

Abstract
13C NMR has many advantages for a metabolomics study, including a large spectral dispersion, narrow singlets at natural abundance, and a direct measure of the backbone structures of metabolites. However, it has not had widespread use because of its relatively low sensitivity compounded by low natural abundance. Here we demonstrate the utility of high-quality 13C NMR spectra obtained using a custom 13C-optimized probe on metabolomic mixtures. A workflow was developed to use statistical correlations between replicate 1D 13C and 1H spectra, leading to composite spin systems that can be used to search publicly available databases for compound identification. This was developed using synthetic mixtures and then applied to two biological samples, Drosophila melanogaster extracts and mouse serum. Using the synthetic mixtures we were able to obtain useful 13C–13C statistical correlations from metabolites with as little as 60 nmol of material. The lower limit of 13C NMR detection under our experimental conditions is approximately 40 nmol, slightly lower than the requirement for statistical analysis. The 13C and 1H data together led to 15 matches in the database compared to just 7 using 1H alone, and the 13C correlated peak lists had far fewer false positives than the 1H generated lists. In addition, the 13C 1D data provided improved metabolite identification and separation of biologically distinct groups using multivariate statistical analysis in the D. melanogaster extracts and mouse serum.
NMR-based metabolomics is most commonly done with 1H detection. Although 13C is often used for compound identification, it is usually recorded indirectly using experiments such as HSQC (heteronuclear single quantum correlation), HMBC (heteronuclear multiple bond correlation), or their variants, all of which employ 1H detection. These indirect methods are powerful but suffer from the unreliable detection of quaternary carbons and correlations that are sometimes difficult to assign, especially with HMBC. Indirect methods also suffer from limited digital resolution along the large 13C chemical shift dimension. Despite its widespread use, 1H NMR metabolomics studies of mixtures are often complicated by resonance overlap from a relatively narrow 10 ppm diamagnetic chemical shift range. This overlap is exacerbated by 1H homonuclear coupling, which creates multiplets that can complicate analysis, although 2D J-resolved experiments can improve that problem. Many 1H chemical shifts are sensitive to pH, temperature, or salts, complicating spectral alignment and database matching. In addition, water reduction can obscure nearby resonances and distort baselines, which can make 1H-based metabolomics analyses difficult.
13C NMR offers many advantages for a metabolomics study, either alone or as a complement to 1H NMR: (1) 13C spectral windows are typically 200 ppm, providing much greater chemical shift dispersion than 1H; (2) at natural abundance, 13C resonances of small metabolites are narrow singlets (with 1H decoupling) resulting in less spectral overlap; (3) biogenic metabolites predominantly contain a carbon backbone, and 13C NMR can detect this directly, including quaternary carbons. These advantages raise the possibility of easier identification of metabolites and better separation between experimental groups using multivariate analyses because of better spectral dispersion.
The major obstacle to 13C NMR metabolomics compared to 1H is low sensitivity due to low natural abundance of 13C (∼1.1%) combined with a decreased gyromagnetic ratio γ (one-quarter of that of 1H). Because the energy of an NMR resonance transition is proportional to γ3, 13C has an intrinsic sensitivity decrease of nearly 64 (43) when compared to 1H. Though solutions such as isotopic labeling exist,1−5 they can be expensive and are often restricted to targeted metabolomics. Untargeted studies with 13C labeling are especially challenging for organisms that cannot be cultured. In addition, isotopic 13C labeling reintroduces the problem of homonuclear couplings, which complicate 13C spectra. The other solution is to improve 13C sensitivity, either through higher magnetic field strengths that improve the Boltzmann polarization,6 a variety of hyperpolarization techniques,7,8 or improved detection with an optimized probe.9 We have designed and built a 1.5 mm high-temperature superconducting (HTS) 13C optimized probe. This probe has a minimal sample volume of 35 μL and over 2× greater 13C mass sensitivity than any commercially available 13C-optimized cryogenic probe, which translates to well over 4× less measurement time for the same sample and same signal-to-noise ratio.9 This new technology makes 13C metabolomics at natural abundance technically feasible.
In the present study we show that 1D 13C data enhance metabolite identification and facilitate better separation of groups for comparison using multivariate statistical analysis when compared to 1D 1H data alone. We analyzed the 13C and 1H 1D data sets with 13C–13C statistical total correlation spectroscopy (STOCSY)10,11 and 13C–1H statistical heterospectroscopy (SHY)12−14 to compile correlated peak lists that were then used in database searches for compound identification. We developed and validated the workflow using synthetic mixtures of 20 common metabolites, defined the limits of detection using current technology, and compared the peak lists generated using both 13C and 1H data to those generated using 1H data alone. We then applied our workflow to comparisons of two types of biological mixtures: extracts of cold hardy and cold susceptible Drosophila melanogaster flies, to demonstrate the superior performance of 1D 13C compared to 1D 1H in multivariate statistical analysis, and mouse serum taken from control and the Duchenne muscular dystrophy mouse model, mdx,(15) to demonstrate the power of 13C metabolomics to not only aid in metabolite identification but also to prevent misidentification.
Experimental Methods
Synthetic Mixtures
Synthetic mixtures with known metabolite compositions were constructed to test the limits of detection of the probe and our ability to correctly identify known compounds. Two groups of mixtures (groups A and B, five replicates each) were each made using 20 common synthetic metabolites ranging in concentration from 1 to 5 mM (Table 1). The first 10 metabolites were at equal concentrations in all mixtures in both groups, and the remaining 10 metabolites differed between groups with half higher in group A and half higher in group B (Table 1). Rather than prepare a uniform batch to eliminate concentration differences between samples, each metabolite was pipetted individually into each mixture. This was to increase the random mixture-to-mixture variation of individual metabolites both within and between groups due to pipetting errors. This random variation between samples allowed us to develop and validate the statistical methods (i.e., STOCSY) used in this study. Variation was not controlled in this experiment and is assumed to be random. Each sample was brought to 50 μL using 99% D2O (Cambridge Isotope Laboratories), of which 40 μL was pipetted into a 1.5 mm (OD) NMR tube (Norell).
