

Abstract
Yeast (Saccharomyces cerevisiae) alcohol dehydrogenase I (ADH1) is the constitutive enzyme that reduces acetaldehyde to ethanol during the fermentation of glucose. ADH1 is a homotetramer of subunits with 347 amino acid residues. A structure for ADH1 was determined by X-ray crystallography at 2.4 Å resolution. The asymmetric unit contains four different subunits, arranged as similar dimers named AB and CD. The unit cell contains two different tetramers made up of “back-to-back” dimers, AB:AB and CD:CD. The A and C subunits in each dimer are structurally similar, with a closed conformation, bound coenzyme, and the oxygen of 2,2,2-trifluoroethanol ligated to the catalytic zinc in the classical tetrahedral coordination with Cys-43, Cys-153, and His-66. In contrast, the B and D subunits have an open conformation with no bound coenzyme, and the catalytic zinc has an alternative, inverted coordination with Cys-43, Cys-153, His-66, and the carboxylate of Glu-67. The asymmetry in the dimeric subunits of the tetramer provides two structures that appear to be relevant for the catalytic mechanism. The alternative coordination of the zinc may represent an intermediate in the mechanism of displacement of the zinc-bound water with alcohol or aldehyde substrates. Substitution of Glu-67 with Gln-67 decreases the catalytic efficiency by 100-fold. Previous studies of structural modeling, evolutionary relationships, substrate specificity, chemical modification, and site-directed mutagenesis are interpreted more fully with the three-dimensional structure.
NAD(P)-dependent oxidoreductases occur in virtually all organisms and catalyze the reversible oxidation of primary and secondary alcohols into aldehydes and ketones, respectively. Medium-chain alcohol dehydrogenases (ADHs, EC 1.1.1.1) contain 327–376 amino acid residues per chain and are usually zinc-dependent.1 The ADHs in higher eukaryotes (plants and animals) are usually dimeric, whereas those in prokaryotes and lower eukaryotes (yeast) are tetrameric. The dimeric horse liver ADH (374 amino acid residues, 80000 Da) was the first of this superfamily to be studied by X-ray crystallography and is a model for the other ADHs.2−4 In yeast, constitutive ADH1 catalyzes the reduction of acetaldehyde to ethanol during the fermentation of glucose. The enzyme was purified and crystallized with ammonium sulfate, and its kinetic, chemical, and physical properties have been studied extensively.5−8 Yeast ADH1 is a tetramer of four identical subunits with 347 amino acid residues each and a calculated mass of 147396 Da. The amino acid and gene sequences of yeast ADH1 were determined.9,10 Molecular modeling shows that yeast ADH is homologous to horse liver ADH,11 but deletion of 21 amino acid residues from the catalytic domain of yeast ADH1 and other gaps and insertions make exact comparisons problematic.
Yeast ADH1 was one of the first enzymes to be crystallized.5 Thin, hexagonal crystals were also found in anaerobically grown yeast, apparently with a tetrahedral arrangement of subunits in the P312 space group.12,13 Crystals that diffracted well for X-ray crystallography were reported previously,14,15 and we collected many data sets; however, most crystals were twinned, and a structure could not be determined by molecular replacement (despite the availability of several ADH structures) or by multiple isomorphous replacement. We found one, only partially twinned, crystal with different cell dimensions and determined the structure by molecular replacement using the structure of the tetrameric Pseudomonas aeruginosa ADH.16 The structure of yeast ADH1 now permits comparisons with other ADHs and a better understanding of previous studies of ADH1.
Experimental Procedures
Protein Preparation
The gene for Saccharomyces cerevisiae ADH1 (adc1, YOL086c; UniProtKB entry P00330), the constitutive, fermentative enzyme from the laboratory strain of baker’s yeast, was expressed from plasmid YEp13 in host yeast that did not express any of the three medium-chain, zinc-containing alcohol dehydrogenases,8,10,17 and the protein was purified to homogeneity as described previously.18
Crystallization
Crystals were obtained by the hanging drop, vapor diffusion method using Fluka polyethylene glycol 5000 monomethyl ether (MPEG-5000) as the precipitating agent. The hanging drop contained 16 μL of 10 mg/mL protein in 125 mM sodium N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid buffer (pH 8.4) (at 25 °C), 1.7 mM nicotinamide 8-iodoadenine dinucleotide (8ID, kindly provided by N. J. Oppenheimer), 0.1 M 2,2,2-trifluoroethanol (TFE), 0.16 mM EDTA, and 7% MPEG-5000 and was equilibrated over 0.73–0.84 mL of a reservoir solution containing 22–26% MPEG-5000 and 0.1 M TFE. Crystals grew in 2 weeks at 4 °C. Crystals were soaked in ∼1 mL of the same buffer with 30% MPEG-5000 and 0.5 M TFE (but no 8ID because no more was available) for 5 days during their shipment on ice from Iowa to Germany before flash vitrification in liquid N2 and data collection.
Data Collection and Structure Determination
X-ray data were collected July 28, 1995, on synchrotron beamline BW7A at the EMBL/DESY Hamburg unit using a single crystal at 100 K and a 300 mm MAR Research imaging plate detector and a wavelength of 0.8570 Å. The data were processed with d*TREK.115 The structure was determined using molecular replacement using AMORE19 and P. aeruginosa ADH [Protein Data Bank (PDB) entry 1LLU, sequence 42% identical to that of yeast ADH1] as the search model. The estimated solvent content of the crystals is 53%, and the Matthews coefficient, VM, is 2.62 Å3/Da. O20 and REFMAC21 were used for model building and refinement, respectively. Refinement was improved by including the twin operator (0.713 for h, k, l and 0.287 for −h, −k, l) and by using eight TLS elements representing the catalytic domains, residues 1–154 and 294–347, and the coenzyme binding domain, residues 155–293, for each of the four subunits. See Table 1 for data collection and refinement statistics.
Table 1. X-ray Data Collection and Refinement Statistics.
| PDB entry | 4W6Z |
| space group | P321 |
| no. of different subunits in the asymmetric unit | 4 |
| cell dimensions (Å) | 144.39, 144.39, 128.20 |
| cell angles (deg) | 90, 90, 120 |
| resolution range (Å) | 28.1–2.4 |
| no. of measured reflections (total, unique) | 229310, 59568 |
| completeness (%) (outer shell) | 98.5 (92.7) |
| Rmeas (outer shell)a | 0.134 (0.484) |
| mean ⟨I⟩/σ⟨I⟩ (outer shell) | 8.0 (3.5) |
| Rvalue, Rfree, test (%)b | 0.178, 0.222, 2.5 |
| rmsdc for bond distances (Å), angles (deg) | 0.015, 1.82 |
| no. of protein atoms | 10328 |
| 8 Zn, 2 NAD(8-Iodo), 4 TFE atoms | 122 |
| no. of waters | 151 |
| mean B value (Å2), Wilson, REFMAC | 39.4, 40.2 |
| estimated coordinate error (Å) | 0.15 |
Results and Discussion
Structure Solution Overview
The diffracting crystals of ADH1 reported previously14 in space group P622 turned out to be twinned,22 and the structure could not be determined. Extensive attempts to prepare other crystal forms and to prepare heavy atom derivatives were also not successful except for one crystal that was prepared in the presence of trifluoroethanol and nicotinamide 8-iodoadenine dinucleotide (8ID in the PDB file, hereafter termed NAD), which we thought might be useful for phasing. This crystal was unusual in that it was shipped in crystallization media that did not contain NAD, but with 0.5 M TFE in an attempt to stabilize the ternary complex.
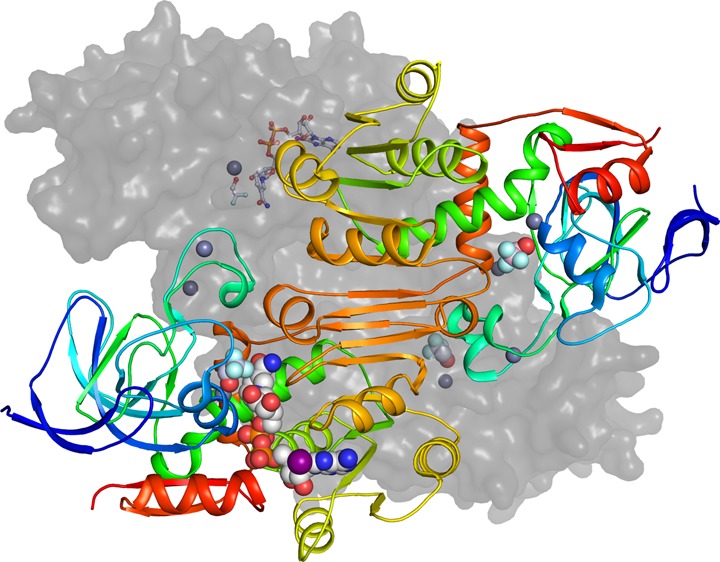
The asymmetric unit contains four crystallographically different, but structurally similar, subunits arranged as two dimers that we named AB and CD and are similar to the dimeric horse liver ADH (Figure 1A). Each subunit in a dimer has a coenzyme binding domain typical of the Rossmann fold (six-stranded parallel β-pleated sheet with two helices on each side of the sheet) to which the coenzyme binds at the carboxyl terminal end.23 Extensive interactions of the two coenzyme binding domains produce an extended β-sheet in a dimer. Each subunit also has a catalytic domain that contains the zinc atom to which the alcohol binds and a structural zinc in a distant loop. The substrates bind in the cleft between the domains. The A and C subunits have a “closed” conformation and contain NAD and TFE bound to the catalytic zinc in the “classic” coordination, whereas subunits B and D have an “open” conformation with TFE in the substrate binding pocket near the catalytic zinc, which has an alternative coordination, and no bound coenzyme. Packing considerations lead to the conclusion that the unit cell contains three biological molecules each of two different tetramers with AB:AB and CD:CD subunits (Figure 1B).
Figure 1.

