

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is considered the etiologic agent of Kaposi's sarcoma and several lymphoproliferative disorders. Recently, the KSHV genome was cloned into a bacterial artificial chromosome and used to construct a recombinant KSHV carrying a deletion of the viral interferon regulatory factor gene (F. C. Zhou, Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao, J. Virol. 76:6185-6196, 2002). The K8.1 glycoprotein is a structural component of the KSHV particle and is thought to facilitate virus entry by binding to heparan sulfate moieties on cell surfaces. To further address the role of K8.1 in virus infectivity, a K8.1-null recombinant virus (BAC36ΔK8.1) was constructed by deletion of most of the K8.1 open reading frame and insertion of a kanamycin resistance gene cassette within the K8.1 gene. Southern blotting and diagnostic PCR confirmed the presence of the engineered K8.1 gene deletion. Transfection of the wild-type genome (BAC36) and mutant genome (BAC36ΔK8.1) DNAs into 293 cells in the presence or absence of the complementing plasmid (pCDNAK8.1A), transiently expressing the K8.1A gene, produced infectious virions in the supernatants of transfected cells. These results demonstrated that the K8.1 glycoprotein is not required for KSHV entry into 293 cells.
Kaposi's sarcoma-associated herpesvirus (KSHV), also referred to as human herpesvirus 8, is a member of the gamma-2-herpesvirus family (genus Rhadinovirus) (36, 48). KSHV is etiologically associated with Kaposi's sarcoma, primary effusion or body cavity-based lymphoma, and multicentric Castleman's disease (6, 18, 49, 50). Recently, it was suggested that KSHV may have a role in the development of primary pulmonary hypertension (14). KSHV can infect a variety of human cell types, including B, T, endothelial, epithelial, fibroblast, and keratinocyte cells, and nonhuman cell types, including owl monkey kidney and baby hamster kidney fibroblast cells (9, 12, 16, 17, 19, 26, 29, 32, 33, 43, 45, 56, 61).
Generally, all herpesviruses initiate infection by means of direct binding to various receptors on cell surfaces that is mediated by several viral glycoproteins embedded in viral envelopes. Viral glycoproteins play important roles in virus attachment to susceptible cells, fusion of the viral envelope with either cellular or endosomal membranes, and virion morphogenesis and egress (reviewed in references 25, 31, and 47). Herpes simplex virus, human cytomegalovirus, and Epstein-Barr virus (EBV) have been shown to enter cells by either pH-independent or pH-dependent pathways, depending on the cell type (8, 13, 30, 37, 41, 47, 51). KSHV has been shown to enter certain cells (human foreskin fibroblast cells and B cells) by endocytosis (1, 4). Regardless of the mode of virus entry, the release of capsids into the cytoplasm is thought to involve fusion of the viral envelope with either plasma or endosomal membranes.
KSHV codes for a number of glycoproteins, some of which have significant homology to glycoproteins of other herpesviruses. These include glycoprotein B (gB) (open reading frame [ORF] 8 [ORF8]), gH (ORF22), gM (ORF39), gL (ORF47) (36, 48), and gN (ORF53) (27). In addition, the K1, K8.1, and vOX2 (K14) glycoproteins are unique to KSHV, with no counterparts in other herpesviruses (10, 11, 36, 49). KSHV gB and glycoprotein K8.1A mediate the initial binding of virions to glycosaminoglycans, e.g., heparan sulfate on cell surfaces (2, 4, 7, 57). In agreement with the strong binding of purified K8.1A to heparan sulfate moieties on cell surfaces, initial studies showed that a soluble form of K8.1A inhibited KSHV attachment to cells (57). However, a later report indicated that a similar soluble form of K8.1A did not block KSHV infectivity (7). In addition, gB binds to integrins, such as α3β1-integrin membrane receptors, through an RGD motif, suggesting that integrins function as cellular receptors for KSHV entry (3, 34). However, soluble integrins or RGD-containing peptides failed to inhibit virus entry into 293 cells (22).
There are two ORFs originating from the K8.1 gene by means of spliced transcripts, K8.1A and K8.1B. The K8.1A cDNA encodes a 228-amino-acid protein containing a signal sequence, transmembrane domain, and four N glycosylation sites. The K8.1B cDNA encodes a 167-amino-acid glycoprotein sharing similar amino and carboxy termini with K8.1A but containing an in-frame deletion (10, 44). K8.1A is the predominant form detected within infected cells and virion envelopes (62). The K8.1 gene has attracted significant interest due to the fact that it is positionally colinear to the EBV major glycoprotein gp350/220 gene (20), the murine gammaherpesvirus 68 gp150 gene (53), the herpesvirus saimiri ORF51 gene (5), and the bovine herpesvirus 4 BOEFD1 gene (36, 48). EBV gp350/220 has been shown to be involved in the binding of the virus to target cells by means of the CD21 receptor on B cells (15, 38-40, 55); however, gp350/220 is not required for virus entry into fibroblasts (24).
Recently, the KSHV genome was cloned into a bacterial artificial chromosome (BAC) and shown to produce infectious virus (61). To resolve whether K8.1A functions in virus infectivity, we used the recombinant KSHV BAC36 as the initial template to construct a K8.1-null virus to address the role of K8.1 glycoproteins in the KSHV life cycle. Our data indicate that both K8.1 glycoproteins are dispensable for virus entry.
MATERIALS AND METHODS
Cells and viruses.
293 cells were grown in Dulbecco's modified Eagle medium (DMEM; GIBCO-BRL, Grand Island, N.Y.) supplemented with 2 mM glutamine, 10% fetal calf serum, and antibiotics. KSHV BAC36 contains the green fluorescent protein (GFP) gene cassette under the control of the human cytomegalovirus immediate-early promoter, constitutively expressing the GFP gene, and inserted between KSHV ORF18 and KSHV ORF19 (61).
Immunofluorescence assays.
The detection of K8.1A was monitored by indirect immunofluorescence with fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G, which detected the expression of the K8.1A plasmid in transiently transfected 293 cells. Transfected cells were incubated for 48 h at 37°C and then fixed with cold methanol for 20 min. Monolayers were blocked for 1 h with 10% normal goat serum in phosphate-buffered saline (PBS), followed by 1 h of incubation with the primary monoclonal antibody (MAb) (60), 19B4, directed against K8.1 proteins, at a dilution of 1:500. Cells were rinsed three times with PBS and then incubated for 1 h with the secondary antibody, a fluorescein-conjugated goat anti-mouse antibody (ICN Pharmaceuticals, Inc., Aurora, Ohio), at a dilution of 1:50. Cells were washed five times with PBS and viewed with a fluorescence microscope.
Immunoblot analysis.
