

Abstract
Certain core and membrane proteins of vaccinia virus undergo proteolytic cleavage at consensus AG/X sites. The processing of core proteins is coupled to morphogenesis and is inhibited by the drug rifampin, whereas processing of the A17 membrane protein occurs at an earlier stage of assembly and is unaffected by the drug. A temperature-sensitive mutant with a lesion in the I7L gene exhibits blocks in morphogenesis and in cleavage of core proteins. We found that the mutant also failed to cleave the A17 membrane protein. To further investigate the role of the putative I7 protease, we constructed a conditional lethal mutant in which the I7L gene was regulated by the Escherichia coli lac repressor. In the absence of an inducer, the synthesis of I7 was repressed, proteolytic processing of the A17 membrane protein and the L4 core protein was inhibited, and virus morphogenesis was blocked. Under these conditions, expression of the wild-type I7 protein in trans restored protein processing. In contrast, rescue did not occur when the putative protease active site residue histidine 241 or cysteine 328 of I7 was converted to alanine. The mutation of an authentic AG/A and an alternative AG/S motif of L4 prevented substrate cleavage. Similarly, when AG/X sites of A17 were mutated, I7-induced cleavages at the N and C termini failed to occur. In conclusion, we provide evidence that I7 is a viral protease that is required for AG/X-specific cleavages of viral membrane and core proteins, which occur at early and late stages of virus assembly, respectively.
Vaccinia virus (VV), the best-studied member of the Poxviridae, is a complex enveloped DNA virus that replicates in the cytoplasm of infected cells (15). Viral DNA replication, transcription, the accumulation of viral structural proteins, and the assembly of virions occur in discrete juxtanuclear viral factories. Morphogenesis begins with the formation of crescent-shaped membranes that engulf dense viroplasm to form spherical immature virions (IV) (6). Maturation of the IV into infectious intracellular mature virions (IMV) involves a poorly understood process in which at least three core proteins, namely P4a, P4b, and P25, encoded by the A10L, A3L, and L4R open reading frames (ORFs), respectively, undergo proteolytic processing (12, 16, 26) at a consensus AG/X motif (21-23, 28). Initial evidence that processing is coupled to virus maturation was obtained by using the drug rifampin, which reversibly blocks an early step in virus assembly and the cleavage of core proteins (12, 17). Subsequently, numerous temperature-sensitive (ts) mutants were shown to have defects in virus assembly and the processing of core proteins under nonpermissive conditions. One IMV membrane protein, encoded by the A17L ORF, also undergoes proteolytic cleavages at AG/X sites (2, 18, 21, 28). The processing of membrane and core proteins appears to be separable, as two studies described no effect of rifampin on the cleavage of A17 (2, 18) but a third study concluded that rifampin blocked cleavage of this protein (28). Differences in the location and regulation of processing of membrane and core proteins raise the possibility that distinct proteases are involved.
Two putative proteases encoded by VV have been identified. The first, encoded by the G1L ORF, contains a motif that is present in metallopeptidases and was found to induce cleavage of the L4 core protein, albeit at an unnatural AG/X site, in a transfection assay (27). The second putative protease, encoded by the I7L ORF, has a motif that is conserved in a family of cellular and viral cysteine proteinases (14). A conditional lethal mutant of VV, ts16 (5), maps to the I7L ORF and has a defect in virus assembly and the processing of core proteins at the nonpermissive temperature (11). I7 is localized within the cores of wild-type mature virions and accumulates in immature and abnormal virions in ts16-infected cells under nonpermissive conditions (9, 11).
The present study was initiated to investigate the relationship between the processing of membrane and core proteins and the role of I7 in these processing events. While our work was in progress, Byrd and coworkers (3, 4) reported their experiments with ts16 which suggested that I7 is the core protease. Using a newly constructed inducible mutant, we show here that I7 and its putative catalytic site are required for the processing of both membrane and core proteins at authentic cleavage sites. Our data indicate that I7 is involved in the cleavage of topologically distinct proteins at different stages of virus assembly.
(Preliminary data were provided at the XIVth International Poxvirus and Iridivirus Workshop in 2002 and at the annual meeting of the American Society of Virology in 2003.)
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 and HeLa S3 cells were grown in minimum essential medium with Earle's salts (EMEM) and in Dulbecco's modified Eagle's medium, respectively, with each being obtained from Quality Biologicals Inc. (Gaithersburg, Md.) and supplemented with 10% fetal bovine serum (FBS). VV strain WR and the recombinant VV strain vT7LacOI were propagated in HeLa cells as described previously (8). Recombinant vI7Li was replicated in HeLa cells in the presence of 20 μM isopropyl-β-d-thiogalactopyranoside (IPTG) and 2.5% FBS. The ts16 mutant, kindly provided by R. Condit (University of Florida, Gainesville), was propagated at 31°C. All virus stocks were stored at −70°C.
Plasmids.
For the construction of pVOTE-I7L, a copy of the I7L ORF flanked by NcoI and PstI sites was generated by a PCR using VV genomic DNA as a template, cloned into pGEM-T (Promega), digested with NcoI and PstI, and transferred to pVOTE.1 (24). To prevent subsequent homologous recombination, we introduced silent mutations at the third nucleotide of each codon within the last 71 nucleotides by two-stage PCR.
pGEM-I7L-GFP was constructed from three DNA segments which were first cloned into separate plasmids. One DNA segment contained the 3′ region of the I8R ORF and the first 729 bp of the I7L ORF, with a frameshift mutation after nucleotide 65, another DNA segment contained the ORF encoding enhanced green fluorescent protein (GFP) regulated by the VV strong late P11 promoter, and the third DNA segment contained the last 78 bp of the I7L ORF and 824 bp of the I6L ORF.
To produce pP11-I7L-HA, we amplified the I7L ORF from viral DNA by a PCR using primers that added the VV late P11 promoter upstream and an influenza virus HA epitope tag at the C terminus and then inserted it into pGEM-T. Single amino acid substitutions were introduced into pP11-I7L-HA by use of a QuickChange mutagenesis kit (Stratagene).
