

Abstract
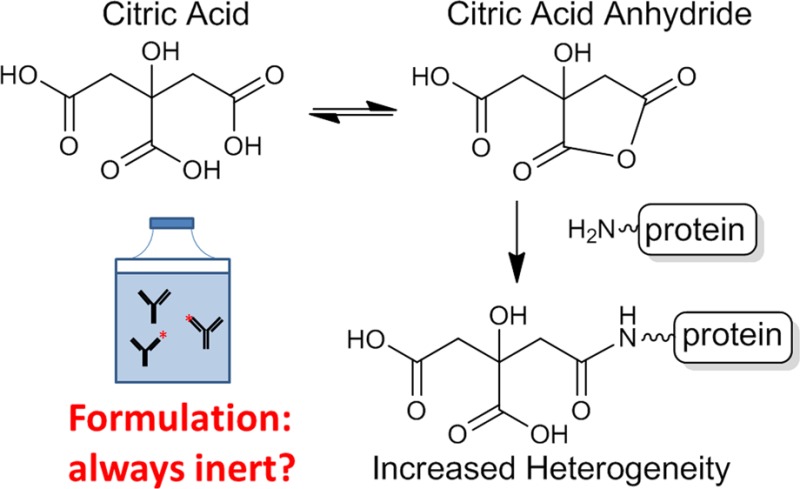
Recombinant therapeutic monoclonal antibodies exhibit a high degree of heterogeneity that can arise from various post-translational modifications. The formulation for a protein product is to maintain a specific pH and to minimize further modifications. Generally Recognized as Safe (GRAS), citric acid is commonly used for formulation to maintain a pH at a range between 3 and 6 and is generally considered chemically inert. However, as we reported herein, citric acid covalently modified a recombinant monoclonal antibody (IgG1) in a phosphate/citrate-buffered formulation at pH 5.2 and led to the formation of so-called “acidic species” that showed mass increases of 174 and 156 Da, respectively. Peptide mapping revealed that the modification occurred at the N-terminus of the light chain. Three additional antibodies also showed the same modification but displayed different susceptibilities of the N-termini of the light chain, heavy chain, or both. Thus, ostensibly unreactive excipients under certain conditions may increase heterogeneity and acidic species in formulated recombinant monoclonal antibodies. By analogy, other molecules (e.g., succinic acid) with two or more carboxylic acid groups and capable of forming an anhydride may exhibit similar reactivities. Altogether, our findings again reminded us that it is prudent to consider formulations as a potential source for chemical modifications and product heterogeneity.
As most protein pharmaceuticals, recombinant monoclonal antibodies have a higher degree of inherent complexity as compared to traditional small molecule drugs. Various protein post-translational modifications (PTMs) have been well documented as major contributors to heterogeneity observed in recombinant monoclonal antibodies.1−6 Some of these processes occur during cell culture, such as modifications by reactive metabolites (e.g., methylglyoxal and homocysteine thiolactone),7,8 glycosylation and sialic acid incorporation,9−17 while others can occur through production, purification, and storage, such as oxidation,18−21 deamidation,22−27 cross-linking,28,29 protein–protein interactions,30 and fragmentation.31−34
An important part of drug development is to optimize formulation for a given biotherapeutic.35,36 The formulation should minimize unwanted modifications or degradation during storage.3,37 For example, polysorbate80 may be added to mitigate aggregation.38−41 Free methionine may reduce the formation of methionine sulfoxide in proteins.42−45 A critical aspect of formulation is the control of pH. One major reason is to minimize the deamidation of asparagine, a spontaneous nonenzymatic process that occurs in all monoclonal antibodies and the vast majority of protein pharmaceuticals.16,22,24−26,46,47 Specifically, mildly acidic pH has been shown to reduce deamidation of asparagine.22−27
While almost all excipients added to the biotherapeutic formulation are Generally Recognized as Safe (GRAS) and considered chemically inert (i.e., free from reactions with the protein products), they may nonetheless display unexpected reactivities. For example, autoxidation of polysorbate 80 generated radicals that in turn increased the oxidative liabilities of the formulation, e.g., increases in methionine sulfoxide.48 Photo-oxidation also induces cleavage, cross-linking, and aggregation.13,29,34,49,50 Glycation has been reported when glucose (a reducing sugar with a hemiacetal or aldehyde group) was added to a lyophilized protein drug.51 As a result of this finding, sucrose (devoid of hemiacetal or aldehyde group) was used instead to reduce aggregation.52 Yet, in other studies, the glycosidic bond of nonreducing sucrose was shown to hydrolyze into glucose and fructose, resulting in glycation during storage.53,54 Pertinent to this work, photochemical degradation of citric acid led to acetonation of therapeutic proteins.55 Therefore, it is important to thoroughly evaluate the protein drug integrity following storage in the defined formulation and to screen for unexpected reactivity and modifications.