Stereoviews of one asymmetric unit, an AB dimer, and of the biologic AB:AB tetramer in a back-to-back orientation. (A) In the AB dimer, the A subunit has bound NAD and TFE (in ball and stick representation) and a closed conformation, whereas the B subunit has only TFE and an open conformation. The zinc atoms are shown as gray spheres. (B) In the tetramer, the catalytic subunits of the two A subunits (blue, with NAD and TFE colored green) are most closely associated with one another in the tetramer, and the catalytic domains of B subunits (magenta) are likewise associated. These figures were made with the PyMOL Molecular Graphics System, version 1.7, from Schrödinger, LLC.
Subunit and Dimer Structures
Superpositioning of the α-carbon atoms of the coenzyme or catalytic domains, or the complete subunits, shows that the conformations of the A and C subunits (containing NAD and TFE) are very similar to one another (average rmsd of 0.30 Å); the B and D subunits (containing only TFE) are also similar to one another [rmsd of 0.54 Å (see Table 1S of the Supporting Information)]. The higher rmsd value for B and D subunits relative to that for the A and C subunits results from disorder, or poor electron density, for residues in some loops of the D subunit: 49–59, 246–251, and 269–274.
The dimers have similar structures, as superposition of dimers AB and CD gives an rmsd value of 0.45 Å. However, the A and C subunits differ somewhat from the B and D subunits in coenzyme and catalytic domain structures (average rmsd of 0.87 Å) and in the overall subunits (average rmsd of 1.97 Å). The conformation of the A and C subunits is more closed than that of the B and D subunits. After the coenzyme binding domains of subunits A and B are superimposed, a rotation of ∼13° around an effective hinge axis24 superimposes the catalytic domains of the A and B subunits. Horse liver ADH was the first enzyme shown in atomic detail by X-ray crystallography to undergo a conformational change when substrates bind; a rotation of ∼10° closes up the substrate binding cleft.3 The extent of conformational change for ADHs from different sources varies; the rotation of the open and closed subunits differs by ∼7° in cod liver ADH and ∼12° in Escherichia coli ADH.25−27 The conformational change is important for coenzyme binding and catalytic efficiency.28
Quaternary Interactions
The packing of the symmetry-related dimers in the unit cell could produce two different tetramers: a “back-to-back” dimer of dimers, AB:AB and CD:CD, in which the active sites are on the “front” sides and fully available to bind substrates; or “front-to-front” dimers, AB:CD, in which the active sites would be opposed and less accessible to solvent. The sensible choice favors the back-to-back arrangement (Figure 1B), and this fits with calculations of buried surface area (Table 2S of the Supporting Information). The formation of the dimers (AB or CD) buries ∼3400 Å2 of the 30000 Å2 of combined area of the monomers. The formation of the AB:AB or CD:CD tetramers buries ∼4400 Å2 of the 54000 Å2 of combined area of the dimers. On the other hand, the formation of an AB:CD tetramer would bury only ∼1700 Å2 of the combined area. Each tetramer contains two different subunits, A1B1:A2B2 or C1D1:C2D2 (1 and 2 being symmetry-related subunits), in a quasi-tetrahedral arrangement, about a noncrystallographic, molecular 2-fold axis (near residues 277 and 290). The back-to-back arrangement of dimers in the tetramer is also found in the other tetrameric ADHs.
The buried surface area is an indicator of the fit between protein subunits, but the specific interactions between subunits in the dimers and tetramers are important for understanding oligomer formation and the heat stability of yeast ADH1 in comparison to those of the more stable yeast ADH2. Most of the interactions between subunits A and B (or C and D) in the dimers involve residues from the coenzyme binding domains of the Rossmann fold (Figure 1S of the Supporting Information). Several hydrogen bonds connect the peptide backbones of the antiparallel β-strands that include residues 274–280 and 288–292 around the dimeric molecular 2-fold axis. A short α-helix links these two β-strands. These interactions produce the extended coenzyme binding domain. Such interactions were also identified in horse liver ADH in the βS and βF elements in horse liver ADH, linked by a short 310-helix.2
Side chains of several residues (101–110) from the structural zinc binding loop of the catalytic domain of one subunit also interact with residues (260–262) of the coenzyme binding domain of the other subunit in a dimer. The carboxylate of Glu-101 forms a salt bridge with the guanidinium group of Arg-260 between subunits of both dimers. The side chain of Asn-110 in subunit A (or C) interacts with side chains of Asn-262 and Ser-287 in subunit B (or D). Because the conformations of the subunits are slightly different, due to the relative rotations of the catalytic and coenzyme binding domains within a subunit, the interactions between subunits are not symmetrical. Some water-mediated hydrogen bonds link Gly-237 and Ala-261 backbone atoms to Tyr-102. Altogether, the intradimer interactions amount to 21–22 hydrogen bonds, 2–4 electrostatic interactions, 14–15 “hydrophobic” interactions (van der Waals interactions, ≤∼4 Å), and one disulfide bond.
The disulfide bond is formed between Cys-277 residues across the molecular 2-fold axis in a dimer (Figure 2S of the Supporting Information). As presented in the Supporting Information, a disulfide bond was also found in the purified and crystallized commercial enzyme. Treatment of ADH1 with 10 mM dithiothreitol at pH 7.0 decreased the t50 (temperature at which 50% of enzyme activity was lost) from ∼59 to ∼38 °C and promoted dissociation to inactive subunits, making the stability of enzyme similar to that of the chimeric ADH1(1–258)–ADH2(259–347) that has Cys-277 substituted with Ser.29 However, this chimeric enzyme also has six other substitutions, which may destabilize the enzyme. [Yeast ADH1 and ADH2 differ in 24 amino acid residues, and ADH2 has a t50 of ∼70 °C (see below also).] The disulfide bond may not be important for heat stability, as the cysteine residue is not conserved in medium-chain ADHs, including thermophilic ADHs.
As illustrated in Figure 1B, the tetramer has the symmetry-related AB:AB dimers with subunit A1 most closely juxtaposed with subunit A2, and B1 with B2, but there are also interactions of A1 with B2 and A2 with B1. The interactions between dimers that produce the tetramers are also extensive (Figure 1S of the Supporting Information). Interactions within the CD:CD tetramer are similar, and we identify approximately 4–6 electrostatic interactions, 16–20 hydrogen bonding interactions, and 30–34 hydrophobic interactions in each tetramer.
The tetrameric enzyme dissociates to enzymatically inactive dimers or monomers upon mild heat treatment, and yeast ADH1 is less stable than ADH2.29 Construction of seven different chimeric ADH1/2 enzymes showed that the heat stability (t50 values) increased from ∼35 to ∼59 °C in the enzyme with the G229S, L232V, D236N, and V242I substitutions and additionally increased to ∼72 °C with the M168R and V173A substitutions. Site-specific substitutions should be used to determine which of these residues are responsible. Introduction of several additional electrostatic interactions between subunits in the mesophilic ADH from Clostridium beijerinckii increased themostability (T1/2) only from 63.8 to 69.5 °C and did not yield the stability found for the homologous thermophilic ADH from Thermoanaerobacter brockii of 93.8 °C.30
Structural Similarities among Medium-Chain ADHs
Comparison of dehydrogenase structures provides information about evolutionary and structure–function relationships.23,31 Progressive sequence alignments showed only ∼24% sequence identity between yeast and horse ADHs,32 but detailed molecular modeling showed that the three-dimensional structures were probably very similar, with conservation of space filling in hydrophobic cores, glycine residues, zinc ligands, and catalytic residues in the active site.11 Nevertheless, 14 deletions (or insertions) in various loops, including the apparent deletion of 21 amino acids in yeast ADH corresponding to residues 119–139 in the liver enzyme, indicated that the structures were different in many places. Comparison of the actual three-dimensional structures (PDB entries 4DXH and 4W6Z, A chains) shows that 312 residues of horse and yeast ADHs are in similar secondary structures and that 11 of the 15 changes we identified agree in most respects with the modeling (Table 1). Good models can be useful approximations for homologous structures, but experimental verification is needed. An automated multiple-sequence alignment located 9 of the 15 differences at positions similar to those found by comparison of the three-dimensional structures.32 Almost all of the insertions or deletions are in the loops connecting α-helices and β-sheet strands and thus do not alter the overall folding of the protein (Table 1). Nevertheless, the differences in the horse and yeast enzymes are reflected in some shifts in helices and sheets so that the subunits do not exactly superimpose.
None of the substitutions in yeast ADH appears to generate interactions that would explain why yeast ADH is a tetramer whereas horse ADH is a dimer. The deletion of 21 amino acid residues from the horse enzyme, which leads to differences in residues 114–122 in yeast ADH, was suggested to be responsible for the tetrameric structure of yeast ADH.33 Multiple-sequence alignments of alcohol dehydrogenases were consistent with this possibility, indicating a distant divergence in evolution of dimers and tetramers.32,34 A recent analysis has labeled this particular sequence as the “quaternary structure-determining loop”,35 but the structural basis needs to be established. Superpositioning the horse liver dimer onto the corresponding dimers in the yeast tetramer shows that the extra loop would probably not prevent formation of a tetrameric horse enzyme, as the subunits could fit together, with a few good interactions and some close contacts that could be accommodated by amino acid substitutions and small structural changes. Because the oligomeric state and the length of the residues in the loop region are not strictly correlated,35,36 it would be interesting to explore the interconversion of dimeric and tetrameric ADHs by protein engineering and to create active yeast ADH dimers and liver ADH tetramers. We do not yet understand the evolutionary basis or significance for the formation of the oligomeric enzymes.
The medium-chain ADHs are homologous, and we identified the common core elements among 13 three-dimensional structures of diverse dimeric and tetrameric ADHs (Table 2), some with <25% sequence identity (Table 3S of the Supporting Information). The common core has 248 amino acid residues in structurally similar elements (Table 4S of the Supporting Information). Although the structures are similar, the enzymes differ in substrate specificity and catalytic activity.