Cell lysates of K8.1A-transfected 293 cells or induced BCBL-1 cells were boiled in loading buffer (125 mM Tris-HCl, 2% sodium dodecyl sulfate, 0.1% bromophenol blue, 1% 2-mercaptoethanol) for 5 min, and the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were electrotransferred to nitrocellulose membranes, blocked with BLOTTO (5% nonfat milk in 0.01 M PBS-0.05% Tween 20), and then reacted for 1 h with primary antibody MAb 19B4 (directed against K8.1A) at a dilution of 1:1,000. A horseradish peroxidase-conjugated secondary antiserum at a dilution of 1:10,000 in PBS containing 10% goat serum was used. The reaction was visualized with Western blotting chemiluminescence detection reagents (Pierce Inc., Rockford, Ill.).
Construction of a KSHV mutant with a deletion of the K8.1 gene (BAC36ΔK8.1).
Mutagenesis of BAC36 DNA was accomplished by using Escherichia coli and the λ gam-recE-recT (GET) recombination system (35, 42). Electrocompetent E. coli DH10B cells harboring BAC36 were transformed with plasmid pGETrec, which contains the recE, recT, and bacteriophage λ gam genes, and grown on plates containing chloramphenicol (12.5 μg/ml) and ampicillin (100 μg/ml). Individual colonies were picked and grown overnight in Luria-Bertani (LB) medium containing chloramphenicol and ampicillin. On the next day, the culture was used to inoculate 250 ml of LB medium containing chloramphenicol and ampicillin until an optical density at 600 nm of 0.4 was reached. The addition of l-arabinose to a final concentration of 0.2% (wt/vol) and further incubation for 40 min induced the expression of the recE, recT, and λ gam genes from plasmid pGETrec. The cells then were harvested and made electrocompetent.
A PCR fragment containing the kanamycin resistance gene flanked by 60 bp of viral sequences on both sides was used for recombination to construct BAC36ΔK8.1, containing a kanamycin resistance gene cassette within the targeted K8.1 genomic region. Briefly, electrocompetent DH10B cells (40 μl), harboring both BAC36 and pGETrec, were electroporated with 200 ng of purified PCR product to delete the target gene (K8.1 glycoprotein) by using standard electroporation parameters (1.8 kV/cm, 200 Ω, and 25 μF). Following electroporation, the cells were grown in 1 ml of LB medium for 60 min and then streaked on LB agar plates containing both chloramphenicol (12.5 μg/ml) and kanamycin (50 μg/ml). Episomal mutant BAC36 DNA containing the deletion in either of the K8.1 genes by means of insertion of the kanamycin resistance gene cassette was isolated from bacterial colonies, a second round of electroporation was performed to remove plasmid pGETrec, and the cells were grown on agar plates with chloramphenicol and kanamycin.
Confirmation of the K8.1 deletion in BAC36ΔK8.1 DNA.
KSHV BAC DNAs (BAC36 and BAC36ΔK8.1) were purified from 1 liter of BAC cultures by using a large-construct kit (Qiagen, Valencia, Calif.). BAC DNA was digested with KpnI and run on 0.8% agarose gels, and the restricted DNA was transferred to charged nylon membranes (Bio-Rad, Richmond, Calif.). Southern blot hybridization was performed with a biotin-labeled kanamycin resistance gene probe by labeling a 1.1-kb kanamycin PCR fragment with biotin (New England Biolabs, Boston, Mass.). Chemiluminescence detection of the DNA was performed by using a North2South chemiluminescence hybridization and detection kit as described by the manufacturer (Pierce Inc., Rockford, Ill.).
Transient transfection of KSHV BAC DNAs.
Transient transfection of 293 cells with BAC DNAs was performed by using Superfect (Qiagen). 293 cells were grown to 80% confluence in six-well plates. Cells were transfected with BAC DNA mixed with Superfect in DMEM as recommended by the manufacturer. After 4 h of incubation at 37°C, the medium was removed from the transfected cells, and the cells were washed with PBS. Next, fresh DMEM with 10% fetal calf serum was added.
Immunohistochemical staining.
293 cells in six-well plates were transfected with BAC36 or BAC36ΔK8.1 DNA either alone or with the regulator of transcription activation (RTA) plasmid (cotransfection) for induction of the lytic cycle. At 48 h after transfection, cells were washed with PBS, fixed with electron microscopy-grade 2% paraformaldehyde (Electron Microscopy Sciences; Fort Washington, Pa.) for 10 min, and then washed twice with PBS-50 mM glycine. Blocking of cell monolayers for 1 h was performed with 5% normal goat serum and 5% bovine serum albumin in PBS. Cells were incubated for 1 h with a primary antibody, washed three times with PBS, and then incubated for 1 h with a secondary antibody conjugated with biotin. Cells were washed three times with PBS and then reacted with a 1:3,000 dilution of horseradish peroxidase-streptavidin in 10% goat serum for 1 h. Cells were washed five times, and Vector VIP substrate was added (Vector Laboratories Inc., Burlingame, Calif.). Cells were examined with a light microscope (Nikon Inc., Garden City, N.Y.). The primary antibodies used in these studies were as follows: anti-K8.1 antibody 19B4 (60), anti-ORF59 antibody, and anti-LANA antibody (Advanced Biotechnologies, Inc., Columbia, Md.).
Production of infectious KSHV particles.
293 cells were transfected with either BAC36, BAC36ΔK8.1, or EGFP-C1 DNA each mixed with ORF50 (RTA) and either pCDNA3.1 (negative control plasmid) or pCDNAK8.1 (complementing plasmid) in six-well plates. Transfected cells were induced with a final concentration of 25 ng of tetradecanoyl phorbol acetate (TPA) (Sigma, St. Louis, Mo.)/ml and 1,000 U of alpha interferon (Sigma)/ml for 5 days. Supernatants were collected from induced cell lines and centrifuged three times at 5,000 × g for 15 min. Supernatants were frozen-thawed three times to eliminate any surviving cells. The viral supernatants were used for infection or quantitative PCR analysis.
Infection of cells with harvested supernatants.
Supernatants from transiently transfected cells were used as viral inocula to infect cells at 80% confluence in a 96-well plate. Infection was performed in triplicate. A 50-μl portion of infectious viral inoculum with Polybrene added to a final concentration of 5 μg/ml (Sigma) was placed on 293 cells for 5 h, 50 μl of fresh medium was added, and the plate was incubated overnight at 37°C. On the next day, the viral inoculum was removed and fresh medium was added. Infectivity was determined 2 days postinfection by counting the number of GFP-expressing cells by fluorescence microscopy as described previously (19).
Sample preparation for KSHV quantitative TaqMan PCR.