To form pSLP-L4R, we amplified a copy of the L4R ORF by a PCR using viral genomic DNA and primers that added an influenza virus HA epitope tag at the C terminus and flanking AvaI and PstI sites and then ligated it into AvaI- and PstI-cleaved pUC19 (pSLP) which had been modified to contain a VV strong late synthetic promoter 10 bp upstream of the AvaI site (7). Mutated forms of this plasmid were constructed by use of a QuickChange mutagenesis kit.
pSPL-A17L was made by inserting a PCR copy of the A17L ORF into the SalI and BamHI sites of pUC-SLP, which contained the VV synthetic late promoter five nucleotides upstream of the SalI site. A modified copy of the A17L gene, in which codons 26 to 37 were replaced by nucleotides encoding a V5 epitope tag, was generated by a standard two-stage PCR using the pSPL-A17L plasmid as a template. The resulting fragment was digested with SalI and BamHI and inserted into the corresponding sites of pUC-SLP to produce pSLP-A17LintV5. Mutated forms of this plasmid, encoding amino acid substitutions in the A17L ORF, were constructed by site-directed mutagenesis with a QuickChange mutagenesis kit. A copy of the A17L ORF containing V5 and HA epitope tags at the N and C termini, respectively, and flanked by SalI and BamHI sites was amplified by PCR with viral genomic DNA and cloned into the corresponding sites of pSLP to produce pSLP-dtA17L. Additional forms of this plasmid were generated by PCR amplification from pSLP-A17L derivatives with codon mutations.
The sequences of relevant portions of all plasmids were determined by fluorescence dideoxy termination sequencing with an Applied Biosynthesis model 3100 genetic analyzer.
Construction of recombinant viruses.
vI7Li was formed in two steps by use of the VOTE system (24). BS-C-1 cells were infected with vT7LacOI at 1 PFU per cell for 1 h at 37°C. The cells were then washed twice with Opti-MEM I reduced medium (Invitrogen) and transfected with 2.5 μg of pVOTE.I7L.2 by the use of Lipofectamine 2000 (Invitrogen). After 5 h, the transfection mixture was removed and replaced with complete EMEM containing 2.5% FBS. The cells were harvested 24 h after infection, and diluted lysates were used to infect BS-C-1 monolayers in the presence of mycophenolic acid, xanthine, and hypoxanthine to select for viruses expressing xanthine-guanine phosphoribosyltransferase. The infected cells were covered with agar, and 48 h later mycophenolic acid-resistant plaques were visualized with neutral red and picked with a Pasteur pipette. Four successive rounds of plaque purification were performed to isolate an intermediate virus (vI7L/I7Li) which contained both the original and the inducible copy of the I7L gene. BS-C-1 cells were infected with vI7L/I7Li at a multiplicity of infection of 1 and were transfected with 2.5 μg of pGEM-I7L.GFP to form vI7Li. The lysates were used to infect BS-C-1 monolayers in the presence of 10 μM IPTG. The infected cells were overlaid with agar and incubated for 2 days at 37°C. Recombinant viruses that formed green fluorescent foci were plaque purified four times.
Antibodies.
A rabbit antiserum to the N-terminal peptide of the mature A17 protein was previously described (2, 29). A rabbit antiserum to the peptide corresponding to the sequence CDKLTTKLNRIVNDDE of the I7L ORF was generated for the present study. Mouse monoclonal antibodies (MAbs) against an influenza virus HA and V5 epitope were purchased from Covance Laboratories and Invitrogen Corp., respectively.
Western blotting.
Cells were lysed in a buffer containing 0.06 M Tris-HCl (pH 6.8), 3% sodium dodecyl sulfate (SDS), 10% (vol/vol) glycerol, and 0.002% bromophenol blue. After the addition of β-mercaptoethanol to a final concentration of 5%, the lysates were heated at 100°C and the proteins were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to a nitrocellulose membrane and blocked overnight with 1% nonfat dried milk and 0.05% Tween 20 in PBS. Membranes were incubated with the indicated antibody (anti-A17L, anti-I7L, or anti-HA.11 at 1:1,000 or anti-V5 at 1:5,000) followed by horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G when the anti-A17L or anti-I7L antibody was used or anti-mouse immunoglobulin G when the anti-HA.11 or anti-V5 antibody was used (Amersham Biosciences). Bound antibodies were detected by chemiluminescence (West Pico; Pierce).
Plaque assay and one-step virus growth.
BS-C-1 cell monolayers in six-well tissue culture plates were infected with 10-fold serial dilutions of virus. After 1 h, the inocula were removed and replaced with complete EMEM containing 2.5% FBS and 0.5% methylcellulose, with or without 20 μM IPTG. The infected cells were incubated at 37°C for 2 days and stained with crystal violet, and the plaques were counted.
For one-step virus growth determinations, BS-C-1 cell monolayers in six-well tissue culture plates were infected with 5 PFU of virus per cell. After 1 h, the inocula were removed and the cell monolayers were washed twice with EMEM containing 2.5% FBS. The cells were then incubated in EMEM containing 2.5% FBS with or without 20 μM IPTG and harvested at various times after infection. The infected cells were subjected to three freeze-thaw cycles, sonicated, and stored at −70°C. Virus titers were determined by plaque assays in the presence of 20 μM IPTG.
Electron microscopy.
BS-C-1 cells were infected with vI7Li at a multiplicity of infection of 5 in the presence or absence of 20 μM IPTG. Twenty-four hours after infection, the cells were fixed with 2% glutaraldehyde, embedded in Epon resin, thin sectioned, and viewed with a Philips CM100 electron microscope.
Complementation assays.
BS-C-1 cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG for 1 h. The cells were washed, overlaid with Opti-MEM I reduced medium (Invitrogen) in the absence of IPTG, and transfected with 1 μg of plasmid DNA(s) in Lipofectamine 2000. After 5 h, the medium was removed and replaced with EMEM containing 2.5% FBS (without IPTG). Sixteen to 24 h after infection, the cells were harvested, freeze-thawed three times, and analyzed by Western blotting.