Table 2. Common Structural Elements in Horse and Yeast ADHs Interrupted by Insertions or Deletionsa.
| similarities |
differences |
|||
|---|---|---|---|---|
| horse | yeast | horse | yeast | location of change |
| 1–6 | 1–3 | amino terminal | ||
| 7–57 | 4–54 | 58 | 55–57 | substrate binding site |
| 59–94 | 58–93 | 95 | 94, 95 | start of structural Zn loop |
| 96–113 | 96–113 | 114–143 | 114–122 | excursionary loop |
| 144–183 | 123–162 | 184, 185 | 163 | connect αA, βA |
| 186–199 | 164–177 | 200 | 178, 179 | connect βA, αB |
| 201–216 | 180–195 | 217, 218 | 196 | connect αB, βB |
| 219–247 | 197–225 | 248, 249 | 226 | connect βC, αCD |
| 250–283 | 227–260 | 284, 285 | 261 | connect αE, βE |
| 286–296 | 262–272 | 297, 298 | 273 | connect βE, βS |
| 299–309 | 274–284 | 310 | 285, 286 | connect βS, βF |
| 311–319 | 287–295 | 320–323 | 296 | connect βF, α3 |
| 324–339 | 297–312 | 340–347 | 313–317 | connect α3, βI:5 |
| 348–366 | 318–336 | 367 | 337–338 | connect α4, βI:6 |
| 368–374 | 339–345 | 346, 347 | carboxyl terminal | |
The structures of the ternary complexes of yeast (PDB entry 4W6Z) and horse liver (PDB entry 4DXH) with NAD and 2,2,2-trifluoroethanol, A chains, were superimposed with O, using 16 common structural elements with 248 residues (Table 4S of the Supporting Information). Inspection of the structures identified the similar structural elements (helices, β-strands, and loops), as described previously,2 and the sites of insertions and deletions. Differences of ∼3 Å in Cα positions were tolerated, as there are shifts in the structural elements, with retention of the conformation. PDBeFold on the EMBL-EBI server aligned 319 residues in 21 structural elements with an rmsd of 2.3 Å, but the structure differed at 11 positions because of shifts of a residue in the alignment.
Zinc Content and Coordination
Each subunit of yeast ADH1 in the crystals contains a “catalytic” zinc and a “structural” zinc, with no known functional role. Determination of the number of zinc atoms in yeast ADH1 has produced differing results (see the Supporting Information), but the crystallography clearly shows that two zincs per subunit can be present. The coordination of these zincs is tetrahedral, and the distances are typical for ADHs; however, the catalytic zincs in subunits A and C ligate TFE, with Glu-67 OE2 in the second sphere, and the zincs in subunits B and D ligate Glu-67 OE2, with TFE in the second sphere (Table 5S of the Supporting Information).
With a closed conformation, one subunit (A or C) in each asymmetric dimer binds NAD and TFE with the catalytic zincs coordinated in the “classical” manner with Cys-43, His-66, Cys-153, and the oxygen of TFE (Figure 2A). This figure also shows that the methylene carbon of TFE would have its pro-R hydrogen directed toward the re-face of the nicotinamide ring. In an open conformation, the other subunit (B or D) coordinates the catalytic zinc in an “alternative” manner, to Cys-43, His-66, Glu-67, and Cys-153 with the oxygen of TFE in the second sphere (Figure 2B). An overlay of the catalytic domains of subunits A and B shows that the zinc coordination is inverted, accompanied by small movements of the zinc ligands, as the zinc moves ∼2.6 Å closer to Glu-67.
Figure 2.

Stereoviews of the two different types of coordination of the catalytic zinc. (A) Classical found in subunits A and C, which has TFE ligated to the zinc, bound NAD, and Glu-67 5 Å from the zinc (gray sphere). (B) “Alternative”, found in subunits B and D, in which TFE interacts with His-66 and not with the zinc, but Glu-67 is ligated to the zinc, and no NAD is bound. The electron density maps are contoured at 1.5σ. These figures were made with the PyMOL Molecular Graphics System, version 1.7.
The classical and alternative coordinations of the catalytic zincs are also found in other ADHs. Of particular interest is the fact that subunits of human ADH3 complexed with NADH (PDB entries 1TEH and 1MC5) or ADP-ribose (PDB entry 2FZE) apparently have both the classical and alternative coordinations, with crystallographic evidence for partial occupancy at two different positions for the zinc separated by 2.3 Å.37 At low resolution, it can appear that the zinc has a bipyramidal coordination.38,39 The structures with human ADH3 can illustrate an intermediate in the mechanism after the coenzyme is bound and before the substrate (e.g., alcohol) binds.40 It is clear that the alternative coordination is common and that the active site zinc coordination is flexible, which leads to a proposal for a mechanism by which zinc-bound water is replaced by an alcohol or aldehyde (see below).
Asymmetry within the Tetrameric Molecule
The asymmetry within the dimeric units of yeast ADH1 raises questions about potential cooperativity in the catalytic mechanism of the molecule, in particular, “half-of-the-sites” reactivity. However, the dimers are the crystallographic asymmetric units, and the subunits can be different because of the crystal lattice contacts. The horse liver ADH1E holoenzyme3 and the cod liver ADH125 also have a dimer as the asymmetric unit, and both subunits bind coenzyme; however, the subunits in the cod enzyme have different extents of closure of the cleft between the coenzyme and catalytic domains. The yeast cinnamyl alcohol dehydrogenase (monoclinic form) also has an asymmetric dimer, and nucleotide is bound to only one subunit.36 If the asymmetry in the crystal of yeast ADH reflects a preexisting state that is present in solution, biochemical studies could provide information about the stoichiometry or cooperativity of coenzyme binding.
Accurate determination of the number of binding sites for coenzyme is uncertain because it depends upon having “pure” enzyme, probably with the full complement of eight zincs per tetramer and sulfhydryl groups in the proper state of oxidation, and knowing the enzyme concentration and the molecular weight of the tetramer (see the Supporting Information). Various studies have given results that range from 2.0 to 3.6 NADH molecules binding per tetramer. In numerous preparations of recombinant yeast ADH1 in our laboratory, we used titration with NAD+ in the presence of 10 or 100 mM pyrazole to determine the concentration of active sites for calculation of turnover numbers. The NAD+–pyrazole complex forms tightly (Kd ∼ 1 μM, too tight to obtain a good value for Kd with micromolar enzyme concentrations) and absorbs light with maxima in the difference spectrum at 285 and 293 nm, with a difference extinction coefficient at 293 nm of ∼12000 M–1 cm–1, so that the concentration of active sites is readily estimated for solutions of ∼1 mg/mL enzyme in split cuvettes with a path length of 0.435 cm.41−43 (A structure of the horse liver enzyme complexed with NAD+ and pyrazole shows a partial covalent bond between C4N of the nicotinamide ring and a N of pyrazole, with the other N of pyrazole ligated to the catalytic zinc; the difference absorbance maximum is relatively flat at 284–296 nm.44) With many batches of purified yeast ADH, we found concentrations of active sites that averaged 72% (range of 50–90%) of the concentration of protein subunits calculated from the ε280 of 1.26 cm–1 mg–1 mL. We suggest that such titrations establish that the enzyme can bind one coenzyme per subunit, but heterogeneity in the enzyme preparations can decrease the observed capacity. Such titrations, however, do not eliminate the possibility that binding of coenzyme is cooperative. Numerous studies show that enzyme activity with ethanol and acetaldehyde also fits Michaelis–Menten kinetics, but such studies do not define the number of sites that are active at any moment. Radiation inactivation studies suggest that each monomer is active.45
Because yeast ADH1 appears to be able to bind one NAD per subunit, can the asymmetry in the crystal structure be explained by the preparation of the crystals? It is relevant that the crystals were soaked for several days and shipped in a mother liquor that contained no added NAD+. The dissociation constant for NAD+ is 920 μM, and the Ki for TFE is 2.8 mM; therefore, most (96% by estimation for an ordered mechanism) of the enzyme should be in the ternary complex in solution during the crystallization.8 However, subsequent soaking of the crystal in buffer without coenzyme could allow most of the coenzyme to dissociate. Nevertheless, if crystal lattice contacts maintained the subunit in the closed conformation, coenzyme dissociation should be prevented (as observed in subunits A and C), and if the crystal lattice contacts held one subunit in the open conformation, the affinity for coenzyme could be greatly diminished. For instance, with horse liver alcohol dehydrogenase, mutations that seem to “lock” the subunits “open” increase the Kd for NAD+ from 27 to 1100 μM (and the Kd for NADH from 0.50 to 320 μM), and the position of the nicotinamide riboside is not clearly defined in the structure.46 Examination of the crystal lattice contacts for yeast ADH1 suggests that the contacts around subunits A and C are somewhat different than those around the B and D subunits, consistent with the asymmetry, and it appears that coenzyme could dissociate from the open B and D subunits. Although the asymmetry in the structure of yeast ADH1 could result from inherent structural differences in solution, there is no evidence of “negative” cooperativity in the binding of coenzymes or the kinetics of catalysis.8,47−50 It seems most likely that the enzyme crystallized in the asymmetric form and that coenzyme dissociated from the subunits in the open conformation when the crystals were soaked without coenzyme. In any case, we believe that the different structures of the subunits represent energetically accessible states that fortuitously provide significant information for the catalytic mechanism. The differences in energy between the open and closed apoenzyme or enzyme–NAD+ complexes of horse liver ADH are calculated to be relatively small,28,51 and this may be true for yeast ADH.
Active Site in Subunits A and C
Coenzyme and the substrate analogue, TFE, bind to one of the two subunits in each dimer, in what appears to be a mimic of a Michaelis complex (Figure 3). The structure is very similar to that found in the holoenzyme complex of horse liver ADH (PDB entry 4DXH).52 The TFE alcohol oxygen is ligated to the zinc, and the methylene carbon is ∼3.7 Å from C4N of the coenzyme ring with the pro-R hydrogen directed toward the re face of the nicotinamide ring as expected for the known stereochemistry.53 (The distance was determined at 1.12 Å resolution in the horse liver enzyme to be 3.44 ± 0.02 Å.54) The oxygen of TFE is hydrogen-bonded to OG1 of Thr-45, which is in turn hydrogen-bonded to O2′ of the nicotinamide ribose. This constitutes the inner part of the proposed proton relay system that links the buried alcohol to solvent, as discovered in the horse liver enzyme.52,55 In contrast to the structures of the horse liver enzyme, where imidazole NE2 of His-51 interacts with the O2′ ribose hydroxyl group and could act directly as a base, imidazole NE2 of His-48 in yeast ADH interacts with the O3′ hydroxyl group of the nicotinamide ribose, while ND1 interacts with OD1 of Asp-53. This conformation puts the imidazole CD2 closest to the O2′ ribose hydroxyl group, but flipping the imidazole group would place ND1 in a hydrogen bond with the O2′ hydroxyl and allow His-48 to act as a base. Alternatively, unhindered rotation about the χ1 angle of His-48 could place NE2 in a hydrogen bond with the O2′ hydroxyl, in position to act as a base, at 2.6 Å.