As reported herein, we observed an early eluting peak (i.e., acidic species) in the weak cation exchange (WCX) chromatogram for an antibody in citric acid formulation. Peptide mapping and mass spectrometric analysis revealed that covalent modifications by citric acid led to the formation of amides (mass increase of 174 Da) and/or imides (mass increase of 156 Da) at the N-terminus of the light chain.56 Furthermore, three additional recombinant monoclonal antibodies displayed a similar susceptibility of the N-termini of both the light and heavy chains. To the best of our knowledge, this is the first report of a citric acid modification of recombinant monoclonal antibodies. By analogy, other molecules (e.g., succinic acid) with two or more carboxylic acid groups and capable of forming an anhydride may exhibit similar reactivities.57,58 Altogether, our findings again remind us that it is prudent to carefully consider formulation excipients as a potential source for chemical modifications.
Materials and Methods
See the Supporting Information.
Results and Discussion
As detailed below, we found that citric acid covalently modified the N-termini of either or both the light and heavy chains in four different antibodies. Our analysis and results are consistent with the mechanism depicted in Scheme 1 with the anhydrides of citric acid as key intermediates.
Scheme 1. (I) Formation of a Citric Acid Anhydride Intermediate from Citric Acid and the Subsequent Reaction of the N-Terminal Amine with the Anhydride and (II) Four Possible Products of the Reaction.
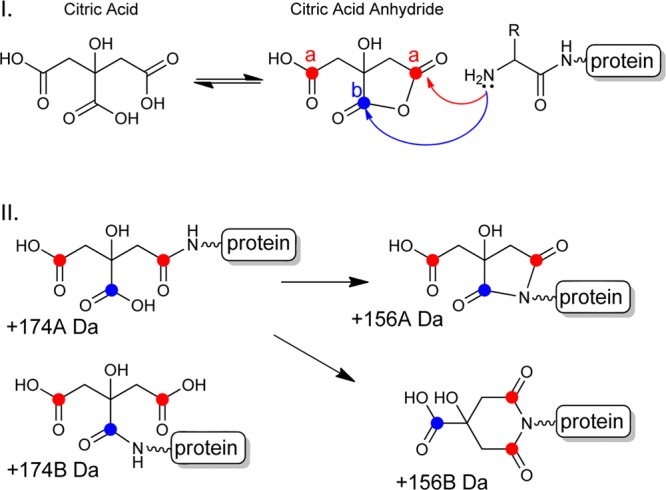
+174A and +174B represent adducts formed between the N-terminal amine and the citric acid anhydride. The +156A and +156B represent the subsequent imide products (5 and 6-membered rings, respectively) resulting from the cyclization of the newly formed amide and another carboxylic acid in citric acid. There are three carboxylic acids in citric acid: two are equivalent as denoted by the red dots and the other by the blue dot.