Table 1. Composition and Summary Results for the Synthetic Metabolite Mixtures Used in This Study.
| target
concentrationa per group (mM) |
database
IDsb/false correlationsc |
|||
|---|---|---|---|---|
| metabolite | A | B | 13Cd | 1He |
| l-alanine | 1.5 | 1.5 | Y/1 | N |
| d-trehalose | 1.0 | 1.0 | N | N |
| l-phenylalanine | 1.0 | 1.0 | N | N |
| malate | 2.5 | 2.5 | N | N |
| l-aspartic acid | 3.0 | 3.0 | Y/0 | N |
| l-asparagine | 2.5 | 2.5 | Y/0 | N |
| l-threonine | 3.5 | 3.5 | Y/0 | N |
| d-fructose | 1.5 | 1.5 | N | N |
| l-glutamine | 2.0 | 2.0 | N | N |
| l-valine | 3.0 | 3.0 | Y/1 | Y/0 |
| l-methionine (Met) | 2.0 | 1.0 | Y/0 | N |
| l-proline (Pro) | 3.0 | 1.5 | Y/0 | N |
| l-glutamate (Glu) | 5.0 | 2.0 | Y/2 | Y/8 |
| d-glucose (Glc) | 2.5 | 1.5 | Y/0 | Y/9 |
| l-isoleucine (Ile) | 3.0 | 1.0 | Y/0 | Y/0 |
| l-arginine (Arg) | 2.0 | 3.0 | Y/2 | Y/0 |
| nicotinamide (NAM) | 2.0 | 3.5 | Y/0 | N |
| l-carnitine (Car) | 1.0 | 2.0 | Y/0 | N |
| l-lysine (Lys) | 1.0 | 3.5 | Y/0 | Y/9 |
| l-tryptophan (Trp) | 1.5 | 2.5 | Y/1 | Y/3 |
| compds identified in BMRB | 15/20 | 7/20 | ||
Compounds were transferred to NMR tubes individually to simulate typical between-sample variation, as described in the Methods.
A database ID is yes (“Y”) when a COLMAR query search of the BMRB returns the known compound in the top 10 matches. Otherwise it is no (“N”).
False correlations are peaks that were in the peak list generated from the correlation maps but not part of the identified compound.
Results obtained using 13C and the entire workflow outlined in this paper.
Results obtained by only using 1H–1H STOCSY data and no 13C data.
Drosophila melanogaster Tissue Extracts
Flies were selected in the laboratory for high (hardy) or low (susceptible) cold hardiness (Williams et al., unpublished results). Flies were lyophilized and weighed prior to metabolite extraction. Polar-phase metabolites were extracted from homogenized fly tissue using an aqueous-optimized metabolite extraction protocol.16 Twenty replicates of each genetic line (cold hardy and cold tolerant) were collected with 40 flies per replicate. Polar phase extracted metabolites were dried and reconstituted in 40 μL of 99% D2O (Cambridge Isotope Laboratories) and pipetted into 1.5 mm NMR tubes (Norell).
Mouse Serum
The Duchenne muscular dystrophy (DMD) mouse model, mdx, has previously been investigated with metabolomics using tissue extracts, but to our knowledge, serum has not been investigated using both 1H and 13C. One technical difficulty is the small quantity of sample available for NMR in mouse serum studies. Mouse measurements were performed on serum from 6 month old C57/B10 control (n = 6) and mdx (n = 8) mice. Two hundred microliters of blood was collected by submandibular puncture, clotted, and centrifuged to extract serum. The mice were then humanely euthanized under 2% isofluorane anesthesia according to approved IACUC protocols. Once the serum was separated from the red blood cells, it was flash frozen in liquid nitrogen and stored at −80 °C. Approximately 100 μL of serum was lyophilized and resuspended in 40 μL of 99% D2O and transferred into 1.5 mm NMR tubes (Norell).
NMR Data Collection and Processing
One-dimensional 1H and 13C spectra were collected on an Agilent VNMRS-600 spectrometer using a custom 1.5 mm 13C high-temperature superconducting (HTS) probe.9 Synthetic mixture and fly extract 1H 1D data were collected in ∼2.5 min using a simple pulse sequence with presaturation of residual water, a spectral width of 12 ppm (7183.9 Hz), an observe frequency of 599.68 MHz, a 2.0 s relaxation delay, a 45° pulse, and 2.3 s acquisition time. Mouse serum 1H data were collected in 18 min using a Carr–Purcell–Meiboom–Gill (CPMG) sequence, to remove the protein signal contribution resulting in a flat baseline. Mouse data were recorded with a spectral width of 16 ppm (9615.4 Hz), a 2.0 s relax delay, a 90° initial pulse, a train of 124 180° pulses with a 1 ms interpulse delay, and a 2.0 s acquisition time. All 13C spectra were collected under conditions that favor nuclei with short T1 relaxation times to maximize overall sensitivity and minimize measurement time: a 60° pulse with a 0.1 s relaxation delay and a 0.8 s acquisition time. The total time for each 13C spectrum was ∼2 h, with a 212 ppm (32051.3 Hz) spectral window and a carrier frequency of 98.0 ppm at a frequency of 150.79 MHz. The mouse serum 13C CPMG sequence used a 90° excitation pulse followed by a train of 18 180° pulses with a 1 ms interpulse delay. All 13C spectra were recorded using continuous 1H decoupling at 599.68 MHz with a power of 37 dB using WALTZ-16. All NMR spectra were processed in NMRPipe.17 All 1H spectra were processed using a cosine-squared window function, zero-filled 2×, Fourier-transformed, and baseline-corrected. 13C spectra were processed using an exponential window function with a 2 Hz line broadening, zero-filled 2×, Fourier-transformed, and baseline-corrected. The synthetic mixture and fly extract 1H spectra were referenced to TSP (0.0 ppm); mouse serum 1H spectra were referenced to the lactate peak at 4.1 ppm, because TSP binds to proteins. 13C spectra were referenced to the anomeric carbon glucose peak at 98.64 ppm.
