

Abstract
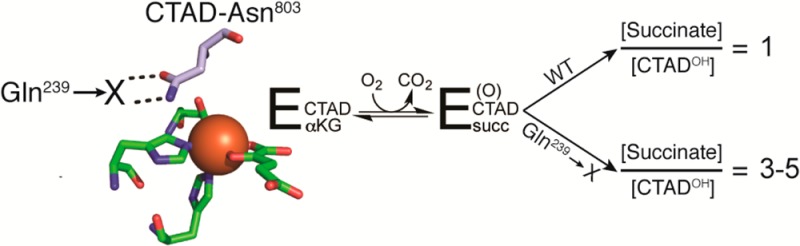
Nonheme Fe(II)/αKG-dependent oxygenases catalyze diverse reactions, typically inserting an O atom from O2 into a C–H bond. Although the key to their catalytic cycle is the fact that binding and positioning of primary substrate precede O2 activation, the means by which substrate binding stimulates turnover is not well understood. Factor Inhibiting HIF (FIH) is a Fe(II)/αKG-dependent oxygenase that acts as a cellular oxygen sensor in humans by hydroxylating the target residue Asn803, found in the C-terminal transactivation domain (CTAD) of hypoxia inducible factor-1. FIH-Gln239 makes two hydrogen bonds with CTAD-Asn803, positioning this target residue over the Fe(II). We hypothesized the positioning of the side chain of CTAD-Asn803 by FIH-Gln239 was critical for stimulating O2 activation and subsequent substrate hydroxylation. The steady-state characterization of five FIH-Gln239 variants (Ala, Asn, Glu, His, and Leu) tested the role of hydrogen bonding potential and sterics near the target residue. Each variant exhibited a 20–1200-fold decrease in kcat and kcat/KM(CTAD), but no change in KM(CTAD), indicating that the step after CTAD binding was affected by point mutation. Uncoupled O2 activation was prominent in these variants, as shown by large coupling ratios (C = [succinate]/[CTAD-OH] = 3–5) for each of the FIH-Gln239 → X variants. The coupling ratios decreased in D2O, indicating an isotope-sensitive inactivation for variants, not observed in the wild type. The data presented indicate that the proper positioning of CTAD-Asn803 by FIH-Gln239 is necessary to suppress uncoupled turnover and to support substrate hydroxylation, suggesting substrate positioning may be crucial for directing O2 reactivity within the broader class of αKG hydroxylases.
Nonheme Fe(II)/αKG-dependent oxygenases make up a large superfamily of enzymes catalyzing diverse reactions, including demethylations, hydroxylations, ring expansions, and epoxidations.1,2 Many of these enzymes have important functions in human health because of their role in O2 sensing,3,4 DNA repair,5,6 histone demethylation,7 and RNA processing,8,9 making this superfamily a growing class of therapeutic targets.10,11 As the consensus chemical mechanism is an ordered sequential one, with O2 reacting at the Fe prior to oxidation of the primary substrate (Scheme 1),12,13 identifying features of the active site by which substrate binding stimulates O2 reactivity is crucial to understanding the chemistry of this superfamily. An ideal enzyme for interrogating these connections is Factor Inhibiting HIF (FIH), because of the extensive contacts with the primary substrate.14
Scheme 1. Proposed Chemical Mechanism of FIH.

Human cells sense O2 through the hypoxia inducible factor (HIF) pathway, which is controlled by a small number of αKG oxygenases, including FIH.15,16 HIF is an αβ dimeric transcription factor that regulates numerous genes involved in tissue development, controlling processes such as glycolysis, erythropoiesis, and angiogenesis.1,17−19 In the presence of O2, FIH hydroxylates the β-carbon of HIF1α-Asn803,20 which is found in the C-terminal activation domain (CTAD) of HIF1α. CTAD-Asn803 hydroxylation blocks recruitment of the cAMP response element-binding protein (CREBP), preventing HIF-dependent gene transcription.4,21 The connection between CTAD-Asn803 positioning and O2 reactivity is critical to understanding how substrate stimulates O2 activation in this enzyme superfamily, as well as illuminating FIH’s role as an O2 sensor.
The consensus chemical mechanism for FIH is based upon an array of kinetic and spectroscopic studies of FIH and other αKG oxygenases. Kinetic studies of thymine hydroxylase, FIH, CAS, and TauD support the ordered, sequential binding of αKG and primary substrate followed by O2.22−25 Although αKG, O2, and an oxidizable compound are all substrates for these enzymes, we will refer to the oxidizable substrate as the “primary substrate”. Spectroscopic studies of CAS,26−28 TfdA,29 FIH,30 and TauD31 revealed that the Fe(II) released an aquo ligand after the primary substrate bound, creating a site for O2 binding. Binding and activation of O2 lead to the oxidative decarboxylation of αKG and the formation of a highly reactive ferryl intermediate (Scheme 1). Although the precise sequence of intermediates is not known, the ferryl intermediate has been observed in TauD32−34 and P4H,35 demonstrating that H atom abstraction by the ferryl intermediate occurs,36 with the next step likely to be •OH rebound to hydroxylate the primary substrate.
The most intriguing feature of the consensus mechanism is that binding the primary substrate stimulates O2 reactivity.22,37,38 Loss of an aquo ligand when the primary substrate is bound opens a coordination site for O2 binding, as observed upon binding of the primary substrate in several αKG-dependent oxygenases, including CytC3, TauD, CAS, and FIH.26,30,31 Although aquo release is central to the widely accepted model for substrate-stimulated O2 activation,39 we note that simple ligand exchange is insufficient for O2 activation in these enzymes. For example, substrate binding to FIH leads to only fractional release of the aquo ligand,30 and mutagenesis suggests that hydrogen bond donors to the αKG are necessary for full activity in this enzyme.40 Computational studies41−44 and mechanistic probes45−47 further point to turnover being limited by steps after O2 binds to the Fe(II). These and related observations lead us to propose that substrate-stimulated O2 reactivity arises from bonding changes throughout the active site, ranging from aquo release at the iron cofactor to altered contacts in the second coordination sphere.