RESULTS
Construction of a conditionally lethal VV mutant with an inducible I7L gene.
To investigate the role of I7 in the proteolytic processing of membrane and core proteins, we constructed a VV mutant with an inducible I7L gene. We started with vT7LacOI, a recombinant VV that contains the Escherichia coli lac repressor gene and an inducible copy of the bacteriophage T7 RNA polymerase gene (24). A copy of the I7L ORF, with silent mutations in the 3′ end segment (discussed below) and regulation by the bacteriophage T7 promoter and the E. coli lac operator, was inserted into the A56R ORF (HA locus) of vT7LacOI by homologous recombination. The resulting virus was named vI7L/I7Li to signify that it has both original and inducible copies of the I7L gene. The next step was to inactivate the original I7L gene. We could not delete the entire I7L ORF, however, without the risk of compromising expression of the neighboring genes. Fathi and Condit (10) have reported that an early I8R transcript initiates 513 bp downstream of the I7L start codon. In addition, the 3′ end of the I7L ORF is likely to contain the promoter of the I6L gene. Therefore, a 464-bp internal segment of the original I7L ORF, which included the region coding for the three amino acids predicted to form the catalytic triad of the I7L protein (14), was deleted and replaced with a GFP reporter gene under the control of a VV late promoter. To avoid the expression of a relatively large N-terminal I7 polypeptide, we inserted a frameshift mutation 65 bp after the start of the I7L ORF. The frame shift generated a premature stop codon which was predicted to allow the synthesis of a 21-amino-acid peptide as well as a unique BclI site that facilitated the screening of recombinant viruses for the desired mutation. A potential problem created by the retention of DNA representing the two ends of the I7L ORF was that the deletion could be repaired by homologous recombination with the complete inducible copy of the I7L ORF. To reduce the likelihood of this, we altered the 78 bp at the 3′ end of the inducible I7L gene by making all possible silent nucleotide mutations. A PCR and DNA sequencing confirmed the fidelity of the insertions in the resulting recombinant VV, vI7Li (Fig. 1A).
FIG. 1.

Construction and initial characterization of an inducible I7L mutant. (A) Diagram of the genome of vI7Li. Important features include the presence of the bacteriophage T7 RNA polymerase gene (T7 pol) regulated by the VV late P11 promoter (PL) and the E. coli lac operator (lacO); the E. coli lac repressor (lacI) regulated by the VV early-late P7.5 promoter (PE/L); the GFP gene regulated by the P11 promoter and replacing a large segment of the original I7L gene; and a new copy of the I7L gene (I7L.2) regulated by the bacteriophage T7 promoter (PT7), the lac operator, and the encephalomyocarditis (EMC) leader. TKL, TKR, HAL, and HAR represent left and right flanking segments of the TK and HA genes. gpt, E. coli guanine phosphoribosyltransferase gene. (B) Effect of IPTG on plaque formation. BS-C-1 cell monolayers were infected with the parental virus vT7LacOI, the intermediate virus vI7L/I7Li, or vI7Li in the presence or absence of 20 μM IPTG. After 48 h, plaques were visualized by staining with crystal violet. (C) Microscopic visualization of cells infected with vI7Li in the presence or absence of IPTG and expressing GFP.
The conditionally lethal phenotype of vI7Li was demonstrated by a plaque assay. The plaque formation of vI7Li was dependent on IPTG, whereas the inducer was not required for the formation of plaques by the parental virus vT7lacOI or the intermediate virus vI7L/I7Li (Fig. 1B). Both vI7Li and vI7L/I7Li induced the formation of syncytia because of the disruption of the VV HA gene. The GFP reporter expressed by vI7Li allowed the detection of infected cells by fluorescence microscopy. In the absence of IPTG, only tiny fluorescent foci were observed, whereas there were large clusters of fluorescent cells due to virus spread in the presence of IPTG (Fig. 1C). This stringent phenotype could result from either an inhibition of virus replication or spread.
Effect of IPTG on virus replication.
Experiments were designed to determine the IPTG requirement for the replication of vI7Li. Cells were infected with vI7Li in the presence of 0 to 250 μM IPTG and harvested 24 h later. A 1.5-log increase in the yield of infectious vI7Li particles occurred with 10 to 25 μM IPTG (Fig. 2A). The virus yield gradually decreased with higher IPTG concentrations (Fig. 2A), which later will be shown to correlate with a higher than normal rate of synthesis of the I7 protein. In cells infected with the intermediate virus vI7L/I7Li, containing two copies of the I7L gene, a decrease in virus yield occurred even with small amounts of IPTG (Fig. 2A). The effect was not due to IPTG per se because the yield of vT7LacOI, which does not overexpress I7, was not affected by excess IPTG (Fig. 2A). Preliminary studies indicated a general inhibition of viral protein synthesis when high levels of I7 were present (data not shown).
FIG. 2.

Effect of IPTG on production of infectious vI7Li. (A) BS-C-1 cells were infected with vT7LacOI (•), vI7L/I7Li (▪), or vI7Li (▴) at a multiplicity of infection of 5 and incubated in the presence of 0 to 250 μM IPTG. Twenty-four hours after infection, virus titers in the presence of 20 μM IPTG were determined by plaque assays. (B) BS-C-1 cells were infected with vT7LacOI (•) or with vI7Li in the presence (▪) or absence (▴) of 20 μM IPTG. Cells were harvested at the indicated times after infection, and virus titers were determined as described above.
The synthesis of I7 was dependent on the expression of the bacteriophage T7 RNA polymerase, which itself was regulated by a VV late promoter. Despite this added step, the time course of vI7Li formation was comparable to that of the parent virus (Fig. 2B). In the absence of IPTG, the production of infectious vI7Li particles remained at the baseline for up to 48 h.
Morphogenesis of vI7Li under nonpermissive conditions.