Fourier-transformed and referenced spectra were imported into an in-house MATLAB Metabolomics Toolbox that has been developed in our laboratory. This Toolbox is a collection of MATLAB scripts that includes various analytical tools for the alignment, normalization, scaling, and multivariate analysis of complex data. The Metabolomics Toolbox has several options for alignment, but we found that the star alignment algorithm where an individual spectrum is chosen as a “star” to which all other spectra are aligned,18 gave the best results in this study. For the statistical correlations, we binned the 1H spectra using an open source optimized bucketing algorithm described by Sousa et al.1913C spectra were peak picked by setting all resonances below 2× the standard deviation of all spectra to zero, finding local maxima, and then merging peaks within 0.2 ppm together. Optimal normalization and scaling methods were specific for each data set. 13C and 1H spectra of the synthetic mixtures and mice serum were normalized using probabilistic quotient normalization20 (PQN) and scaled using pareto scaling.21 PQN can be used with NMR spectra in the absence of a reliable internal standard. Pareto scaling is similar to autoscaling except that it keeps more to the original data set and is less susceptible to noise. This prevents the high-intensity peaks from dominating our models. 1H fly spectra were normalized to TSP (an internal standard) and scaled using range scaling.21 Range scaling is also similar to autoscaling by allowing all metabolites to become equally important. Unlike autoscaling, it uses the biological range as a scaling factor. This is useful for exploratory analysis as in our experiments and multivariate statistical analysis were performed with no previous knowledge of the contents of our fly mixtures. Range scaling has been shown to give biologically sensible results.21 Principal component analysis (PCA) was conducted on the 1D 1H and 13C data sets of the synthetic mixtures and fly samples. Partial least-squares-discriminant analysis (PLS-DA) was conducted on the 1D 1H and 13C data sets of the both the fly samples and mouse serum. All NMR raw data, processing scripts, and MATLAB code were deposited in the Metabolomics Workbench database (http://www.metabolomicsworkbench.org/) supported by the NIH Common Fund.
Data Analysis
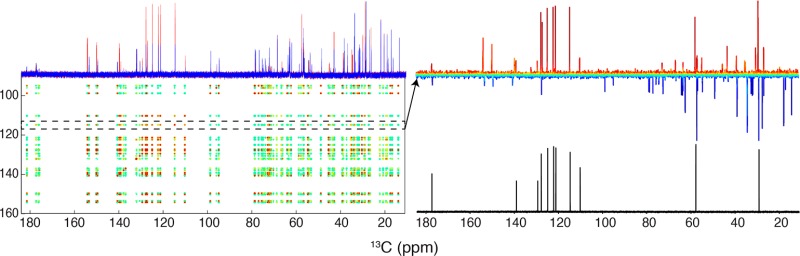
Our workflow consisted of computing statistical correlations to compile peak lists, followed by database searching to identify metabolites (Figure 1). All of the steps described below were implemented in an in-house MATLAB Metabolomics Toolbox22 that was used to analyze all the data sets.
Figure 1.

Overview of approach to 1H and 13C NMR statistical analysis of synthetic mixtures. The 1D 13C (A) and 1H (B) spectra were processed, aligned, normalized, and scaled. Compounds below 1.5 mM (indicated by * in (A)) showed no significant correlations. 13C–13C STOCSY (C) and 13C–1H SHY (D) maps were then made from all of the data in (A) and (B). An expansion of the aromatic region of the 13C–13C STOCSY (C) is shown in H. 13C–13C STOCSY traces of resonances (i.e., driver peak (↓) at 114.8 ppm in (E)) were used to generate peak lists as described in the text. Negatively correlated peaks were removed (F), and positively correlated peaks with significant correlations as described in Methods (G) were used as inputs for COLMAR query searches, which for example, found tryptophan (Trp). Tryptophan 1D 1H and 13C reference spectra from the BMRB (black) are overlaid on both correlation matrices (C and D) and the expansion region in (H).
1. Statistical Correlations
We calculated 2D statistical correlation maps for 13C–13C and 1H–1H homonuclear (STOCSY:10,11 Figure 1C) and 1H–13C heteronuclear spectra (SHY:12−14 Figure 1D). In 1D versions of STOCSY and SHY, correlations and covariance are determined for a specific “driver” peak of interest. In the 2D implementation used here, each peak serves as a driver for all other resonances in the spectra, as shown in panels C and D in Figure 1.
2. Generation of Peak Lists
The STOCSY and SHY maps were analyzed by an in-house script written in MATLAB according to the following steps. We first systematically evaluated all 13C Pearson correlations and covariances from the STOCSY and removed any that were not greater than 0. Thus, all peak lists were constructed from the positive 13C 2D correlation map, as shown in Figure 1C. We systematically examined all 13C frequencies along the vertical STOCSY axis—now considered “driver peaks”—for every 13C and 1H resonance along the horizontal axes of the STOCSY and SHY, respectively (Figure 1E). A starting point for peak lists for database searching was generated by compiling list of peaks that covary with the driver peak by ±half the covariance of the driver peak with itself. This was done to eliminate resonances that have a high correlation but a low covariance with the driver peak. In addition, if the significance (q) of a correlation was less than 0.05 after FDR correction,23 that peak was removed to further eliminate resonances that are unlikely to be part of the same molecule (Figure 1F). The remaining peaks (Figure 1G) had both significantly high correlation and a strong covariance with the driver peak. Peak lists from all driver peaks were compared, and those sharing more than three peaks (both 1H and 13C) were combined and classified as part of the same metabolite. We found that lists sharing at least three peaks improved our metabolite coverage. Fewer shared peaks resulted in fewer but very large peak lists, and more shared peaks (>3) essentially excludes compounds with fewer carbons.