A focus of this research in our lab is to identify those active site features that change upon substrate binding to stimulate O2 activation in αKG oxygenases. Although the precise sequence of intermediates formed during turnover is not known, we define O2 activation as the steps between O2 binding and oxidative decarboxylation (Scheme 1) by virtue of the irreversible chemistry; this step is depicted as the nucleophilic attack of the putative ferric superoxide on the α-keto position of αKG. On the basis of known crystal structures of FIH,14,48,49 we have used point mutagenesis to identify several essential second-coordination sphere interactions in FIH, including those hydrogen bonding to Fe(II) ligands, as well as FIH-Gln239, an anchor residue that forms two hydrogen bonds with the target residue, CTAD-Asn803 (Figure 1).14 Intriguingly, disruption of this two-point hydrogen bond in the FIH-Gln239 → Asn point mutant led to a decrease in kcat of 250-fold, but a negligible change in KM(CTAD).40 This was attributed to a combination of steric hindrance near the open coordination site on Fe(II) and incorrect CTAD positioning for the HAT step. Subsequently, it was shown that an irreversible step associated with O2 activation was rate-limiting in wild-type FIH (WT-FIH),25 suggesting that the slower turnover for the Gln239 → Asn variant could arise from slower O2 activation. This suggests the intriguing possibility that target residue position may stimulate O2 activation and that the overall structure of the active site is crucial for O2 activation.
Figure 1.
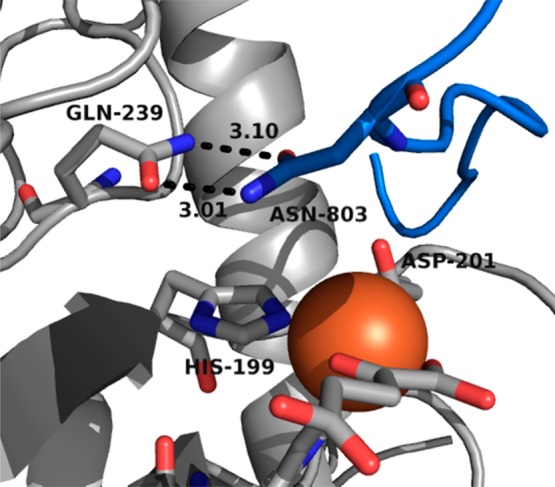
CTAD-Asn803 (CTAD, blue) positioned by FIH-Gln239 (FIH, gray) over the active site. Hydrogen bonding distances are given in angstroms (Protein Data Bank entry 1H2L(14)).
This study tests the role of Gln239 in substrate hydroxylation in αKG oxygenases. As FIH hydroxylates a specific target residue within a large peptide (CTAD-Asn803), our focus was directed at this target residue pocket, formed by the side chains of FIH residues Tyr102, His199, Arg238, and Gln239 (Figure 1). Five FIH-Gln239 → X variants were prepared (X = Ala, Asn, Glu, His, and Leu) to vary the bulk and hydrogen bonding potential within the target residue pocket. Although these variants exhibited significantly reduced steady-state rate constants that decreased monotonically with increasing residue bulk, CTAD binding affinity was unaffected by mutation. In contrast to the case in WT-FIH, O2 activation was appreciably uncoupled from CTAD hydroxylation in the variants; uncoupled O2 activation was partially suppressed in D2O. These data establish that the proper orientation of CTAD-Asn803 by FIH-Gln239 is required for substrate hydroxylation, most likely because of the need for the proper target residue positioning during steps after O2 activation.
Experimental Procedures
Materials
All reagents were purchased from commercial vendors and were not further purified, with the exception of the 39-mer CTAD peptide. The 39-mer CTAD peptide corresponding to the C-terminal activation domain of human HIF1α, (HIF1α788–826) contained a Cys800 → Ala change (underlined) (DESGLPQLTSYDAEVNAPIQGSRNLLQGEELLRALDQVN). This was purchased as a desalted peptide from EZBiolab (Carmel, IN) with free N- and C-termini. The peptide was purified as previously described using reverse-phase high-performance liquid chromatography (RP-HPLC) to obtain >95% pure CTAD.25 The 19-mer CTAD peptide corresponding to HIF1α788–806 also contained a Cys800 → Ala change and was purchased at >95% purity from EZBiolab with free N- and C-termini. The CTAD-Asn803 → Gln peptide was the 19-mer sequence but contained the Asn803 → Gln change (DESGLPQLTSYDAEVQAPI).
FIH Mutations
The Stratagene QuikChange mutagenesis kit was used to introduce the mutations into the pET28a-FIH construct.50 All mutations were sequenced (Genewiz) to confirm that the DNA sequence contained only the desired point mutation. Sequenced plasmids were transformed into BL21(DE3) cells for protein expression.
Protein Expression and Purification
WT-FIH and all variants were overexpressed in Escherichia coli with an N-terminal His6 tag and purified as previously described.25 Three additional residues (NH2-GlySerHis-) from the fusion protein remained on the N-terminus following thrombin cleavage. Purified FIH was buffer-exchanged into 50 mM HEPES (pH 7.00). The purity (>95%) of each variant was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Steady-State Kinetics Assays
All assays used saturating concentrations of FeSO4 (25 μM) and ascorbate (2 mM) and an ambient O2 concentration (217 μM at 37.0 °C) and were performed in 50 mM HEPES (pH 7.00) at 37.0 °C unless specifically noted otherwise. DTT (200 μM) was added to assays for FIH-Gln239 → His and FIH-Gln239 → Glu when these variants were tested in D2O, as nonlinear progress curves were observed otherwise. Assays in which CTAD was the varied substrate (from 15 to 300 μM) utilized a saturating αKG concentration (500 μM). Assays with αKG as the varied substrate (from 2.5 to 100 μM) utilized a fixed CTAD concentration of 100 μM [∼KM(CTAD)] to conserve on the use of the peptide. Assay reagents were mixed and incubated for 2 min at 37.0 °C before the addition of enzyme ([E]T = 1.5–10 μM). Reaction aliquots (5 μL) were quenched with a 75% acetonitrile/0.2% TFA mixture (20 μL) saturated with 3,5-dimethoxy-4-hydroxycinnamic acid and analyzed for peptide hydroxylation using a Bruker Daltonics Omniflex matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) instrument. Initial rates were determined from five to seven quenched time points (0 to ∼15% fractional conversion). The nonlinear least-squares fitting of initial rate data to the Michaelis–Menten equation yielded the apparent steady-state rate constants, kcat and kcat/KM. All assays were replicated a minimum of three times.