Figure 3.
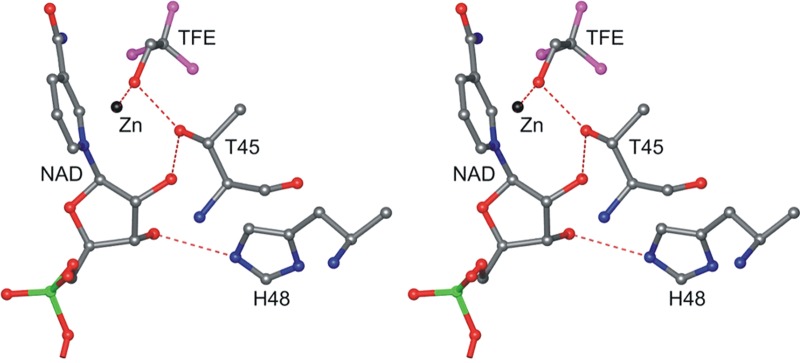
Active site in subunit A showing the proton relay system, including the alcohol ligated to the catalytic zinc. His-48 forms a hydrogen bond with nicotinamide ribose O3′ but can readily swing to form a hydrogen bond with ribose O2′ and complete the proton relay system from the bound alcohol and His-48. This figure was made with the Molray web interface in Uppsala, Sweden.117
Modification of approximately one histidine per subunit with diethyl pyrocarbonate inactivates the enzyme, with a rate constant increasing with pH and a pK value of 7.1, apparently because a histidine is involved in ternary complex formation rather than binding of coenzyme.56−58 The H48Q substitution decreases the catalytic efficiency for oxidation of ethanol by 11-fold and alters the pH dependence.59,60 The change in activity is consistent with a role for His-48 in catalysis, similar to that found for the horse liver enzyme with the H51Q substitution, which affects both coenzyme binding and subsequent catalytic reactions.61,62
The two cysteines at the active site that chelate the catalytic zinc, Cys-43 and Cys-153, are chemically modified by different reagents with loss of activity.7,63 The structure shows that the modifications would disrupt binding of substrates and affect ternary complex formation with coenzymes and substrate analogues.
Coenzyme Binding
The coenzyme is bound in the extended conformation typical of ADHs. The pyrophosphate oxygen atoms interact with several NH groups: O1A interacts with Gly-181 N, O2A with His-44 ND1, O1N with Leu-182 N, and O2N with Arg-340 NH1. O1A also interacts with Gly-339 O and Arg-340 NH1 via a water. O1N also interacts with a water that interacts with Ala-245 O, Gly-177 O, and Gly-183 N. These interactions are similar to those found for the horse liver structures.52,64 Water molecules in the coenzyme binding interface are also found in many dinucleotide binding domains and may be important for specificity and modulating affinity.65
The H44R substitution in yeast ADH1 decreased the dissociation constants for NAD+ and NADH by 2–4-fold and decreased turnover numbers by 4–6-fold.18 Although these effects are small, they are physiologically significant because the decreased rate of reduction of acetaldehyde apparently leads to a more reduced intracellular state during fermentation and protects the yeast against toxicity due to oxidation of allyl alcohol to acrolein.66 Aerobic growth of yeast in the presence of allyl alcohol is inversely correlated with the catalytic efficiency of six different ADH mutants, whereas anaerobic growth in the absence of allyl alcohol is positively correlated with the catalytic efficiency.
As compared to horse liver ADH1E, yeast ADH1 has 40–100-fold faster turnover and 30–60-fold weaker binding of coenzymes, and it is of interest to identify the responsible structural features (in addition to His-44). Enzymes binding NAD or FAD have the signature sequence GXGXXG within the ADP binding βαβ-fold that connects the first β-strand to the first α-helix and makes a tight turn to accommodate the coenzyme.67,68 The sequence in horse liver ADH consists of residues 199–204, GLGGVG, whereas in yeast ADH1 it consists of residues 177–183, GAAGGLG. Converting the Ala-Ala sequence in yeast ADH1 to Leu with the A178Deletion:A179L substitution (AA:L) decreased by 5–23-fold the affinity for NAD+ (Kia) and the turnover number and catalytic efficiency for ethanol oxidation.69 Comparison of the yeast and horse ADH structures suggests that the AA:L substitution should have been readily accommodated, which perhaps fits with the small changes in kinetics. The GXGXXG motif sometimes has Ala instead of the final Gly. In yeast ADH1, the G183A substitution decreased the affinity for coenzymes and adenosine nucleotides, and turnover numbers and catalytic efficiencies by 10–400-fold.69 Addition of the methyl group (with G183A) would create steric hindrance with Gly-177 and Ala-178 that would distort the structure and affect activity.
At the end of the second β-strand of the βαβ-fold in NAD-dependent ADHs is usually a conserved aspartic acid residue, Asp-201 in yeast ADH (Asp-223 in horse), which forms hydrogen bonds with O2′ and O3′ of the adenosine ribose and interferes with binding of NADP. Lys-206 (Lys-228 in horse ADH) also interacts with O3′ of the adenosine ribose. The critical role of Asp-201 was demonstrated with the D201G substitution, which eliminates the specificity for NAD+, and allows the enzyme to use NADP+ about as well as NAD+; however, the affinity for NAD+ decreases by ∼15-fold, and the turnover number for ethanol oxidation decreases by ∼8-fold.70 The conserved aspartate is replaced with a neutral residue (Gly, Ala, Val, Ser, Thr, Asn, and Leu), and one of the next two residues is usually an Arg in NADP-dependent ADHs. In an attempt to improve the catalytic activity of yeast ADH with NADP, the G203R and D201G:G203R enzymes were created. The G203R enzyme had kinetic constants quite similar to those of the wild-type enzyme, whereas the kinetic constants for the D201G:G203R enzyme with NAD+ or NADP+ were very similar to those for the D201G enzyme.69 Apparently, the introduced Arg-203 is exposed to solvent and does not swing into place to interact with the 2′-phosphate of NADP.
The adenine ring of the NAD+ bound to yeast ADH is sandwiched between Phe-221 on one side and Ser-246, Ser-248, and Ala-251 on the other side, as compared to the more hydrophobic residues in horse liver ADH (Phe-198, Ile-224, and Ile-269). [NAD and 8-I-NAD should bind similarly, as the 8-iodo substituent of 8-I-NAD is exposed to solvent, but close (4.1–4.2 Å) to CB of Val-247.] Attempting to increase the affinity of yeast ADH for coenzymes, we introduced the S176F, G202I, and S246I substitutions, which could have increased the hydrophobic character near the adenine ring, but for all three enzymes, the affinity for coenzymes decreased!69 The kinetics of the G202I enzyme were similar to those of wild-type ADH1; however, turnover numbers for the S176F enzyme decreased ∼10-fold, and turnover numbers for the S246I enzyme decreased ∼350-fold. Inspection of the yeast ADH1 structure suggests that Ile-202 might interact with Phe-221 (in contact with the adenine ring) but not generate new interactions with the adenine ring. Phe-176 would apparently fit into the adenine binding pocket if Tyr-258 could swing out of its position, so there might be some small structural rearrangements that account for the relatively large changes in kinetics. The S246I change is the most enigmatic because it would seem that Ile-246 could interact well with the adenine ring, but the S246I enzyme exhibited very large decreases in catalytic efficiency. For comparison, I224G (Gly-202 in yeast) and I269S (Ser-246 in yeast) substitutions in horse liver ADH decreased the affinity for both coenzymes 60- and 350-fold, respectively, and increased the turnover number for ethanol by 7- and 26-fold, respectively, but did not change the catalytic efficiency for ethanol oxidation or acetaldehyde reduction.71 The increases in turnover numbers are due to faster release of NADH, which is rate-limiting for the horse enzyme. It is satisfying that the horse I224G ADH has binding constants for coenzymes that are similar to those for wild-type yeast ADH, but the reverse G202I substitution in yeast ADH also has dissociation constants (lower affinity) larger (5–9-fold) than those of wild-type yeast ADH.69 Comparing the results for the mutagenesis studies on the yeast and liver enzymes suggests that it is easier to diminish catalytic activity than to enhance it. More extensive, and multiple, substitutions that are designed on the known structures may more successfully explain the differences in activity of the yeast and horse ADHs.
Substrate Binding Site
The substrate binding site is illustrated in Figure 4. Several large hydrophobic residues, Trp-54, Trp-92, Met-270, and Tyr-294, produce a cavity that accommodates ethanol as the best substrate.6 Longer, branched, or secondary alcohols are poorer substrates for ADH1.72,73 The catalytic efficiencies (V/Km substrate) of the three recombinant ADHs from S. cerevisiae on primary alcohols decrease with chain length, but the M270L substitution in ADH1 (yeast ADH numbering, Val-294 in horse ADH) increases activity on pentanol and hexanol by 10-fold, without affecting activity on ethanol or propanol, and reproduces the pattern of specificity found in ADH2 and ADH3, which have Leu-270.8 However, ADH2 has 8-fold higher activity than ADH1 on ethanol, which must be due to substitutions of residues that are not in the active site and probably affect dynamics of catalysis. Early mutagenesis studies showed surprisingly modest effects (∼5-fold) of T45S, W92F, W92A, T45S:W92F, and T45S:W92A substitutions on specificities of oxidation of the series of primary alcohols from ethanol to hexanol.74 Subsequent studies showed that the single T45S and W54M substitutions and the triple T45S:W54M:W92A (to mimic human75 and monkey ADH1A, αα-isoenzyme) substitution decrease activity on primary alcohols relative to that of wild-type ADH1, and activity decreases as chain length increases.72 In contrast, the W92A and T45S:W92A substitutions decrease activity on ethanol by ∼400-fold but increase activity by 3–9-fold on hexanol (an ∼1000-fold inversion of activity), with a pattern of activity that resembles that observed with monkey ADH1A.72 The W92A substitution also confers weak activity on cyclohexanol (no detectable activity with wild-type yeast ADH1), but the activity is less than 1/105 of that of monkey ADH1A. Increasing the size of the binding pocket with these several substitutions and the W54L substitution also increases activity on branched-chain alcohols.76
Figure 4.