Unexpected Covalent Modifications by Citric Acid
A recombinant monoclonal antibody (Antibody A) was stored at 40 °C for 6 months in two different formulations to determine if there were any major differences in the protein stability. One formulation had 1 mM sodium citrate, 6.5 mM citric acid and the other formulation was without sodium citrate/citric acid; both had mannitol and polysorbate 80 and were at pH 5.2. Analysis of the samples by weak cation exchange (WCX) chromatography revealed that significant degradation and accumulation of multiple acidic species in both samples (see Figure 1). Most noticeably, the citrate formulation induced a very early eluting and well-defined peak (peak A in Figure 1) that was absent in the other sample. This finding prompted us to perform subsequent analysis in order to determine the nature of these species.
Figure 1.

Weak cation exchange chromatogram of the recombinant monoclonal antibody formulated with and without citrate. The top trace shows an early eluting acidic peak (Peak A) which is significantly smaller in the formulation without citrate. The control represents Antibody A in the citrate formulation stored at 4 °C.
Reduced LC/MS Analysis
Peak A fractions were examined by reduced LC/MS (see Figure S-1 in the Supporting Information). The major peak (observed mass 23 408 Da) corresponded to the native light chain (theoretical mass 23 408 Da). Two other masses of 23 564 and 23 582 were observed, increases of 156 and 174 Da, respectively. Pertinent to the mechanism of formation discussed later, these two masses differ by 18 Da and are likely due to the loss of a water molecule. In addition, a mass of 23 570 Da was determined to be a glycation product (+162 Da).
Peptide Mapping and Determination of Sites of Modifications in Antibody A
Peptide mapping with mass spectrometric detection revealed three tryptic peptides present in the formulation with citrate but were absent in the formulation without citrate. These peaks corresponded to doubly charged ions of peptides with masses of 2051.90 Da (Peptide B) and 2033.88 Da (Peptides C and D), respectively (Figure S-2 in the Supporting Information). Peptides C and D were isobaric yet chromatographically resolved on the C18 RP-HPLC column and both exhibited greater retention and thus greater hydrophobicity than Peptide B. The analysis of the MS/MS spectra of Peptides B, C, and D were in good agreement to each other with the exception of the 18 Da mass shift between some of the b ions but clearly all three spectra were from the same fragmentation series. Manual de novo sequencing (Figure S-5 in the Supporting Information) performed on these peptides revealed high homology to the predicted y ion series of the N-terminal peptide of the light chain (Peak A). A comparison of these MS/MS spectra against the experimental MS/MS spectrum of the N-terminus of the light chain peptide showed high similarity between the fragmentation patterns as shown in Figure S-3 in the Supporting Information. The y ion series between Peak A (Native), Peak B (+174 Da), Peak C and Peak D (+156 Da) covered all residues in the peptide with the exception of the N-terminal aspartate. The b ion series, although limited, showed strong signal with coverage among the first three residues. Consequently, we were able to assign the observed mass increases to the N-terminus of the light chain. Thus, the data confirmed that the mass increases of 156 or 174 Da were from modifications on the light chain N-terminal amine (i.e., Asp1). Subsequent analysis of the heavy chain N-terminal peptide of Antibody A did not show any modification.