293 cells were cotransfected with various combinations of KSHV BAC genomes and plasmids pRTA, pCDNA3.1, and pCDNAK8.1. All transfections contained the same amount of total DNA transfected. The amount of BAC36 or BACΔK8.1 remained constant, while various amounts of pCDNA3.1 (control), RTA-expressing plasmid pRTA, and complementing plasmid pCDNAK8.1 were used. TPA (25 ng/ml) and alpha interferon (1,000 U/ml) were added 24 h posttransfection to enhance viral induction. Supernatants were centrifuged three times at 5,000 × g for 15 min to remove floating cells. Supernatants collected after centrifugation were subsequently treated with TurboDNase (Ambion Inc., Austin, Tex.) for 3 h at 37°C to ensure that nonencapsidated genomes were not present during the real-time PCR assay. In these experiments, 1 U of TurboDNase was used per 100 μl of sample. In control experiments, 1 U of TurboDNase reduced the level of BAC36 DNA by more than 1,000-fold, as determined by TaqMan PCR. Viral DNA from supernatants was extracted in triplicate by using a standard proteinase K-phenol-chloroform protocol.
KSHV TaqMan PCR quantitation.
Reagents and enzymes used for TaqMan PCR were obtained from PE Applied Biosystems (Foster City, Calif.). The sequences for the TaqMan FAM probe and ORF37 primers used for the quantitative detection of KSHV molecules were published previously (52) and are listed in Table 1. Each 25-μl PCR mixture contained TaqMan universal PCR master mix and 0.25 μl of 20 μM primer stock for both forward and reverse primers. The ORF37-FAM probe (100 μM) was diluted 1:50, and 0.25 μl of the probe was used per reaction. The reaction conditions were as follows: 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 s and 60°C for 1 min (two-step PCR). During amplification, an ABI-Prism 7700 sequence detector monitored real-time PCR amplification by quantitatively analyzing fluorescence emissions. Samples were analyzed in triplicate in three independent runs. The Ct value was defined as the cycle number in which the fluorescence detected exceeded an established threshold level that was kept constant in all experiments (21).
TABLE 1.
Synthetic oligonucleotide primers and TaqMan probe
| Primer
|
Primer sequence | Purpose and product size | |
|---|---|---|---|
| Designation | Name | ||
| A | 5′K8.1/KanF | 5′-CAATATTAAAGGGACCCAAGTTAATCCCTTAATCCTCTGGGATTAATAACCATGAGTTAGCCACGTTGTGTCTCAAAATCTCTGATGTTA-3′ | GET recombination (1.2 kb) |
| B | 3′K8.1/KanR | 5′-CTAGCACAGGTAAAGTATAAGGACAAGTCCCAGCAATAAACCCACAGCCCATAGTATGTACGGTTGATGA-3′ | |
| C | 5′K8.1/up | 5′-CATGCTGATGCGAATGTGCCA-3′ | Diagnostic PCR for BAC36 (wild type) (964 bp) and BAC36ΔK8.1 (1.4 kb) |
| D | 3′K8.1/nostop | 5′-CACTATGTAGGGTTTCTTACG-3′ | |
| E | 5′KanFwd | 5′-ATGAGCCATATTCAACGG-3′ | Kanamycin probe (1.1 kb) |
| F | 3′KanRev | 5′-CTCATCGAGCATCAAATG-3′ | |
| G | 5′ORF37-Fwd | 5′-TCGGTGGCGATGCTTTAGAC-3′ | TaqMan PCR product (99 bp) |
| H | 3′ORF37-Rev | 5′-TGAAGCAGACGATGCTTTGC-3′ | |
| I | ORF37-FAM | 5′-TCGTAACCCCCGTCTACTTTCCCCG-3′ | TaqMan FAM probe |
| J | K8.1F | 5′-TAACCATGAGTTCCACACAGATTC-3′ | K8.1A PCR product (763 bp) |
| K | K8.1R | 5′-GGTTTTGTGTTACACTATGTAGG-3′ | |
The concentration of BAC36 DNA after purification with the large-construct kit was determined by measuring the optical density at 260 nm and by comparative gel electrophoresis with known amounts of molecular markers. Serial 10-fold dilutions of BAC36 DNA were used as controls to construct a standard curve based on the Ct value and logarithmic amounts of diluted BAC36. The number of KSHV genomes in each specific supernatant sample was determined by comparison of the obtained Ct value to the corresponding value of the standard curve reflecting a specific amount of KSHV DNA. The number of genomes within this specific amount of viral DNA was obtained by using the following relationship: one KSHV genome was approximately equal to 0.197 fg.
RESULTS
Construction of KSHV BAC36ΔK8.1.
The complete KSHV genome was recently cloned into a BAC, yielding BAC36 and enabling the genetic manipulation of the KSHV genome in E. coli. BAC36 constitutively expresses the GFP gene, allowing the detection of eukaryotic cells containing KSHV genomes (61). The BAC-based GET homologous recombination system was used to construct a large deletion within the K8.1 gene in E. coli. Deletion of most of the K8.1 gene was accomplished by targeting the K8.1 ORF with specific primers as detailed in Materials and Methods. The genomic region encompassing the K8.1 gene codes for K8.1A and K8.1B by means of spliced transcripts. The 5′-most primer (primer A; Table 1 and Fig. 1A) used for homologous recombination overlaps the K8.1 ATG and extends into the K8.1 ORF by 6 nucleotides. Insertion of the kanamycin resistance gene cassette produced multiple stop codons in different frames immediately downstream of the ATG codon, preventing the expression of any aberrant proteins. The 3′ primer (primer B; Table 1 and Fig. 1A) is located approximately 90 nucleotides upstream of the TAA stop codon of K8.1 (Fig. 1B).
FIG. 1.

(A) Schematic illustration of the GET homologous recombination system BAC36ΔK8.1 DNA. A PCR fragment containing the kanamycin resistance gene flanked by 60 bp of K8.1 upstream and 3′-end sequences was used to construct BAC36ΔK8.1 DNA recombinants containing the kanamycin resistance gene cassette within the targeted genomic site. The primer binding sites of the majority of the primers (Table 1) used to create the BAC36ΔK8.1 DNA are shown. Seq., sequence. (B) Schematic representation of the ORF50 gene locus and the relevant transcript for ORF50 as described previously (58). Vertical broken lines indicate the deleted genomic region encompassing most of the K8.1 ORF. The relative location of each gene and the locations of splice donor (SD) and splice acceptor (SA) sites are indicated.