Solvent Kinetic Isotope Effects (SKIEs)
Steady-state assays for SKIEs were performed under the same conditions reported above, with the exception that all reagents were prepared in D2O. Deuterium oxide (D, 99.9%) was purchased from Cambridge Isotope Laboratories (Andover, MA) and used as received. Working FIH stock solutions were made by diluting high-concentration stocks from H2O into D2O containing 50 mM HEPES (pD 7.00). Assays were performed in 50 mM HEPES (pD 7.00), with a final D2O percentage estimated to be 96%. SKIEs were calculated from the direct comparison of kinetic parameters observed in buffers containing H2O and D2O; e.g., D2Okcat = kcat(H2O)/kcat(D2O).
Succinate Quantification
The coupling between the two half-reactions was determined by monitoring the production of succinate and CTADOH concentrations in several quench points from a common reaction. Reactions of αKG (500 μM), FeSO4 (25 μM), CTAD (350 μM), and FIH (5–10 μM) were conducted at 37.0 °C and analyzed similarly using previously reported procedures.25,40,47 As HEPES interfered with the succinate analysis, the reaction buffer consisted of 50 mM Tris (pL 7.00). A Hamilton PRP-X300 anion exclusion column was used to separate the succinate produced from the quenched reactions, and UV detection at 210 nm was used to determine the succinate concentration. Using aliquots from the same quenched assay, a Bruker Daltonics Omniflex MALDI-TOF MS was used to determine the CTADOH concentration. The coupling ratio (C) was determined by taking the ratio of the rate of succinate formation and the rate of CTADOH formation from matched time points.
Fluorescence Spectroscopy
The FIH–CTAD binding constants were measured through quenching of the intrinsic tryptophan fluorescence of (Co + αKG)FIH upon CTAD binding at room temperature (∼20 °C). The fluorescence cuvette contained FIH (1.5 μM), CoSO4 (25 μM), αKG (500 μM), and 50 mM HEPES (pH 7.05). This solution was titrated with 50 mM HEPES (pH 7.05) containing CTAD (1 mM), FIH (1.5 μM), CoSO4 (25 μM), and αKG (500 μM). All titrations were performed aerobically. After each addition of titrant, samples were gently mixed and allowed to equilibrate for 5 min before being excited at 295 nm. The fluorescence intensities at 330 nm were plotted versus the total CTAD concentration and fit using eq 1
| 1 |
where I is the measured fluorescence intensity, [E] is the protein concentration, [S] is the total CTAD concentration, n is the number of binding sites, and KD is the binding affinity. The initial intensity (I0) and final intensity (If) were obtained from measured spectra.
Results
Variants of FIH-Gln239 were used to test the effect of target residue positioning on substrate hydroxylation in FIH. The variants were designed to vary hydrogen bonding potential (Gln239 → Glu and Gln239 → His) and cavity size (Gln239 → Ala, Gln239 → Asn, and Gln239 → Leu) in the target residue pocket of FIH. The Gln239 variants were kinetically characterized in the steady state with CTAD as the varied substrate, giving the apparent rate constants kcat and kcat/KM(CTAD). The kinetic characterization revealed significantly diminished rate constants for turnover, leading us to determine the binding affinity of CTAD as well as the coupling ratio of the two half-reactions for each point mutant.
Kinetic Characterization of Gln239 → X Variants
We hypothesized the positioning of CTAD-Asn803 by FIH-Gln239 was necessary to support turnover and therefore focused our studies on steady-state characterization by monitoring CTADOH formation via MALDI-TOF. Although O2 uptake was the first method that we considered, the slow turnover for FIH makes high-precision kinetic determinations by this method challenging. Assays using fixed concentrations of αKG (500 μM) and O2 (217 μM) and varied concentrations of CTAD (15–300 μM) were used to measure initial rates, which where then fit to the Michaelis–Menten equation to obtain the apparent steady-state rate constants, kcat and kcat/KM(CTAD). The Michaelis constant for αKG was determined for each variant [KM(αKG) = 4–7 μM], which was slightly lower than that for WT-FIH [KM(αKG) = 16 μM]. Because of the O2 concentration is subsaturating, the apparent kcat encompasses all steps after CTAD binding, including those involved in O2 binding and activation. All of the Gln239 → X variants exhibited a significant decrease in kcat (Figure 2). The Gln239 → Ala (kcat = 1.27 ± 0.10 min–1) variant was most active, as the kcat decreased 20-fold relative to that of WT-FIH, whereas the kcat for Gln239 → Asn (0.14 ± 0.02 min–1) decreased 200-fold. The kcat for variants capable of one-point hydrogen bonding decreased >1200-fold: Gln239 → His (0.023 ± 0.003 min–1) and Gln239 → Glu (0.024 ± 0.002 min–1). We were unable to observe hydroxylation from the Gln239 → Leu variant.
Figure 2.

Steady-state kinetics of Q239A in H2O. FIH (1.5 μM), ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and CTAD (0–300 μM) were in 50 mM HEPES (pH 7.00). The inset shows the steady-state kinetics of Q239N (▲),Q239H (■), and Q239E (◆) in H2O. FIH (1.5–30 μM), ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and CTAD (0–300 μM) were in 50 mM HEPES (pH 7.00).
Steps from CTAD binding through the first irreversible step (decarboxylation) comprise kcat/KM(CTAD) (Scheme 2). The effect of each point variant on kcat/KM(CTAD) was nearly identical to their effect on kcat, indicating that the variants affected a step that was separate from CTAD binding.
Scheme 2. Minimal Chemical Scheme for Uncoupling.

We tested the activity of FIH-Gln239 → Asn using a 19-mer CTAD peptide containing the complementary CTAD-Asn803 → Gln point mutation, which switched the residues at this interface. This switch mutation was designed to restore the bulk and hydrogen bonds observed between WT-FIH and WT-CTAD. However, the activity level was below our detection limit (0.002 min–1), as hydroxylated CTAD-Asn803 → Gln was not detected upon being incubated with FIH-Gln239 → Asn. WT-FIH was similarly unreactive toward this variant CTAD, as WT-FIH hydroxylated the 19-mer WT-CTAD with an appreciable rate, but did not hydroxylate the variant CTAD (Table 1).