Elucidation of the Chemical Nature of the Modifications
No protein modifications listed in either the ABRF Delta Mass database (www.abrf.org/index.cfm/dm.home) or the Unimod database (www.unimod.org) could give rise to the three observed species. As previously stated, citric acid was only present in the formulation of the sample where these modifications were found. The molecular weight of citric acid is 192 Da; therefore, the difference between the observed variants of +174 Da and +156 Da suggests two successive losses of water from citric acid. As illustrated in Scheme 1, we propose a mechanism that involves the initial formation of citric anhydride, the subsequent formation of an amide with an amino group in the protein (e.g., the N-terminus) that results in a molecular weight increase of 174 Da. Further condensation of the resulting amide and another carboxylic group in covalently attached citric acid leads to the formation of imides (either five- or six-membered), which confers a molecular weight increase of 156 Da. In addition, it is reasonable to expect that these two products would form at different rates favoring the 5-membered product and further supported by the two +156 isobaric peptides we observed (156A and 156B shown in Figure S-2 in the Supporting Information). This mechanism is consistent with the results reported on citrate modification of peptides and the propensity of citric acid to form an anhydride under acidic conditions.56−60
Reactions in Citrate Buffers (As Compared to Formulation)
To isolate and narrow down the factors involved in the modification, antibody A was incubated in citric acid buffer at the same pH (5.2) but without other formulation excipients (e.g., without mannitol and polysorbate 80) at 40 °C for 1 month. Similar to the sample from citrate formulation, the weak cation exchange chromatogram (Figure S-4 in the Supporting Information) shows a clear time-dependent increase in the amount of acidic species. In addition, the formation of the distinct early eluting peak has a comparable retention time to peak A from the sample formulated in citrate. Similarly, the reduced LC/MS analysis of the light chain showed a major peak in good agreement with the theoretical mass and also showed two higher molecular weight masses with increases of +156 Da and +174 Da but at a higher abundance than the citrate formulation (Figure S-1 in the Supporting Information). And again, tryptic mapping confirmed on the same adducts localized to the N-terminus of the light chain (data not shown). Thus, these experiments supported our hypotheses that the citric acid was indeed the modifying agent causing the heterogeneity on the N-terminus of the light chain. In addition, we searched for the same modifications on the heavy chain N-terminal peptide and found trace levels of the +174 Da adduct and no detection of the +156 Da adduct, thus we saw similar susceptibility as our stability sample (see Table 1).
Table 1. Percentage of Citric Acid Modification Found in the N-Terminus of Each Chain in Different Antibodiesa.
| recombinant IgG1 | LC N-terminus | +174 Da | +156 Daa | +156 Dab | HC N-terminus | +174 Da | +156 Daa | +156 Dab |
|---|---|---|---|---|---|---|---|---|
| Antibody A (1×, 6 M) | DIQMTQSPSS | 1.3 | 1.2 | 0.4 | EVQLVESGGG | n.d. | n.d. | n.d. |
| Antibody A (20×, 1 M) | DIQMTQSPSS | 7.0 | 2.0 | 1.4 | EVQLVESGGG | 0.02 | n.d. | n.d. |
| Antibody A-S (20×, 1 M) | EIQMTQSPSS | 8.7 | 1.3 | 0.6 | DVQLVESGGG | 0.8 | n.d. | n.d. |
| Antibody B (20×, 1 M) | EIVLTQSPDF | 4.8 | 0.1 | 0.1 | EVQLVQSGAE | 6.4 | 0.4 | 0.6 |
| Antibody C (20×, 1 M) | DVLVTQSPLS | 1.8 | 0.2 | 0.2 | EVKLVESGGG | 3.2 | 0.2 | 0.3 |
Antibody A in citrate formulation for 6 months at 40 °C; and Antibodies A, A-S, B and C in 20× citrate buffer for 30 days at 40 °C. The +156 Daa and +156 Dab refer to the two products formed after the second anhydride formation. The first 10 residues of the N-terminal framework of the heavy chains and light chains of antibodies are also listed (n.d. denotes not detected).
Prevalence of the Citrate Modification
To better understand the scope of this modification, several additional antibodies were examined (see Table 1). One was a variant of Antibody A (Antibody A-S) in which the N-terminus of the light chain had an aspartate substituted with a glutamate and the N-terminus of the heavy chain had a glutamate substituted with an aspartate; in essence, the two termini were swapped. LC/MS analysis of the light and heavy chains of Antibody A-S showed the same site of modification and similar susceptibility as Antibody A (see Table 1), suggesting protein structures (such as solvent accessibility) perhaps play a more dominant role than specific amino acid residues.61−63 Additionally, as shown in Table 1, Antibody B and Antibody C were also modified by citric acid at the N-termini of both the light chain and heavy chain. The modification was also observed on a heavy chain N-terminal alanine residue (data not shown), suggesting that this modification may occur on other residues at the N-terminus and the N-terminal acidic side chains (Asp or Glu) are not obligatory. Thus, the modification of the N-terminal primary amine by citrate appears to be common among recombinant IgG1 monoclonal antibodies but may be influenced by other factors such as the antibody structure and microenvironment.7,8,15,61,62 Furthermore, in all cases, the +174 Da species were more prominent than the +156 Da species, indicating the former are likely the initial products as proposed in Scheme 1.
