

Abstract
Cysteine dioxygenase (CDO) is a mononuclear, non-heme iron-dependent enzyme that converts exogenous cysteine (Cys) to cysteine sulfinic acid using molecular oxygen. Although the complete catalytic mechanism is not yet known, several recent reports presented evidence for an Fe(III)-superoxo reaction intermediate. In this work, we have utilized spectroscopic and computational methods to investigate the as-isolated forms of CDO, as well as Cys-bound Fe(III)CDO, both in the absence and presence of azide (a mimic of superoxide). An analysis of our electronic absorption, magnetic circular dichroism, and electron paramagnetic resonance data of the azide-treated as-isolated forms of CDO within the framework of density functional theory (DFT) computations reveals that azide coordinates directly to the Fe(III), but not the Fe(II) center. An analogous analysis carried out for Cys-Fe(III)CDO provides compelling evidence that at physiological pH, the iron center is six coordinate, with hydroxide occupying the sixth coordination site. Upon incubation of this species with azide, the majority of the active sites retain hydroxide at the iron center. Nonetheless, a modest perturbation of the electronic structure of the Fe(III) center is observed, indicating that azide ions bind near the active site. Additionally, for a small fraction of active sites, azide displaces hydroxide and coordinates directly to the Cys-bound Fe(III) center to generate a low-spin (S = 1/2) Fe(III) complex. In the DFT-optimized structure of this complex, the central nitrogen atom of the azide moiety lies within 3.12 Å of the cysteine sulfur. A similar orientation of the superoxide ligand in the putative Fe(III)-superoxo reaction intermediate would promote the attack of the distal oxygen atom on the sulfur of substrate Cys.
Cysteine dioxygenase (CDO) is a mononuclear non-heme iron enzyme that catalyzes the first step in oxidative cysteine metabolism, namely, the oxidation of l-cysteine (Cys) to cysteine sulfinic acid (CSA) via the incorporation of both oxygen atoms of O2.1,2 In certain organisms, including mammals such as mice and humans, CSA has then been shown to be further metabolized to pyruvate and sulfate through deamination and subsequent C–S bond cleavage, or to taurine through decarboxylation, followed by further oxidation of the sulfur.3 The malfunctioning of CDO, and thus the accumulation of Cys and deficiencies of sulfate and taurine, have been implicated in a variety of neurological diseases.4−8 Additionally, recent investigations with both knockdown and knockout mice have revealed that a complete lack of CDO activity is correlated with severe developmental problems, as well as drastically (∼90%) lower hepatic levels of taurine.9
When CDO was characterized by X-ray crystallography in 2006,10 it was discovered that this enzyme contained two distinct structural features. The first was the existence of a neutral three histidine (3His) facial triad that coordinated the ferrous iron center, as opposed to the much more common, anionic 2-His-1-carboxylate facial triad motif.11,12 The 3His motif has been observed in only two other iron enzymes to date (namely, diketone cleaving dioxygenase13 and gentisate 1,2 dioxygense14), though it has been postulated to also occur in cysteamine dioxygenase.15 The second unusual feature revealed by the structure of CDO was the presence of a thioether cross-link in the active site, with the thiol of C93 being covalently bonded to an ortho carbon of Y157. Much like the 3His triad, this feature of the CDO structure has only been observed in a relatively small number of enzymes (including galactose oxidase16 and NirA17). The exact role this cross-link plays in catalysis is unknown. A potential role in proper substrate Cys positioning and gating of solvent access to the active site has been proposed on the basis of kinetic and EPR spectroscopic measurements,18 while participation in a tyrosyl radical-based mechanism has been ruled out.19 Although eukaryotic forms of CDO are catalytically competent if the cross-link is not formed, enzymatic activity increases drastically (>20-fold) when the cross-link is present.18 Notably, prokaryotic versions of CDO lack this cross-link, yet they maintain a similar level of activity as eukaryotic, cross-linked CDO.20
Several reaction mechanisms for CDO have been proposed in the literature, but experimental data providing conclusive support for any one of these mechanisms is still missing. Cys has been shown both spectroscopically21 and crystallographically22,23 to coordinate to the iron center in a bidentate fashion through its thiolate and amino groups and to activate the Fe(II) center for the reaction with dioxygen.19 Previous spectroscopic studies by our group have revealed that only the Fe(II)CDO state is catalytically active, though substrate Cys as well as is its close analogue selenocysteine (Sec) are also capable of binding to the oxidized Fe(III) ion. Magnetic circular dichroism (MCD), resonance Raman (rR), and density functional theory (DFT) data indicated that the Cys-Fe(III)CDO adduct is structurally similar to the Cys-Fe(II)CDO species, and as such the oxidized complex provides an excellent starting point for generating models of putative reaction intermediates featuring an Fe(III) center.21 This strategy has recently been employed by exposing Cys-Fe(III)CDO to cyanide, which led to the formation of an S = 1/2 species whose electronic structure was found to be sensitive to the absence or presence of the C–Y cross-link.18 Furthermore, an electron paramagnetic resonance (EPR)-based investigation of a superoxide- and Cys-bound Fe(III)CDO complex generated in situ demonstrated that this species is capable of turning over, albeit slowly, strongly suggesting that a (superoxo/Cys)-Fe(III)CDO intermediate is formed in the catalytic cycle.24 Additional support for the involvement of such an intermediate has been obtained in a recent investigation of the NO adducts of Cys- and Sec-Fe(II)CDO.25 After validating the computational methodology through the successful prediction of key spectroscopic parameters of the nitrosyl complexes, O2-bound Cys- and Sec-Fe(II)CDO adducts and likely reaction intermediates were investigated computationally. These studies revealed that a low-energy, S = 2 (superoxo/Cys)-Fe(III)CDO adduct featuring an intermediate spin Fe(III) ion coupled ferromagnetically to a superoxo-based radical likely participates in the CDO reaction mechanism.
Prompted by the likely involvement of a superoxo-Fe(III) complex in the CDO reaction mechanism, we have engaged in a detailed characterization of Cys-Fe(III)CDO both in the absence and presence of azide. Having similar frontier orbitals and the same charge as superoxide (O2•–), azide has frequently been utilized to prepare models of putative Fe(III)- and Mn(III)-superoxide intermediates that are amenable to a wide range of spectroscopic techniques. In the present study, the different Cys-Fe(III)CDO species were characterized by MCD and EPR spectroscopic techniques, in conjunction with quantum mechanics/molecular mechanics (QM/MM) and DFT computations. The results obtained in this study provide unprecedented insights into the active-site properties of Cys-Fe(III)CDO and afford important clues regarding the nature of the putative superoxo-Fe(III)CDO intermediate.
1. Experimental Procedures
1.1. Gene Expression and Protein Purification
The expression of the cdo gene for Mus musculus and isolation of the wild-type protein were performed as described previously.25 In brief, the cdo gene was inserted into a pVP16 vector with an N-terminal maltose binding protein (MBP) tag, and CDO was expressed in Escherichia coli BL21(DE3) cells after induction with IPTG. Cells were collected, lysed using pulsed sonication in a buffer containing 25 mM HEPES (pH 7.5), and pelleted by centrifugation. The cell-free extract was loaded onto a pre-equilibrated DEAE column, and a gradient of 0–400 mM NaCl was applied. The appropriate fractions were collected and loaded onto an amylose column, to which the MBP-CDO fusion protein binds. Bound protein was released from the column by washing with buffer containing 25 mM maltose. Cleavage of the MBP protein from CDO was achieved by incubation with tobacco etch virus protease at 4 °C overnight.26 The resultant protein solution was applied to a Sephacryl 100 size exclusion column, and fractions containing the CDO protein were identified by SDS-PAGE, pooled, and concentrated. Consistent with other isolations reported in the literature, in the as-isolated protein only ∼60% of the active sites contained iron, ∼85% of which were in the Fe(II) oxidation state.
1.2. Sample Preparation
Protein concentration and active-site iron occupancy were determined as described previously.25 Samples used for spectroscopic measurements were ∼2 mM in Fe-bound CDO, as well as 2 mM in dithionite, 5 mM in Cys and/or ∼200 mM in azide, where appropriate. All samples for Abs and MCD experiments were prepared anaerobically, with solid reagents (namely, l-Cys, sodium azide, and sodium dithionite) being degassed separately on a Schlenk line before they were dissolved in degassed buffer under an N2 atmosphere. Samples for MCD studies also contained 55% glycerol as a glassing agent.