Cells infected with vI7Li were examined by electron microscopy. At 24 h, cells infected in the presence of 20 μM IPTG contained abundant immature and mature forms which were indistinguishable from those in cells infected with the wild-type virus (not shown). In contrast, mature virions were not found in cells infected with vI7Li in the absence of the inducer. Instead, there was an accumulation of immature particles, including crescents and IV, some of which had nucleoids (Fig. 3A). In addition, there were large numbers of dense virus particles that were generally more spherical than IMV and in some cases were irregularly shaped (Fig. 3A). At a higher magnification, the cores of such particles appeared to be poorly formed (Fig. 3B). Nevertheless, many of the defective particles went on to the next stage of wrapping and exocytosis (Fig. 3B).
FIG. 3.

Electron microscopy of cells infected with vI7Li in the absence of IPTG. BS-C-1 cells were infected with vI7Li at a multiplicity of infection of 5. Twenty-four hours after infection, cells were fixed and embedded in Epon, and ultrathin sections were prepared for transmission electron microscopy. (A) Clusters of IV, some with nucleoids (n), are shown. In addition, there are dense particles, many of which are irregularly shaped (arrows). (B) Cluster of intracellular enveloped virions and one cell-associated extracellular enveloped virion (on the right). The arrows marked “c” point to poorly formed cores.
Expression of I7 is required for proteolytic processing of the A17 membrane protein.
Cells were infected for 24 h with vI7Li in the presence of 0 to 250 μM IPTG, and after 24 h the cells were harvested and the extracts were analyzed by Western blotting with antibodies to I7 and A17. In the absence of IPTG, the I7 protein was detected as a faint 46-kDa band (Fig. 4A). This band was first detected 12 h after infection (data not shown), indicating a slight leakage at late times. Approximately the same low level of I7 was detected with 2.5 μM IPTG. The synthesis of I7 increased with higher IPTG concentrations, equaling that of cells infected with the wild-type virus at 10 to 25 μM and exceeding wild-type levels at higher concentrations (Fig. 4A).
FIG. 4.

Expression and processing of viral proteins. (A) Effect of IPTG on the expression of I7 protein. Cells were left uninfected (UN) or were infected with vI7Li in the presence of 0 to 250 μM IPTG or with wild-type VV (WR) at a multiplicity of infection of 5. The cells were harvested after 18 h, and the proteins in the cellular extracts were separated by SDS-PAGE and detected by Western blotting with a polyclonal antibody to I7 (α-I7). The apparent molecular mass, in kilodaltons, is indicated on the right. (B) Effect of IPTG on processing of A17 protein. BS-C-1 cells were infected and analyzed as described for panel A, except that Western blotting was performed with a polyclonal antibody to an N-terminal peptide of the mature A17 protein (α-A17). The apparent masses of precursor and product proteins are shown on the right. (C) Effect of temperature on processing of A17 protein expressed by VV ts16 mutant. BS-C-1 cells were left uninfected or infected with WR or the ts16 mutant and were maintained at the permissive temperature of 31°C or the nonpermissive temperature of 39°C. The cells were analyzed as described for panel B. (D) Effect of rifampin on processing of A3 and A17 proteins. BS-C-1 cells were infected with WR in the absence (−) or presence (+) of 100 μg of rifampin per ml and were analyzed as described for the other panels, except for the use of a polyclonal antibody to the A3 (α-A3) or A17 protein. The apparent masses of the precursor and product proteins are shown on the right.
In the absence of the inducer, A17 migrated predominantly as a 23-kDa uncleaved protein (Fig. 4B). With increasing concentrations of IPTG, there was an increase in the intensity of the 21-kDa processed form and a corresponding decrease in the 23-kDa precursor (Fig. 4B). At an IPTG concentration of 25 μM or higher, the ratio of the unprocessed form to the processed form was similar to that of wild-type VV WR. The processing of core proteins was also inhibited in the absence of IPTG (see below).
Cells infected with the I7L ts16 mutant were previously shown to be defective in the processing of core proteins, but the effect on the processing of the A17 membrane protein had not been examined. We found that the processing of A17 occurred in cells infected with the ts16 mutant at 31°C but was severely inhibited at 39°C (Fig. 4C). In contrast, the processing of A17 by wild-type VV WR was unaffected by the temperature elevation (Fig. 4C). Thus, both inducible and ts I7L mutants exhibited a similar defect in the processing of the A17 membrane protein.
Our data suggesting that I7 is required for the processing of membrane and core proteins made it important to reexamine the effects of rifampin, which interrupts VV assembly at a very early stage. As noted above, one report concluded that rifampin inhibited cleavage of the A17 membrane protein as well as that of core proteins (28), whereas two others found that cleavage of the A17 protein occurred in the presence of the drug (2, 18). As shown in Fig. 4D, rifampin inhibited cleavage of the A3 core protein but not of the A17 protein. There was, however, a slight difference in the mobility of the upper A17 band which was likely due to differences in phosphorylation (20). Taken together, our results suggest that although I7 is required for the processing of membrane and core proteins, the two events are not coupled.
Putative catalytic site residues of I7 are required for cleavage of substrate proteins.
The putative catalytic protease motif of I7 contains conserved cysteine and histidine residues (14). Trans-complementation experiments were performed to investigate the importance of these amino acids for the processing of A17. Cells were infected with vI7Li in the absence of IPTG and cotransfected with a plasmid expressing A17 and either wild-type I7 with a C-terminal influenza HA epitope tag or I7 that also had cysteine 328 mutated to serine or histidine 241 mutated to arginine. After 24 h, the cells were harvested and the extracts were analyzed by Western blotting with an antibody to the A17 protein. The cleaved 21-kDa band was dominant when the wild-type I7L expression plasmid was transfected (Fig. 5A). However, when the transfected plasmid expressed I7 with either of the point mutations, the cleavage of A17 was inhibited even more than in cells that were not transfected with an I7-expressing plasmid (Fig. 5A), consistent with a dominant-negative effect of the mutated proteins.
FIG. 5.