Influence of pH
As shown in Scheme 1, a key intermediate for the modification is citric acid anhydride.57−60 Citric acid has three pKas of 3.14, 4.75, and 6.39, so at pH 5.2, one of the carboxylic acids will be predominantly protonated, a first step for anhydride formation. As reported, formation of citric acid anhydride occurs between pH 3.0 to 6.0, with the maximum at pH 4.0 to 4.5.56 At pH 5.2 for our formulation, citric acid anhydride can still accumulate to a significant degree and modify the antibodies. Increasing the pH to neutral conditions, however, would markedly diminish the formation of the anhydride, thus little modification of the antibodies (see Figure S-1 in the Supporting Information, pH 7 data).
Selectivity of Amines
We investigated whether there were any citrate modifications to the primary amines of lysine residues following the accelerated storage conditions. We searched the peptide mapping data using the Sequest algorithm (ThermoFisher Scientific) and did not find any modification to lysine residues. In general, N-terminal amines have a lower pKa (around 8) than those on the side chains of lysine (around 10). Under mildly acidic conditions (e.g., pH 5.2), though the vast majority of the N-terminal and lysyl amines are protonated, significant higher percentage of the N-terminal amines are deprotonated, thereby nucleophilic, and can react with anhydride. Therefore, the observed selectivity of amines are consistent with the generally observed reactivities of N-terminal amines.64,65
Conclusions
Our results suggest the general susceptibility of the N-terminal amines to modifications by citric acid. The reactivity is likely influenced by multiple factors, including pH, pKa at the N-terminal amines, and structural features, therefore the sites and extent of modification cannot be precisely predicted and thus should be investigated experimentally. In addition, formulations with elevated concentrations of citric acid would likely cause a greater extent of the modification; therefore, it would be prudent to consider other excipients which may be better suited for the desired pH range. In particular, other molecules containing two or more juxtaposed carboxylic acid groups may exhibit analogous reactivities (via the formation of anhydrides). Examples from the Generally Recognized as Safe (GRAS) list include adipic acid, malic acid, succinic acid, and tartaric acid. Altogether, our findings are yet a reminder that the unexpected reactivity of excipients and formulation, though generally considered chemically inert and safe, should be carefully scrutinized.
Acknowledgments
The authors would like to thank David Lee for his assistance with the chemical pathway and mechanism and Shanshan Liu for assisting us with a detailed literature search. This activity is partially supported by a grant from NIH NIGMS (Grant 1R01GM101396 to Z.S.Z.). This is contribution number 1051 from the Barnett Institute. The design, study conduct, and financial support for the study was provided by AbbVie (formerly the proprietary pharmaceutical division of Abbott Laboratories, now an independent biopharmaceutial company). AbbVie participated in the interpretation of data, review, and approval of the publication. C.C., L.L.Z., Y.S., J.W., E.F., and C.R. are AbbVie employees. R.B. is a former AbbVie employee. Z.S.Z. serves as the doctoral advisor to C.C. and has received AbbVie support for this role.
Supporting Information Available
Materials and methods, mass spectra from LC/MS analysis, weak cation exchange chromatograms, de novo sequencing of doubly charged peptides, and MS/MS analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors contributed to the development of the publication and maintained control over the final content.
The authors declare no competing financial interest.