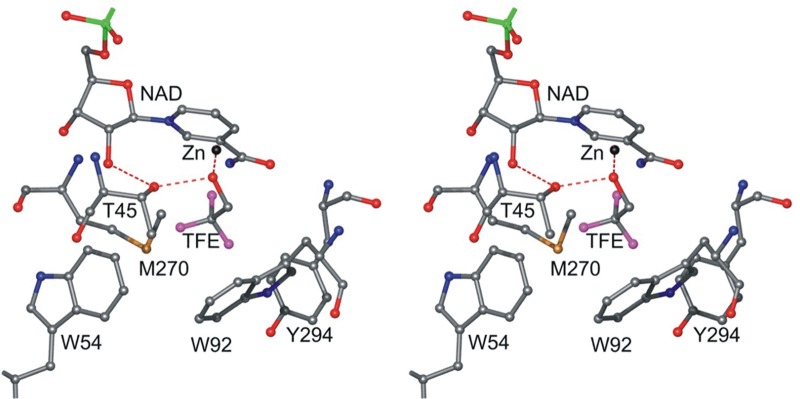
Substrate binding pocket, in subunits A and C. This figure was made with the Molray web interface in Uppsala, Sweden.117
Engineering the substrate specificity of yeast ADH based on a model constructed from the horse liver enzyme structure gave some patterns of activity that were expected, but changes in catalytic efficiencies are difficult to predict. Comparison of the yeast and horse enzyme active sites suggests that this is due in part to the insertions or deletions (Trp-54 and Tyr-119) near the active site and to differences in rotamers (Trp-92), the peptide backbone, and the particular amino acid residues at and near the active site.
Yeast ADH1 is highly specific for transfer of the (4R)-nicotinamide hydrogen of NADH to aldehydes and the pro-R hydrogen from ethanol or octanol to NAD+.77−79 However, it has weak activity (1/800 of that on ethanol) on 2-propanol, which indicates that the enzyme can accommodate (poorly) a methyl substituent on the oxidizable carbon.72 ADH1 has highly preferential activity on (S)-2-butanol as compared to (R)-2-butanol, and it appears that the methyl group of (S)-2-butanol must be accommodated near Trp-92.72,80 Yeast ADH does not measurably oxidize secondary alcohols with both substituents larger than a methyl group, so that there are limits to the flexibility of the active site.81
Mechanistic Implications of Asymmetry and Alternative Coordination of Catalytic Zinc
The two different conformational states of the subunits can represent two relevant structures in the mechanism, the apoenzyme and the ternary complex. The kinetic mechanism for the yeast enzyme acting on ethanol appears to be ordered (or preferred ordered) in which coenzyme binds to free apoenzyme subunits with the open conformation and then isomerizes to form the closed conformation.8,82 The isomerization step has been described for the horse liver enzyme.83,84 Transient kinetic evidence is lacking for an enzyme–coenzyme isomerization for the yeast enzyme, but effects of pressure on V/K for benzyl alcohol oxidation led to the suggestion that the E–NAD+ ↔ E*–NAD+ equilibrium constant is 75 ± 13.85 Hydrogen–deuterium exchange studies also suggest that binding of NAD+ results in a conformational change.86
The next step in the mechanism, the binding of alcohol to the enzyme–NAD+ complex, may occur by direct displacement of Glu-67 by the alcohol or by a double displacement of a water that could bind to the zinc in the enzyme–NAD+ complex. The homologous E. coli enzyme complexed with NAD has water bound to the catalytic zinc.26 The mechanism for ligand exchange in ADHs is controversial. The observation of the alternative zinc coordination (Figure 2B) and the many examples of such coordination in other ADHs (Table 6S of the Supporting Information) are significant because they indicate that the zinc coordination can change during catalysis and can lead to a reasonable mechanism for the exchange of ligands. Computational studies suggest that the carboxyl group of Glu-68 in liver ADH (Glu-67 in yeast ADH) could transiently displace a zinc-bound water and then allow the alcohol to bind by displacing the carboxyl group.87 This ligand exchange mechanism is more energetically feasible than associative (tight, intermediate five coordination) or dissociative (intermediate three coordination) mechanisms. The exchange would occur as the zinc moved closer to Glu-67, to form a loose, transient intermediate, trigonal–bipyramidal pentacoordinate zinc with the water and glutamate in axial positions.40 The configuration of the zinc is essentially inverted, and then alcohol displaces the Glu to restore the configuration.
The alternative zinc locations in human ADH3 structures provide evidence of an exchange mechanism.37−40 Moreover, the substitution of the highly conserved Glu32 with Leu in ADH3 decreased catalytic efficiency by 3000-fold for oxidation of (S)-hydroxymethylglutathione, without large changes in the protein structure or binding of the coenzyme.37 Likewise, for yeast ADH1, the E67Q substitution decreased the catalytic efficiency for ethanol oxidation or acetaldehyde reduction by ∼100-fold and shifted pH dependencies.88 The results from the site-directed substitutions are consistent with the proposed mechanism, but electrostatic effects and local, small structural changes could also affect the dynamics of catalysis.
Although a structure for the binary yeast ADH1–coenzyme complex is not available, we propose, by analogy to human ADH3 structures (PDB entries 1TEH and 1MC5), that the binding of Glu-67 to the zinc may be weakened when the coenzyme binds because Glu-67 interacts with Arg-340, which in turn interacts with a phosphate oxygen of the coenzyme. Such an interaction is common in most tetrameric ADHs complexed with NAD(P)(H). In apoenzyme subunits (B and D) of yeast ADH1, Glu-67 is ligated to the zinc and does not interact with Arg-340, but in the holoenzyme subunits (A and C), Glu-67 interacts with Arg-340. In the proposed binary enzyme–coenzyme complex, ligand exchange of water, alcohol, and aldehyde would be facilitated by a modulated interaction of Glu-67 with the zinc.
The binding of alcohol to the zinc to form ternary complexes (of horse liver ADH) appears to require deprotonation of the zinc-bound water in the enzyme–NAD+ complex before ligands bind to the zinc.84 This deprotonation appears to be facilitated by a proton relay system (see Figure 3 for yeast ADH) in the hydrogen-bonded system that includes the zinc-bound water, the hydroxyl group of Ser-48, the 2′-hydroxyl group of the nicotinamide ribose, and the imidazole group of His-51.55,62 Dissociation of hydroxide from zinc is likely to be less favorable than release of water, but displacement by the glutamate carboxylate could be energetically favorable. After a neutral alcohol binds to the zinc and displaces the glutamate, the alcohol would be deprotonated via the proton relay system to form the zinc alkoxide that is the ground state for the hydride transfer step.28,46,83 The dissociation of aldehyde and binding of water could also occur by an inversion of the zinc coordination, which is illustrated by the complexes of human ADH3 with NADH.
An alternative mechanism for ligand exchange could involve a transient pentacoordinated zinc.89 Pentacoordinate zinc was observed in the binary complex of horse liver ADH with 1,10-phenanthroline, indicating some flexibility of the zinc coordination.90 However, the first structures of ternary complexes (coenzyme and substrate analogue) showed that no water remained bound to the zinc.3,55 Nevertheless, such a pentacoordinated zinc could be a transient species during the replacement of the water with the alcohol. A pentacoordinated zinc has not been observed in any ternary complexes of ADHs, except for the complex of sorbitol dehydrogenase with a chelating (“transition state analogue”) inhibitor that mimics the substrate sorbitol (PDB entry 1P16).91
Another version of a mechanism involving a pentacoordinate zinc was proposed to account for alternative waters bound to the catalytic zinc and to C6N of the nicotinamide ring in binary complexes with NADH, but this mechanism is not supported by structures of ternary complexes.92,93
Time-resolved X-ray absorption studies of the transient reaction of T. brockii ADH with NADP+ and the substrate 2-propanol showed that two different pentacoordinate species formed at 5 and 15 ms (during the “burst” phase) but then perhaps reverted (to tetracoordination) during the steady state phase.94 These results could fit with the X-ray structure of the complex with NADPH, where the zinc is essentially bipyramidal with the oxygens of Ser-39 and Glu-60 ∼3.7 Å from the zinc, and 2-propanol displaces the Ser oxygen. A mechanism that involves displacement of Glu-60 with water and then binding of alcohol to form the reactive complex was proposed, but the water was not assigned a catalytic role. The proposed mechanism seems to be more elaborate than chemically necessary. Nevertheless, evidence of transient species is required to describe the exchange mechanism. Experimental support for a catalytic role of Glu-60 in this enzyme is limited as the substitution with Ala or Asp decreases the catalytic efficiency by only 4- or 8-fold, respectively.95 The mechanisms for ligand exchange may differ in the enzymes with two carboxylates bound to the zinc instead of two cysteine sulfhydryl groups.
Transition State Studies
The transition state of yeast ADH has been extensively characterized. Structure–activity correlations for para-substituted benzyl alcohols and benzaldehydes suggested a transition state with little change in charge relative to that of the alcohol, whereas α-secondary isotope effects suggested a transition state that resembled aldehyde.96−99 Other studies provided values for intrinsic isotope effects.100−103 In a landmark study, yeast ADH was used to show that the transfer of hydrogen from benzyl alcohol to NAD+ occurs with quantum mechanical tunneling.104 Subsequent studies with horse liver and bacterial ADHs show that the tunneling, along with coupled motions, is a general phenomenon that can explain the results with yeast ADH.105−109 Pressure diminishes substrate and solvent isotope effects, perhaps by affecting a “mechanical” component involving vibrational dynamics.110−113 These early studies used benzyl alcohol as a substrate (where hydrogen transfer is rate-limiting for catalytic turnover because of small “commitment factors”) and commercial yeast ADH, which contains mostly ADH1, but which has ∼40000-fold more activity on ethanol than on benzyl alcohol.73 In contrast, yeast ADH2 is ∼100-fold more active than ADH1 on benzyl alcohol; fortunately, the structure–activity relationships for ADH1 and ADH2 with the substituted benzyl alcohols and benzaldehydes are similar.73 Subsequently, the secondary and equilibrium isotope effects for reduction of substituted benzaldehydes were determined for yeast ADH2, and a model that describes the “tunneling ready state”, explains the isotope and substituent effects, and fits the three-dimensional structure was created.114 Interestingly, the distance between the methylene carbon of the alcohol and C4N of the nicotinamide ring was calculated to be 3.2 Å, which is close to the distance of 3.8 Å for the structure with TFE. Benzyl alcohol can fit with some close contacts into the active site, in particular with the M270L substitution,73 and local equilibrium motions could bring the donor–acceptor distance to 3.2 Å. The structure of yeast ADH1 provides a framework for further calculations to explain the catalysis of hydride transfer.