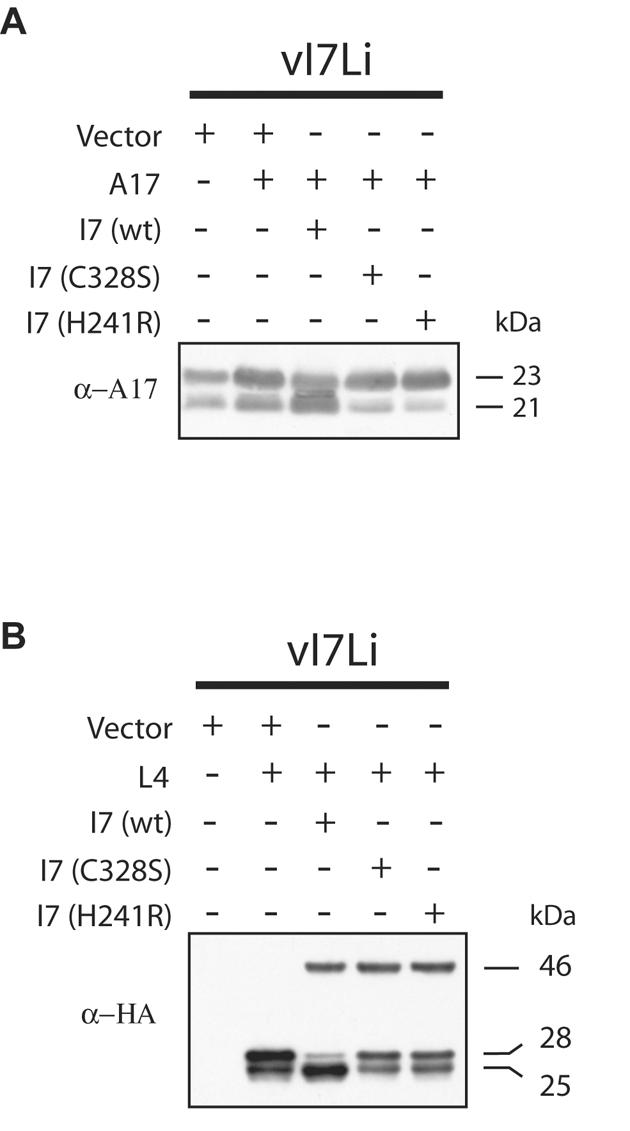
The putative catalytic site of I7 is required for cleavage of the A17 membrane protein and the L4 core protein. (A) I7 requirement for cleavage of A17. Cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG and transfected with the indicated vector plasmid, a plasmid expressing A17, or a wild type (wt) or mutated (C328S or H241R) form of I7 regulated by the VV late P11 promoter and containing a C-terminal influenza virus HA epitope tag. Eighteen hours after infection, cells were harvested and proteins were separated by SDS-PAGE and detected by Western blotting with an antibody to the N-terminal peptide of the mature A17 protein (α-A17). The apparent molecular masses of the precursor and cleaved forms of A17 are shown on the right. (B) I7 requirement for cleavage of L4. Infection and transfection were performed as described above, with an additional plasmid that expressed L4 regulated by a synthetic late promoter and containing an influenza virus HA epitope tag. The Western blot was analyzed with MAb HA.11 to the HA epitope tag. The apparent molecular masses of the I7 protein (46 kDa) and the precursor (28 kDa) and cleaved (25 kDa) forms of the L4 protein are shown on the right.
The L4R ORF encodes a core protein of 28 kDa that is cleaved to a 25-kDa product (26). Infections and transfections were performed in the absence of IPTG, as described above, except that a plasmid expressing an influenza virus HA epitope-tagged version of the L4 protein was cotransfected instead of one expressing A17. In this case, the Western blots were probed with a MAb to the HA epitope tag which detected both the wild-type and mutated forms of the 46-kDa I7 protein as well as the unprocessed and processed forms of L4. When the wild-type I7 expression plasmid was transfected, the cleaved 25-kDa protein was dominant (Fig. 5B). When plasmids expressing either of the mutated forms of I7 were transfected, the 28-kDa precursor was dominant (Fig. 5B). The wild-type and mutated forms of the 46-kDa I7 protein were present at equivalent levels, indicating no difference in their stabilities (Fig. 5B).
I7-dependent cleavage of A17 at N- and C-terminal AG/X sites.