Table 1. Initial Rates for 19-mer Peptides CTAD and CTAD-N803Qa.
Assays contained ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and 19-mer CTAD (400 μM) in 50 mM HEPES (pH 7.00) at 37.0 °C. The CTAD peptide used contained 19 residues.
No activity detected; estimated detection limit, if active.
Binding Affinity of CTAD for Gln239 → X Variants
The binding affinity of each FIH variant for CTAD was measured by titration using the intrinsic tryptophan fluorescence of FIH. A solution containing CTAD (1 mM) was titrated into a solution containing FIH (1.5 μM) while the fluorescence at 330 nm was monitored (λex = 295 nm); both solutions were anaerobic and contained CoSO4 (25 μM) and αKG (500 μM). The change in fluorescence intensity (330 nm) was plotted as a function of CTAD concentration and fit to eq 1. The experimentally determined KD for each point mutant (Table 2) was similar to that of WT-FIH (78 ± 7 μM), indicating the thermodynamics of CTAD binding was not affected by point mutation.
Table 2. Apparent Kinetic Parameters for FIH and Its Variantsa.
| kcat (min–1)b | kcat/KM(CTAD) (μM–1 min–1)b | KM(CTAD) (μM)b | KD(CTAD) (μM)c | KM(αKG) (μM)d | |
|---|---|---|---|---|---|
| WT | 30 ± 2.5e | 0.4 ± 0.1e | 70 ± 20e | 78 ± 7f | 16 ± 3.0 |
| Q239A | 1.3 ± 0.10 | 0.021 ± 0.002 | 61 ± 10 | 100 ± 16 | 5.0 ± 0.5 |
| Q239N | 0.14 ± 0.02 | 2.0 × 10–3 ± 8 × 10–4 | 74 ± 30 | 98 ± 10 | 4.0 ± 0.4 |
| Q239H | 0.023 ± 0.003 | (3.4 ± 1) × 10–4 | 68 ± 18 | 64 ± 14 | 7.0 ± 1.4 |
| Q239E | 0.024 ± 0.002 | 3.4 × 10–4 ± 7 × 10–5 | 71 ± 10 | 75 ± 15 | 4.7 ± 2.0 |
| Q239L | <0.005g | <8 × 10–5g | NDh | 80 ± 8 | NDh |
In 50 mM HEPES (pH 7.00) at 37.0 °C.
Assays in which CTAD was the varied substrate, in ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and CTAD (0–300 μM).
Determined using intrinsic tryptophan fluorescence with Co-substituted enzyme.
Assays in which αKG was the varied substrate, in ascorbate (2 mM), αKG (2–200 μM), FeSO4 (25 μM), and CTAD (100 μM).
From ref (25).
From ref (30).
No activity detected; estimated detection limits as reported.
Not determined.
Uncoupled Turnover in the Gln239 → X Variants
The kinetic parameters of the Gln239 → X mutations led us to explore the coupling of O2 activation to substrate hydroxylation. We hypothesized that if the conformational state of CTAD-Asn803 were incorrect for HAT, then the two half-reactions would uncouple to produce more succinate than hydroxylated product (CTADOH). Quenched aliquots from reaction mixtures containing saturating concentrations of αKG (500 μM) and CTAD (350 μM) in 50 mM Tris (pH 7.00) were analyzed for CTADOH via MALDI-TOF MS and succinate via HPLC. Tris buffer was used for these assays to minimize the background signal in the HPLC chromatograms that arose due to buffer components.
The coupling values for the Gln239 → X (X = Ala, Asn, Glu, and His) variants were obtained by taking the ratio of the rates of formation for succinate and CTADOH. Variants produced three to five succinates per equivalent of CTADOH; succinate formation was observed for the Gln239 → Leu variant [kobs(suc) = 0.08 min–1], indicating O2 activation occurred even though CTAD hydroxylation was not detected for this variant (Table 3). This uncoupling is similar to the values found previously for second-coordination sphere variants of FIH.40
Table 3. Coupling of Succinate and CTADOH Concentrations for FIH and Variantsa.
| kobs(H2O) ([succinate] min–1 [FIH]−1) | CH2Ob | CD2Ob | |
|---|---|---|---|
| WT | 28 ± 2 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| Q239A | 5.5 ± 0.3 | 4 ± 1 | 1.4 ± 0.2 |
| Q239N | 0.49 ± 0.08 | 3.3 ± 0.3 | 2.2 ± 0.2 |
| Q239H | 0.06 ± 0.02 | 3 ± 1 | NDc |
| Q239E | 0.07 ± 0.03 | 5 ± 1 | NDc |
| Q239L | 0.08 ± 0.02 | NDc | NDc |
Reaction mixtures contained FIH (5–10 μM), αKG (500 μM), FeSO4 (25 μM), and CTAD (350 μM) in 50 mM Tris (pL 7.00) at 37 °C.
C = (moles of succinate per minute)/(moles of CTADOH per minute).
Not determined.
The coupling of WT, Gln239 → Ala, and Gln239 → Asn in deuterated buffer was used to determine if the coupling ratio changed between protonated and deuterated buffers. The coupling for WT FIH in H2O (C = 1.0 ± 0.1) and D2O (C = 1.0 ± 0.1) was in agreement with our previous work,25 showing WT remains tightly coupled under all tested conditions. However, the coupling ratio in D2O for Gln239 → Ala (C = 1.4 ± 0.2) and Gln239 → Asn (C = 2.2 ± 0.2) approached unity, indicating that solvent deuteration led to more tightly coupled turnover for these variants.
Solvent Kinetic Isotope Effects (SKIEs)
SKIEs on both kcat and kcat/KM(CTAD) were used to test the importance of solvent-dependent steps during turnover. Initial rates from steady-state assays using saturating αKG concentrations (500 μM), ambient O2 concentrations (217 μM), and varied concentrations of CTAD (15–300 μM) were fit to the Michaelis–Menten equation (Figure 3). Turnover was faster in D2O with both WT-FIH and each variant, leading to an inverse SKIE on kcat and kcat/KM(CTAD) (Table 4). However, these SKIEs must be considered in the context of the solvent-dependent uncoupling observed for the variants.51
Figure 3.
