit from inoculation site to the central nervous system. Lab. Investig. 28:361-376. [PubMed] [Google Scholar]
- 54.New, D. R., S. B. Maggirwar, L. G. Epstein, S. Dewhurst, and H. A. Gelbard. 1998. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-d-aspartate receptors by a NF-κB-independent mechanism. J. Biol. Chem. 273:17852-17858. [DOI] [PubMed] [Google Scholar]
- 55.Nicholson, D. W., A. Ali, N. A. Thornberry, J. P. Vaillancourt, C. K. Ding, M. Gallant, Y. Gareau, P. R. Griffin, M. Labelle, Y. A. Lazebnik, N. A. Munday, S. M. Raju, M. E. Smulson, T.-T. Yamin, V. L. Yu, and D. K. Miller. 1995. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376:37-43. [DOI] [PubMed] [Google Scholar]
- 56.Nitsch, R., I. Bechmann, R. A. Deisz, D. Haas, T. N. Lehmann, U. Wending, and F. Zipp. 2000. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 356:827-828. [DOI] [PubMed] [Google Scholar]
- 57.Petersen, J. M., L. S. Her, V. Varvel, E. Lund, and J. E. Dahlberg. 2000. The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol. Cell. Biol. 20:8590-8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petersen, J. M., L. S. Her, and J. E. Dahlberg. 2001. Multiple vesiculoviral matrix proteins inhibit both nuclear export and import. Proc. Natl. Acad. Sci. USA 98:8590-8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prehaud, C., S. Lay, B. Dietzschold, and M. Lafon. 2003. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis. J. Virol. 77:10537-10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salvesen, G. S., and V. M. Dixit. 1997. Caspases: intracellular signalling by proteolysis. Cell 91:443-446. [DOI] [PubMed] [Google Scholar]
- 61.Scaffidi, C., S. Fulda, A. Srinivasan, C. Friesen, F. Li, K. J. Tomaselli, K. M. Debatin, P. H. Krammer, and M. E. Peter. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sedger, L. M., D. M. Shows, R. A. Blanton, J. J. Peschon, R. G. Goodwin, D. Cosman, and S. R. Wiley. 1999. IFN-γ mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 163:920-926. [PubMed] [Google Scholar]
- 63.Sheridan, J. P., S. A. Marsters, R. M. Pitti, A. Gurney, M. Skubatch, D. Baldwin, L. Ramakrishnan, C. L. Gray, K. Baker, W. I. Wood, A. D. Goddard, P. Godowski, and A. Ashkenazi. 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277:818-821. [DOI] [PubMed] [Google Scholar]
- 64.Srinivasula, S. M., T. Fernandes-Alnemri, J. Zangrilli, N. Robertson, R. C. Armstrong, L. Wang, J. A. Trapani, K. J. Tomaselli, G. Litwack, and E. S. Alnemri. 1996. The Ced-3/interleukin 1β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J. Biol. Chem. 271:27099-27106. [DOI] [PubMed] [Google Scholar]
- 65.Steller, H. 1995. Mechanisms and genes of cellular suicide. Science 267:1445-1449. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi, H., M. Kinouchi, and H. Iizuka. 1997. Interleukin-1β-converting enzyme and CPP32 are involved in ultraviolet B-induced apoptosis of SV40-transformed human keratinocytes. Biochem. Biophys. Res. Commun. 236:194-198. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka, M., T. Itai, M. Adachi, and S. Nagata. 1998. Downregulation of Fas ligand by shedding. Nat. Med. 4:31-36. [DOI] [PubMed] [Google Scholar]
- 68.Teodoro, J. G., and P. E. Branton. 1997. Regulation of apoptosis by viral gene products. J. Virol. 71:1739-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thongcharoen, P., P. Sureau, C. Wasi, H. Bourhy, P. Chaiprasithikul, and P. Puthavathana. 1990. Monoclonal antibody studies of rabies viruses isolated from Thailand. Southeast Asian J. Trop. Med. Public Health 21:129-133. [PubMed] [Google Scholar]
- 70.Trabzuni, D., K. S. Famulski, and M. Ahmad. 2000. Functional analysis of tumour necrosis factor-alpha-related apoptosis-inducing ligand (TRAIL): cysteine-230 plays a critical role in the homotrimerization and biological activity of this novel tumoricidal cytokine. Biochem. J. 350:505-510. [PMC free article] [PubMed] [Google Scholar]
- 71.von Kobbe, C., J. M. van Deursen, J. P. Rodrigues, D. Sitterlin, A. Bachi, X. Wu, M. Wilm, M. Carmo-Fonseca, and E. Izaurralde. 2000. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol. Cell 6:1243-1252. [DOI] [PubMed] [Google Scholar]
- 72.Walczak, H., M. A. Degli-Esposti, R. S. Johnson, P. J. Smolak, J. Y. Waugh, N. Boiani, M. S. Timour, M. J. Gerhart, K. A. Schooley, C. A. Smith, R. G. Goodwin, and C. T. Rauch. 1997. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 16:5386-5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walczak, H., R. E. Miller, K. Ariail, B. Gliniak, T. S. Griffith, M. Kubin, W. Chin, J. Jones, A. Woodward, T. Le, C. Smith, P. Smolak, R. G. Goodwin, C. T. Rauch, J. C. Schuh, and D. H. Lynch. 1999. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5:157-163. [DOI] [PubMed] [Google Scholar]
- 74.Westendorp, M. O., R. Frank, C. Ochsenbauer, K. Stricker, J. Dhein, H. Walczak, K. M. Debatin, and P. H. Krammer. 1995. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375:497-500. [DOI] [PubMed] [Google Scholar]
- 75.Willey, S. R., K. Schooley, P. J. Smolak, W. S. Din, C. P. Huang, J. K. Nicholl, G. R. Sutherland, T. D. Smith, C. Rauch, C. A. Smith, and R. G. Goodwin. 1995. Identification and characterisation of a new member of the TNF family that induces apoptosis. Immunity 3:673-682. [DOI] [PubMed] [Google Scholar]
- 76.Wunner, W. H., and B. Dietzschold. 1987. Rabies virus infection: genetic mutations and the impact on viral pathogenicity and immunity. Contrib. Microbiol. Immunol. 8:103-124. [PubMed] [Google Scholar]
- 77.Zhao, X., B. Bausano, B. R. Pike, J. K. Newcomb-Fernandez, K. K. Wang, E. Shohami, N. C. Ringger, S. M. DeFord, D. K. Anderson, and R. L. Hayes. 2001. TNF-α stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J. Neurosci. Res. 64:121-131. [DOI] [PubMed] [Google Scholar]