The BAC36ΔK8.1 construct was tested for the presence of the engineered insertion by diagnostic PCR and Southern blotting (Fig. 2). Primers C and D (Table 1 and Fig. 1A), located immediately upstream of the K8.1 gene and within the 3′ nondeleted portion of the K8.1 ORF, respectively, were used to amplify a diagnostic DNA fragment from KSHV DNA purified from BCBL-1 cells, BAC36 DNA, and BAC36ΔK8.1 DNA (Fig. 2A). DNA from BCBL-1 cells and BAC36 DNA produced a predicted DNA fragment of 964 bp. In contrast, amplification of the targeted region of BAC36ΔK8.1 DNA produced a DNA fragment of approximately 1.4 kbp, as predicted after insertion of the kanamycin resistance gene cassette. Further confirmation of the genetic content of BAC36ΔK8.1 was obtained by restriction endonuclease fragment analysis and Southern blotting (Fig. 2B). Restriction enzyme analysis of BAC36 DNA and BAC36ΔK8.1 DNA with KpnI revealed similar DNA fragmentation patterns, with the exception that a DNA fragment of approximately 4.6 kbp was absent from the BAC36ΔK8.1 profile but was present in the BAC36 restriction pattern. In addition, the BAC36ΔK8.1 pattern included a new DNA fragment migrating with an apparent size of approximately 5 kbp, in agreement with the theoretical size of a new DNA fragment produced after insertion of the kanamycin resistance gene cassette within the K8.1 gene (Fig. 2B, panel 1). Southern blotting with a biotinylated kanamycin resistance gene probe for KpnI-digested BAC36 DNA and BAC36ΔK8.1 DNA revealed the presence of a unique kanamycin insertion within a DNA KpnI fragment of approximately 5 kbp, while there was no reaction of the probe with the BAC36 DNA. A prebiotinylated molecular weight ladder was used for DNA fragment size determination (Fig. 2B, panel 2). These results are consistent with the insertion of the kanamycin resistance gene cassette within the K8.1 gene and with the deletion of most of the K8.1 ORF.
FIG. 2.
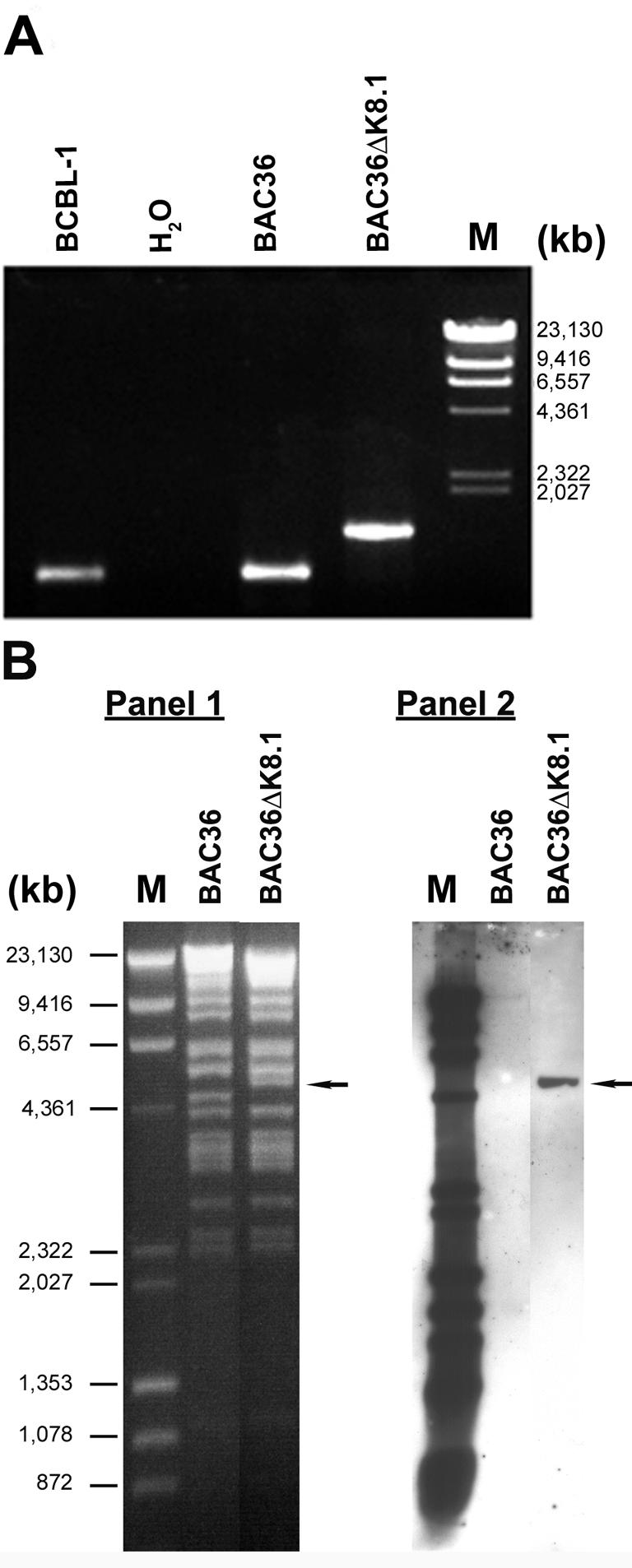
Genomic analysis of BAC36ΔK8.1 DNA. (A) PCR assay to confirm the deletion of the K8.1A gene in BAC36. Amplification of the K8.1 region produced a 964-bp band from both BCBL-1 and BAC36 DNAs; the mutant BAC36ΔK8.1 DNA showed a band of 1,405 bp due to the size difference from deletion or insertion mutagenesis. Lane M, molecular size markers. (B, panel 1) Restriction fragment analysis of BAC36ΔK8.1 DNA and wild-type BAC36 DNA. The KpnI restriction pattern of BAC36ΔK8.1 DNA was compared to that of wild-type BAC36 DNA. As predicted from the KpnI restriction pattern, the introduction of the kanamycin resistance gene into BAC36 led to an increase in size from an estimated 4.5-kb fragment to a 4.9-kb fragment (arrow). (B, Panel 2). Southern blot analysis of BAC36ΔK8.1 DNA. The KpnI restriction fragment from panel 1 was hybridized with a probe derived from a kanamycin PCR product that was labeled with biotin. A biotinylated kanamycin probe hybridized to a 4.9-kb KpnI DNA fragment that was absent in wild-type BAC36 DNA (arrow).
Transient expression of K8.1A.
The K8.1 gene was cloned into the transient expression plasmid pCDNA3.1 and expressed in 293 cells by transfection. Detection of the transiently expressed K8.1A glycoprotein was achieved by indirect immunofluorescence with anti-K8.1 MAb 19B4 (Fig. 3A, panel A) (60). Approximately, 50% of the transfected cells reacted strongly with anti-K8.1 MAb 19B4, while mock-transfected cells failed to react in immunofluorescence assays (Fig. 3A, panels B and D). Similar results were produced by immunohistochemical staining of 293 cells transfected with plasmid pCDNAK8.1A (Fig. 3A, panel C). To further characterize the expression of K8.1A under transient expression conditions, lysates of 293 transfected cells were electrophoretically separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and tested by immunoblotting with MAb 19B4, and the expression of K8.1 was compared to that in BCBL-1 cells after TPA induction (Fig. 3B). Transiently expressed K8.1A migrated with an apparent molecular mass of 37 kDa, while BCBL-1 cells expressed K8.1 migrating with molecular masses ranging from 34 to 37 kDa (Fig. 3B, lanes 2 and 3). The 37-kDa glycoprotein species has been shown to represent fully glycosylated K8.1 glycoprotein (44). MAb 19B4 reacted with proteins of higher apparent molecular masses presumably corresponding to higher-order multimers of K8.1 (60).