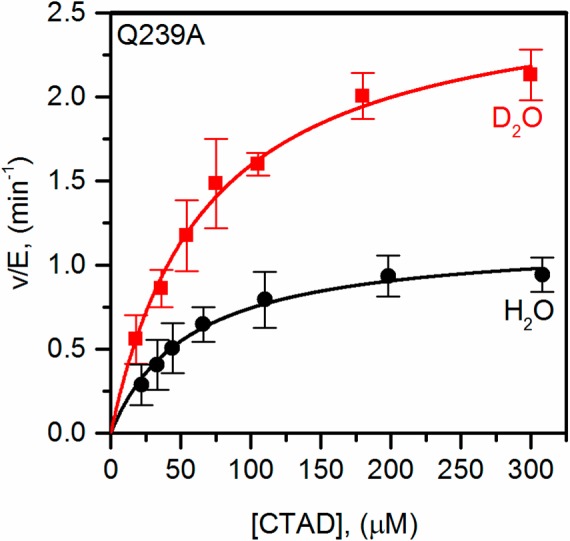
Steady-state kinetics of Q239A in H2O (●) and 96% D2O (■) buffers. FIH (1.5–30 μM), ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and CTAD (0–300 μM) were in 50 mM HEPES (pL 7.00).
Table 4. Apparent Kinetic Parameters in D2O and SKIEs for FIH and Its Variantsa.
| kcat (min–1)b | kcat/KM(CTAD) (μM–1 min–1)b | D2Okcatc | D2Okcat/KM(CTAD)d | |
|---|---|---|---|---|
| WTe | 59 ± 2 | 1.09 ± 0.11 | 0.51 ± 0.07 | 0.40 ± 0.07 |
| Q239A | 2.55 ± 0.21 | 0.044 ± 0.011 | 0.50 ± 0.05 | 0.48 ± 0.15 |
| Q239N | 0.27 ± 0.02 | 0.005 ± 0.001 | 0.50 ± 0.06 | 0.41 ± 0.18 |
| Q239H | 0.050 ± 0.003 | 2.0 × 10–3 ± 8 × 10–4 | 0.46 ± 0.07 | 0.17 ± 0.06 |
| Q239E | 0.046 ± 0.002 | (8 ± 3) × 10–4 | 0.52 ± 0.05 | 0.41 ± 0.16 |
| Q239L | NDf | NDf | NDf | NDf |
In 50 mM HEPES (pD 7.00) at 37.0 °C.
Determined from assays with CTAD as the varied substrate, in ascorbate (2 mM), αKG (500 μM), FeSO4 (25 μM), and CTAD (0–250 μM); χD2O = 0.96.
D2Okcat = kcat(H2O)/kcat(D2O).
D2Okcat/KM(CTAD) = [kcat/KM(CTAD) in H2O]/[kcat/KM(CTAD) in D2O].
From ref (25).
Not determined.
Discussion
The ordered sequential consensus mechanism for αKG oxygenases leads to coupled turnover when primary substrate binding stimulates reactivity toward O2, a phenomenon termed substrate-induced activity enhancement,52 priming,53 or triggering54 by different groups. As primary substrate does not directly bind to the Fe(II), altered local contacts within the active site likely stimulate O2 activation. Although the idea of stimulated O2 activation refers to the empirical observation of increased turnover rates induced by substrate binding, the dominant model used to explain this focuses on aquo release, which creates an open coordination site for O2 binding.39 In our opinion, broader changes within the active site are correlated with this effect, such as the position of the primary substrate.
FIH is notable in that enzyme–substrate contacts are quite extensive because the substrate is a large peptide (CTAD), with the target residue positioned above the Fe by a two-point hydrogen bond to the side chain of an anchoring residue, FIH-Gln239.14,55 This study varied the sterics and H-bonding potential of this anchor residue to test its role in hydroxylating CTAD-Asn803.
CTAD Hydroxylation Is Slowed by Gln239 Variants
Each of the Gln239 → X variants (X = Ala, Asn, Glu, His, or Leu) altered the hydrogen bond potential and/or bulk of the target residue pocket, disrupting the positioning of the target residue. This incorrect positioning could have impacted any one of several steps within the kinetic mechanism, which may be distinguished through analysis of steady-state kinetic parameters and coupling ratios. The significant reduction in kcat and kcat/KM(CTAD) relative to those of WT-FIH indicated that the anchor residue played a prominent role in supporting turnover. Keeping in mind the observation that the binding affinity of CTAD was unchanged from that of WT-FIH (Table 2), we are led to conclude that the predominant role of FIH-Gln239 is to position substrate for a chemical step rather than to bind CTAD.
Although it may seem surprising that the anchor residue FIH-Gln239 contributes very little to the CTAD binding affinity, this is consistent with prior studies of CTAD variants. As the length of the CTAD has been shown to have a significant affect on the KM,56 and the binding affinity of WT-FIH for CTAD is indistinguishable from the Michaelis constant, it appears that the dominant factor in CTAD binding is the surface contact with FIH, with only minor contributions from the target residue pocket. Alanine scanning point mutagenesis of CTAD revealed that CTAD-Val802 was the most significant residue for CTAD binding, with a 2-fold increase in the KM(CTAD) for the CTAD-Val802 → Ala variant.57 Molecular dynamics studies suggested that this mutation led to reorientation of Asn803, perhaps because of disruption of the tight turn conformation in residues 801–803 of CTAD. Further support for a minimal impact of FIH-Gln239 on CTAD binding is the observation that FIH hydroxylates substrates with target residues other than asparagine.58,59 The structural features of these substrates suggest the overall contact between FIH and the CTAD peptide is important in determining substrate binding to FIH.59,60
Inverse SKIEs and Coupling
We recently reported inverse SKIEs for WT FIH, on both kcat and kcat/KM(CTAD).25 This was due to the isotopically sensitive metal–aquo fractionation prior to a rate-limiting step for WT-FIH. Importantly, WT-FIH exhibited fully coupled turnover, such that O2 activation always led to substrate hydroxylation. Consequently, it was deduced that the rate-limiting step for kcat and kcat/KM(CTAD) was an irreversible step immediately after aquo release. This step is depicted as the oxidative decarboxylation of αKG in Scheme 1.