1.3. Spectroscopy
Room-temperature electronic absorption (RT Abs) spectra were collected using a Varian Cary 5e spectrophotometer. Low-temperature (LT) Abs and MCD spectra were collected with a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM4000-8T magnetocryostat. To reduce contributions from the CD background signal and glass strain, the MCD spectra presented herein were obtained by taking the difference between spectra obtained with the magnetic field aligned parallel and antiparallel to the light-propagation axis. Iterative Gaussian deconvolutions of all absorption and MCD spectra were conducted using IGOR version 6.22a.27
X-band EPR spectra were collected using a Bruker ESP 300E spectrometer equipped with a Varian EIP model 625A continuous-wave frequency counter. The sample temperature was maintained at 20 K by an Oxford ESR 900 continuous-flow liquid He cryostat that was regulated by an Oxford ITC temperature controller. Spectra were fit using the EasySpin program by Stoll and Schweiger.28 Double integration of the signals was conducted using IGOR version 6.22a.27
1.4. Computations
The initial coordinates for the QM/MM whole-protein optimizations were generated as described previously.25 In brief, two published crystal structures of mouse/rat CDO (2ATF10 and 3ELN(29)) were modified appropriately and used to build the initial coordinates for Cys-bound Fe(III)CDO, as at the time this study was initiated no high-resolution crystal structure of Cys-bound mouse CDO was available. The optimized structure for this species was used as a starting point for generating the initial coordinates for all other CDO models included in this study, with the appropriate atoms for the various adducts being added manually to this structure.
All QM/MM optimizations were conducted in Gaussian09,30 utilizing DFT in conjunction with the B3LYP31,32 functional for the QM region, and the Amber force field33 for the MM portion of the calculation. The QM region included residues R60, H86, H88, C93, H140, H155, and Y157, in addition to six crystallographically defined water molecules in the active site, the iron atom, and all exogenous ligands at the iron center. The boundary between the QM and MM regions was placed between the alpha and beta carbons of the protein residues, and hydrogen was used as the link atom. The 6-31G34 basis set was used for all QM region atoms except the iron, the atoms immediately ligated to it, and bound azide, hydroxide, or water, for which TZVP35 was used. During the optimization process, the alpha carbons of the QM residues were kept frozen. Note that well after this study was initiated, Driggers et al. deposited several crystal structures of mouse CDO in the presence of Cys at a variety of pH values at a reasonably high (1.45–2.00 Å) resolution.23 Importantly, our QM/MM optimized protein models reproduce the key geometric features of the new crystal structures very well (vide infra).
In generating small active-site models for single-point calculations, residues H86, H88, and H140 were excised from the protein matrix and capped with a methyl group at the beta carbon, while all exogenous ligands to the Fe center were retained. Spin-unrestricted single-point DFT calculations were conducted using Orca 2.9.136 with the same basis sets and functional as described above for the QM/MM optimizations. EPR parameters for these models were computed by solving the coupled-perturbed self-consistent field equations as implemented in Orca 2.9.1,37,38 using the B3LYP hybrid functional and the TZVP basis set on all atoms except iron, for which CP(PPP)39,40 was used. These calculations included all orbitals within ±100 hartree of the HOMO–LUMO gap, and the origin of the g-tensor was set at the center of the electronic charge. A high-resolution radial grid with an integration accuracy of 7.0 was used for the Fe atom. Both spin–spin and spin–orbit contributions to the D-tensor were considered. Because previous attempts to utilize time-dependent DFT (TDDFT) for predicting the absorption spectra of CDO species proved unsuccessful,25 in part because this method tends to underestimate the energies of charge transfer transitions,41 electronic transition energies and absorption intensities were computed by utilizing the semiempirical INDO/S-CI module, as implemented in Orca 2.9.1. For each model the orbitals obtained from the single-point DFT calculation were used as the initial orbital guess. PyMOL version 1.5.0.442 was used to generate isosurface plots of the molecular orbitals (MOs) and electron density difference maps (EDDMs), with isodensity values of 0.05 and 0.005 au, respectively.
2. Results
2.1. Spectroscopy
2.1.1. Fe(III)CDO Resting State
CDO is isolated with iron occupying only 60% of the active sites. Of the occupied sites, the majority (typically ∼85%) contain catalytically active Fe(II) with the remaining sites containing Fe(III). We have previously demonstrated that the Fe(III) sites can be quantitatively reduced to the Fe(II) state by the addition of a slight stoichiometric excess of sodium dithionite under anaerobic conditions.21 Thus, the Abs and MCD spectroscopic features associated with the Fe(III)CDO fraction can be readily resolved by removing any spectral contributions from fully reduced CDO. No signal subtraction is necessary when using perpendicular mode EPR spectroscopy, as any adduct of Fe(II)CDO with a closed-shell substrate (analogue) will necessarily possess an integer spin ground state.
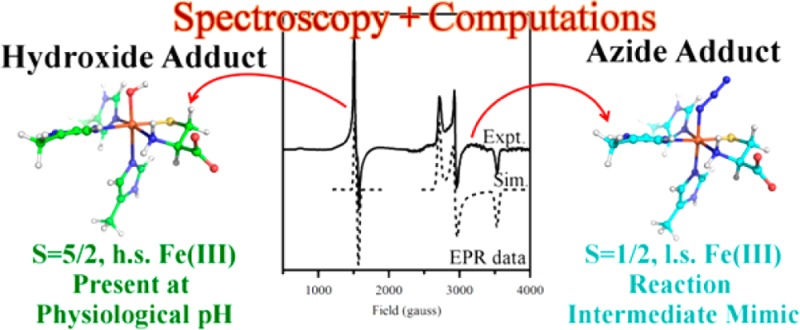
The Abs spectrum of as-isolated CDO exhibits a shoulder at ∼21 750 cm–1 on a slowly rising background absorption above 12 000 cm–1 (see Figure 1). As these features disappear upon the addition of sodium dithionite, they can be attributed to ligand-to-metal charge transfer (LMCT) transitions involving the Fe(III) center. While neither the CD nor the MCD spectra of as-isolated CDO display any features of note (data not shown), the perpendicular mode EPR spectrum exhibits a broad, distorted signal at gobs ≈ 4.27 (see Figure 2), consistent with previous reports. Though the coordination number of the Fe(III) centers contributing to this spectrum is ambiguous (with one to three water molecules potentially completing the first coordination sphere), a recent crystal structure of reduced CDO at low (≤5) pH revealed a tetrahedral coordination of the iron(II) center.23
Figure 1.
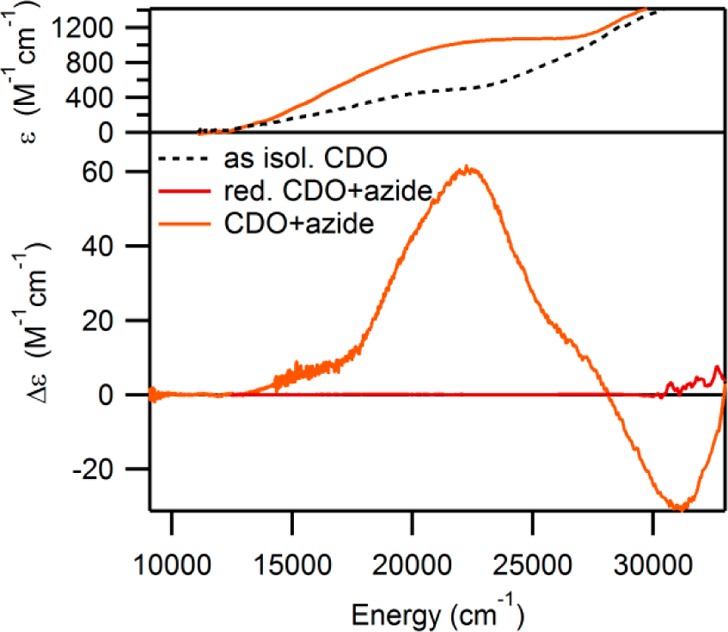
RT Abs spectra of as-isolated CDO in the absence and presence of azide (top) and 4.5 K MCD spectra of as-isolated and fully reduced CDO in the presence of azide (bottom).