FIG. 3.

Detection of K8.1A gene expression in transiently transfected cells. (A) Immunofluorescence (panel A) and immunohistochemical staining (panel C) of 293 cells transiently transfected with K8.1A. Immunofluorescence (panel B) and immunohistochemical reactivities (panel D) of mock-transfected 293 cells. (B) Immunoblot of lysates from mock-transfected cells (lane 1), cells transiently transfected with K8.1A (lane 2), and induced BCBL-1 cells (lane 3).
Characterization of latent and lytic gene expression.
Log-phase 293 cells were transfected with BAC36 or BAC36ΔK8.1 DNA, and the profiles of expression of the LANA latent gene and lytic genes ORF59 and K8.1 were assessed by immunohistochemical staining with MAbs specific for each protein (Fig. 4). Typically, BAC36ΔK8.1 DNA transfected more cells (20 to 30%) than did BAC36 DNA (10 to 15%). In the absence of TPA induction, the anti-LANA antibody reacted with cells transfected with either BAC36 or BAC36ΔK8.1 DNA (Fig. 4F and L). Similar results were obtained after transfected 293 cells were induced with TPA (Fig. 4I and O). Immunostaining of transfected 293 cells with the anti-ORF59 antibody detected the expression of ORF59 (lytic gene) in either the absence (Fig. 4E and K) or the presence (Fig. 4H and N) of TPA induction, although after TPA induction, approximately twofold more cells stained positively for ORF59. Similar results were obtained with anti-K8.1 MAb immunostaining, with the exception that K8.1A was not expressed in 293 cells transfected with BAC36ΔK8.1 DNA (Fig. 4J and M). No reactivity was observed between the anti-K8.1, anti-ORF59, or anti-LANA antibody and mock-transfected 293 cells (negative control) (Fig. 4A, B, and C).
FIG. 4.

Latent and lytic gene expression profiles for uninduced and induced 293 cells transfected with either BAC36 DNA or BAC36ΔK8.1 DNA. Transfections with BAC36ΔK8.1 DNA did not result in any expression of a K8.1A gene product after RTA and TPA induction (J and M). As a positive control, 293 cells transiently transfected with BAC36 DNA were reacted with an anti-K8.1A antibody (D and G). The anti-ORF59 MAb reacted with uninduced or induced (TPA plus RTA) 293 cells transfected with BAC36ΔK8.1 DNA (K and N) or BAC36 DNA (E and H). Similarly, the anti-LANA MAb reacted with uninduced or induced 293 cells transfected with BAC36 DNA (panels F and I) or BAC36ΔK8.1 DNA (L and O). Mock-transfected 293 cells failed to show any reactivity with the anti-K8.1, anti-ORF59, and anti-LANA MAbs (A, B, and C, respectively). Arrows indicate K8.1, ORF59, or LANA expression in 293 cells.
Production of infectious KSHV by BAC36 DNA- and BAC36ΔK8.1 DNA-transfected 293 cells.
To assess the role of K8.1 in virus infectivity, purified BAC36 DNA and BAC36ΔK8.1 DNA were cotransfected with a plasmid transiently expressing the RTA (ORF50) gene and either pCDNA3.1 (control plasmid) or pCDNAK8.1 (complementing plasmid). The number of transfected cells expressing GFP remained constant in the presence or absence of the RTA-expressing plasmid (data not shown). RTA is the major KSHV transactivator protein and is capable of inducing the lytic replication of KSHV (54). KSHV lytic replication was further enhanced through the addition of TPA and alpha interferon for 5 days, at which point supernatants of transfected cells were clarified of cellular debris and tested for the presence of infectious KSHV. The relative infectivities of supernatant viruses were determined by monitoring the number of GFP-expressing cells (see Materials and Methods) after infection of 293 cells (19) (Fig. 5A, panels A, B, and C).
FIG. 5.

Relative infectivities of KSHV mutants. 293 cells were infected with viruses obtained from supernatants of 293 cells transfected with BAC36 DNA and BAC36ΔK8.1 DNA. (A) Approximately 104 293 cells per well in a 96-well plate were incubated with 50 μl of supernatant containing virus obtained from transfections of 293 cells with BAC36, pRTA, and pCDNA3.1 (panel A2), BAC36ΔK8.1, pRTA, and pCDNA3.1 (panel B2), BAC36ΔK8.1, pRTA, and pCDNAK8.1 (panel C2), or pEGFP-C1, pRTA, and pCDNA3.1 (panel D2). Matching phase-contrast images are shown in panels A1, B1, C1, and D1. (B) Virus infectivity was determined by measuring the number of GFP-expressing cells. Error bars indicate standard deviations. All experiments were performed in triplicate. Infectious virions were obtained from 293 cells transfected with DNA mixtures as shown.
Control experiments were also performed in parallel to determine whether cellular debris from transfected cells carried over GFP during the infection process. 293 cells were transfected with plasmids pRTA, pCDNA3.1, and pEGFP-C1 (constitutively expressing GFP). Extracellular fluids were treated in a manner similar to those containing infectious viruses and were tested for their ability to produce GFP-expressing cells under similar infection conditions. These control experiments revealed no GFP carryover from ruptured cells (Fig. 5A, panels D).
Transfection experiments performed in triplicate revealed that BAC36 DNA and BAC36ΔK8.1 DNA consistently produced, on average, 260 and 460 GFP-expressing cells/ml, respectively. Cotransfection of BAC36ΔK8.1 DNA with complementing plasmid pCDNAK8.1 seemed to enhance infectious virus production by approximately twofold (Fig. 5B). Cotransfection of BAC36 DNA with pCDNAK8.1 or other control plasmids did not have an effect on infectious virus production. Furthermore, different isolates of BAC36 or BAC36ΔK8.1 exhibited similar infectivities (data not shown).
Quantitation of KSHV by TaqMan PCR.
To better quantify the amount of virus produced by transfection of 293 cells, a real-time PCR assay targeting the ORF37 gene was used essentially as described previously (52). The relative amounts of BAC36 DNA and BAC36ΔK8.1 DNA found in supernatants of transfected 293 cells were obtained from a standard curve based on known amounts of BAC36 DNA (see Materials and Methods). All BAC-transfected 293 cells produced relatively large numbers of genome equivalents in supernatants (Fig. 6). The number of BAC36ΔK8.1 genomes found in supernatants of transfected cells was consistently higher than that of BAC36 genomes, in part due to the higher transfection efficiency of BAC36ΔK8.1 DNA than of BAC36 DNA (data not shown). Cotransfection of plasmid pCDNAK8.1A with BAC36ΔK8.1 produced approximately two- to threefold more viral genomes in 293 cell supernatants than did transfection with BAC36ΔK8.1 alone (Fig. 6). This effect was pCDNAK8.1 specific, since cotransfection of BAC36ΔK8.1 DNA with vector plasmid pCDNA3.1 did not increase the number of viral genomes found in supernatants (Fig. 5B).