For each of the Gln239 → X variants, inverse SKIEs were measured on both kcat and kcat/KM(CTAD) when determined from the rate of CTADOH formation (Table 4). Although the SKIE data resembled those reported for WT-FIH, turnover for these variants was significantly uncoupled, which precluded the use of SKIEs to diagnose rate-limiting steps in the steady state. Nevertheless, uncoupling in the variants depended on solvent isotopic composition (Table 3), suggesting that the ferryl intermediate could form even when CTAD was improperly positioned. The fact that C approached unity in D2O for these variants suggested that the main effect of the Gln239 → X change was to perturb the hydroxylation step.
Sterics and H-Bonding Impact Substrate Hydroxylation
A simple model to explain how the target residue pocket impacts productive turnover is one in which multiple conformational states of the target residue are adopted but only one conformation supports hydroxylation. X-ray crystal structures of (M+αKG)FIH bound to CTAD14 or Notch-derived peptides55 revealed that the target residue adopted a specific rotameric conformation, with a side chain torsional angle (HN–Cα–Cβ–Cγ) of −71°. This is observed for both Notch target residues Notch-Asn210 and Notch-Asn1945 [Protein Data Bank (PDB) entries 3P3P and 3P3N, respectively]. As a good deal of flexibility near Gln239 was observed crystallographically for (Fe+αKG)FIH when CTAD was absent (PDB entry 1MZF),48 changing the hydrogen bonding potential and packing density of this anchor residue should alter the target residue position above the Fe(II). The significant reduction in catalytic efficiency for each point mutant strongly suggests that the major role of FIH-Gln239 is to stabilize the proper rotamer of CTAD-Asn803 that can undergo hydroxylation during turnover.
The kinetic data further suggest that packing near the target residue may also impact O2 activation in FIH. The overall trend in the kinetic parameters measured by coupled turnover (Table 2) was dominated by bulk, as the kinetic parameters for the Gln239 → X point variants decreased monotonically in a series: X = (Ala > Asn > Glu and His ≫ Leu). As the kinetic parameters of the variants listed in Table 2 are functions of all steps leading to CTAD hydroxylation, it is not possible to separately identify the impact of the variants on O2 activation. However, the coupling data directly measured succinate production (Table 4), which reports directly on O2 activation. The rates of succinate production clearly showed that each variant produced succinate much more slowly that WT-FIH, suggesting that O2 activation was slowed in these variants. As the packing about CTAD-Asn803 is quite tight in WT-FIH,14 it is possible that the Gln239 → X variants excluded CTAD-Asn803 from the proper conformation, which could impede access of O2 to the Fe(II) as well as hydroxylation by the putative ferryl intermediate (Scheme 2).
Conclusion
This work establishes that proper positioning of the primary substrate (CTAD) is required to support coupled turnover by FIH. We conclude that proper positioning of substrate is crucial for the hydroxylation of CTAD as well as for stimulation of O2 activation. Mispositioned CTAD impedes O2 activation, suggesting that the environment near the Fe(II) cofactor plays a marked role in O2 activation.
Acknowledgments
We thank Dr. Evren Saban for the purification of the Gln239 → Asn variant and the late Professor Robert Weiss for the use of his differential scanning calorimeter.
Glossary
Abbreviations
- αKG
α-ketoglutarate
- CAS
clavaminate synthase
- CD
circular dichroism
- CREBP
cAMP response element-binding protein
- CTAD
C-terminal transactivation domain
- DFT
density functional theory
- DTT
dithiothreitol
- FIH
factor-inhibiting HIF
- HAT
hydrogen atom transfer
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HIF
hypoxia inducible factor-1α
- MCD
magnetic circular dichroism
- NOG
N-oxalyl glycine
- P4H
prolyl-4-hydroxylase
- SKIE
solvent kinetic isotope effect
- TauD
taurine dioxygenase.
Supporting Information Available
Control experiments showing the thermal stability of the Gln239 → X variant and intrinsic tryptophan fluorescence titrations with CTAD. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Research reported in this publication was supported by National Institutes of Health Grant R01-GM077413 and National Institutes of Health Chemistry-Biology Interface Predoctoral Training Fellowship T32-GM008515.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hausinger R. P. (2004) Fe(II)/α-Ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68. [DOI] [PubMed] [Google Scholar]
- Purpero V.; Moran G. R. (2007) The diverse and pervasive chemistries of the α-keto acid dependent enzymes. JBIC, J. Biol. Inorg. Chem. 12, 587–601. [DOI] [PubMed] [Google Scholar]
- Jaakkola P.; Mole D. R.; Tian Y. M.; Wilson M. I.; Gielbert J.; Gaskell S. J.; von Kriegsheim A.; Hebestreit H. F.; Mukherji M.; Schofield C. J.; Maxwell P. H.; Pugh C. W.; Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- Lando D.; Peet D. J.; Whelan D. A.; Gorman J. J.; Whitelaw M. L. (2002) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861. [DOI] [PubMed] [Google Scholar]
- Duncan T.; Trewick S. C.; Koivisto P.; Bates P. A.; Lindahl T.; Sedgwick B. (2002) Reversal of DNA alkylation damage by two human dioxygenases. Proc. Natl. Acad. Sci. U.S.A. 99, 16660–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewick S. C.; Henshaw T. F.; Hausinger R. P.; Lindahl T.; Sedgwick B. (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; Warren M. E.; Borchers C. H.; Tempst P.; Zhang Y. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Dahl J. A.; Niu Y.; Fedorcsak P.; Huang C.-M.; Li C. J.; Vagbo C. B.; Shi Y.; Wang W.-L.; Song S.-H.; Lu Z.; Bosmans R. P. G.; Dai Q.; Hao Y.-J.; Yang X.; Zhao W.-M.; Tong W.-M.; Wang X.-J.; Bogdan F.; Furu K.; Fu Y.; Jia G.; Zhao X.; Liu J.; Krokan H. E.; Klungland A.; Yang Y.-G.; He C. (2013) ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G.; Fu Y.; Zhao X.; Dai Q.; Zheng G.; Yang Y.; Yi C.; Lindahl T.; Pan T.; Yang Y.-G.; He C. (2011) N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose N. R.; McDonough M. A.; King O. N. F.; Kawamura A.; Schofield C. J. (2011) Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 40, 4364–4397. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Schofield C. J. (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 36, 7–18. [DOI] [PubMed] [Google Scholar]