Figure 2.

X-band EPR spectrum (at 20 K) of as-isolated CDO in the absence (top) and presence (bottom) of azide. No features attributable to Fe(III)CDO species are observed at higher fields.
2.1.2. N3–-CDO Adduct
It has previously been demonstrated that dioxygen is incapable of binding to the Fe(II)- and Fe(III)-containing CDO active sites in the absence of pre-bound substrate (analogue).19 However, upon the addition of azide to as-isolated CDO, a new Abs feature is observed centered at ∼24 000 cm–1, and several additional features appear in the corresponding LT MCD spectrum (see Figure 1 for spectra and Supporting Information, Figure S3, for Gaussian deconvolutions). As no such features are observed when azide is added to fully reduced CDO (which does not necessarily rule out the formation of an N3–-Fe(II)CDO species), these data clearly indicate that an inner-sphere N3–-Fe(III)CDO complex is formed. As evidenced by the EPR spectrum of as-isolated CDO in the presence of azide (with resonances at geff ≈ 4.20, 4.35, and 4.45 see Figure 2), the azide-bound Fe(III) center remains high-spin (S = 5/2). To the best of our knowledge, this represents the first time a small molecule has been shown to be capable of binding to the Fe center in CDO in the absence of Cys (or one of its analogues).
2.1.3. Cys-Fe(III)CDO Adduct
Consistent with previous investigations into the electronic structure of Cys-Fe(III)CDO,21,24 the addition of Cys to a solution containing as-isolated CDO leads to the appearance of a new Abs feature in the visible region at ∼15 875 cm–1 that is absent when a complete reduction of the enzyme is performed prior to Cys addition. This Abs feature coincides with two positively signed bands in the corresponding MCD spectrum (see Figure 3). An iterative Gaussian deconvolution of the Abs and MCD spectra of Cys-Fe(III)CDO was performed to determine the energies of the underlying transitions (see Table 1). These transitions have previously been assigned as SCys → Fe(III) charge transfer (CT) transitions on the basis of resonance Raman data.21
Figure 3.
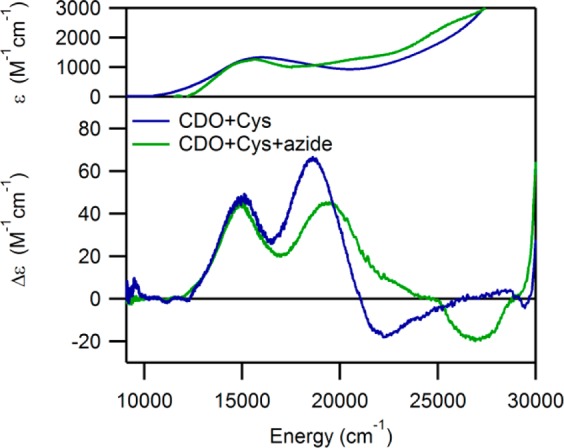
RT Abs (top) and 4.5 K MCD (bottom) spectra of Cys-Fe(III)CDO in the absence and presence of azide.
Table 1. Band Positions (in Wavenumbers) Derived from Iterative Gaussian Deconvolutions of the RT Abs and 4.5 K MCD Spectra Shown in Figures 1 and 3a.
| species | band 1 | band 2 | band 3 | band 4 | band 5 | band width |
|---|---|---|---|---|---|---|
| N3–-Fe(III)CDO | 16 300 | 20 150 | 22 825 | 26 100 | 30 775 | 3300 |
| Cys-Fe(III)CDO | 15 050 | 18 650 | 22 475 | 2650 | ||
| (N3–/Cys)-Fe(III)CDO | 14 950 | 19 300 | 22 000 | 2850 |
The values shown are for the 4.5 K MCD spectra, differing very slightly from those obtained for the RT Abs spectra. See the Supporting Information for overlays of the experimental and Gaussian-fit spectra.
The 20 K X-band EPR spectrum of the Cys-Fe(III)CDO adduct exhibits a sharp signal with effective g-values of 4.24, 4.32, and 4.43 (see Figure 4), as well as a significantly weaker feature at geff ≈ 9. No features attributable to this species are discernible at higher magnetic fields (note that the small signal at geff ≈ 2 is due to a contaminant in the cavity). The EPR feature centered at geff ≈ 4.3 is readily distinguishable from that exhibited by Fe(III)CDO in the absence of substrate, as the latter is significantly broader in nature (see Figure 2). Together, the Abs, MCD, rR, and EPR data of Cys-Fe(III)CDO obtained in this study and reported previously21 provide compelling evidence that a high-spin Fe(III) complex with direct sulfur ligation is formed upon the addition of Cys to as-isolated CDO.
Figure 4.

X-band EPR spectra at 20 K of Cys-Fe(III)CDO in the absence (top) and presence (bottom) of azide, as well as simulations of these spectra where S2 and S3 refer to two distinct species (center). No features attributable to these Fe(III) species were observed at higher fields. For simulation parameters, see Supporting Information, Table S1.
2.1.4. Cys-Fe(III)CDO in the Presence of Azide
Upon the addition of a 100-fold excess of sodium azide to Cys-bound as-isolated CDO (corresponding to a >500-fold excess of azide over Cys-Fe(III)CDO), modest changes are observed in the Abs, MCD, and EPR spectra of the Fe(III)-bound fraction. Specifically, the broad, asymmetric Abs feature at 15 875 cm–1 undergoes a modest (∼725 cm–1) red-shift, while the higher energy component of the pair of MCD features blue-shifts by ∼650 cm–1 (see Figure 3). An additional shoulder is also observed at 25 500 cm–1, though the corresponding negatively signed feature in the MCD spectrum is temperature independent and potentially the result of residual glass strain. Although it may seem possible that these shifts are merely caused by the appearance of an underlying set of features attributable to a new species, the Gaussian deconvolution of the MCD spectrum of Cys-bound Fe(III)CDO in the presence of azide for the region in question is readily completed using only three peaks with band widths and intensities similar to those in the original spectrum. Additionally, the trace obtained by subtracting the MCD spectrum collected in the absence of azide from that in the presence of azide exhibits residual features that are unreasonably intense when taking into account the low concentration of the (N3–/Cys)-bound species as established by EPR spectroscopy (vide infra). Therefore, the close resemblance of the Abs and MCD data obtained for Cys-Fe(III)CDO in the absence and presence of azide indicates that the N3– ion does not bind directly to the Cys-bound Fe(III) ion in the majority of active sites. Instead, azide must bind somewhere within or near the active-site pocket, resulting in only a modest perturbation of the electronic structure of the iron center. These findings suggest that a sixth ligand is already present in the Cys-Fe(III)CDO species, making the Fe(III) center coordinately saturated.
The low-field regions of the X-band EPR spectra of Cys-Fe(III)CDO in the absence and presence of azide are strikingly similar to each other (see Figure 4), both exhibiting a sharp feature centered around geff ≈ 4.3. However, an additional weak signal is observed at higher magnetic fields only when azide is present, suggesting that a minor fraction (∼15%, based on an approximate spin quantitation by double integration of both signals) of active sites directly bind azide when a >500-fold excess of azide is used over Cys-Fe(III)CDO. This additional signal is rhombic in nature with effective g-values of 2.47, 2.29, and 1.90, and with no resolvable hyperfine structure, consistent with a low-spin (S = 1/2) Fe(III) complex. Thus, our EPR data indicate that a small population of (N3–/Cys)-Fe(III)CDO is formed in the presence of a large molar excess of azide, even though we were are unable to identify features associated with this species in our MCD spectra (which are dominated by (OH–/Cys)-Fe(III)CDO-based features).