Acknowledgments
We thank Kristine B. Berst for protein chemistry and The University of Iowa Molecular Analysis Facility for amino acid and molecular mass analyses. We also appreciated the numerous efforts by Darla Ann Kratzer, Susan Souhrada, Andrew D. Hershey, Fan Fan, Doo-Hong Park, Suresh Pal, Henry A. Charlier, Jr., and Mark Hermes to prepare crystals of yeast ADH. We thank Lokesh Gakhar in The University of Iowa Crystallography Facility for advice. We also thank the staff and support at beamline BW7A at the EMBL/DESY Hamburg synchrotron. S.R. collected the data while working in the Department of Molecular Biology at the Swedish University of Agricultural Sciences (Uppsala, Sweden) and determined the structure at The University of Iowa.
Glossary
Abbreviations
- ADH
alcohol dehydrogenase
- TFE
2,2,2-trifluoroethanol (ETF in the PDB entry, with the methylene carbon labeled “2”)
- rmsd
root-mean-square deviation
- amino acid substitutions, e.g., D236N
Asp-236 substituted with Asn.
Supporting Information Available
Additional figures and tables illustrating the packing interactions of yeast ADH, crystallographic and chemical evidence of a disulfide bond, structural homology of 13 diverse medium-chain ADHs, zinc content and coordination in yeast ADH and other ADHs, and a summary of the methods and results used to determine protein concentrations, molecular weights, and stoichiometries of coenzyme binding sites. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The X-ray coordinates and structure factors have been deposited in the Protein Data Bank as entry 4W6Z (making the earlier deposition of 2HCY obsolete). Coordinates for the biological tetramers can be generated by applying the rotation matrix found in the header of the PDB file.
Author Present Address
† S.B.R.: Lawson State Community College, Birmingham, AL 35216.
Author Present Address
‡ S.R.: Institute for Stem Cell Biology and Regenerative Medicine (inSTEM), National Center for Biological Sciences, GKVK Post, Bellary Road, Bangalore 560065, India.
This work was supported by NSF grant MCB 9118657 and NIH grant AA00279.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jörnvall H.; Hedlund J.; Bergman T.; Kallberg Y.; Cederlund E.; Persson B. (2013) Origin and evolution of medium chain alcohol dehydrogenases. Chem.-Biol. Interact. 202, 91–96. [DOI] [PubMed] [Google Scholar]
- Eklund H.; Nordström B.; Zeppezauer E.; Söderlund G.; Ohlsson I.; Boiwe T.; Söderberg B. O.; Tapia O.; Brändén C.-I.; Åkeson Å. (1976) Three-dimensional structure of horse liver alcohol dehydrogenase at 2.4 Å resolution. J. Mol. Biol. 102, 27–59. [DOI] [PubMed] [Google Scholar]
- Eklund H.; Samama J. P.; Wallén L.; Brändén C. I.; Åkeson Å.; Jones T. A. (1981) Structure of a triclinic ternary complex of horse liver alcohol dehydrogenase at 2.9 Å resolution. J. Mol. Biol. 146, 561–587. [DOI] [PubMed] [Google Scholar]
- Eklund H.; Ramaswamy S. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: Three-dimensional structures of MDR alcohol dehydrogenases. Cell. Mol. Life Sci. 65, 3907–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negelein E.; Wulff H.-J. (1937) Diphosphopyridinproteid. Alkohol, Acetaldehyd. Biochem. Z. 293, 351–389. [Google Scholar]
- Sund H., and Theorell H. (1963) Alcohol Dehydrogenases. In The Enzymes, 2nd ed., Vol. 7, pp 25–83, Academic Press, New York. [Google Scholar]
- Brändén C.-I., Jörnvall H., Eklund H., and Furugren B. (1975) Alcohol Dehydrogenases. In The Enzymes, 3rd Ed., Vol. 11, pp 103–190, Academic Press, New York. [Google Scholar]
- Ganzhorn A. J.; Green D. W.; Hershey A. D.; Gould R. M.; Plapp B. V. (1987) Kinetic characterization of yeast alcohol dehydrogenases. Amino acid residue 294 and substrate specificity. J. Biol. Chem. 262, 3754–3761. [PubMed] [Google Scholar]
- Jörnvall H. (1977) The primary structure of yeast alcohol dehydrogenase. Eur. J. Biochem. 72, 425–442. [DOI] [PubMed] [Google Scholar]
- Bennetzen J. L.; Hall B. D. (1982) The primary structure of the Saccharomyces cerevisiae gene for alcohol dehydrogenase. J. Biol. Chem. 257, 3018–3025. [PubMed] [Google Scholar]
- Jörnvall H.; Eklund H.; Brändén C.-I. (1978) Subunit conformation of yeast alcohol dehydrogenase. J. Biol. Chem. 253, 8414–8419. [PubMed] [Google Scholar]
- Künkel W.; Hädrich H.; Damaschun H.; Damaschun G. (1980) Alkoholdehydrogenase (ADH) in Hefezellen. III. Strukturuntersuchungen an zellulären ADH-Kristallen von Saccharomyces carlsbergensis mit Hilfe der Elektronenmikroskopie und Röntgenkleinwinkelstreuung. Mikroskopie 36, 81–92. [PubMed] [Google Scholar]
- Lange R. H. (1981) Alkoholdehydrogenase (ADH) aus Hefe. Eine Krystallographische Interpretation. Mikroskopie 38, 78–80. [PubMed] [Google Scholar]
- Ramaswamy S.; Kratzer D. A.; Hershey A. D.; Rogers P. H.; Arnone A.; Eklund H.; Plapp B. V. (1994) Crystallization and preliminary crystallographic studies of Saccharomyces cerevisiae alcohol dehydrogenase I. J. Mol. Biol. 235, 777–779. [DOI] [PubMed] [Google Scholar]
- Kim K. J.; Howard A. J. (2002) Crystallization and preliminary X-ray diffraction analysis of the trigonal crystal form of Saccharomyces cerevisiae alcohol dehydrogenase I: Evidence for the existence of Zn ions in the crystal. Acta Crystallogr. D58, 1332–1334. [DOI] [PubMed] [Google Scholar]
- Levin I.; Meiri G.; Peretz M.; Burstein Y.; Frolow F. (2004) The ternary complex of Pseudomonas aeruginosa alcohol dehydrogenase with NADH and ethylene glycol. Protein Sci. 13, 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson V. M.; Bennetzen J.; Young E. T.; Nasmyth K.; Hall B. D. (1980) Isolation of the structural gene for alcohol dehydrogenase by genetic complementation in yeast. Nature 283, 214–216. [DOI] [PubMed] [Google Scholar]
- Gould R. M.; Plapp B. V. (1990) Substitution of arginine for histidine-47 in the coenzyme binding site of yeast alcohol dehydrogenase I. Biochemistry 29, 5463–5468. [DOI] [PubMed] [Google Scholar]
- Navaza J. (1994) AMoRe: An automated package for molecular replacement. Acta Crystallogr. A50, 157–163. [Google Scholar]
- Jones T. A.; Zou J. Y.; Cowan S. W.; Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47(Part 2), 110–119. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates T. O. (1997) Detecting and overcoming crystal twinning. Methods Enzymol. 276, 344–358. [PubMed] [Google Scholar]
- Rossmann M. G., Liljas A., Brändén C.-I., and Banaszak L. J. (1975) Evolutionary and Structural Relationships among Dehydrogenases. The Enzymes, 3rd ed., Vol. 11, pp 62–102, Academic Press, New York. [Google Scholar]
- Hayward S.; Berendsen H. J. (1998) Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30, 144–154. [PubMed] [Google Scholar]
- Ramaswamy S.; El-Ahmad M.; Danielsson O.; Jörnvall H.; Eklund H. (1996) Crystal structure of cod liver class I alcohol dehydrogenase: Substrate pocket and structurally variable segments. Protein Sci. 5, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A.; el-Ahmad M.; Johansson K.; Shafqat J.; Jörnvall H.; Eklund H.; Ramaswamy S. (2003) Tetrameric NAD-dependent alcohol dehydrogenase. Chem.-Biol. Interact. 143–144, 239–245. [DOI] [PubMed] [Google Scholar]
- Ceccarelli C.; Liang Z. X.; Strickler M.; Prehna G.; Goldstein B. M.; Klinman J. P.; Bahnson B. J. (2004) Crystal structure and amide H/D exchange of binary complexes of alcohol dehydrogenase from Bacillus stearothermophilus: Insight into thermostability and cofactor binding. Biochemistry 43, 5266–5277. [DOI] [PubMed] [Google Scholar]
- Plapp B. V. (2010) Conformational changes and catalysis by alcohol dehydrogenase. Arch. Biochem. Biophys. 493, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bolle X.; Vinals C.; Prozzi D.; Paquet J. Y.; Leplae R.; Depiereux E.; Vandenhaute J.; Feytmans E. (1995) Identification of residues potentially involved in the interactions between subunits in yeast alcohol dehydrogenases. Eur. J. Biochem. 231, 214–219. [DOI] [PubMed] [Google Scholar]
- Bogin O.; Levin I.; Hacham Y.; Tel-Or S.; Peretz M.; Frolow F.; Burstein Y. (2002) Structural basis for the enhanced thermal stability of alcohol dehydrogenase mutants from the mesophilic bacterium Clostridium beijerinckii: Contribution of salt bridging. Protein Sci. 11, 2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plapp B. V. (1982) Origins of Structure and Function of Proteins. In Perspectives in Evolution (Milkman R., Ed.) pp 129–147, Sinauer Associates, Sunderland, MA. [Google Scholar]
- Sun H. W.; Plapp B. V. (1992) Progressive sequence alignment and molecular evolution of the Zn-containing alcohol-dehydrogenase family. J. Mol. Evol. 34, 522–535. [DOI] [PubMed] [Google Scholar]
- Jörnvall H. (1977) Differences between alcohol dehydrogenases. Structural properties and evolutionary aspects. Eur. J. Biochem. 72, 443–452. [DOI] [PubMed] [Google Scholar]