The 1H–1H STOCSY results (Supporting Information Figure S1) were not as useful as the 13C–13C STOCSYs, but we used these with the synthetic mixture to compare the results with 13C to traditional approaches with just 1H.
3. Database Searching
At this stage in the analysis, the peak lists from step 2 could be analyzed several ways including de novo compound identification using chemical knowledge or, for known compounds, matched to a database. We chose to use the COLMAR query24 search engine and the BMRB database,25 because the BMRB has a large number of metabolites with assigned 13C spectra. However, any search protocol and database with both 1H and 13C NMR data could be used for this step. We note that our database searches in this study were sequential; namely, we first searched the 13C data and then searched for 1H data. It would be advantageous in the future to simultaneously search using all the data. Like all database searches, several possible matches for each query were returned, and the final step in our analysis was manual inspection of BMRB spectra overlaid onto our primary data and covariance maps for final identification.
Results and Discussion
The goal of this paper was to determine the extent to which 1D 13C NMR can significantly enhance an untargeted metabolomics study. The specific algorithms—STOCSY, SHY, COLMAR query, and PCA or PLS-DA—were all known and used with little or no modification but were combined into a workflow that incorporates the 13C data. We used three different test cases to evaluate the workflow and approach. To develop and validate the method, we first used a simple synthetic mixture of known compounds. This allowed us to optimize parameters and define the steps of the overall workflow, as well as allowing us to estimate a lower practical limit for the amount of material required with current technology. In order to demonstrate the robustness of this approach, we applied the workflow to two very different biological systems, comparing genetically distinct fruit fly lines with different phenotypic responses to cold and comparing control mice to those with a known mutation in a gene that causes muscular dystrophy. The fly data provided an example of several relatively abundant metabolites that change between groups, and similar animals have recently been studied using traditional 1H-based NMR methods, allowing for some comparisons. The mouse serum data validates that this technique can be used when only very limited quantities of starting material are available and shows the value of 13C measurements to confirm or reject assignments from 1H data alone.
Workflow Development
We collected and analyzed 1D 13C (Figure 1A) and 1H (Figure 1B) spectra of two groups of synthetic mixtures to determine the limit of detection for this method and to test our ability to identify metabolites that we knew were in the samples. The concentration ranges of metabolites in these mixes were between about 1 and 5 mM; thus, the total quantity of compounds in each experiment ranged from 40 to 200 nmol. The metabolites at the lower concentrations (indicated with an asterisk* in Figure 1A) were detectable, but only with relatively low S/N. However, we were unable to obtain 13C correlations for the resonances with the lowest S/N and were only able to obtain reliable correlations from compounds at or above about 1.5 mM. Thus, we conclude that 60 nmol is the lower limit of material that works for this workflow with the current 13C probe, measurement time, and other NMR parameters. Several ideas to improve this overall sensitivity are given below.
The overall workflow is illustrated in Figure 1 with the identification of tryptophan (Trp) in the synthetic mixtures. The 13C–13C STOCSY and 1H–13C SHY maps were used to generate peak lists, as described above. One of the correlated peak lists had a good match to Trp, following COLMAR query of the BMRB database. The 1D BMRB reference spectra of Trp are shown in black on the axes of plots in Figure 1 to illustrate how the statistical correlations were used to match the database. A closer look at the 13C–13C STOCSY region between 109.5 and 141.5 ppm showed the strong correlations between the aromatic tryptophan peaks (red cross-peaks). The trace through the 13C–13C STOCSY at one of the aromatic Trp resonances (114.8 ppm) illustrates the correlations with other Trp peaks (Figure 1E).
To evaluate the extent to which 13C data improved our identification of compounds, we compared results using the entire workflow outlined in Figure 1 with results that just used 1H–1H STOCSY (Supporting Information Figure S1). Using all the data (13C and 1H), our workflow returned 36 peak lists but using the 1H data alone returned only 26 peak lists. It must be noted that a peak list is defined as the set of correlated peaks that pass a given information-quality threshold, as described above in Methods. Peak lists can be fragments of molecules, so it is not surprising to have a greater number of peak lists than detected metabolites in the sample. When we searched the appropriate BMRB database using COLMAR query (i.e., 13C or 1H), using all of the data we found 15 of the correct compounds in the list of the top 10 hits from the query, but using just the 1H data we only found 7 of the correct matches (Table 1). Because the 1H and 13C data were collected from the same samples and the 1H data had better overall signal-to-noise, we believe that the major factor underlying the better performance of the 13C data for compound identification was that the 13C workflow generated fewer false correlations—defined as a peak in the generated peak list that is not in the database-matched molecule. In the 1H–1H STOCSY 60% of the peaks were falsely correlated, but the 13C–13C STOCSY had only 20% false correlations, as indicated in columns 4 and 5 of Table 1. This large difference in the false correlations results from the improved resolution in the 13C 1D data. PCA performed on both 1D 13C (Supporting Information Figure S2A) and 1H (Supporting Information Figure S2B) data sets gave predictably good separation considering these were artificially made groups. However, it can also be seen that the loadings from the 13C PCA are much better resolved than those from the 1H PCA (Supporting Information Figure S2). This is demonstrated more clearly below in the fly data. In general, for the synthetic test data set, we found that the 13C data were much more useful than 1H in generating comprehensive peak lists leading to metabolite identification. In most instances, the peaks generated from 13C data alone were enough to successfully match metabolites in the database, and the 1H data were reserved primarily for verification of results.