2.2. Computations
2.2.1. Geometric and Electronic Structures of N3–-Bound Fe(III)CDO
As evidenced by our EPR and MCD spectra presented above, azide readily binds to Fe(III)CDO in the absence of Cys to form an inner-sphere coordination complex. In an effort to determine the identity of the resulting species, several viable models of azide-bound Fe(III)CDO were generated via QM/MM whole-protein geometry optimizations. Initially, three related models were investigated with azide and two water molecules completing an octahedral coordination environment of the Fe center, which differed with respect to the positioning of the azide ligand relative to the three facially ligating His residues. All three models remained six-coordinate at the Fe center and converged to similar relative geometries with nearly identical total energies for the QM region (ΔE ≈ 5.7 kcal/mol). However, one model (with the azide moiety trans to His140) optimized with the distal nitrogen of azide within 2.13 Å of a hydrogen bond donor on Arg60’s guanidyl group, whereas a similar stabilizing interaction could not be found with either of the other two models. Thus, moving forward in the investigation of the azide-bound species, only models with azide in this coordination site were considered. Given the low pKa (∼2.2)43 associated with Fe(III)-bound water, we optimized three additional models possessing either one hydroxide and one water (two permutations, with hydroxide initially bound trans to either His86 or His88) or two hydroxides also bound to the Fe center. Depending on which site was originally occupied by hydroxide, the two (N3–/OH–/H2O)-Fe(III)CDO models converged to distinct geometries, yielding either a five-coordinate, trigonal bipyramidal (N3–/OH–) species or a six-coordinate, octahedral (N3–/OH–/H2O) species.
As all QM/MM-optimized models converged to reasonable geometries (see Table S2), their Abs spectra and EPR parameters were calculated (Figure S1) to determine which model best reproduces our experimental data for azide-bound Fe(III)CDO. On the basis of a comparison between the computationally predicted and experimentally derived EPR parameters (see Table S1 and Figure S1), we propose that a single hydroxide and a single azide ligate to the Fe center of azide-bound Fe(III)CDO, with both the five-coordinate model and the six-coordinate model (featuring an additional water ligand) yielding nearly identical results. The same conclusion is reached from a comparison of the INDO-calculated Abs spectrum for each of the four models investigated with the experimentally determined Abs spectrum (see Figure S1). Although for all models the computed energies and intensities of the electronic transitions are overestimated, both the (N3–/OH–)- and (N3–/OH–/H2O)-Fe(III)CDO models are predicted to exhibit transitions with significant N3– → Fe CT character in the visible spectral region. In contrast, the EDDMs calculated for the relevant transitions of the (N3–/2H2O)- and (N3–/2OH–)-Fe(III)CDO models reveal substantial contributions from His → His and N3–→N3– intraligand excitations, respectively. Hence, these transitions would be expected to carry very little intensity in the corresponding MCD spectrum, which is inconsistent with our experimental data.
As the computed Abs spectra and EPR parameters for both the (N3–/OH–)- and (N3–/OH–/H2O)-Fe(III)CDO models are in reasonable agreement with our experimental data obtained for azide-bound Fe(III)CDO, a closer examination of the electronic structures of these models is warranted. In each case, all of the Fe 3d-based spin-up (α) MOs are filled, while their spin-down (β) counterparts are unoccupied (see Figure S2), as expected for high-spin Fe(III) species. Moreover, because the water molecule in the (N3–/OH–/H2O)-Fe(III)CDO model only weakly interacts with the Fe center, the dominant N3–/OH––Fe(III) bonding interactions in this and the five-coordinate model are nearly identical. Both σ- and π-type OH––Fe(III) bonding interactions are present. Alternatively, the azide in-plane (i.p., where the plane in question is defined by the Fe–NAz,prox-NAz,med fragment) π-nonbonding orbital engages in a strong σ-type bonding interaction with the Fe center, whereas the out-of-plane (o.p.) π-nonbonding orbital has only a very weak π-type interaction with the Fe center.
2.2.2. Geometric and Electronic Structures of Cys-Bound Fe(III)CDO in the Absence of Azide
The low affinity of the active site of Cys-Fe(III)CDO for azide suggests that a sixth, semi-labile ligand is present at the iron center of this species. To evaluate this possibility computationally, we performed whole-protein QM/MM geometry optimizations of Cys-bound Fe(III)CDO with either water or hydroxide occupying the open coordination site, as well as a model featuring a five-coordinate iron center. The relevant structural parameters for these models (see Table 2) all agree well with those derived from the 2.00 Å resolution crystal structure of Cys-Fe(II)CDO published recently.23 The orientation and relative position of the key outer-sphere residues (including R60, Y157, and C93) are remarkably similar for all structures considered here, where in each case the carboxylate of the substrate Cys serves as a hydrogen-bond acceptor from the guanidyl group of R60 and the phenol moiety of Y157.
Table 2. Relevant Active-Site Bond Lengths (in Å) and Angles (in Degrees) As Derived from the QM/MM-Optimized Models and the 2.00 Å Resolution Crystal Structure of Cys-Bound Fe(II)CDO.
| species | Fe–His86 | Fe–His88 | Fe–His140 | Fe–SCys | Fe–NCys | Fe–Xa | His86–Fe–X |
|---|---|---|---|---|---|---|---|
| Cys-Fe(II)CDO, 4JTOb | 1.893 | 2.199 | 2.110 | 2.291 | 2.262 | n.a. | n.a. |
| (OH–/Cys)-Fe(III)CDO | 2.347 | 2.135 | 2.238 | 2.436 | 2.252 | 1.899 | 168.6 |
| (H2O/Cys)-Fe(III)CDO | 2.148 | 2.221 | 2.100 | 2.446 | 2.175 | 2.219 | 169.1 |
| Cys-Fe(III)CDO | 2.116 | 2.147 | 2.109 | 2.436 | 2.170 | n.a. | n.a. |
| (N3–/Cys)-Fe(III)CDOc | 2.073 | 2.154 | 1.994 | 2.261 | 2.069 | 1.930 | 177.6 |
X refers to the ligating atom of the sixth ligand (hydroxide, water, or azide) bound to iron.
Crystal structure at 2.00 Å resolution, ref (23).
Optimized as an S = 1/2 species.
Despite having a higher iron oxidation state, our QM/MM-optimized models of Cys-Fe(III)CDO feature a somewhat (∼0.15 Å) longer Fe–SCys bond than is observed in the Cys-Fe(II)CDO crystal structure, while the Fe–NCys bond length is predicted to be shorter in the computational models. This unexpectedly long Fe–SCys bond in the Cys-Fe(III)CDO models may be attributed to partial electron donation from the Cys sulfur to the Fe center, which reduces the effective nuclear charge of the Fe(III) ion. Consistent with this prediction, all three models contain significant unpaired spin density on the sulfur, as determined by a Loewdin spin population analysis (vide infra).
In both the crystal structure and our five-coordinate model, the iron center resides in a distorted trigonal bipyramidal coordination environment, with a clearly visible open coordination site trans to the His86 ligand. Upon the binding of a hydroxide or water to this site, the coordination about the Fe center becomes very nearly octahedral, though in the case of the (OH–/Cys) adduct the Fe–NHis88 bond lengthens considerably as a consequence of the trans influence exerted by the hydroxide ligand. In contrast, the binding of water to the Fe center causes only a modest perturbation to the original Fe–ligand bonds, as expected considering that water is a weak field ligand.
As all of our QM/MM optimized models converged to reasonable geometries, the corresponding active sites were evaluated further using DFT and semiempirical INDO/S-CI computations to generate quantitative bonding descriptions and predict their EPR parameters and Abs spectra (see Figures 5 and 6). Of the DFT-computed EPR spectra for the various Cys-Fe(III)CDO models examined, the one obtained for the six-coordinate, hydroxide-bound species provides by far the closest match to the experimental EPR spectrum with regards to the overall shape, rhombicity, and g-values. Likewise, the INDO/S-CI-computed Abs spectrum for this model agrees best with the experimental Abs spectrum of Cys-Fe(III)CDO (see Figure 6). EDDMs for the two intense transitions predicted at 18 500 and 21 050 cm–1 show predominant SCys → Fe(III) CT character, consistent with published resonance Raman data obtained with laser excitation into the dominant Abs feature at ∼15 875 cm–1.21 Although the INDO/S-CI computation overestimates the energies and intensities of these two features by ∼4000 cm–1 and about an order of magnitude, respectively, these errors are within the expected range for this type of calculation.44
Figure 5.