- Nordling E.; Persson B.; Jörnvall H. (2002) Differential multiplicity of MDR alcohol dehydrogenases: Enzyme genes in the human genome versus those in organisms initially studied. Cell. Mol. Life Sci. 59, 1070–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll M.; Pleiss J. (2008) The medium-chain dehydrogenase/reductase engineering database: A systematic analysis of a diverse protein family to understand sequence-structure-function relationship. Protein Sci. 17, 1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia E.; Larroy C.; Ochoa W. F.; Parés X.; Fita I.; Biosca J. A. (2004) Apo and holo structures of an NADPH-dependent cinnamyl alcohol dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 341, 1049–1062. [DOI] [PubMed] [Google Scholar]
- Sanghani P. C.; Davis W. I.; Zhai L.; Robinson H. (2006) Structure-function relationships in human glutathione-dependent formaldehyde dehydrogenase. Role of Glu-67 and Arg-368 in the catalytic mechanism. Biochemistry 45, 4819–4830. [DOI] [PubMed] [Google Scholar]
- Yang Z. N.; Bosron W. F.; Hurley T. D. (1997) Structure of human chi chi alcohol dehydrogenase: A glutathione-dependent formaldehyde dehydrogenase. J. Mol. Biol. 265, 330–343. [DOI] [PubMed] [Google Scholar]
- Sanghani P. C.; Bosron W. F.; Hurley T. D. (2002) Human glutathione-dependent formaldehyde dehydrogenase. Structural changes associated with ternary complex formation. Biochemistry 41, 15189–15194. [DOI] [PubMed] [Google Scholar]
- Sanghani P. C.; Robinson H.; Bosron W. F.; Hurley T. D. (2002) Human glutathione-dependent formaldehyde dehydrogenase. Structures of apo, binary, and inhibitory ternary complexes. Biochemistry 41, 10778–10786. [DOI] [PubMed] [Google Scholar]
- Theorell H.; Yonetani T. (1963) Liver alcohol dehydrogenase-DPN-pyrazole complex: A model of a ternary intermediate in the enzyme reaction. Biochem. Z. 338, 537–553. [PubMed] [Google Scholar]
- Twu J. S.; Chin C. C.; Wold F. (1973) Studies on the active-site sulfhydryl groups of yeast alcohol dehydrogenase. Biochemistry 12, 2856–2862. [DOI] [PubMed] [Google Scholar]
- Karlović D.; Amiguet P.; Bonner F. J.; Luisi P. L. (1976) Spectroscopic investigation of binary and ternary coenzyme complexes of yeast alcohol dehydrogenase. Eur. J. Biochem. 66, 277–284. [DOI] [PubMed] [Google Scholar]
- Rubach J. K.; Plapp B. V. (2003) Amino acid residues in the nicotinamide binding site contribute to catalysis by horse liver alcohol dehydrogenase. Biochemistry 42, 2907–2915. [DOI] [PubMed] [Google Scholar]
- Suarez M. D.; Ferguson Miller S. (1987) Yeast and horse liver alcohol dehydrogenases: Potential problems in target size analysis and evidence for a monomer active unit. Biochemistry 26, 3340–3347. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S.; Park D. H.; Plapp B. V. (1999) Substitutions in a flexible loop of horse liver alcohol dehydrogenase hinder the conformational change and unmask hydrogen transfer. Biochemistry 38, 13951–13959. [DOI] [PubMed] [Google Scholar]
- Dickenson C. J.; Dickinson F. M. (1975) A study of the pH- and temperature-dependence of the reactions of yeast alcohol dehydrogenase with ethanol, acetaldehyde and butyraldehyde as substrates. Biochem. J. 147, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson C. J.; Dickinson F. M. (1975) A study of the oxidation of butan-1-ol and propan-2-ol by nicotinamide-adenine dinucleotide catalysed by yeast alcohol dehydrogenase. Biochem. J. 147, 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wratten C. C.; Cleland W. W. (1963) Product inhibition studies on yeast and liver alcohol dehydrogenases. Biochemistry 2, 935–941. [DOI] [PubMed] [Google Scholar]
- Klinman J. P. (1975) Acid-base catalysis in the yeast alcohol dehydrogenase reaction. J. Biol. Chem. 250, 2569–2573. [PubMed] [Google Scholar]
- Colonna-Cesari F.; Perahia D.; Karplus M.; Eklund H.; Brändén C.-I.; Tapia O. (1986) Interdomain motion in liver alcohol dehydrogenase. Structural and energetic analysis of the hinge bending mode. J. Biol. Chem. 261, 15273–15280. [PubMed] [Google Scholar]
- Plapp B. V.; Ramaswamy S. (2012) Atomic-resolution structures of horse liver alcohol dehydrogenase with NAD+ and fluoroalcohols define strained Michaelis complexes. Biochemistry 51, 4035–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher H. F.; Conn E. E.; Vennesland B.; Westheimer F. H. (1953) The enzymatic transfer of hydrogen. I. The reaction catalyzed by alcohol dehydrogenase. J. Biol. Chem. 202, 687–697. [PubMed] [Google Scholar]
- Yahashiri A.; Rubach J. K.; Plapp B. V. (2014) Effects of cavities at the nicotinamide binding site of liver alcohol dehydrogenase on structure, dynamics and catalysis. Biochemistry 53, 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund H.; Plapp B. V.; Samama J. P.; Brändén C.-I. (1982) Binding of substrate in a ternary complex of horse liver alcohol dehydrogenase. J. Biol. Chem. 257, 14349–14358. [PubMed] [Google Scholar]
- Leskovac V.; Pavkov-Peričin D. (1975) Evidence for a histidine and a cysteine residue in the substrate-binding site of yeast alcohol dehydrogenase. Biochem. J. 145, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson C. J.; Dickinson F. M. (1975) The role of an essential histidine residue of yeast alcohol dehydrogenase. Eur. J. Biochem. 52, 595–603. [DOI] [PubMed] [Google Scholar]
- Dickenson C. J.; Dickinson F. M. (1977) A study of the ionic properties of the essential histidine residue of yeast alcohol dehydrogenase in complexes of the enzyme with its coenzymes and substrates. Biochem. J. 161, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould R. M. (1988) Histidines in the Mechanism of Yeast Alcohol Dehydrogenase. Ph.D. Thesis, The University of Iowa, Iowa City, IA. [Google Scholar]
- Plapp B. V.; Ganzhorn A. J.; Gould R. M.; Green D. W.; Jacobi T.; Warth E.; Kratzer D. A. (1991) Catalysis by yeast alcohol dehydrogenase. Adv. Exp. Med. Biol. 284, 241–251. [DOI] [PubMed] [Google Scholar]
- LeBrun L. A.; Plapp B. V. (1999) Control of coenzyme binding to horse liver alcohol dehydrogenase. Biochemistry 38, 12387–12393. [DOI] [PubMed] [Google Scholar]
- LeBrun L. A.; Park D. H.; Ramaswamy S.; Plapp B. V. (2004) Participation of histidine-51 in catalysis by horse liver alcohol dehydrogenase. Biochemistry 43, 3014–3026. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Welsh K. M.; Hogue Angeletti R. (1977) Epoxide inhibition of alcohol dehydrogenases. Identification of modified cysteines in yeast alcohol dehydrogenase and demonstration of reversible and irreversible inhibition of liver alcohol dehydrogenase by styrene oxide. Biochemistry 16, 5521–5527. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S.; Eklund H.; Plapp B. V. (1994) Structures of horse liver alcohol dehydrogenase complexed with NAD+ and substituted benzyl alcohols. Biochemistry 33, 5230–5237. [DOI] [PubMed] [Google Scholar]
- Bottoms C. A.; Smith P. E.; Tanner J. J. (2002) A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 11, 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plapp B. V.; Lee A. T.; Khanna A.; Pryor J. M. (2013) Bradykinetic alcohol dehydrogenases make yeast fitter for growth in the presence of allyl alcohol. Chem.-Biol. Interact. 202, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K.; De Maeyer M. C. H.; Hol W. G. J. (1985) Interaction of pyrophosphate moieties with α-helixes in dinucleotide binding-proteins. Biochemistry 24, 1346–1357. [Google Scholar]
- Wierenga R. K.; Terpstra P.; Hol W. G. J. (1986) Prediction of the occurrence of the ADP-binding β-α-β-fold in proteins, using an amino-acid-sequence fingerprint. J. Mol. Biol. 187, 101–107. [DOI] [PubMed] [Google Scholar]
- Fan F.; Plapp B. V. (1999) Probing the affinity and specificity of yeast alcohol dehydrogenase I for coenzymes. Arch. Biochem. Biophys. 367, 240–249. [DOI] [PubMed] [Google Scholar]
- Fan F.; Lorenzen J. A.; Plapp B. V. (1991) An aspartate residue in yeast alcohol dehydrogenase I determines the specificity for coenzyme. Biochemistry 30, 6397–6401. [DOI] [PubMed] [Google Scholar]
- Fan F.; Plapp B. V. (1995) Substitutions of isoleucine residues at the adenine binding site activate horse liver alcohol dehydrogenase. Biochemistry 34, 4709–4713. [DOI] [PubMed] [Google Scholar]
- Green D. W.; Sun H. W.; Plapp B. V. (1993) Inversion of the substrate specificity of yeast alcohol dehydrogenase. J. Biol. Chem. 268, 7792–7798. [PubMed] [Google Scholar]
- Pal S.; Park D.-H.; Plapp B. V. (2009) Activity of yeast alcohol dehydrogenases on benzyl alcohols and benzaldehydes. Characterization of ADH1 from Saccharomyces carlsbergensis and transition state analysis. Chem.-Biol. Interact. 178, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creaser E. H.; Murali C.; Britt K. A. (1990) Protein engineering of alcohol dehydrogenases: Effects of amino acid changes at positions 93 and 48 of yeast ADH1. Protein Eng. 3, 523–526. [DOI] [PubMed] [Google Scholar]