13C NMR Data Yields Improved Group Discrimination
To test our workflow on samples from a biological system, we utilized two isogenic lines of D. melanogaster, one hardy and one susceptible to cold exposure. D. melanogaster is a model organism and the subject of many NMR-based metabolomics studies,26−28 making it desirable to optimize the performance of NMR-based metabolomics for this species. The lines used here originated from the same population as the Drosophila Genetic Reference Panel29 but were selected for either rapid (hardy) or slow (susceptible) recovery from a cold-induced coma (a common metric of cold tolerance in insects and other ectotherms). We used these strains because they have a well-annotated metabolome as well as robust and stable differences in the metabolome based on previous 1H NMR measurements (Williams et al., unpublished results), against which we can evaluate the power of 13C NMR to identify (A) NMR-visible metabolites and (B) metabolites that discriminate the lines (i.e., a biomarker approach). All data were independently analyzed and subsequently cross-checked with the previously annotated 1D and 2D 1H NMR measurements. Detailed physiological analysis of metabolic changes upon cold exposure will be presented elsewhere.
The 13C (Figure 2A) and 1H (Figure 2B) spectra are plotted in red (cold hardy) and blue (cold susceptible). As described above, peak lists defining spin systems from the 13C–13C STOCSY (Figure 2E) and 1H–13C-SHY (Figure 2F) correlations were used to search the BMRB, which yielded the 13 metabolites given in Supporting Information Table S1. As above, the 13C data were more useful than 1H for database matching. As an example, a slice of the 2D 13C–1H SHY with 13C driver peak 31.69 ppm (Figure 2D), which is the β carbon of proline, shows correlations with many 1H resonances other than the reference proline resonances (black), making unambiguous proline identification using 1H alone difficult. In contrast, the slice from the same (31.69 ppm) trace in the 13C–13C STOCSY (Figure 2C) showed very strong correlations with all other proline peaks and matched those from the BMRB perfectly. Metabolites such as glutamine and glutamate could only be distinguished using the improved resolution and chemical shift differences in 13C and not with the highly overlapped 1H spectra. In the absence of 13C, many of these metabolites would have been ambiguous using 1D 1H alone. Prior to our study, methionine sulfoxide was not annotated in the 1D or 2D 1H spectra of the fly metabolome and was found here by statistically correlating carbons through heteroatoms such as sulfur using 13C–13C STOCSY. Metabolites identified in Supporting Information Table S1 were consistent with previously collected and analyzed data using conventional methods (Chenomx), 2D NMR, and spiking experiments (Williams et al., unpublished results).
Figure 2.

Identification of D. melanogaster metabolites using 1D 13C (A) and 1H NMR (B) data. 13C–13C STOCSY (E) and 2D 13C–1H SHY (F) correlation maps were generated as described in the text. Slices at 31.69 ppm from the 13C–13C STOCSY (B) and 13C–1H SHY (D) show the correlations and covariances with that resonance, which yielded proline in a database search. Proline spectra from the BMRB (black lines in (C) and (D)) are overlaid onto the 1D 13C STOCSY and 13C–1H SHY to confirm identity.
PLS-DA performed on the fly 13C 1D spectra yielded clear separation of cold-hardy flies and cold-susceptible flies on component 1 (Figure 3A). The 1H spectra failed to yield separation on either component 1 or 2. Here we show that the 13C data provided much better separation between hardy and susceptible flies (Q2 = 0.77 and R2 = 0.85) and had stronger loadings than those from the 1H data in Figure 3B (Q2 = 0.24 and R2 = 0.34.). Annotated 13C and 1H loadings of component 1 are given in Figure 3. In addition, PCA performed on the fly 13C 1D spectra gave better separation along PC1 (Supporting Information Figure S3A), while the 1H spectra gave some separation along PC2 (Supporting Information Figure S3B). The PC1 loadings for the 13C 1D spectra and PC2 loadings for the 1H 1D spectra gave similar results as the PLS-DA. histidine, AMP, phosphocholine, taurine, and methionine sulfoxide all loaded with the hardy flies, and glucose (possibly also other sugars), proline, alanine, and arginine loaded with the susceptible flies. All of these compounds were identified and verified as altered by selection in a parallel study using 1D and 2D 1H NMR, and the direction of alteration was confirmed in 78% of cases (all but AMP and taurine, which were higher in susceptible flies in the parallel study: Williams et al., unpublished results). The differences in the direction of the effect may be due to using only a subset of lines in the present study, and the flies in the present study were also not exposed to cold. These findings support the primary conclusions that hardy flies have alterations to pathways involved in membrane lipid metabolism, oxidative stress, and energy balance. These alterations are consistent with the hypothesis that cold-hardy flies maintain aerobic metabolism in the cold, preventing the (potentially deleterious) accumulation of alanine and sugars.
Figure 3.

PLS-DA of 13C (A) and 1H (B) 1D spectra from cold hardy and cold susceptible flies. PLS-DA produced better separation of cold hardy (red) and susceptible flies (blue) in 13C data with a Q2 value of 0.77 and an R2 value of 0.85 when compared to 1H data with a Q2 value of 0.24 and an R2 value of 0.34. Loadings plots of component 1 for both 13C and 1H are given in (A) and (B) respectively. 13C component 1 loadings gave the compounds contributing to the differences between the hardy and susceptible flies. 1H component 1 loadings plot (B) showed little differences between the groups, though sugars seemed to load better with the cold susceptible flies. Red and positive peaks indicate resonances that were correlated with cold hardy flies, and blue and negative resonances indicate resonances that correlated well with the cold susceptible. Annotations are given for 1D 13C and 1H loadings plot (A and B, respectively).