Comparison between the DFT-computed EPR spectra for various Cys-Fe(III)CDO models and the experimental 20 K EPR spectrum of Cys-Fe(III)CDO (bottom). The EPR parameters for each model are given in the Supporting Information, Table S1.
Figure 6.
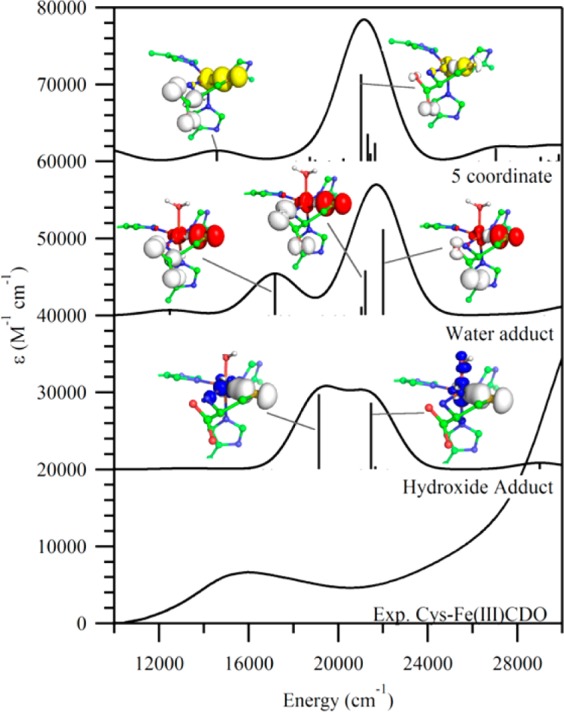
Comparison between the INDO/S-CI-predicted Abs spectra for various Cys-Fe(III)CDO models and the experimental RT Abs spectrum of Cys-Fe(III)CDO (bottom). EDDMs are shown for the major transitions for each species, where loss and gain of electron density are denoted by white and colored lobes, respectively.
An examination of the EDDMs computed for the other two Cys-Fe(III)CDO models [namely, six-coordinate (H2O/Cys)-Fe(III)CDO and five-coordinate Cys-Fe(III)CDO] reveals that the major electronic transitions predicted to occur in the region between 13 000 and 25 000 cm–1 involve electronic excitations from MOs localized on the carboxylic acid moiety of the substrate Cys to MOs with mixed Fe/S orbital character, likely because the thiolate ligand possesses partial radical character (see below). This prediction is not supported by the resonance Raman-based assignment of the Abs feature in the visible region as SCys → Fe(III) CT transitions.21 For this reason, as well as the poor match between the experimentally derived vs computationally predicted EPR parameters for the water-bound and five-coordinate Cys-Fe(III)CDO models along with the inability of azide to readily coordinate to Cys-bound Fe(III)CDO, we conclude that the six-coordinate model with hydroxide occupying the sixth position provides the most realistic description of the active site of Cys-bound Fe(III)CDO.
The drastically different EPR parameters predicted for the three models considered for Cys-Fe(III)CDO have prompted us to carry out further investigations into the DFT-computed electronic structures for these species. Because in general the Fe 3d-based spin-up MOs of high-spin Fe(III) complexes are strongly mixed with ligand-based orbitals due to spin polarization, only the compositions of the spin-down MOs will be examined further. In the (OH–/Cys) adduct, all of the Fe 3d-based α-MOs are filled, whereas the spin-down counterparts are unoccupied, as expected for a high spin Fe(III) species. The highest occupied β-MO (β-HOMO) is a sulfur-based lone pair, whereas the β-HOMO–1 contains contributions from the S 3py and Fe 3d orbitals, making it σ-bonding with respect to the Fe–SCys bond (see Figure 7). Both π- and σ-bonding interactions between the Fe(III) center and the hydroxide ligand are also present, though at substantially lower energies (HOMO–2 and HOMO–3, respectively). The Fe 3dxz-, 3dxy-, and 3dyz-based unoccupied MOs (the t2g set of orbitals in the parent octahedral symmetry) are lower in energy than the Fe 3dx2–y2- and 3dz2-derived MOs (the eg set). Both the Fe 3dyz- and Fe 3dxz-based MOs are π*-antibonding with respect to the Fe–OH bond, while the Fe 3dxy orbital is essentially nonbonding. In contrast, the Fe 3dx2–y2- and 3dz2-based MOs are strongly σ*-antibonding with respect to the Fe–SCys and Fe–OH bonds, respectively.
Figure 7.

DFT-computed energies of relevant spin-down MOs for the active sites of (from left to right) the (OH–/Cys)-Fe(III)CDO, (H2O/Cys)-Fe(III)CDO, and five-coordinate Cys-Fe(III)CDO models, with lines connecting analogous MOs between different species. Boundary surface plots of key MOs for the (OH–/Cys) and five-coordinate Cys models (far left and far right, respectively) are also shown. Note that the orientations of the molecular axes differ between these models, and orbitals lacking significant Fe, S, hydroxide, or water character are omitted for clarity. The orbital energies for each species were adjusted such that α-LUMO (a ligand-based orbital with essentially identical compositions for all species) lies at 0 eV.
Interestingly, the computed electronic structures for the (H2O/Cys)- and five-coordinate Fe(III)CDO species do not support a +3 oxidation state assignment for the iron center. Although in both cases all five Fe 3d-based spin-up MOs are occupied, a single Fe 3d-based spin-down orbital is also occupied (note that due to extensive mixing among the filled orbitals, two separate β-MOs actually contain substantial Fe 3dxz orbital character, see Figure 7). Accordingly, both the (H2O/Cys) and five-coordinate models can be described as possessing a high-spin Fe(II) center, with the remaining unpaired spin density residing on the cysteine moiety. This description is corroborated by the Loewdin spin densities obtained from the single-point DFT calculation for each species. In the case of the (OH–/Cys) model, the total spin population on the cysteine moiety is 0.52, due to the high covalency of the Fe(III)–SCys bond in this species, as opposed to 0.95 and 1.05, respectively, in the (H2O/Cys) and five-coordinate models.
Note that in an earlier computational investigation, the five-coordinate Cys-Fe(III)CDO complex was described as possessing a high-spin Fe(III) center.21 However, this earlier study only used a truncated first sphere model in the optimization of the crystal structure coordinates, and thus did not account for any steric or electrostatic constraints imposed by outer-sphere residues. Since the QM/MM approach employed in the present study does account for these effects, we expect our computed electronic structure description to be more accurate, and thus conclude that the active site of Cys-bound Fe(III)CDO binds hydroxide to yield a six-coordinate species in which the Cys sulfur retains partial radical character.
2.2.3. Geometric and Electronic Structures of Putative (N3–/Cys)-Fe(III)CDO Species
In addition to investigating a variety of potential Cys-Fe(III)CDO adducts in the absence of azide, we also performed whole-protein QM/MM geometry optimizations for viable S = 1/2 and S = 5/2 (N3–/Cys)-Fe(III)CDO complexes. Although both spin states converged to reasonable geometries, the S = 1/2 species is predicted to be more stable by ∼18 kcal/mol (corresponding to the difference in single-point energies). As an S = 1/2 minority species is indeed formed upon the addition of a large excess of azide to Cys-Fe(III)CDO according to our EPR analysis (see Figure 4), the computational results for the S = 1/2 adduct were investigated further. For this species, the bond lengths and angles about the iron center are consistent with those reported for our other models (see Table 2), with slightly shorter Fe–NHis bonds trans to the cysteinyl amide and azide moieties. Azide binds in a bent, end-on fashion with an Fe–NAz distance of 1.93 Å and an Fe–N–N bond angle of 131.5°. The azide moiety is oriented such that the NAz–NAz–NAz bond vector lies directly above the Fe–SCys bond vector, with an S–Fe–NAz,prox–NAz,med dihedral angle of just 0.1°. This places the medial nitrogen atom of the azide moiety (which mimics the position of the distal oxygen atom of superoxide) within 3.12 Å of the cysteine sulfur.