FIG. 6.

Quantitative, real-time PCR assay for determination of KSHV genomes in supernatants of transfected 293 cells. Supernatants were either treated (gray bars) or not treated (black bars) with DNase I. Viral DNA extraction and real-time PCR were performed in triplicate with supernatants obtained from cells transfected with either BAC36, BAC36ΔK8.1, or BAC36ΔK8.1 DNA and various plasmid mixtures. Supernatants from 293 cells transfected with pEGFP-C1, pCDNA3.1, and pRTA were included as a negative control. Error bars indicate standard deviations.
Viral genomes in supernatants were protected from TurboDNase treatment, indicating that most of the viral genomes in 293 cell supernatants were encapsidated (Fig. 6). These results were most likely not due to differences in transfection efficiencies, since the number of transfected cells expressing the viral GFP gene remained approximately constant in the presence or absence of complementing plasmid pCDNAK8.1A (data not shown). Therefore, the two- to threefold increase in the number of encapsidated KSHV genomes with a deletion of K8.1 in supernatants from cells transfected with pCDNAK8.1A, compared to the number seen with transfection with BAC36ΔK8.1 alone, was probably due to the production of a larger number of encapsidated KSHV virions (Fig. 6).
DISCUSSION
Viral glycoproteins encoded by herpesviruses are important determinants of viral infectivity and spread. Although there are significant differences among herpesviruses belonging to the three herpesvirus subfamilies—alpha, beta, and gamma—there are important similarities among all herpesviruses with regard to virus entry into cells and egress from infected cells. Generally, all herpesviruses require initial attachment to cell surfaces, often mediated by binding to ubiquitous glycosaminoglycans, such as heparan sulfate. Virus penetration is achieved by means of either direct fusion of viral envelopes with cellular membranes or virion endocytosis and subsequent fusion of viral envelopes with endosomal membranes, releasing capsids into the cytoplasm of infected cells. For KSHV, initial attachment to cell surfaces is mediated by the binding of gB and glycoprotein K8.1 to heparan sulfate moieties (2, 4, 7, 57). In addition, gB binds to the α3β1-integrin receptor, which is thought to facilitate virus penetration into certain cells by means of endocytosis (3). The role of K8.1A in virus infectivity is not immediately apparent, since an initial report indicated that soluble K8.1A substantially inhibited virion attachment to cells (57), while a later report suggested that a similar soluble form of K8.1A did not inhibit virus infectivity. In this report, we directly addressed the role of K8.1 in virus infectivity by generating a KSHV recombinant with a deletion of both spliced variants of the K8.1 gene. Characterization of this mutant virus in 293 cell culture assays revealed that K8.1 genes were not essential for virus entry.
The BAC36ΔK8.1 virus was made by using the GET homologous recombination system to insert a gene cassette coding for kanamycin resistance within the BAC36K8.1 genomic region. The K8.1 gene deletion spanned the K8.1 ORF immediately after the initiation codon (ATG) and extended to approximately 90 bases upstream of the K8.1 termination codon. KSHV ORF50, specifying the major lytic transactivator RTA, and the K8 ORF specifying K-bZIP, which has been suggested to act either as a repressor of lytic replication (23, 28) or as a replication activator protein (59), lie immediately 5′ to the K8.1 gene. All three genes code for mRNAs that utilize the same polyadenylation signal immediately downstream of K8.1. The engineered deletion of K8.1 removes the last splice acceptor site and, consequently, prevents splicing of the 3′-most exon. It is possible that the resultant 3′ modification of the RTA and K8 mRNAs adversely affects the expression of these proteins. To ensure that the transfected BAC36ΔK8.1 DNA could enter lytic replication, a plasmid capable of the transient expression of RTA was cotransfected with BAC36ΔK8.1 DNA. Consistently, BAC36ΔK8.1 DNA produced three- to fivefold more infectious virus than BAC36 DNA. A possible explanation for this result is that deletion of the terminal exon of K-bZIP (K8) reduced its expression, allowing for improved trans-activation by the exogenously provided RTA.
KSHV genomes remain latent in BCBL-1 cells and could be activated for lytic replication, as evidenced by the expression of lytic proteins, such as those encoded by ORF59 and K8.1 after induction with TPA and/or exogenously provided RTA. Transfection experiments with 293 cells revealed that both BAC36 and BAC36ΔK8.1 genomes expressed a low level of ORF59, indicating that under transient transfection conditions, some viral genomes may be activated for lytic replication in the absence of exogenously provided TPA or RTA; this notion is consistent with previous observations (61). These results are also in agreement with recent observations that infection of primary human umbilical vein endothelial cell cultures occurs in two phases: (i) a permissive phase, in which the cultures undergo active viral lytic replication and infectious virus production; and (ii) a latent phase, in which the surviving cells from the lytic phase switch to latent infection, with a small number of cells undergoing spontaneous viral lytic replication (19). However, in most experiments, trans-activation by RTA produced at least two- to threefold more cells in which KSHV replicated in a lytic phase, as evidenced by the expression of the ORF59 and K8.1 genes. As expected, BAC36ΔK8.1 failed to express any K8.1 under either induced or uninduced conditions.
Transfection of either BAC36 DNA or BAC36ΔK8.1 DNA into 293 cells produced infectious virions in the supernatants of the transfected cells. The number of infectious virions produced by the BAC36ΔK8.1 DNA transfection in triplicate (Fig. 5B) was higher than that obtained with BAC36 DNA. This difference was largely due to the lower transfection efficiency obtained with BAC36 DNA (approximately 15% of 293 cells expressed GFP) than with BAC36ΔK8.1 DNA (approximately 30% of 293 cells expressed GFP), as determined by measuring the number of GFP-expressing cells (data not shown). Cotransfection of BAC36ΔK8.1 DNA with plasmid pCDNAK8.1A, transiently expressing the K8.1 glycoprotein, consistently produced approximately twofold larger numbers of infectious virions in extracellular fluid than did transfections with BAC36ΔK8.1 DNA and pCDNA3.1 (control). This result was not due to differences in transfection efficiencies between BAC36ΔK8.1 DNA-pCDNA3.1 and BAC36ΔK8.1 DNA-pCDNAK8.1 tranfections, because both transfections produced equivalent numbers of GFP-expressing 293 cells. In addition, supernatants from 293 cells transfected with a plasmid expressing GFP (pEGFP-C1) and pCDNA3.1 did not produce any GFP-positive cells, indicating that there was no GFP carryover by potentially ruptured cells. These results indicate that K8.1 is not needed for virus entry, since BAC36ΔK8.1 virions remain infectious in the absence of K8.1.