- Stone C. L.; Li T. K.; Bosron W. F. (1989) Stereospecific oxidation of secondary alcohols by human alcohol dehydrogenases. J. Biol. Chem. 264, 11112–11116. [PubMed] [Google Scholar]
- Weinhold E. G.; Benner S. A. (1995) Engineering yeast alcohol dehydrogenase. Replacing Trp54 by Leu broadens substrate specificity. Protein Eng. 8, 457–461. [DOI] [PubMed] [Google Scholar]
- Popják G. (1970) Stereospecificity of Enzymatic Reactions. In The Enzymes, 3rd ed., Vol. 2, pp 115–157, Academic Press, New York. [Google Scholar]
- Weinhold E. G.; Glasfeld A.; Ellington A. D.; Benner S. A. (1991) Structural determinants of stereospecificity in yeast alcohol dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 88, 8420–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro S.; Arunachalam T.; Caspi E. (1983) Equilibration of 1-octanol with alcohol dehydrogenase. Evidence for horse liver alcohol dehydrogenase responsibility for exchange of the 1-pro-S hydrogen atom. J. Am. Chem. Soc. 105, 1642–1646. [Google Scholar]
- Dickinson F. M.; Dalziel K. (1967) Substrate specificity and stereospecificity of alcohol dehydrogenases. Nature 214, 31–33. [DOI] [PubMed] [Google Scholar]
- Dickinson F. M.; Dalziel K. (1967) The specificities and configurations of ternary complexes of yeast and liver alcohol dehydrogenases. Biochem. J. 104, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson F. M.; Dickenson C. J. (1978) Estimation of rate and dissociation constants involving ternary complexes in reactions catalysed by yeast alcohol dehydrogenase. Biochem. J. 171, 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar V. C.; Plapp B. V. (1990) Rate constants for a mechanism including intermediates in the interconversion of ternary complexes by horse liver alcohol dehydrogenase. Biochemistry 29, 4289–4295. [DOI] [PubMed] [Google Scholar]
- Kovaleva E. G.; Plapp B. V. (2005) Deprotonation of the horse liver alcohol dehydrogenase-NAD+ complex controls formation of the ternary complexes. Biochemistry 44, 12797–12808. [DOI] [PubMed] [Google Scholar]
- Cho Y. K.; Northrop D. B. (1999) Effects of pressure on the kinetics of capture by yeast alcohol dehydrogenase. Biochemistry 38, 7470–7475. [DOI] [PubMed] [Google Scholar]
- DeWeck Z.; Pande J.; Kägi J. H. R. (1987) Interdependence of coenzyme-induced conformational work and binding potential in yeast alcohol and porcine heart lactate dehydrogenases: A hydrogen-deuterium exchange study. Biochemistry 26, 4769–4776. [DOI] [PubMed] [Google Scholar]
- Ryde U. (1995) On the role of Glu-68 in alcohol dehydrogenase. Protein Sci. 4, 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganzhorn A. J.; Plapp B. V. (1988) Carboxyl groups near the active site zinc contribute to catalysis in yeast alcohol dehydrogenase. J. Biol. Chem. 263, 5446–5454. [PubMed] [Google Scholar]
- Dworschack R. T.; Plapp B. V. (1977) pH, isotope, and substituent effects on the interconversion of aromatic substrates catalyzed by hydroxybutyrimidylated liver alcohol dehydrogenase. Biochemistry 16, 2716–2725. [DOI] [PubMed] [Google Scholar]
- Boiwe T.; Brändén C.-I. (1977) X-ray investigation of the binding of 1,10-phenanthroline and imidazole to horse-liver alcohol dehydrogenase. Eur. J. Biochem. 77, 173–179. [DOI] [PubMed] [Google Scholar]
- Pauly T. A.; Ekstrom J. L.; Beebe D. A.; Chrunyk B.; Cunningham D.; Griffor M.; Kamath A.; Lee S. E.; Madura R.; McGuire D.; Subashi T.; Wasilko D.; Watts P.; Mylari B. L.; Oates P. J.; Adams P. D.; Rath V. L. (2003) X-ray crystallographic and kinetic studies of human sorbitol dehydrogenase. Structure 11, 1071–1085. [DOI] [PubMed] [Google Scholar]
- Meijers R.; Morris R. J.; Adolph H. W.; Merli A.; Lamzin V. S.; Cedergren-Zeppezauer E. S. (2001) On the enzymatic activation of NADH. J. Biol. Chem. 276, 9316–9321. [DOI] [PubMed] [Google Scholar]
- Meijers R.; Adolph H. W.; Dauter Z.; Wilson K. S.; Lamzin V. S.; Cedergren-Zeppezauer E. S. (2007) Structural evidence for a ligand coordination switch in liver alcohol dehydrogenase. Biochemistry 46, 5446–5454. [DOI] [PubMed] [Google Scholar]
- Kleifeld O.; Frenkel A.; Martin J. M. L.; Sagi I. (2003) Active site electronic structure and dynamics during metalloenzyme catalysis. Nat. Struct. Biol. 10, 98–103. [DOI] [PubMed] [Google Scholar]
- Kleifeld O.; Shi S. P.; Zarivach R.; Eisenstein M.; Sagi I. (2003) The conserved Glu-60 residue in Thermoanaerobacter brockii alcohol dehydrogenase is not essential for catalysis. Protein Sci. 12, 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P. (1972) The mechanism of enzyme-catalyzed reduced nicotinamide adenine dinucleotide-dependent reductions. Substituent and isotope effects in the yeast alcohol dehydrogenase reaction. J. Biol. Chem. 247, 7977–7987. [PubMed] [Google Scholar]
- Klinman J. P. (1976) Isotope effects and structure-reactivity correlations in the yeast alcohol dehydrogenase reaction. A study of the enzyme-catalyzed oxidation of aromatic alcohols. Biochemistry 15, 2018–2026. [DOI] [PubMed] [Google Scholar]
- Welsh K. M.; Creighton D. J.; Klinman J. P. (1980) Transition-state structure in the yeast alcohol dehydrogenase reaction: The magnitude of solvent and α-secondary hydrogen isotope effects. Biochemistry 19, 2005–2016. [DOI] [PubMed] [Google Scholar]
- Klinman J. P. (1981) Probes of mechanism and transition-state structure in the alcohol dehydrogenase reaction. CRC Crit. Rev. Biochem. 10, 39–78. [DOI] [PubMed] [Google Scholar]
- Cook P. F.; Cleland W. W. (1981) Mechanistic deductions from isotope effects in multireactant enzyme mechanisms. Biochemistry 20, 1790–1796. [DOI] [PubMed] [Google Scholar]
- Cook P. F.; Cleland W. W. (1981) pH variation of isotope effects in enzyme-catalyzed reactions. 1. Isotope- and pH-dependent steps the same. Biochemistry 20, 1797–1805. [DOI] [PubMed] [Google Scholar]
- Cook P. F.; Cleland W. W. (1981) pH variation of isotope effects in enzyme-catalyzed reactions. 2. Isotope-dependent step not pH dependent. Kinetic mechanism of alcohol dehydrogenase. Biochemistry 20, 1805–1816. [DOI] [PubMed] [Google Scholar]
- Scharschmidt M.; Fisher M. A.; Cleland W. W. (1984) Variation of transition-state structure as a function of the nucleotide in reactions catalyzed by dehydrogenases. 1. Liver alcohol dehydrogenase with benzyl alcohol and yeast aldehyde dehydrogenase with benzaldehyde. Biochemistry 23, 5471–5478. [DOI] [PubMed] [Google Scholar]
- Cha Y.; Murray C. J.; Klinman J. P. (1989) Hydrogen tunneling in enzyme reactions. Science 243, 1325–1330. [DOI] [PubMed] [Google Scholar]
- Bahnson B. J.; Park D.-H.; Kim K.; Plapp B. V.; Klinman J. P. (1993) Unmasking of hydrogen tunneling in the horse liver alcohol-dehydrogenase reaction by site-directed mutagenesis. Biochemistry 32, 5503–5507. [DOI] [PubMed] [Google Scholar]
- Kohen A.; Cannio R.; Bartolucci S.; Klinman J. P. (1999) Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 399, 496–499. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Meadows C. W.; Dong M.; Bahnson B. J.; Klinman J. P. (2012) Active site hydrophobic residues impact hydrogen tunneling differently in a thermophilic alcohol dehydrogenase at optimal versus nonoptimal temperatures. Biochemistry 51, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. (2010) Update 1 of: Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 110, PR41–PR67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. (2013) Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop D. B.; Cho Y. K. (2000) Effect of pressure on deuterium isotope effects of yeast alcohol dehydrogenase: Evidence for mechanical models of catalysis. Biochemistry 39, 2406–2412. [DOI] [PubMed] [Google Scholar]
- Northrop D. B.; Cho Y. K. (2000) Effects of high pressure on solvent isotope effects of yeast alcohol dehydrogenase. Biophys. J. 79, 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidman G.; Park H.; Northrop D. B. (2004) Pressure stability of proteins at their isoelectric points. Protein Pept. Lett. 11, 543–546. [DOI] [PubMed] [Google Scholar]
- Park H.; Girdaukas G. G.; Northrop D. B. (2006) Effect of pressure on a heavy-atom isotope effect of yeast alcohol dehydrogenase. J. Am. Chem. Soc. 128, 1868–1872. [DOI] [PubMed] [Google Scholar]
- Roston D.; Kohen A. (2010) Elusive transition state of alcohol dehydrogenase unveiled. Proc. Natl. Acad. Sci. U.S.A. 107, 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrath J. W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- Brunger A. T. (1992) Free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Harris M.; Jones T. A. (2001) Molray: A web interface between O and the POV-Ray ray tracer. Acta Crystallogr. D57, 1201–1203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.