Consistent with our EPR results, the S = 1/2 (N3–/Cys)-Fe(III)CDO species is predicted to feature a low-spin iron(III) center. The Fe 3dyz- and 3dxz-based MOs are occupied in both the α and β manifolds, whereas the Fe 3dxy-based MO is occupied only in the α manifold (see Figure 8, right side). The occupied Fe 3d-based orbitals contain considerable contributions from the azide πnb orbitals and, in the case of the Fe 3dyz-based MO, a strong σ-bonding interaction with one of the SCys 3p-based orbitals. The unoccupied Fe 3d-based β-MOs show π-antibonding (Fe 3dxy) or σ-antibonding (Fe 3dx2–y2 and Fe 3dz2) interactions with the ligands. The Fe–NAz bond is very covalent, having contributions from both σ- and π-type bonding interactions between the Fe 3d orbitals and the azide in-plane and out-of-plane πnb orbitals, respectively. Of particular importance with respect to the orientation of the azide ligand within the CDO active site is the composition of the Fe 3dyz-based β-MO. To maximize its σ- and π-bonding interactions with the thiolate and azide ligands, respectively, this orbital forces the NAz–NAz–NAz and Fe–SCys bond vectors to lie directly above each other. This particular configuration is predicted to be stabilized further by a short (∼2.35 Å) hydrogen bond between residue H155 and the distal nitrogen of the azide ligand.
Figure 8.

DFT-computed energies of relevant spin-down MOs for the active sites of the (OH–/Cys)-Fe(III)CDO and (N3–/Cys)-Fe(III)CDO models, with lines connecting analogous MOs between different species. Boundary surface plots of key MOs for each model are also shown. Note that the orientations of the molecular axes differ between these models, and orbitals lacking significant Fe, S, hydroxide, or azide character are omitted for clarity. The orbital energies for each species were adjusted such that the α-HOMO (a ligand-based orbital with essentially identical compositions for both species) lies at 0 eV.
Although extensive mixing between the Fe 3dxy and ligand orbitals in the α manifold precludes a direct analysis of the “singly occupied MO” (SOMO), the Fe 3dxy β-MO does not undergo such orbital mixing and thus provides an excellent representation of the unpaired spin density distribution for the (N3–/Cys)-Fe(III)CDO species. As required by the low-spin Fe(III) description for this species, the Fe 3dxy β-MO possesses predominantly (71%) Fe 3d character, with modest contributions from the sulfur (11%) and azide (6%) moieties. A similar composition of the “SOMO” was recently predicted for a related S = 1/2 adduct, namely the (CN–/Cys)-Fe(III)CDO species. This species exhibits a g-spread similar to, though slightly smaller than, that of the (N3–/Cys)-Fe(III)CDO adduct (Δg = 0.442 for the cyanide adduct, as compared to Δg = 0.547 for the azide-bound species).18 The small differences in g-spread likely reflect a more delocalized “SOMO” and/or smaller splitting of the “t2g set” of Fe 3d-based MOs (due to π-backbonding) in the (CN–/Cys)-Fe(III)CDO species.
3. Discussion
All recently proposed mechanisms for CDO invoke a (superoxo/Cys)-Fe(III)CDO intermediate,24,25,45 thus making this species a particularly attractive target to mimic. To this end, we initiated a detailed investigation of the Cys-bound Fe(III)CDO active site both in the absence and in the presence of azide, which serves as a mimic for superoxide. Azide mimics not only the electronic structure of superoxide (having similar frontier MOs) but also its charge and size, yet it does not support catalytic turnover and instead forms a stable adduct.
To the best of our knowledge, the binding of azide to Fe(III)CDO represents the first time a small molecule has been unequivocally observed to bind to the active site of CDO in the absence of Cys (or one of its analogues). An evaluation of QM/MM-optimized viable models for azide-bound Fe(III)CDO on the basis of our spectroscopic data reveals that a hydroxide also binds to the Fe center in the presence of azide, giving iron an overall dianionic ligand environment. We cannot, however, determine whether this species is five- or six-coordinate at the Fe center. Regardless, these results suggest that superoxide may also be capable of binding to Fe(III)CDO, though it has been previously reported that superoxide is incapable of reducing the Fe(III)CDO active site.24
Several factors indicate that the Cys-Fe(III)CDO complex is coordinately saturated, including the ease by which azide binds to Fe(III)CDO in the absence of Cys and the large super-stoichiometric excess of azide over Cys-Fe(III)CDO required to observe the formation of any inner-sphere complex. Because both the EPR and Abs spectra collected for the Cys adduct of Fe(III)CDO are in much better agreement with those computed for our (OH–/Cys)-Fe(III)CDO model than for the (H2O/Cys)-Fe(III)CDO and five-coordinate Cys-Fe(III)CDO models, we conclude that a hydroxide ion occupies the sixth coordination site of the Cys-bound Fe(III) center. It is this hydroxide ligand that must be displaced for azide (or any other potential ligand) to coordinate to the Fe(III) ion in the presence of Cys.
The slight red-shifts of the Abs and MCD spectral features observed upon exposure of Cys-Fe(III)CDO to azide suggest that only a minor fraction of active sites form an inner-sphere complex. The majority of sites appear to bind azide in an outer-sphere pocket, such that this anion perturbs the electronic structure of the (OH–/Cys)-Fe(III)CDO complex without actually displacing the hydroxide ligand from the iron center. Being a member of the cupin family, CDO possess a relatively open active site, with at least two potential substrate tunnels to the active site (see Figure 9). However, there are few residues lining the tunnels with which a negatively charged small molecule would favorably associate. The most obvious candidate is Arg60, which has previously been proposed to play a key role in the proper positioning of the substrate Cys.18 With one side of the guanidyl group participating in a salt bridge with the carboxylate moiety of the bound substrate Cys, the other side would be free to associate with azide. The presence of azide in this position could conceivably subtly modulate the H-bonding abilities of Arg60’s guanidyl group, and thus the positioning of Cys and the electronic structure of the active site. Due to its cationic nature, this same R60 side chain may also help form a pre-binding site for the Cys substrate, as well as potentially gate access to the Fe center. On the opposite side of the active site, another access tunnel can be identified that may serve to provide O2 (or analogues thereof) access to the Fe center.
Figure 9.
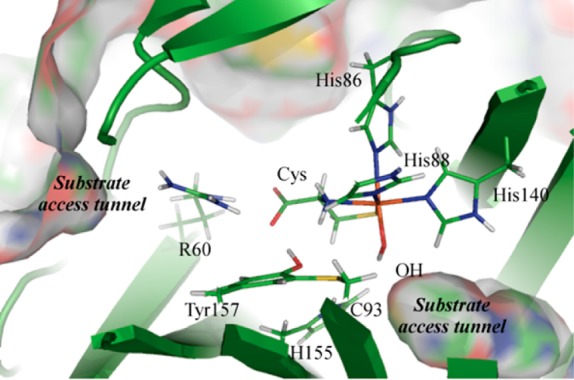
Interior view of the QM/MM-optimized structure of (OH–/Cys)-Fe(III)CDO, with the protein surface (as determined by PyMOL) shown. For clarity, only atoms that were included in the QM region are shown.