13C NMR Offers Improved Confidence in Metabolite Identification
The mdx mouse is an animal model for Duchenne muscular dystrophy (DMD)—a muscle degenerating disease found in young males. The inability to produce dystrophin results in a pathology in mice similar to that seen in humans.30 Tracking disease progression in children requires difficult and painful muscle biopsies31 and effort-based functional testing.32 Importantly, current methods may be insensitive to therapeutic intervention. Identifying biomarkers of disease progression for DMD using an easily obtained sample such as serum or urine is essential for improving our ability to treat and diagnose this debilitating disease.
Although the overall sensitivity with the small volume of mouse serum prevents the detection of large numbers of metabolites, we were able to obtain high-quality NMR data on the most concentrated compounds, suggesting that 13C NMR metabolomics using isotopically labeled compounds for flux studies on survival bleeds is also possible with this probe. Similar to the fly study, the mouse serum revealed that 13C produced more robust PLS-DA results than 1H data, separating along PC1 with a cross-validation (Q2) value of 0.67 and R2 of 0.79 compared to the 1H results of a Q2 value of 0.46 and R2 value of 0.66 (Supporting Information Figure S5). Thus, for at least the two biological data sets analyzed in this paper, 13C NMR data improves group separation using PLS-DA. Using the 13C workflow developed here, we have found that the addition of 13C helps in making specific identifications and, perhaps more important, in preventing the misidentification of metabolites suggested by 1H NMR alone. For example, 1H resonances at 0.9, 1.3, and 2.75 ppm are often associated with fatty acids, but the specific fatty acid species is typically not annotated. Here, with the addition of the 13C–13C STOCSY and 13C–1H SHY correlation maps, we were able to identify linoleic acid as the specific fatty acid abundant in our 1H and 13C spectra (Supporting Information Figure S4).
Figure 4 shows representative 1H and 13C 1D spectra from the mouse serum. Previous NMR studies in the muscle of mdx vs control mice have reported a decrease in muscle creatine that varies with age and tissue, thought to result from increased inflammation and degeneration of the muscle.33,34 It is hypothesized that this leads to an in increase in serum creatine of mdx mice through dystrophin deficient “leaky” muscle membranes.34 In our data, the 1H PLS-DA results indicated a resonance at 1.30 ppm that separates the control from mdx. Analysis of the 1H STOCSY using 1.30 ppm as a driver peak indicated a correlating resonance or overlapping resonances at 3.02 ppm, often assigned to creatine. However, other creatine 1H resonances were overlapped, making it difficult to analyze. To further investigate whether creatine is associated with disease development in the mdx mouse model, we examined the 13C–1H SHY data and found correlations with the 3.02 ppm resonance to the 13C peaks at 56.5 and 41.2 ppm; the correlation at 38.9 ppm we would expect to see for creatine was not present (Figure 4). Creatine is often assumed to be the largest contributor to the peak at 1H 3.02 ppm.35,36 To determine if the absence of the creatine peak at 38.9 ppm in the 13C 1D was due to sensitivity limitations, we compared the integrals of the 1H resonance at the 3.02 ppm (−CH3) to the succinate at peak 2.39 ppm (CH2–CH2) (Figure 4A). Given the relative abundance of the creatine compared to the succinate resonance, we expect to see a 13C peak at 38.9 ppm comparable to the succinate resonance seen at 36.5 ppm in the 13C spectra. Therefore, using 13C data we conclude that creatine is not present at concentrations that we can measure in this experiment. This was verified by a spiking experiment with synthetic creatine, which did not correspond to resonances observed from the mice (data not shown). Despite extensive precedent in the literature assigning 1H resonances at 3.02 ppm in metabolomics mixtures, the 13C data clearly shows that creatine would have been an incorrect assignment in our case.
Figure 4.

Analysis of specific resonances in mouse serum for metabolite identification or error detection. The mdx data are in red and the control in blue. All the spectra are overlaid in (A), with the average data for each group shown as a bold line. Resonances at 3.02 ppm (unknown) and 2.39 ppm (succinate) in the 1H (A) are expanded, and the relative intensities are given in the box plot. Metabolites indicated in (B) are overlaid onto the 1D spectra (A and B). Expansion of the carbon 1D from ∼20 to 60 ppm shows the absence of creatine peak at 38.9 ppm, but the presence of a peak at 36.5 ppm confirms the possibility of succinate. Using just the 1H data, the peak at 3.02 ppm would likely be assigned to creatine, which we can rule out with the 13C data.
Citrulline (Figure 4B) has a resonance near 3.02 ppm and is another possible assignment of this peak. 13C–1H SHY and 13C–13C STOCSY data show correlations between the resonance at 3.02 ppm in the 1H spectra and suspected citrulline resonances 56.5 and 41.2 ppm. We do not, however, observe strong correlations to other citrulline 13C peaks. In addition, the 1H chemical shifts do not agree well with citrulline (Figure 4A). Thus, by using both 1H and 13C spectra of the same sample, we were able to rule out two previously associated compounds as unlikely to be contributing to our NMR spectra. Further analysis is needed to identify the compound(s) associated with these peaks, but this result further demonstrates the utility of 13C NMR in metabolomics studies.