Notes
⊥ A portion of this work was conducted by C.C. as a doctoral student in the Department of Chemistry and Chemical Biology at Northeastern University.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Awdeh Z. L.; Williamson A. R.; Askonas B. A. Biochem. J. 1970, 116, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Gaza-Bulseco G.; Faldu D.; Chumsae C.; Sun J. J. Pharm. Sci. 2008, 97, 2426–2447. [DOI] [PubMed] [Google Scholar]
- Manning M.; Chou D.; Murphy B.; Payne R.; Katayama D. Pharm. Res. 2010, 27, 544–575. [DOI] [PubMed] [Google Scholar]
- Vlasak J.; Ionescu R. Curr. Pharm. Biotechnol. 2008, 9, 468–481. [DOI] [PubMed] [Google Scholar]
- Fenselau C.; Vestling M. M.; Cotter R. J. Curr. Opin. Biotechnol. 1993, 4, 14–19. [DOI] [PubMed] [Google Scholar]
- Costello C. E. Curr. Opin. Biotechnol. 1999, 10, 22–28. [DOI] [PubMed] [Google Scholar]
- Chumsae C.; Gifford K.; Lian W.; Liu H.; Radziejewski C. H.; Zhou Z. S. Anal. Chem. 2013, 85, 11401–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang T.; Dai S.; Chen D.; Lee B. W. K.; Liu S.; Karger B. L.; Zhou Z. S. Anal. Chem. 2009, 81, 9065–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuochi T.; Taniguchi T.; Shimizu A.; Kobata A. J. Immunol. 1982, 129, 2016–2020. [PubMed] [Google Scholar]
- Parekh R. B.; Dwek R. A.; Sutton B. J.; Fernandes D. L.; Leung A.; Stanworth D.; Rademacher T. W.; Mizuochi T.; Taniguchi T.; Matsuta K.; Takeuchi F.; Nagano Y.; Miyamoto T.; Kobata A. Nature 1985, 316, 452–457. [DOI] [PubMed] [Google Scholar]
- Jefferis R. Biotechnol. Prog. 2005, 21, 11–16. [DOI] [PubMed] [Google Scholar]
- Matthews R. G.; Smith A. E.; Zhou Z. S.; Taurog R. E.; Bandarian V.; Evans J. C.; Ludwig M. Helv. Chim. Acta 2003, 86, 3939–3954. [Google Scholar]
- Zhou Z. S.; Smith A. E.; Matthews R. G. Bioorg. Med. Chem. Lett. 2000, 10, 2471–2475. [DOI] [PubMed] [Google Scholar]
- Wan W.; Zhao G.; Al-Saad K.; Siems W. F.; Zhou Z. S. Rapid Commun. Mass Spectrom. 2004, 18, 319–324. [DOI] [PubMed] [Google Scholar]
- Gui S.; Wooderchak-Donahue W. L.; Zang T.; Chen D.; Daly M. P.; Zhou Z. S.; Hevel J. M. Biochemistry 2012, 52, 199–209. [DOI] [PubMed] [Google Scholar]
- Chen T.; Nayak N.; Majee S. M.; Lowenson J.; Schäfermeyer K. R.; Eliopoulos A. C.; Lloyd T. D.; Dinkins R.; Perry S. E.; Forsthoefel N. R.; Clarke S. G.; Vernon D. M.; Zhou Z. S.; Rejtar T.; Downie A. B. J. Biol. Chem. 2010, 285, 37281–37292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biastoff S.; Teuber M.; Zhou Z. S.; Draeger B. Planta Med. 2006, 72, 1136–1141. [DOI] [PubMed] [Google Scholar]
- Chumsae C.; Gaza-Bulseco G.; Sun J.; Liu H. J. Chromatogr., B 2007, 850, 285–294. [DOI] [PubMed] [Google Scholar]
- Liu H.; Gaza-Bulseco G.; Zhou L. J. Am. Soc. Mass Spectrom. 2009, 20, 525–528. [DOI] [PubMed] [Google Scholar]
- Gaza-Bulseco G.; Faldu S.; Hurkmans K.; Chumsae C.; Liu H. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2008, 870, 55–62. [DOI] [PubMed] [Google Scholar]