- Holme E. (1975) Kinetic study of thymine 7-hydroxylase from Neurospora crassa. Biochemistry 14, 4999–5003. [DOI] [PubMed] [Google Scholar]
- De Carolis E.; De Luca V. (1993) Purification, characterization, and kinetic analysis of a 2-oxoglutarate-dependent dioxygenase involved in vindoline biosynthesis from Catharanthus roseus. J. Biol. Chem. 268, 5504–5511. [PubMed] [Google Scholar]
- Elkins J. M.; Hewitson K. S.; McNeill L. A.; Seibel J. F.; Schlemminger I.; Pugh C. W.; Ratcliffe P. J.; Schofield C. J. (2003) Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α. J. Biol. Chem. 278, 1802–1806. [DOI] [PubMed] [Google Scholar]
- Ozer A.; Bruick R. K. (2007) Non-heme dioxygenases: Cellular sensors and regulators jelly rolled into one?. Nat. Chem. Biol. 3, 144–153. [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2004) Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 19, 176–182. [DOI] [PubMed] [Google Scholar]
- Iyer N. V.; Kotch L. E.; Agani F.; Leung S. W.; Laughner E.; Wenger R. H.; Gassmann M.; Gearhart J. D.; Lawler A. M.; Yu A. Y.; Semenza G. L. (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 12, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–32. [DOI] [PubMed] [Google Scholar]
- Metzen E.; Ratcliffe P. J. (2004) HIF hydroxylation and cellular oxygen sensing. Biol. Chem. 385, 223–230. [DOI] [PubMed] [Google Scholar]
- McNeill L. A.; Hewitson K. S.; Claridge T. D.; Seibel J. F.; Horsfall L. E.; Schofield C. J. (2002) Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the β-carbon of asparagine-803. Biochem. J. 367, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D.; Peet D. J.; Gorman J. J.; Whelan D. A.; Whitelaw M. L.; Bruick R. K. (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Hoffart L. M.; Krebs C.; Bollinger J. M. (2005) Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 44, 8138–8147. [DOI] [PubMed] [Google Scholar]
- Salowe S. P.; Marsh E. N.; Townsend C. A. (1990) Purification and characterization of clavaminate synthase from Streptomyces clavuligerus: An unusual oxidative enzyme in natural product biosynthesis. Biochemistry 29, 6499–6508. [DOI] [PubMed] [Google Scholar]
- Thornburg L. D.; Lai M. T.; Wishnok J. S.; Stubbe J. (1993) A non-heme iron protein with heme tendencies: An investigation of the substrate specificity of thymine hydroxylase. Biochemistry 32, 14023–14033. [DOI] [PubMed] [Google Scholar]
- Hangasky J. A.; Saban E.; Knapp M. J. (2013) Inverse Solvent Isotope Effects Arising from Substrate Triggering in the Factor Inhibiting Hypoxia Inducible Factor. Biochemistry 52, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Kelly W. L.; Bachmann B. O.; Gunsior M.; Townsend C. A.; Solomon E. I. (2001) Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional α-KG-dependent non-heme iron enzyme: Correlation with mechanisms and reactivities. J. Am. Chem. Soc. 123, 7388–7398. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Gunsior M.; Bachmann B. O.; Townsend C. A.; Solomon E. I. (1998) Substrate Binding to the α-Ketoglutarate-Dependent Non-Heme Iron Enzyme Clavaminate Synthase 2: Coupling Mechanism of Oxidative Decarboxylation and Hydroxylation. J. Am. Chem. Soc. 120, 13539–13540. [Google Scholar]
- Pavel E. G.; Zhou J.; Busby R. W.; Gunsior M.; Townsend C. A.; Solomon E. I. (1998) Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with α-ketoglutarate cosubstrate. J. Am. Chem. Soc. 120, 743–753. [Google Scholar]
- Whiting A. K.; Que L.; Saari R. E.; Hausinger R. P.; Fredrick M. A.; McCracken J. (1997) Metal Coordination Environment of a Cu(II)-Substituted α-Keto Acid-Dependent Dioxygenase That Degrades the Herbicide 2,4-D. J. Am. Chem. Soc. 119, 3413–3414. [Google Scholar]
- Light K. M.; Hangasky J. A.; Knapp M. J.; Solomon E. I. (2013) Spectroscopic Studies of the Mononuclear Non-Heme Fe-II Enzyme FIH: Second-Sphere Contributions to Reactivity. J. Am. Chem. Soc. 135, 9665–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidig M. L.; Brown C. D.; Light K. M.; Fujimori D. G.; Nolan E. M.; Price J. C.; Barr E. W.; Bollinger J. M.; Krebs C.; Walsh C. T.; Solomon E. I. (2007) CD and MCD of CytC3 and taurine dioxygenase: Role of the facial triad in α-KG-dependent oxygenases. J. Am. Chem. Soc. 129, 14224–14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M.; Krebs C. (2003) The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- Riggs-Gelasco P. J.; Price J. C.; Guyer R. B.; Brehm J. H.; Barr E. W.; Bollinger J. M.; Krebs C. (2004) EXAFS spectroscopic evidence for an Fe=O unit in the Fe(IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 126, 8108–8109. [DOI] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Henshaw T. F.; Monterosso G. R.; Ryle M. J.; Hausinger R. P. (2004) Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 126, 1022–1023. [DOI] [PubMed] [Google Scholar]
- Hoffart L. M.; Barr E. W.; Guyer R. B.; Bollinger J. M.; Krebs C. (2006) Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 103, 14738–14743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Glass T. E.; Krebs C.; Bollinger J. M. (2003) Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc. 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Liu A.; Muthukumaran R. B.; Ho R. Y. N.; Koehntop K. D.; McCracken J.; Que L.; Hausinger R. P. (2003) O2- and α-Ketoglutarate-Dependent Tyrosyl Radical Formation in TauD, an α-Keto Acid-Dependent Non-Heme Iron Dioxygenase. Biochemistry 42, 1854–1862. [DOI] [PubMed] [Google Scholar]