Another Fe-dependent enzyme with a coordination environment similar to that of Cys-bound Fe-CDO is superoxide reductase (SOR). This enzyme features a 4His1Cys ligand set at the Fe center, with a sixth, monoanionic ligand (a glutamate residue) also binding when the metal center is in the +3 oxidation state. The addition of a 45-fold molar excess of azide over Fe(III)SOR results in the complete conversion to an azide-bound enzyme complex, where azide binds trans to the cysteine sulfur at the position originally occupied by the glutamate ligand. EPR studies revealed that this complex possesses an S = 5/2 ground state, as opposed to the S = 1/2 ground state observed for the (N3–/Cys)-Fe(III)CDO complex described above.46 However, a model complex of SOR with the azide and sulfur ligands arranged in a cis configuration features essentially the same binding mode of the azide (bent, and positioned directly over the Fe–S bond), other key geometric parameters (e.g., a relatively short Fe–S bond), and spin ground state (S = 1/2) as our computational model for (N3–/Cys)-Fe(III)CDO.47
The present investigation was motivated, in part, by the fact that three separate studies have recently provided evidence for the formation of a superoxo-Fe(III) intermediate in the catalytic cycle. In one study this intermediate was described as an S = 3 species [featuring a high-spin Fe(III) ion coupled ferromagnetically to a superoxide radical] on the basis of parallel-mode EPR data obtained for a complex that was prepared by reacting superoxide with Cys-bound Fe(III)-CDO. However, the authors pointed out that their data could also be fit with parameters describing a species with an S = 2 ground state.24 The second study predicted an S = 2 ground state [formally due to ferromagnetic coupling between an intermediate spin Fe(III) ion and a superoxide radical] on the basis of a spectroscopically validated computational methodology.25 Finally, a purely computational, QM/MM study suggested that this adduct is best described as possessing an S = 1 ground state (with a low-spin Fe(III) center coupled ferromagnetically to a superoxide radical45); however, this prediction is inconsistent with the EPR signal displayed by the species obtained by reacting superoxide with Cys-bound Fe(III)-CDO.24
The (N3–)-Fe(III)CDO and (N3–/Cys)-Fe(III)CDO species investigated in this work have provided us with a valuable opportunity to evaluate further a computational methodology suitable for modeling the putative (superoxo/Cys)-Fe(III)CDO intermediate, and thus to distinguish between the three proposed spin states for this species. Importantly, by using the same computational methodology we have successfully employed in previous investigations of (NO/Cys)-Fe(II)CDO25 and (CN–/Cys)-Fe(III)CDO,24 the spin ground states and key spectroscopic signatures of both the (N3–)-Fe(III)CDO and (N3–/Cys)-Fe(III)CDO species could be adequately reproduced. Hence, we are quite confident that our computational approach is capable of successfully predicting key geometric and electronic structures that are relevant to the CDO catalytic cycle. As such, favor is lent toward the involvement of an S = 2 superoxo-Fe(III) intermediate in this reaction mechanism, as we previously proposed.25
In our preferred CDO mechanism, the initial step in the reaction of Fe(II)CDO with O2 is to generate a (superoxo/Cys)-Fe(III)CDO adduct. In the next step (Scheme 1), a bond forms between the SCys and distal superoxo oxygen, yielding a four-membered Fe–O–O–S ring. Our present computational investigation of the (H2O/Cys)- and five-coordinate Cys-Fe(III)CDO adducts suggests that sulfur can act as a redox non-innocent ligand in the absence of a sixth, strongly donating ligand at the metal center. This finding lends indirect support for our previously proposed mechanism in which the accumulation of radical character on sulfur was invoked to avoid the formation of a Fe(IV)-oxo species, and to direct the attack of the putative “oxyl-Fe(III)” intermediate toward the SCys atom.25
Scheme 1. Partial Mechanism for Oxidation of Cys by CDO, Where the Formation of the First S–O Bond Is Shown.

Accumulation of partial SCys radical character may also play an important role in directing the formation of the first S–O bond in the CDO catalytic cycle. A resonance structure of our proposed Fe(III)-superoxo intermediate exists that features a reduced Fe(II) ion linked to a cysteine radical cation (see Scheme 1). With unpaired spin density located on both the sulfur and the superoxide moiety, the formation of the first S–O bond could occur through a radical recombination mechanism. Such a thiolate-based stabilization of an Fe(III)-superoxo species [by imposing partial Fe(II)-superoxo character] has previously been invoked in the catalytic mechanism of isopenicillin-N-synthase.48 While our previous computational investigation of viable intermediates in the CDO catalytic cycle revealed that the Fe(III)-superoxo intermediate possesses little unpaired spin density on the SCys atom, indicating that the Fe(II)/cysteine radical cation resonance structure is a relatively minor contributor, the accumulation of unpaired spin density on SCys is expected to become more pronounced on approaching the transition state for S–O bond formation.
4. Conclusions
Fe(III)CDO is capable of binding azide in the absence of pre-bound Cys substrate to produce a five- or six-coordinate complex featuring one additional anionic ligand (hydroxide). The addition of Cys to a solution containing Fe(III)CDO results in the formation of a six-coordinate complex with a hydroxide ion completing the ligand sphere. The presence of hydroxide makes the Fe(III) ion coordinatively saturated, disfavoring the binding of an alternative exogenous ligand. Consequently, azide occupies a position within the active site (likely near the guanidyl group of R60) without displacing the hydroxide ligand, resulting in a slightly perturbed electronic structure of the active site. However, in a small subset of active sites, azide does bind directly to the iron center, which results in the formation of an S = 1/2 species possessing a low-spin Fe(III) ion. Because the spin ground states and key spectroscopic features of the species investigated in this work could be adequately reproduced by using the same computational methodology we have previously employed to evaluate key steps in the CDO catalytic cycle, our results lend further support to the involvement of an S = 2 superoxo-Fe(III)CDO intermediate featuring an intermediate spin Fe(III) ion.
Acknowledgments
E.J.B. thanks the Fox laboratory for providing extensive access to their facilities.
Glossary
Abbreviations
- CDO
cysteine dioxygenase
- Cys
cysteine
- Sec
selenocysteine
- EPR
electron paramagnetic resonance
- MCD
magnetic circular dichroism
- Abs
electronic absorption
- DFT
density functional theory
- RT
room temperature
- LT
low temperature
- QM/MM
quantum mechanics/molecular mechanics
- MO
molecular orbital
- EDDM
electron density difference map
- INDO/S-CI
intermediate neglect of differential overlap/singles-configuration interaction
Supporting Information Available
Table of geometric parameters for converged QM/MM models, EPR parameters obtained from fits of experimental and computationally predicted spectra, results from iterative Gaussian deconvolutions of Abs and MCD spectra obtained for (N3–)-, (Cys)-, and (N3–/Cys)-Fe(III)CDO, and Cartesian coordinates for all active-site atoms derived from the QM/MM-optimized models. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the National Institutes of Health grant GM 64631 (T.C.B.), the National Science Foundation grant MCB-0843239 (B.G.F.), and the National Science Foundation grant CHE-0840494 (computational resources).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yamaguchi K.; Hosokawa Y. (1987) Cysteine Dioxygenase. Methods Enzymol. 143, 395–403. [DOI] [PubMed] [Google Scholar]
- Lombardini J. B.; Singer T. P.; Boyer P. D. (1969) Cysteine Oxygenase. 2. Studies on Mechanism of Reaction with 18Oxygen. J. Biol. Chem. 244, 1172–1175. [PubMed] [Google Scholar]
- Stipanuk M. H.; Ueki I. (2011) Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J. Inherited Metab. Dis. 34, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivka A.; Cohen G. (1993) Brain Ischemia Markedly Elevates Levels of the Neurotoxic Amino-Acid, Cysteine. Brain Res. 608, 33–37. [DOI] [PubMed] [Google Scholar]
- Pean A. R.; Parsons R. B.; Waring R. H.; Williams A. C.; Ramsden D. B. (1995) Toxicity of Sulfur-Containing-Compounds to Neuronal Cell-Lines. J. Neurol. Sci. 129, 107–108. [DOI] [PubMed] [Google Scholar]