To differentiate the complementation effect of transiently expressed K8.1 on BAC36ΔK8.1 infectious virus production, the number of KSHV genomes contained in extracellular fluid of transfected 293 cells was determined by real-time PCR. Cotransfection of BAC36ΔK8.1 with complementing plasmid pCDNAK8.1 consistently produced larger numbers of viral genome equivalents in supernatants than did cotransfection of BAC36ΔK8.1 with pCDNA3.1. These genomes represented encapsidated DNA, since treatment with DNase I did not appreciably reduce the number of KSHV genomes, suggesting that K8.1 enhanced infectious virus production.
Previous studies addressing the role of K8.1A in virus attachment showed that a soluble version of K8.1A reduced KSHV attachment by as much as 70% (57). In a latter study, a similar soluble form of K8.1A did not affect virus infectivity (7). In this report, we conclusively show that K8.1 is not necessary for virus infectivity, in agreement with the latter study mentioned above. The relative increase in the number of encapsidated KSHV virions after transient expression of K8.1 suggests that K8.1 may facilitate virion morphogenesis. A similar scenario may occur for EBV gp350/220, since deletion of the gp350/220 gene caused a decrease in infectivity which was complemented in trans by exogenously supplied gp350/220 (24).
Characteristically, KSHV produces a large number of noninfectious virions in supernatants of BCBL-1 cells after induction to enter lytic replication. In one study, it was calculated that approximately 107 genome equivalents were produced per 1 ml of BCBL-1 cell suspension culture at 48 h after TPA induction (46). However, typically, a very small portion of these viruses enter susceptible cells and initiate a viral infection, as evidenced by the expression of either latent or lytic genes (A. Baghian, R. E. Luna, and K. G. Kousoulas, unpublished data). Similarly, BAC36 and BAC36ΔK8.1 transfections produced a relatively large number of encapsidated genomes in the supernatants of transfected cells; however, a very small percentage of these virions were infectious, as evidenced by the expression of the virus-encoded GFP gene after infection of 293 cells. It is unclear at this time why KSHV produces such a large number of noninfectious viruses, which are presumably defective in one or more functions. One possibility is that 293 cells do not support efficient cytoplasmic morphogenesis and egress, resulting in a high percentage of partially defective virions in supernatant fluids. The production of high-titer infectious KSHV should enable ultrastructural visualization of egressing KSHV, visualization of the intracellular sites used by the virus, and dissection of the roles of individual glycoproteins in efficient morphogenesis and egress.
Acknowledgments
This work was supported by NIH grant R01AI43000 to K. G. Kousoulas and by NIH grant R01CA096512 to S.-J. Gao. R. Luna was supported by a supplement to R01AI43000 and by NIH/NCRR grant P20 RR16456 (principal investigator, E. Wischusen). We acknowledge financial support given to BIOMMED by the School of Veterinary Medicine, Louisiana State University.
We thank R. Sun for providing the RTA-expressing plasmid.
REFERENCES
- 1.Akula, S. M., P. P. Naranatt, N. S. Walia, F. Z. Wang, B. Fegley, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 77:7978-7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2001. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284:235-249. [DOI] [PubMed] [Google Scholar]
- 3.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407-419. [DOI] [PubMed] [Google Scholar]
- 4.Akula, S. M., F. Z. Wang, J. Vieira, and B. Chandran. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282:245-255. [DOI] [PubMed] [Google Scholar]
- 5.Albrecht, J. C., J. Nicholas, D. Biller, K. R. Cameron, B. Biesinger, C. Newman, S. Wittmann, M. A. Craxton, H. Coleman, B. Fleckenstein, et al. 1992. Primary structure of the herpesvirus saimiri genome. J. Virol. 66:5047-5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antman, K., and Y. Chang. 2000. Kaposi's sarcoma. N. Engl. J. Med. 342:1027-1038. [DOI] [PubMed] [Google Scholar]
- 7.Birkmann, A., K. Mahr, A. Ensser, S. Yaguboglu, F. Titgemeyer, B. Fleckenstein, and F. Neipel. 2001. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol. 75:11583-11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodaghi, B., M. E. Slobbe-van Drunen, A. Topilko, E. Perret, R. C. Vossen, M. C. van Dam-Mieras, D. Zipeto, J. L. Virelizier, P. LeHoang, C. A. Bruggeman, and S. Michelson. 1999. Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosis. Investig. Ophthalmol. Vis. Sci. 40:2598-2607. [PubMed] [Google Scholar]
- 9.Cerimele, F., F. Curreli, S. Ely, A. E. Friedman-Kien, E. Cesarman, and O. Flore. 2001. Kaposi's sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 75:2435-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandran, B., C. Bloomer, S. R. Chan, L. Zhu, E. Goldstein, and R. Horvat. 1998. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology 249:140-149. [DOI] [PubMed] [Google Scholar]
- 11.Chung, Y. H., R. E. Means, J. K. Choi, B. S. Lee, and J. U. Jung. 2002. Kaposi's sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 76:4688-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciufo, D. M., J. S. Cannon, L. J. Poole, F. Y. Wu, P. Murray, R. F. Ambinder, and G. S. Hayward. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Compton, T., R. R. Nepomuceno, and D. M. Nowlin. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387-395. [DOI] [PubMed] [Google Scholar]
- 14.Cool, C. D., P. R. Rai, M. E. Yeager, D. Hernandez-Saavedra, A. E. Serls, T. M. Bull, M. W. Geraci, K. K. Brown, J. M. Routes, R. M. Tuder, and N. F. Voelkel. 2003. Expression of human herpesvirus 8 in primary pulmonary hypertension. N. Engl. J. Med. 349:1113-1122. [DOI] [PubMed] [Google Scholar]
- 15.Fingeroth, J. D., J. J. Weis, T. F. Tedder, J. L. Strominger, P. A. Biro, and D. T. Fearon. 1984. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. USA 81:4510-4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flore, O., S. Rafii, S. Ely, J. J. O'Leary, E. M. Hyjek, and E. Cesarman. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588-592. [DOI] [PubMed] [Google Scholar]
- 17.Foreman, K. E., J. Friborg, Jr., W. P. Kong, C. Woffendin, P. J. Polverini, B. J. Nickoloff, and G. J. Nabel. 1997. Propagation of a human herpesvirus from AIDS-associated Kaposi's sarcoma. N. Engl. J. Med. 336:163-171. [DOI] [PubMed] [Google Scholar]
- 18.Ganem, D. 1998. Human herpesvirus 8 and its role in the genesis of Kaposi's sarcoma. Curr. Clin. Top. Infect. Dis. 18:237-251. [PubMed] [Google Scholar]