- Houde D.; Kauppinen P.; Mhatre R.; Lyubarskaya Y. J. Chromatogr., A 2006, 1123, 189–198. [DOI] [PubMed] [Google Scholar]
- Alfaro J. F.; Gillies L. A.; Sun H. G.; Dai S.; Zang T.; Klaene J. J.; Kim B. J.; Lowenson J. D.; Clarke S. G.; Karger B. L.; Zhou Z. S. Anal. Chem. 2008, 80, 3882–3889. [DOI] [PubMed] [Google Scholar]
- Huang L.; Lu J.; Wroblewski V. J.; Beals J. M.; Riggin R. M. Anal. Chem. 2005, 77, 1432–1439. [DOI] [PubMed] [Google Scholar]
- Ni W.; Dai S.; Karger B. L.; Zhou Z. S. Anal. Chem. 2010, 82, 7485–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Cheetham J.; Cauchon N.; Ostovic J.; Ni W.; Ren D.; Zhou Z. S. Anal. Chem. 2011, 84, 1056–1062. [DOI] [PubMed] [Google Scholar]
- Dai S.; Ni W.; Patananan A. N.; Clarke S. G.; Karger B. L.; Zhou Z. S. Anal. Chem. 2013, 85, 2423–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B.; Steen S.; Hambly D.; Valliere-Douglass J.; Bos T. V.; Smallwood S.; Yates Z.; Arroll T.; Han Y.; Gadgil H.; Latypov R. F.; Wallace A.; Lim A.; Kleemann G. R.; Wang W.; Balland A. J. Pharm. Sci. 2009, 98, 3509–3521. [DOI] [PubMed] [Google Scholar]
- Liu M.; Zhang Z.; Zang T.; Spahr C.; Cheetham J.; Ren D.; Zhou Z. S. Anal. Chem. 2013, 85, 5900–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Zhang Z.; Cheetham J.; Ren D.; Zhou Z. S. Anal. Chem. 2014, 86, 4940–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngounou Wetie A. G.; Sokolowska I.; Woods A. G.; Roy U.; Loo J. A.; Darie C. C. Proteomics 2013, 13, 538–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba A. J.; Shyong B.-J.; Breen D.; Harris R. J. J. Chromatogr., B 2005, 818, 115–121. [DOI] [PubMed] [Google Scholar]
- Liu H.; Gaza-Bulseco G.; Lundell E. J. Chromatogr., B 2008, 876, 13–23. [DOI] [PubMed] [Google Scholar]
- Vlasak J.; Ionescu R. MAbs 2011, 3, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Rejtar T.; Zhou Z. S.; Karger B. L. Rapid Commun. Mass Spectrom. 2010, 24, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Singh S.; Zeng D. L.; King K.; Nema S. J. Pharm. Sci. 2007, 96, 1–26. [DOI] [PubMed] [Google Scholar]
- Park J.; Nagapudi K.; Vergara C.; Ramachander R.; Laurence J.; Krishnan S. Pharm. Res. 2013, 30, 968–984. [DOI] [PubMed] [Google Scholar]
- Patel J.; Kothari R.; Tunga R.; Ritter N. M.; Tunga B. S. BioProc. Int. 2011, 9, 20–31. [Google Scholar]
- Srinivasan C.; Weight A.; Bussemer T.; Klibanov A. Pharm. Res. 2013, 30, 1749–1757. [DOI] [PubMed] [Google Scholar]
- Feng Y. W.; Ooishi A.; Honda S. J. Pharm. Biomed. Anal. 2012, 57, 143–152. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Kita Y. J. Pharm. Sci. 2000, 89, 646–651. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Hao Y.; Chu J.; Wang Y.; Zhang S.; Zhuang Y. Huaxue Yu Shengwu Gongcheng 2008, 25, 49–52. [Google Scholar]
- Lam X. M.; Yang J. Y.; Cleland J. L. J. Pharm. Sci. 1997, 86, 1250–1255. [DOI] [PubMed] [Google Scholar]