Future Improvements
There are several possible improvements that could be made in the future. First, a larger volume NMR probe with 13C optimization would provide greater absolute sensitivity. This, however, would result in lower mass sensitivity because the sensitivity of NMR probes is roughly inversely proportional to the diameter of the coils.37 A 5 mm NMR tube requires a volume that is approximately 600 μL compared with 40 μL for our 1.5 mm probe used here, a 15-fold difference. A commercial 5 mm 13C optimized probe would provide about 7-fold greater absolute sensitivity with 15-fold more sample than our small volume probe.9 Therefore, for situations that are not sample-limited such as human urine or serum, a commercial large volume 13C probe would provide up to about 7× better sensitivity than we can achieve using our small volume probe. However, for mass-limited samples such as mouse serum or D. melanogaster extracts, our 40 μL probe would provide about 2× better results than the same quantity of sample diluted to 600 μL. Second, a T1 relaxation agent could be added to the samples to further reduce the relaxation time and also better recover slowly relaxing quaternary carbons that we attenuated in this study. Finally, dissolution dynamic nuclear polarization (DNP) is an emerging technique that can transiently enhance the 13C signal of a sample by >10 000 fold for about 1 or 2 min.6 If some or all of these techniques could be used to improve sensitivity of the NMR measurement even more, then the benefits of 13C metabolomics that we have demonstrated could be fully realized.
Conclusion
We have shown that by using a 13C-optimized NMR probe,9 we were able to acquire high-quality 13C NMR data in a reasonable amount of time and with relatively small sample volumes. The overall workflow allowed for more accurate identification of compounds through database matching by utilizing both the standard 1D 13C and 1H NMR data. There is a cost of about 2 h per sample in terms of extra measurement time, but inspection of the NMR spectra clearly shows the increased spectral dispersion of the 13C compared to the 1H data from the same sample (e.g., Figure 1A and B) that in turn leads to many desirable results in database matching and ultimately better separation of groups using multivariate analysis. Using the synthetic mixture, we determined that the limit of analysis through correlations was about 60 nmol, although this could be improved even more with more sensitive NMR measurements.
When statistically combined with 1H NMR data on the same samples, the 13C spectra enable new approaches that produce the following: (1) peak lists for better spectral annotation, (2) improved group separation using multivariate statistical analysis because of reduced spectral overlap, and (3) improved confidence in identified metabolites and the ability to reject identifications based on 1H data alone. We developed and validated our approach using a synthetic mixture and then applied it to two different biological examples: D. melanogaster cold tolerance models in which metabolites that significantly changed between hardy and susceptible lines of flies were easily identified and mass-limited mouse serum where metabolite identification using 13C spectral analysis could not be done with 1H analysis alone. In the case of the mouse serum, the addition of 13C data not only enhanced our ability to identify specific compounds but it also prevented mistakes that could have been made with 1H data alone.
Acknowledgments
We are grateful to Bill Brey, Jerris Hooker, Vijay Ramaswamy, and colleagues at Agilent Technologies for the HTS NMR probe and continued support during this project. Steve Robinette provided the initial MATLAB toolbox. Jim Rocca provided support in the NMR data collection. Travis Crossett, Ravneet Vohra, and Stephen Chrzanowski helped with mouse serum collection. Data were collected at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida. Funding for this study was from the NIH (1U24DK097209-01A1 and R01EB009772 to A.S.E.). D.A.H. and C.M.W. were supported by NSF (IOS-1051890) to D.A.H. and A.S.E.. G.A.W and B.L-M were supported by NIH (U54AR052646) to G.A.W.
Supporting Information Available
Supporting figures (spectra) and tables (chemical shifts) are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Integrative Biology, University of California, Berkeley, CA 94720.
Author Contributions
MATLAB scripts were written by C.S.C. with substantial contributions from G.S.S., B.L-M., and A.S.E.; C.M.W. provided Drosophila extracts as well as help with data interpretation; C.S.C. collected and analyzed the synthetic mixtures and Drosophila extracts; B.L-M. prepared, collected, and analyzed mouse serum samples; C.S.C., B.L-M., and A.S.E designed the experiment; C.S.C., B.L-M., C.M.W. and A.S.E. wrote the paper with significant contributions from all authors.
Author Contributions
▲ These authors contributed equally to this study.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- An Y. J.; Xu W. J.; Jin X.; Wen H.; Kim H.; Lee J.; Park S. ACS Chem. Biol. 2012, 7, 2012–2018. [DOI] [PubMed] [Google Scholar]
- Bingol K.; Zhang F.; Bruschweiler-Li L.; Bruschweiler R. Anal. Chem. 2012, 84, 9395–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardi R.; Traaseth N. J.; Masterson L. R.; Vostrikov V. V.; Veglia G. Adv. Exp. Med. Biol. 2012, 992, 35–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. L.; Bruschweiler-Li L.; Bruschweiler R. J. Magn. Reson. 2012, 225, 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyperski T.; Glaser R. W.; Hochuli M.; Fiaux J.; Sauer U.; Bailey J. E.; Wuthrich K. Metab. Eng. 1999, 1, 189–197. [DOI] [PubMed] [Google Scholar]