- 19.Gao, S. J., J. H. Deng, and F. C. Zhou. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738-9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong, M., and E. Kieff. 1990. Intracellular trafficking of two major Epstein-Barr virus glycoproteins, gp350/220 and gp110. J. Virol. 64:1507-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 22.Inoue, N., J. Winter, R. B. Lal, M. K. Offermann, and S. Koyano. 2003. Characterization of entry mechanisms of human herpesvirus 8 by using an Rta-dependent reporter cell line. J. Virol. 77:8147-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izumiya, Y., S. F. Lin, T. Ellison, L. Y. Chen, C. Izumiya, P. Luciw, and H. J. Kung. 2003. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol. 77:1441-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Hammerschmidt, and H. J. Delecluse. 2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 74:10142-10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kieff, E., and A. B. Rickinson. 2001. Epstein-Barr virus and its replication, p. 2511-2574. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 26.Kliche, S., E. Kremmer, W. Hammerschmidt, U. Koszinowski, and J. Haas. 1998. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J. Virol. 72:8143-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koyano, S., E. C. Mar, F. R. Stamey, and N. Inoue. 2003. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 84:1485-1491. [DOI] [PubMed] [Google Scholar]
- 28.Liao, W., Y. Tang, S. F. Lin, H. J. Kung, and C. Z. Giam. 2003. K-bZIP of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 77:3809-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesri, E. A., E. Cesarman, L. Arvanitakis, S. Rafii, M. A. Moore, D. N. Posnett, D. M. Knowles, and A. S. Asch. 1996. Human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med. 183:2385-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller, N., and L. M. Hutt-Fletcher. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mocarski, E. S., and C. T. Courcelle. 2001. Cytomegaloviruses and their replication, p. 2629-2674. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 32.Moore, P. S., S. J. Gao, G. Dominguez, E. Cesarman, O. Lungu, D. M. Knowles, R. Garber, P. E. Pellett, D. J. McGeoch, and Y. Chang. 1996. Primary characterization of a herpesvirus agent associated with Kaposi's sarcoma. J. Virol. 70:549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moses, A. V., K. N. Fish, R. Ruhl, P. P. Smith, J. G. Strussenberg, L. Zhu, B. Chandran, and J. A. Nelson. 1999. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 73:6892-6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naranatt, P. P., S. M. Akula, C. A. Zien, H. H. Krishnan, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narayanan, K., R. Williamson, Y. Zhang, A. F. Stewart, and P. A. Ioannou. 1999. Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 6:442-447. [DOI] [PubMed] [Google Scholar]
- 36.Neipel, F., J. C. Albrecht, and B. Fleckenstein. 1997. Cell-homologous genes in the Kaposi's sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity? J. Virol. 71:4187-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nemerow, G. R., and N. R. Cooper. 1984. Early events in the infection of human B lymphocytes by Epstein-Barr virus: the internalization process. Virology 132:186-198. [DOI] [PubMed] [Google Scholar]
- 38.Nemerow, G. R., R. A. Houghten, M. D. Moore, and N. R. Cooper. 1989. Identification of an epitope in the major envelope protein of Epstein-Barr virus that mediates viral binding to the B lymphocyte EBV receptor (CR2). Cell 56:369-377. [DOI] [PubMed] [Google Scholar]
- 39.Nemerow, G. R., C. Mold, V. K. Schwend, V. Tollefson, and N. R. Cooper. 1987. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 61:1416-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nemerow, G. R., R. Wolfert, M. E. McNaughton, and N. R. Cooper. 1985. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor (CR2). J. Virol. 55:347-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicola, A. V., A. M. McEvoy, and S. E. Straus. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orford, M., M. Nefedov, J. Vadolas, F. Zaibak, R. Williamson, and P. A. Ioannou. 2000. Engineering EGFP reporter constructs into a 200 kb human beta-globin BAC clone using GET recombination. Nucleic Acids Res. 28:E84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panyutich, E. A., J. W. Said, and S. A. Miles. 1998. Infection of primary dermal microvascular endothelial cells by Kaposi's sarcoma-associated herpesvirus. AIDS 12:467-472. [DOI] [PubMed] [Google Scholar]
- 44.Raab, M. S., J. C. Albrecht, A. Birkmann, S. Yaguboglu, D. Lang, B. Fleckenstein, and F. Neipel. 1998. The immunogenic glycoprotein gp35-37 of human herpesvirus 8 is encoded by open reading frame K8.1. J. Virol. 72:6725-6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 47.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2460. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 48.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulz, T. F., Y. Chang, and P. S. Moore. 1998. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8), p. 87-134. In D. J. McCance (ed.), Human tumor viruses. American Society for Microbiology, Washington, D.C.
- 50.Schulz, T. F., J. Sheldon, and J. Greensill. 2002. Kaposi's sarcoma associated herpesvirus (KSHV) or human herpesvirus 8 (HHV8). Virus Res. 82:115-126. [DOI] [PubMed] [Google Scholar]
- 51.Spear, P. G. 1993. Membrane fusion induced by herpes simplex virus, p. 201-232. In J. Bentz (ed.), Viral fusion mechanisms. CRC Press Inc., Boca Raton, Fla.
- 52.Stamey, F. R., M. M. Patel, B. P. Holloway, and P. E. Pellett. 2001. Quantitative, fluorogenic probe PCR assay for detection of human herpesvirus 8 DNA in clinical specimens. J. Clin. Microbiol. 39:3537-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart, J. P., N. J. Janjua, S. D. Pepper, G. Bennion, M. Mackett, T. Allen, A. A. Nash, and J. R. Arrand. 1996. Identification and characterization of murine gammaherpesvirus 68 gp150: a virion membrane glycoprotein. J. Virol. 70:3528-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanner, J., J. Weis, D. Fearon, Y. Whang, and E. Kieff. 1987. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 50:203-213. [DOI] [PubMed] [Google Scholar]
- 56.Vieira, J., P. O'Hearn, L. Kimball, B. Chandran, and L. Corey. 2001. Activation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 75:1378-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, F. Z., S. M. Akula, N. P. Pramod, L. Zeng, and B. Chandran. 2001. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 75:7517-7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.West, J. T., and C. Wood. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150-5163. [DOI] [PubMed] [Google Scholar]
- 59.Wu, F. Y., J. H. Ahn, D. J. Alcendor, W. J. Jang, J. Xiao, S. D. Hayward, and G. S. Hayward. 2001. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J. Virol. 75:1487-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu, L., R. Renne, D. Ganem, and B. Forghani. 2000. Human herpesvirus 8 glycoprotein K8.1: expression, post-translational modification and localization analyzed by monoclonal antibody. J. Clin. Virol. 17:127-136. [DOI] [PubMed] [Google Scholar]
- 61.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu, L., V. Puri, and B. Chandran. 1999. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 262:237-249. [DOI] [PubMed] [Google Scholar]