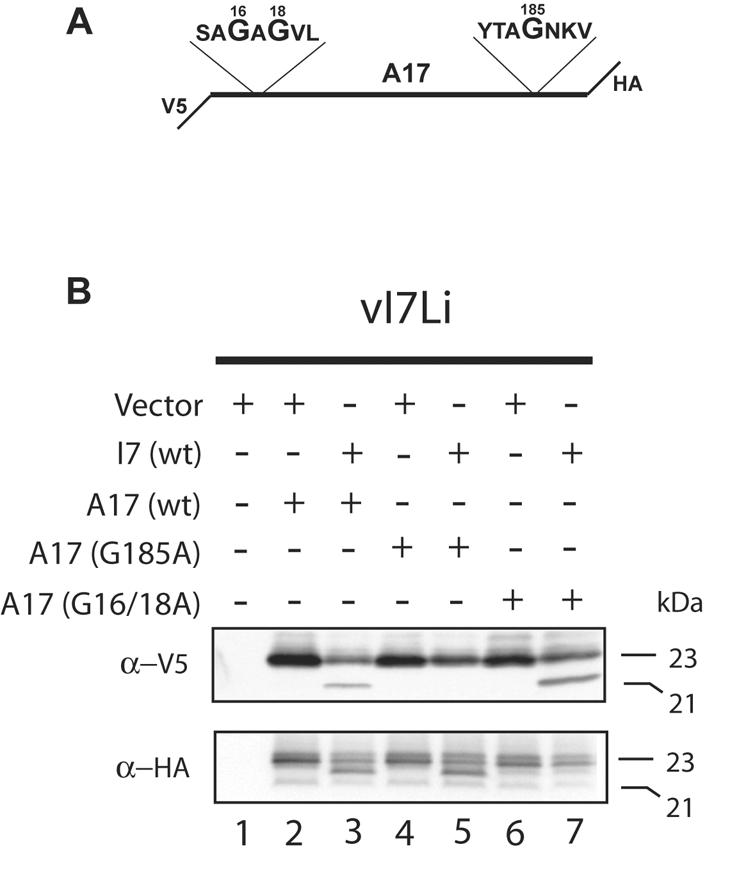
The A17 protein contains overlapping AG/X consensus sites near its N terminus and another site near its C terminus (Fig. 6A). Trans-complementation experiments were designed to determine whether I7-dependent processing of A17 occurs at these positions. Plasmids expressing V5 epitope-tagged forms of the A17 protein were constructed in order to discriminate the mutated forms from the wild-type endogenous protein. Because cleavage of A17 occurs at both the N and C termini, the epitope tag was inserted internally (Fig. 6A), replacing a similar length of peptide to which polyclonal antibodies had been made previously (2). Cells were infected with vI7Li in the absence of IPTG and cotransfected with plasmids expressing I7 and wild-type or mutated forms of A17. After 24 h, the cells were harvested and extracts were analyzed by Western blotting using a V5 MAb. As shown in Fig. 6B, the internal epitope tag did not interfere with the processing of A17, which was dependent on cotransfection of the plasmid expressing wild-type I7. Processing of A17 to the 21-kDa form did not occur, however, when the glycine at position 185 was mutated to alanine, either individually or in combination with other mutations (Fig. 6B). In contrast, a mutation of the glycine at position 16 or 18 or at both of those sites did not have an apparent effect on the processing of A17 (Fig. 6B). We interpreted these data as indicating that the expression of I7 resulted in the cleavage of the AG/N site near the C terminus of the A17 protein. In this system, cleavage near the N terminus did not occur or was not detected because the mobility of the protein was not sufficiently changed after cleavage.
FIG. 6.

Processing of mutated A17 proteins containing an internal V5 tag. (A) Diagram of A17 substrate showing the location of the internal V5 tag and the sequence around the three glycines that were mutated to alanine. (B) Analysis of A17 cleavage. Cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG and were transfected with a vector plasmid or a plasmid expressing A17 with an internal V5 tag containing no other mutations (wt) or a mutation by which a glycine(s) was replaced with an alanine(s). Eighteen hours after infection, cells were harvested and proteins were separated by SDS-PAGE in a Tris-glycine-4 to 20% polyacrylamide gel and detected by Western blotting with a MAb to the V5 tag. The apparent molecular masses of the precursor and cleaved A17 are shown on the right.
Because of the ambiguity regarding the N-terminal cleavage, we expressed another version of A17 with a V5 epitope tag at the N terminus and an influenza HA epitope tag at the C terminus (Fig. 7A). With this substrate, products cleaved only at the C terminus would be detected with an antibody to the N-terminal V5 tag and products cleaved only at the N terminus would be detected with an antibody to the C-terminal HA tag. The tag at the N terminus also served to increase the difference between the masses of the uncleaved and cleaved forms of the A17 protein to allow for a higher level of resolution. Cells were infected with vI7Li in the absence of IPTG and transfected with plasmids expressing wild-type I7 or wild-type or mutated forms of the double-tagged A17 or with the vector plasmid. We first considered the products detected with the V5 antibody. For lane 2 of Fig. 7B, the cells were transfected with a plasmid expressing wild-type A17 and with the vector plasmid. As expected, the V5 antibody detected the 23-kDa form of A17 and also a slightly retarded incompletely resolved band that represents a phosphorylated species (20). The V5 antibody also detected a 21-kDa band that must represent a product that is cleaved only at the C terminus when I7 was also expressed by transfection (Fig. 7B, lane 3). This band was not detected when A17 was mutated at glycine 185 (Fig. 7B, lane 5) or when I7L was not expressed (Fig. 7B lanes 2, 4, and 6), but it was detected when glycines 16 and 18 were mutated (Fig. 7B, lane 7), confirming this interpretation. Next, we considered the products detected with the HA antibody. Again, when the wild-type A17 and vector plasmids were transfected, 23-kDa bands were detected (Fig. 7B, lane 2). We also detected a faint rapidly migrating band that was present in all lanes, was not dependent on the expression of I7, and was not further investigated. When both I7 and A17 were coexpressed, however, a unique band migrating slightly faster than the 23-kDa species was detected (Fig. 7B, lane 3). This band represents an N-terminally processed form that was still detected when glycine 185 was mutated (Fig. 7B, lane 5) but was not detected when I7 was not expressed (Fig. 7B, lanes 2, 4, and 6) or when glycines 16 and 18 were mutated (Fig. 7B, lane 7). Thus, cleavages at both the N- and C-terminal AG/X consensus sites of A7L were dependent on I7.
FIG. 7.
Processing of mutated A17 proteins containing an N-terminal V5 tag and a C-terminal HA tag. (A) Diagram of A17 showing the locations of the V5 and HA tags and the sequences around the three glycines that were mutated to alanine. (B) Analysis of A17 cleavage. Cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG and were transfected with a vector plasmid or a plasmid expressing A17 with an N-terminal V5 tag and a C-terminal HA tag and containing no other mutations (wt) or a mutation by which the indicated glycine(s) was replaced with an alanine(s). Eighteen hours after infection, cells were harvested and proteins were separated by SDS-PAGE in a Tris-glycine-4 to 20% polyacrylamide gel (upper panel) or a Tricine-10% polyacrylamide gel (lower panel) and detected by Western blotting with a MAb to the V5 tag (α-V5) or the HA tag (α-HA). The apparent molecular masses of the precursor and cleaved A17 are shown on the right.
I7-dependent cleavage of L4 at two AG/X sites.