- Saban E.; Flagg S.; Knapp M. (2011) Uncoupled O2-activation in the human HIF-asparaginyl hydroxylase, FIH, does not produce reactive oxygen species. JBIC, J. Biol. Inorg. Chem. 105, 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S.-K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y.-S.; Zhou J. (2000) Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 100, 235–350. [DOI] [PubMed] [Google Scholar]
- Saban E.; Chen Y.-H.; Hangasky J.; Taabazuing C.; Holmes B.; Knapp M. (2011) The second coordination sphere of FIH controls hydroxylation. Biochemistry 50, 4733–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski T.; Bassan A.; Siegbahn P. E. M. (2004) Mechanism of dioxygen activation in 2-oxoglutarate-dependent enzymes: A hybrid DFT study. Chem.—Eur. J. 10, 1031–1041. [DOI] [PubMed] [Google Scholar]
- De Visser S. P. (2007) Can the peroxosuccinate complex in the catalytic cycle of taurine/α-ketoglutarate dioxygenase (TauD) act as an alternative oxidant?. Chem. Commun. 171–173. [DOI] [PubMed] [Google Scholar]
- Diebold A. R.; Brown-Marshall C. D.; Neidig M. L.; Brownlee J. M.; Moran G. R.; Solomon E. I. (2011) Activation of α-Keto Acid-Dependent Dioxygenases: Application of an {FeNO}7/{FeO2}8 Methodology for Characterizing the Initial Steps of O2 Activation. J. Am. Chem. Soc. 133, 18148–18160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.; Riplinger C.; Hansen A.; Krebs C.; Bollinger J. M.; Neese F. (2012) Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII- and α-Ketoglutarate (αKG)-Dependent Dioxygenases. Chem.—Eur. J. 18, 6555–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirica L. M.; McCusker K. P.; Munos J. W.; Liu H.; Klinman J. P. (2008) 18O kinetic isotope effects in non-heme iron enzymes: Probing the nature of Fe/O2 intermediates. J. Am. Chem. Soc. 130, 8122–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryle M. J.; Padmakumar R.; Hausinger R. P. (1999) Stopped-Flow Kinetic Analysis of Escherichia coli Taurine/α-Ketoglutarate Dioxygenase: Interactions with α-Ketoglutarate, Taurine, and Oxygen. Biochemistry 38, 15278–15286. [DOI] [PubMed] [Google Scholar]
- Flashman E.; Hoffart L. M.; Hamed R. B.; Bollinger J. M.; Krebs C.; Schofield C. J. (2010) Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 277, 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann C. E.; Bruick R. K.; Deisenhofer J. (2002) Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 15351–15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Kim S. J.; Jeong D. G.; Lee S. M.; Ryu S. E. (2003) Structure of Human FIH-1 Reveals a Unique Active Site Pocket and Interaction Sites for HIF-1 and von Hippel-Lindau. J. Biol. Chem. 278, 7558–7563. [DOI] [PubMed] [Google Scholar]
- Chen Y. H.; Comeaux L. M.; Herbst R. W.; Saban E.; Kennedy D. C.; Maroney M. J.; Knapp M. J. (2008) Coordination changes and auto-hydroxylation of FIH-1: Uncoupled O2-activation in a human hypoxia sensor. J. Inorg. Biochem. 102, 2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantom P. A.; Fitzpatrick P. F. (2003) Uncoupled forms of tyrosine hydroxylase unmask kinetic isotope effects on chemical steps. J. Am. Chem. Soc. 125, 16190–16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb K. E.; Westre T. E.; Kappock T. J.; Mitic N.; Glasfeld E.; Caradonna J. P.; Hedman B.; Hodgson K. O.; Solomon E. I. (1997) Spectroscopic characterization of the catalytically competent ferrous site of the resting, activated, and substrate-bound forms of phenylalanine hydroxylase. J. Am. Chem. Soc. 119, 1901–1915. [Google Scholar]
- Hegg Whiting A. K.; Saari R. E.; McCracken J.; Hausinger R. P.; Que L. Jr. (1999) Herbicide degrading α-keto acid dependent enzyme Tfda: Metal coordination environment and mechanistic insights. Biochemistry 38, 16714–16726. [DOI] [PubMed] [Google Scholar]
- Bollinger J. M.; Price J. C.; Hoffart L. M.; Barr E. W.; Krebs C. (2005) Mechanism of Taurine:α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 2005, 4245–4254. [DOI] [PubMed] [Google Scholar]
- Coleman M. L.; McDonough M. A.; Hewitson K. S.; Coles C.; Mecinović J.; Edelmann M.; Cook K. M.; Cockman M. E.; Lancaster D. E.; Kessler B. M.; Oldham N. J.; Ratcliffe P. J.; Schofield C. J. (2007) Asparaginyl Hydroxylation of the Notch Ankyrin Repeat Domain by Factor Inhibiting Hypoxia-inducible Factor. J. Biol. Chem. 282, 24027–24038. [DOI] [PubMed] [Google Scholar]
- Koivunen P.; Hirsilä M.; Günzler V.; Kivirikko K. I.; Myllyharju J. (2004) Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904. [DOI] [PubMed] [Google Scholar]
- Linke S.; Stojkoski C.; Kewley R. J.; Booker G. W.; Whitelaw M. L.; Peet D. J. (2004) Substrate requirements of the oxygen-sensing asparaginyl hydroxylase factor-inhibiting hypoxia-inducible factor. J. Biol. Chem. 279, 14391–14397. [DOI] [PubMed] [Google Scholar]
- Yang M.; Chowdhury R.; Ge W.; Hamed R. B.; McDonough M. A.; Claridge T. D. W.; Kessler B. M.; Cockman M. E.; Ratcliffe P. J.; Schofield C. J. (2011) Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 278, 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Hardy A. P.; Chowdhury R.; Loik N. D.; Scotti J. S.; McCullagh J. S. O.; Claridge T. D. W.; McDonough M. A.; Ge W.; Schofield C. J. (2013) Substrate Selectivity Analyses of Factor Inhibiting Hypoxia-Inducible Factor. Angew. Chem., Int. Ed. 52, 1700–1704. [DOI] [PubMed] [Google Scholar]
- Wilkins S. E.; Karttunen S.; Hampton-Smith R. J.; Murchland I.; Chapman-Smith A.; Peet D. J. (2012) Factor Inhibiting HIF (FIH) Recognizes Distinct Molecular Features within Hypoxia-inducible Factor-α (HIF-α) versus Ankyrin Repeat Substrates. J. Biol. Chem. 287, 8769–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.