- Heafield M. T.; Fearn S.; Steventon G. B.; Waring R. H.; Williams A. C.; Sturman S. G. (1990) Plasma Cysteine and Sulphate Levels in Patients with Motor Neurone, Parkinson’s and Alzheimer’s Disease. Neurosci. Lett. 110, 216–220. [DOI] [PubMed] [Google Scholar]
- Gordon C.; Bradley H.; Waring R. H.; Emery P. (1992) Abnormal Sulfur Oxidation in Systemic Lupus-Erythematosus. Lancet 339, 25–26. [DOI] [PubMed] [Google Scholar]
- Emery P.; Bradley H.; Gough A.; Arthur V.; Jubb R.; Waring R. (1992) Increased prevalence of poor sulphoxidation in patients with rheumatoid arthritis: effect of changes in the acute phase response and second line drug treatment. Ann. Rheum. Dis. 51, 318–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki I.; Roman H. B.; Valli A.; Fieselmann K.; Lam J.; Peters R.; Hirschberger L. L.; Stipanuk M. H. (2011) Knockout of the Murine Cysteine Dioxygenase Gene Results in Severe Impairment in Ability to Synthesize Taurine and an Increased Catabolism of Cysteine to Hydrogen Sulfide. Am. J. Physiol. Endocrinol. Metab. 301, E668–E684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy J. G.; Bailey L. J.; Bitto E.; Bingman C. A.; Aceti D. J.; Fox B. G.; Phillips G. N. (2006) Structure and Mechanism of Mouse Cysteine Dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehntop K. D.; Emerson J. P.; Que L. Jr. (2005) The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-heme Iron(II) Enzymes. J. Biol. Inorg. Chem. 10, 87–93. [DOI] [PubMed] [Google Scholar]
- Hegg E. L.; Que L. (1997) The 2-His-1-carboxylate facial triad—An emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 250, 625–629. [DOI] [PubMed] [Google Scholar]
- Straganz G. D.; Diebold A. R.; Egger S.; Nidetzky B.; Solomon E. I. (2010) Kinetic and CD/MCD Spectroscopic Studies of the Atypical, Three-His-Ligated, Non-Heme Fe2+ Center in Diketone Dioxygenase: The Role of Hydrophilic Outer Shell Residues in Catalysis. Biochemistry 49, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Li W.; Wang M. Z.; Zhu G. Y.; Liu D. Q.; Sun F.; Hao N.; Li X. M.; Rao Z. H.; Zhang X. C. (2008) Crystal structure and mutagenic analysis of GDOsp, a gentisate 1,2-dioxygenase from Silicibacter Pomeroyi. Protein Sci. 17, 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy J.; Simmons C. R.; Hirschberger L. L.; Hwang J.; Coloso R. M.; Stipanuk M. H. (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem. 282, 25189–25198. [DOI] [PubMed] [Google Scholar]
- Ito N.; Phillips S. E. V.; Stevens C.; Ogel Z. B.; McPherson M. J.; Keen J. N.; Yadav K. D. S.; Knowles P. F. (1991) Novel Thioether Bond Revealed by a 1.7-Å Crystal-Structure of Galactose-Oxidase. Nature 350, 87–90. [DOI] [PubMed] [Google Scholar]
- Schnell R.; Sandalova T.; Hellman U.; Lindqvist Y.; Schneider G. (2005) Siroheme- and Fe-4-S-4 -dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. J. Biol. Chem. 280, 27319–27328. [DOI] [PubMed] [Google Scholar]
- Li W.; Blaesi E. J.; Pecore M. D.; Crowell J. K.; Pierce B. S. (2013) Second-Sphere Interactions between the C93–Y157 Cross-Link and the Substrate-Bound Fe Site Influence the O2 Coupling Efficiency in Mouse Cysteine Dioxygenase. Biochemistry 52, 9104–9119. [DOI] [PubMed] [Google Scholar]
- Pierce B. S.; Gardner J. D.; Bailey L. J.; Brunold T. C.; Fox B. G. (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- Dominy J. E.; Simmons C. R.; Karplus P. A.; Gehring A. M.; Stipanuk M. H. (2006) Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J. Bacteriol. 188, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J. D.; Pierce B. S.; Fox B. G.; Brunold T. C. (2010) Spectroscopic and Computational Characterization of Substrate-Bound Mouse Cysteine Dioxygenase: Nature of the Ferrous and Ferric Cysteine Adducts and Mechanistic Implications. Biochemistry 49, 6033–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C. R.; Liu Q.; Huang Q. Q.; Hao Q.; Begley T. P.; Karplus P. A.; Stipanuk M. H. (2006) Crystal Structure of Mammalian Cysteine Dioxygenase: A Novel Mononuclear Iron Center for Cysteine Thiol Oxidation. J. Biol. Chem. 281, 18723–18733. [DOI] [PubMed] [Google Scholar]
- Driggers C. M.; Cooley R. B.; Sankaran B.; Hirschberger L. L.; Stipanuk M. H.; Karplus P. A. (2013) Cysteine Dioxygenase Structures from pH 4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J. Mol. Biol. 425, 3121–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. A.; Li W.; Pierce B. S. (2011) Single Turnover of Substrate-Bound Ferric Cysteine Dioxygenase with Superoxide Anion: Enzymatic Reactivation, Product Formation, and a Transient Intermediate. Biochemistry 50, 10241–10253. [DOI] [PubMed] [Google Scholar]
- Blaesi E. J.; Gardner J. D.; Fox B. G.; Brunold T. C. (2013) Spectroscopic and Computational Characterization of the NO Adduct of Substrate-Bound Fe(II) Cysteine Dioxygenase: Insights into the Mechanism of O2 Activation. Biochemistry 52, 6040–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommel P. G.; Fox B. G. (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr. Purif. 55, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igor, Version 6.22a (2011), Wavemetrics, Inc., Lake Oswego, OR.
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Simmons C. R.; Krishnamoorthy K.; Granett S. L.; Schuller D. J.; Dominy J. E.; Begley T. P.; Stipanuk M. H.; Karplus P. A. (2008) A Putative Fe2+-Bound Persulfenate Intermediate in Cysteine Dioxygenase. Biochemistry 47, 11390–11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L.; Hada J. L., Ehara M., Toyota M., Fukuda K., Hasegawa R., Ishida J., Nakajima M., Honda T., Kitao Y., Nakai O., Vreven H., Montgomery T. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J. (2010) Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT.
- Lee C. T.; Yang W. T.; Parr R. G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Becke A. D. (1993) Density-Functional Thermochemistry.3 . The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. (1995) A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, And Organic-Molecules. J. Am. Chem. Soc. 117, 5179–5197. [Google Scholar]
- Hehre W. J.; Ditchfie R.; Pople J. A. (1972) Self-Consistent Molecular-Orbital Methods. 12. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 56, 2257–2261. [Google Scholar]
- Schafer A.; Horn H.; Ahlrichs R. (1992) Fully Optimized Contracted Gaussian-Basis Sets for Atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. [Google Scholar]
- Neese F. (2012) Orca 2.9.1, An Ab Initio, DFT and Semiempirical Electronic Structure Package.
- Neese F. (2001) Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree-Fock and Kohn-Sham theory. J. Chem. Phys. 115, 11080–11096. [Google Scholar]
- Neese F. (2003) Metal and ligand hyperfine couplings in transition metal complexes: The effect of spin-orbit coupling as studied by coupled perturbed Kohn-Sham theory. J. Chem. Phys. 118, 3939–3948. [Google Scholar]
- Neese F. (2002) Prediction and interpretation of the Fe-57 isomer shift in Mossbauer spectra by density functional theory. Inorg. Chim. Acta 337, 181–192. [Google Scholar]
- Wachters A. J. (1970) Gaussian Basis Set for Molecular Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 52, 1033–1036. [Google Scholar]
- Neese F. (2006) A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry. J. Biol. Inorg. Chem. 11, 702–711. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, 1.5.0.4 ed., Schrodinger LLC.
- Flynn C. M. (1984) Hydrolysis of Inorganic Iron(III) Salts. Chem. Rev. 84, 31–41. [Google Scholar]
- Gonzalez L.; Escudero D.; Serrano-Andres L. (2012) Progress and Challenges in the Calculation of Electronic Excited States. ChemPhysChem 13, 28–51. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Thiel W.; de Visser S. P. (2011) Theoretical Study on the Mechanism of the Oxygen Activation Process in Cysteine Dioxygenase Enzymes. J. Am. Chem. Soc. 133, 3869–3882. [DOI] [PubMed] [Google Scholar]
- Clay M.; Jenney F.; Hagedoorn P.; George G.; Adams M.; Johnson M. (2002) Spectroscopic studies of Pyrococcus furiosus superoxide reductase: Implications for active-site structures and the catalytic mechanism. J. Am. Chem. Soc. 124, 788–805. [DOI] [PubMed] [Google Scholar]
- Shearer J.; Fitch S. B.; Kaminsky W.; Benedict J.; Scarrow R. C.; Kovacs J. A. (2003) How does cyanide inhibit superoxide reductase? Insight from synthetic (FeN4S)-N-III model complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 3671–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. D.; Neidig M. L.; Neibergall M. B.; Lipscomb J. D.; Solomon E. I. (2007) VTVH-MCD and DFT studies of thiolate bonding to {FeNO}(7)/{FeO2}(8) complexes of isopenicillin N synthase: Substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes. J. Am. Chem. Soc. 129, 7427–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.