L4 contains an AG/S and an AG/A site near the N terminus, of which only the latter was shown by N-terminal sequencing to be cleaved naturally (21). Cleavage exclusively at the AG/S site, however, occurred when plasmids expressing L4 and G1 were cotransfected (13). To investigate whether I7-induced cleavage occurs at one or both of these sites, we constructed three plasmids, with each expressing L4 with a C-terminal influenza virus HA epitope tag. Two of the plasmids had either glycine 18 or 32 changed to alanine and the third had both changed (Fig. 8A). Cells were cotransfected with the vector or I7 plasmid and with a plasmid expressing wild-type or mutated L4. When the plasmid containing I7 was not transfected, there was a major 28-kDa band and a minor one of 25 kDa, presumably due to a slight leakage of I7 expression (Fig. 8B, lane 2). When I7 was expressed, the 28-kDa band was reduced and a strong 25-kDa doublet was detected, suggesting that there was cleavage at each of the two glycines (Fig. 8B, lane 3). When glycine 18 was mutated, the faster migrating band remained (Fig. 8B, lane 4). The slower migrating band was present when glycine 32 was mutated (Fig. 8B, lane 5). These results indicate that I7 is required for the cleavage of L4 and that cleavage occurs at both AG/X sites.
FIG. 8.
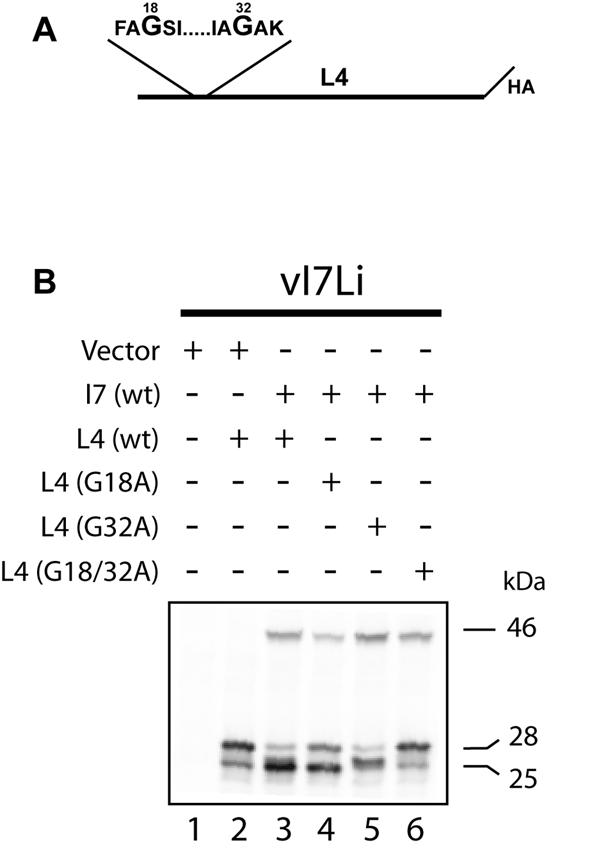
Processing of mutated L4 proteins. (A) Diagram of L4 showing the C-terminal location of the HA epitope tag and the sequences around the two glycines that were mutated to alanine. (B) Analysis of L4 cleavage. Cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG and were transfected with a vector plasmid, a plasmid expressing wild-type I7, or a plasmid expressing L4 with a C-terminal HA tag and containing no other mutations (wt) or a mutation by which the indicated glycine(s) was replaced with an alanine(s). Eighteen hours after infection, cells were harvested and proteins were separated by SDS-PAGE and detected by Western blotting with a MAb to the HA epitope tag (HA.11). The apparent molecular masses of I7 (46 kDa) and the precursor and cleaved forms of L4 (28 and 25 kDa, respectively) are shown on the right.
DISCUSSION
VV was the first DNA virus that was shown to proteolytically process its structural proteins (12, 16). The present study and the recent studies of Byrd et al. (3, 4) have provided strong evidence that the I7L ORF encodes a protease that performs this function. Our work was motivated by sequence similarities between the I7 protein and a family of cysteine proteinases (14; E. Koonin, personal communication) and by the phenotype of a mutant, ts16, that was mapped to the I7L ORF (11). This mutant exhibited defects in the processing of core proteins and in virus morphogenesis. Nevertheless, it was unknown whether the block in protein cleavage was the primary defect or was secondary to the arrest in morphogenesis. To answer this question, we examined the processing of the A17 membrane protein because that event was reported to be independent of morphogenesis (2, 18), which was confirmed in the present study by the use of the drug rifampin. Our initial experiments indicated that processing of the A17 protein was inhibited in cells infected with the ts16 mutant at the nonpermissive temperature, strongly suggesting that the I7 protein was directly involved in processing.
We constructed a conditionally lethal mutant, which had repressed expression of I7 in the absence of an inducer, for two reasons. First, previous reports on the role of I7 were based on experiments with the ts16 mutant, which still expresses a mutated I7 protein under nonpermissive conditions that is detectable by Western blotting (9, 11). The possibility remained that the mutated I7 protein retained some functions that would be revealed by comparison with a null mutant. Second, we planned transfection experiments using mutated I7 which would be best performed with cells infected with a null mutant. The phenotype of the new mutant, vI7Li, was very similar to that of the ts16 mutant; processing of the A17 membrane protein as well as that of core proteins was inhibited and morphogenesis was blocked at a step between IV and IMV. Aberrant particles formed that had grossly abnormal cores. The viral membrane, however, appeared unremarkable, and some of the particles were wrapped by cisternal membranes and exocytosed.
The null mutant allowed us to perform a series of transfection experiments in which we demonstrated that the predicted active site amino acids Cys328 and His241 of I7 are required for the processing of both the A17 membrane protein and the L4 core protein. Furthermore, the I7-dependent processing of the membrane and core proteins occurred at AG/X sites. The L4 protein was cleaved at AG/S and AG/A sites, although only the latter was previously identified by N-terminal sequencing of core proteins. Cleavage of the L4 protein was not indiscriminate, however, as it was not detected at an AG/I sequence. The A17 protein was cleaved at N-terminal AG/A and C-terminal AG/N sites, consistent with previous data.