- Raijmakers M. T. M.; Schilders G. W.; Roes E. M.; van tits L. J. H.; Hak-Lemmers H. L. M.; Steegers E. A. P.; Peters W. H. M. Clin. Sci. 2003, 105, 173–180. [DOI] [PubMed] [Google Scholar]
- Luo S.; Levine R. L. FASEB J. 2009, 23, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thordstein M.; Baagenholm R.; Thiringer K.; Kjellmer I. Pediatr. Res. 1993, 34, 23–26. [DOI] [PubMed] [Google Scholar]
- Pace A. L.; Wong R. L.; Zhang Y. T.; Kao Y.-H.; Wang Y. J. J. Pharm. Sci. 2013, 102, 1712–1723. [DOI] [PubMed] [Google Scholar]
- Shire S. J. Curr. Opin. Biotechnol. 2009, 20, 708–714. [DOI] [PubMed] [Google Scholar]
- Yao J.; Dokuru D.; Noestheden M.; Park S.; Kerwin B.; Jona J.; Ostovic D.; Reid D. Pharm. Res. 2009, 26, 2303–2313. [DOI] [PubMed] [Google Scholar]
- Mozziconacci O.; Kerwin B. A.; Schöneich C. Chem. Res. Toxicol. 2010, 23, 1310–1312. [DOI] [PubMed] [Google Scholar]
- Torosantucci R.; Schöneich C.; Jiskoot W. Pharm. Res. 2014, 31, 541–553. [DOI] [PubMed] [Google Scholar]
- Li S.; Patapoff T. W.; Overcashier D.; Hsu C.; Nguyen T. H.; Borchardt R. T. J. Pharm. Sci. 1996, 85, 873–877. [DOI] [PubMed] [Google Scholar]
- Andya J. D.; Hsu C. C.; Shire S. J. AAPS PharmSci 2003, 5, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadgil H. S.; Bondarenko P. V.; Pipes G.; Rehder D.; McAuley A.; Perico N.; Dillon T.; Ricci M.; Treuheit M. J. Pharm. Sci. 2007, 96, 2607–2621. [DOI] [PubMed] [Google Scholar]
- Banks D. D.; Hambly D. M.; Scavezze J. L.; Siska C. C.; Stackhouse N. L.; Gadgil H. S. J. Pharm. Sci. 2009, 98, 4501–4510. [DOI] [PubMed] [Google Scholar]
- Valliere-Douglass J. F.; Connell-Crowley L.; Jensen R.; Schnier P. D.; Trilisky E.; Leith M.; Follstad B. D.; Kerr J.; Lewis N.; Vunnum S.; Treuheit M. J.; Balland A.; Wallace A. Protein Sci. 2010, 19, 2152–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole R. A.; Kasper P. T.; Jiskoot W. J. Pharm. Sci. 2011, 100, 3018–3022. [DOI] [PubMed] [Google Scholar]
- Higuchi T.; Miki T.; Shah A. C.; Herd A. K. J. Am. Chem. Soc. 1963, 85, 3655–3660. [Google Scholar]
- Higuchi T.; Eberson L.; McRae J. D. J. Am. Chem. Soc. 1967, 89, 3001–3004. [Google Scholar]
- Higuchi T.; Miki T. J. Am. Chem. Soc. 1961, 83, 3899–3901. [Google Scholar]
- Higuchi T.; Eberson L.; Herd A. K. J. Am. Chem. Soc. 1966, 88, 3805–3808. [DOI] [PubMed] [Google Scholar]
- Zhou Z. S.; Flohr A.; Hilvert D. J. Org. Chem. 1999, 64, 8334–8341. [DOI] [PubMed] [Google Scholar]
- Zhou Z. S.; Jiang N.; Hilvert D. J. Am. Chem. Soc. 1997, 119, 3623–3624. [Google Scholar]
- Zhao G.; Zhou Z. S. Bioorg. Med. Chem. Lett. 2001, 11, 2331–2335. [DOI] [PubMed] [Google Scholar]
- Gilmore J. M.; Scheck R. A.; Esser-Kahn A. P.; Joshi N. S.; Francis M. B. Angew. Chem., Int. Ed. 2006, 45, 5307–5311. [DOI] [PubMed] [Google Scholar]
- Witus L. S.; Moore T.; Thuronyi B. W.; Esser-Kahn A. P.; Scheck R. A.; Iavarone A. T.; Francis M. B. J. Am. Chem. Soc. 2010, 132, 16812–16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.