- Ardenkjær-Larsen J. H.; Fridlund B.; Gram A.; Hansson G.; Hansson L.; Lerche M. H.; Servin R.; Thaning M.; Golman K. Proc. Natl. Acad. Sci. U.S.A. 2011, 100, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze K. H.; Wu Q.; Tse H. S.; Zhu G. Top. Curr. Chem. 2012, 326, 215–242. [DOI] [PubMed] [Google Scholar]
- Keshari K. R.; Wilson D. M. Chem. Soc. Rev. 2014, 43, 1627–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V.; Hooker J. W.; Withers R. S.; Nast R. E.; Brey W. W.; Edison A. S. J. Magn. Reson. 2013, 235C, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloarec O.; Dumas M. E.; Craig A.; Barton R. H.; Trygg J.; Hudson J.; Blancher C.; Gauguier D.; Lindon J. C.; Holmes E.; Nicholson J. Anal. Chem. 2005, 77, 1282–1289. [DOI] [PubMed] [Google Scholar]
- Alves A. C.; Rantalainen M.; Holmes E.; Nicholson J. K.; Ebbels T. M. D. Anal. Chem. 2009, 81, 2075–2084. [DOI] [PubMed] [Google Scholar]
- Crockford D. J.; Holmes E.; Lindon J. C.; Plumb R. S.; Zirah S.; Bruce S. J.; Rainville P.; Stumpf C. L.; Nicholson J. K. Anal. Chem. 2006, 78, 363–371. [DOI] [PubMed] [Google Scholar]
- Coen M.; Hong Y.-S.; Cloarec O.; Rhode C. M.; Reily M. D.; Robertson D. G.; Holmes E.; Lindon J. C.; Nicholson J. K. Anal. Chem. 2007, 79, 8956–8966. [DOI] [PubMed] [Google Scholar]
- Keun H. C.; Athersuch T. J.; Beckonert O.; Wang Y.; Saric J.; Shockcor J. P.; Lindon J. C.; Wilson I. D.; Holmes E.; Nicholson J. K. Anal. Chem. 2008, 80, 1073–1079. [DOI] [PubMed] [Google Scholar]
- Pourfathi M.; Kuzma N. N.; Kara H.; Ghosh R. K.; Shaghaghi H.; Kadlecek S. J.; Rizi R. R. J. Magn. Reson. 2013, 235, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Southam A. D.; Hines A.; Viant M. R. Anal. Biochem. 2008, 372, 204–212. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Robinette S. L.; Ajredini R.; Rasheed H.; Zeinomar A.; Schroeder F. C.; Dossey A. T.; Edison A. S. Anal. Chem. 2011, 83, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa S.; Magalhaes A.; Ferreira M. M. C. Chemom. Intell. Lab. Syst 2013, 122, 93–102. [Google Scholar]
- Dieterle F.; Ross A.; Schlotterbeck G.; Senn H. Anal. Chem. 2006, 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- van den Berg R. A.; Hoefsloot H. C. J.; Westerhuis J. A.; Smilde A. K.; van der Werf M. J. BMC Genomics 2006, 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette S. L.; Zhang F.; Bruschweiler-Li L.; Bruschweiler R. Anal. Chem. 2008, 80, 3606–3611. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. J. R. Stat. Soc., Ser. B: Method. 1995, 57, 289–300. [Google Scholar]
- Zhang F.; Robinette S. L.; Bruschweiler-Li L.; Bruschweiler R. Magn. Reson. Chem: MRC 2009, 47Suppl. 1S118–S122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Kent Wenger R.; Yao H.; Markley J. L. Nucleic Acids Res. 2008, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmendal A.; Sørensen J. G.; Overgaard J.; Holmstrup M.; Nielsen N. C.; Loeschcke V. Naturwissenschaften 2013, 100, 417–427. [DOI] [PubMed] [Google Scholar]
- Sarup P.; Pedersen S. M. M.; Nielsen N. C.; Malmendal A.; Loeschcke V. PloS One 2012, 7, e47461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard J.; Malmendal A.; Sørensen J. G.; Bundy J. G.; Loeschcke V.; Nielsen N. C.; Holmstrup M. J. Insect Physiol. 2007, 53, 1218–1232. [DOI] [PubMed] [Google Scholar]
- Mackay T. F. C.; Richards S.; Stone E. A.; Barbadilla A.; Ayroles J. F.; Zhu D.; Casillas S.; Han Y.; Magwire M. M.; Cridland J. M.; Richardson M. F.; Anholt R. R. H.; Barrón M.; Bess C.; Blankenburg K. P.; Carbone M. A.; Castellano D.; Chaboub L.; Duncan L.; Harris Z.; Javaid M.; Jayaseelan J. C.; Jhangiani S. N.; Jordan K. W.; Lara F.; Lawrence F.; Lee S. L.; Librado P.; Linheiro R. S.; Lyman R. F.; Mackey A. J.; Munidasa M.; Muzny D. M.; Nazareth L.; Newsham I.; Perales L.; Pu L.-L.; Qu C.; Ràmia M.; Reid J. G.; Rollmann S. M.; Rozas J.; Saada N.; Turlapati L.; Worley K. C.; Wu Y.-Q.; Yamamoto A.; Zhu Y.; Bergman C. M.; Thornton K. R.; Mittelman D.; Gibbs R. A. Nature 2012, 482, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A.; Jockusch H. Nature 1991, 349, 69–71. [DOI] [PubMed] [Google Scholar]
- Khurana T. S.; Davies K. E. Nat. Rev. Drug Discovery 2003, 2, 379–390. [DOI] [PubMed] [Google Scholar]
- McDonald C. M.; Henricson E. K.; Abresch R. T.; Han J. J.; Escolar D. M.; Florence J. M.; Duong T.; Arrieta A.; Clemens P. R.; Hoffman E. P.; Cnaan A.; Muscle Nerve 2013, 48, 32–54.23677550 [Google Scholar]
- McIntosh L. M.; Garrett K. L.; Megeney L.; Rudnicki M. A.; Anderson J. E. Anat. Rec. 1998, 252, 311–324. [DOI] [PubMed] [Google Scholar]
- McClure W. C.; Rabon R. E.; Ogawa H.; Tseng B. S. Neuromuscular Disord. 2007, 17, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckonert O.; Coen M.; Keun H. C.; Wang Y. L.; Ebbels T. M. D.; Holmes E.; Lindon J. C.; Nicholson J. K. Nat. Protoc 2010, 5, 1019–1032. [DOI] [PubMed] [Google Scholar]
- Gulston M. K.; Rubtsov D. V.; Atherton H. J.; Clarke K.; Davies K. E.; Lilley K. S.; Griffin J. L. J. Proteome Res. 2008, 7, 2069–2077. [DOI] [PubMed] [Google Scholar]
- Ramaswamy V.; Hooker J. W.; Withers R. S.; Nast R. E.; Edison A. S.; Brey W. W. eMagRes. 2013, 2, 215–228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.