The simplest interpretation of the results is that I7 directly cleaves both membrane and core proteins, albeit at different stages of virus assembly. However, thus far we have been unable to demonstrate a proteolytic activity of I7 in vitro and therefore cannot eliminate the possibility that the role of I7 is to activate another protease. Nevertheless, we think that this is unlikely because the sequence-related proteases encoded by adenovirus and African swine fever virus cleave their substrates at similar GG/X sites and both have been shown to be active in vitro (1, 19, 25). Interestingly, the adenovirus protease is activated by a peptide, raising the possibility that I7 needs to be activated by an as yet undetermined mechanism. Whitehead and Hruby (27) described transfection experiments suggesting that the G1 protein cleaved the L4 protein at the AG/S site. However, the G1 protein was predicted to be a metallopeptidase, and those enzymes usually have complex substrate specificities. Moreover, in a separate paper (1a) we will provide evidence that G1 is not required for the cleavage of either membrane or core proteins at consensus AG/X sites.
Acknowledgments
We thank Norman Cooper for cells, Andrea Weisberg for electron microscopy, Richard Condit for the VV ts16 mutant, and Brian Ward and Patricia Szajner for their assistance with protocols.
C.A.-S. received partial support from the Special Program for Microbiology of the Brazilian Council for Scientific Technological Development (CNPq).
REFERENCES
- 1.Andres, G., A. Alejo, C. Simon-Mateo, and M. L. Salas. 2001. African swine fever virus protease, a new viral member of the SUMO-1-specific protease family. J. Biol. Chem. 276:780-787. [DOI] [PubMed] [Google Scholar]
- 1a.Ansarah-Sobrinho, C., and B. Moss. Vaccinia virus G1 protein, a predicted metalloprotease, is essential for morphogenesis of infectious virions but not for cleavage of major core proteins. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 2.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L protein kinase. J. Virol. 73:3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2003. Molecular dissection of the vaccinia virus I7L core protein proteinase. J. Virol. 77:11279-11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76:8973-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Condit, R. C., A. Motyczka, and G. Spizz. 1983. Isolation, characterization, and physical mapping of temperature-sensitive mutants of vaccinia virus. Virology 128:429-443. [DOI] [PubMed] [Google Scholar]
- 6.Dales, S., and L. Siminovitch. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davison, A. J., and B. Moss. 1990. New vaccinia virus recombination plasmids incorporating a synthetic late promoter for high level expression of foreign proteins. Nucleic Acids Res. 18:4285-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earl, P. L., and B. Moss. 1991. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.16. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Wiley Interscience, New York, N.Y.
- 9.Ericsson, M., S. Cudmore, S. Shuman, R. C. Condit, G. Griffiths, and J. K. Locker. 1995. Characterization of ts16, a temperature-sensitive mutant of vaccinia virus. J. Virol. 69:7072-7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fathi, Z., and R. C. Condit. 1991. Genetic and molecular biological characterization of a vaccinia virus temperature sensitive complementation group affecting a virion component. Virology 181:258-272. [DOI] [PubMed] [Google Scholar]
- 11.Kane, E. M., and S. Shuman. 1993. Vaccinia virus morphogenesis is blocked by a temperature-sensitive mutation in the I7 gene that encodes a virion component. J. Virol. 67:2689-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz, E., and B. Moss. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. USA 6:677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee, P., and D. E. Hruby. 1993. Trans processing of vaccinia virus core proteins. J. Virol. 67:4252-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li, S. J., and M. Hochstrasser. 1999. A new protease required for cell-cycle progression in yeast. Nature 398:246-251. [DOI] [PubMed] [Google Scholar]
- 15.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 16.Moss, B., and E. N. Rosenblum. 1973. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol. 81:267-269. [DOI] [PubMed] [Google Scholar]
- 17.Moss, B., E. N. Rosenblum, E. Katz, and P. M. Grimley. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280-1284. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez, D., J. R. Rodriguez, and M. Esteban. 1993. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 67:3435-3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubio, D., A. Alejo, I. Rodriguez, and M. L. Salas. 2003. Polyprotein processing protease of African swine fever virus: purification and biochemical characterization. J. Virol. 77:4444-4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szajner, P., A. S. Weisberg, and B. Moss. 2004. Physical and functional interactions between vaccinia virus F10 protein kinase and virion assembly proteins A30 and G7. J. Virol. 78:266-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi, T., M. Oie, and Y. Ichihashi. 1994. N-terminal amino acid sequences of vaccinia virus structural proteins. Virology 202:844-852. [DOI] [PubMed] [Google Scholar]
- 22.VanSlyke, J. K., C. A. Franke, and D. E. Hruby. 1991. Proteolytic maturation of vaccinia virus core proteins—identification of a conserved motif at the N termini of the 4b and 25K virion proteins. J. Gen. Virol. 72:411-416. [DOI] [PubMed] [Google Scholar]
- 23.VanSlyke, J. K., S. S. Whitehead, E. M. Wilson, and D. E. Hruby. 1991. The multistep proteolytic maturation pathway utilized by vaccinia virus P4a protein: a degenerate conserved cleavage motif within core proteins. Virology 183:467-478. [DOI] [PubMed] [Google Scholar]
- 24.Ward, G. A., C. K. Stover, B. Moss, and T. R. Fuerst. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. USA 92:6773-6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webster, A., R. T. Hay, and G. Kemp. 1993. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 72:97-104. [DOI] [PubMed] [Google Scholar]
- 26.Weir, J. P., and B. Moss. 1985. Use of a bacterial expression vector to identify the gene encoding a major core protein of vaccinia virus. J. Virol. 56:534-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitehead, S. S., and D. E. Hruby. 1994. A transcriptionally controlled trans-processing assay: putative identification of a vaccinia virus-encoded proteinase which cleaves precursor protein P25K. J. Virol. 68:7603-7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitehead, S. S., and D. E. Hruby. 1994. Differential utilization of a conserved motif for the proteolytic maturation of vaccinia virus proteins. Virology 200:154-161. [DOI] [PubMed] [Google Scholar]
- 29.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]