

Abstract
Direct insertion of amino acid sequences into the adeno-associated virus type 2 (AAV) capsid open reading frame (cap ORF) is one strategy currently being developed for retargeting this prototypical gene therapy vector. While this approach has successfully resulted in the formation of AAV particles that have expanded or retargeted viral tropism, the inserted sequences have been relatively short, linear receptor binding ligands. Since many receptor-ligand interactions involve nonlinear, conformation-dependent binding domains, we investigated the insertion of full-length peptides into the AAV cap ORF. To minimize disruption of critical VP3 structural domains, we confined the insertions to residue 138 within the VP1-VP2 overlap, which has been shown to be on the surface of the particle following insertion of smaller epitopes. The insertion of coding sequences for the 8-kDa chemokine binding domain of rat fractalkine (CX3CL1), the 18-kDa human hormone leptin, and the 30-kDa green fluorescent protein (GFP) after residue 138 failed to lead to formation of particles due to the loss of VP3 expression. To test the ability to complement these insertions with the missing capsid proteins in trans, we designed a system for producing AAV vectors in which expression of one capsid protein is isolated and combined with the remaining two capsid proteins expressed separately. Such an approach allows for genetic modification of a specific capsid protein across its entire coding sequence leaving the remaining capsid proteins unaffected. An examination of particle formation from the individual components of the system revealed that genome-containing particles formed as long as the VP3 capsid protein was present and demonstrated that the VP2 capsid protein is nonessential for viral infectivity. Viable particles composed of all three capsid proteins were obtained from the capsid complementation groups regardless of which capsid proteins were supplied separately in trans. Significant overexpression of VP2 resulted in the formation of particles with altered capsid protein stoichiometry. The key finding was that by using this system we successfully obtained nearly wild-type levels of recombinant AAV-like particles with large ligands inserted after residue 138 in VP1 and VP2 or in VP2 exclusively. While insertions at residue 138 in VP1 significantly decreased infectivity, insertions at residue 138 that were exclusively in VP2 had a minimal effect on viral assembly or infectivity. Finally, insertion of GFP into VP1 and VP2 resulted in a particle whose trafficking could be temporally monitored by using confocal microscopy. Thus, we have demonstrated a method that can be used to insert large (up to 30-kDa) peptide ligands into the AAV particle. This system allows greater flexibility than current approaches in genetically manipulating the composition of the AAV particle and, in particular, may allow vector retargeting to alternative receptors requiring interaction with full-length conformation-dependent peptide ligands.
Adeno-associated virus type 2 (AAV) (32) requires the assembly of 60 individual structural proteins into a nonenveloped, T-1 icosahedral lattice capable of protecting a 4.7-kb single-stranded DNA genome (24, 63). Purified infectious AAV particles contain three major structural proteins designated VP1, VP2, and VP3 (87, 73, and 62 kDa, respectively) in an approximate ratio of 1:1:18 (4). The antiparallel β-barrel topology of these capsid proteins results in a particle with a defined tropism (23, 34, 38, 50, 51) that is highly resistant to degradation.
The three AAV capsid proteins are produced in an overlapping fashion from the cap open reading frame (ORF) by using alternative mRNA splicing of the transcript and alternative translational start codon usage (1, 2, 6, 22, 29, 42, 53, 56). A common stop codon is employed for all three proteins (48). Correct capsid protein stoichiometry is maintained by translating VP1 from an ATG start codon (amino acid M1) on the 2.4-kb mRNA (1, 6, 53), while VP2 and VP3 arise from the 2.3-kb mRNA, using a weaker ACG start codon for VP2 production and readthrough translation to the next available ATG codon for the production of the most abundant capsid protein, VP3 (amino acids T138 and M203, respectively) (2, 31).
The specific roles for the individual capsid proteins in the assembly process and the absolute requirements for each in the formation of a functional virus particle are unclear. Studies of the viral life cycle in the absence of capsid protein expression (18, 47, 52, 55) and reports of capsid intermediates that accumulate during AAV infection (9, 21, 25, 37, 57, 58) indicate that these proteins are required for the accumulation of single-stranded genomes and clearly show that the assembly process occurs in the nucleus. Absence of the largest capsid protein, VP1, or deletion of the N-terminal sequence unique to VP1 leads to assembly of low-infectivity particles (lip phenotype) (18, 52, 60). This phenotype has been shown to be due to the absence of a phospholipase activity in the amino acid sequence unique to VP1 (12, 65). Some evidence also suggests that expression of either of the less abundant proteins, VP1 or VP2, is necessary for assembly of empty or full (genome-containing) particles (20, 44, 49, 57). Site-directed missense mutagenesis of the individual capsid protein start codons or the expression of separate capsid protein genes suggests that empty or full particles are obtained only if VP3 is coexpressed with VP1 or VP2 (20, 31, 49, 57). AAV capsid protein expression in SF9 cells (44) also suggests an essential role for VP2 in particle formation. The requirement for either VP1 or VP2 for capsid assembly seems to correlate with a lower nuclear localization of VP3, the most abundant capsid protein (20, 44, 49). However, a more recent insertional mutagenesis analysis of the cap ORF (40) has indicated the formation of a particle composed only of VP3, and studies in the absence of adenovirus (Ad) helper function and packageable AAV genomes have shown that intact virus-like particles can be formed with VP3 alone provided that the VP3 is fused to a nuclear localization signal (20). Finally, studies of capsid assembly in insect cells, in which the three capsid proteins were expressed from separate constructs in the absence of viral DNA or helper virus, suggest that VP1 plus VP3, VP1 plus VP2, or VP2 alone can form virus-like particles (44), while similar studies with HeLa cells suggest that VP1 or VP2 alone, but not VP3, can form intact particles (49). Thus, the absolute requirement for each capsid protein in the formation of intact particles has not been completely resolved.
Interest in the composition, assembly, and atomic structure of the AAV particle stems in part from its promise as a recombinant gene delivery vehicle in vivo. However, further clinical development of AAV for gene therapy will require the ability to target specific tissue types. A better understanding of the particle's surface architecture has been obtained through systematic alanine scanning (60) and insertional mutagenesis (11, 40, 45) of the AAV cap ORF and determination of the atomic structure of AAV (24, 63). These studies have identified several regions on the particle surface that tolerate the insertion of foreign sequences. Thus far, small changes in size, sequence, and/or position of the insertion have resulted in unpredictable changes in the mutant particle phenotype. Nevertheless, direct insertion of targeting sequences into the cap ORF has resulted in the successful production of AAV vectors with both expanded and retargeted tropisms (5). In particular, the insertion of targeting sequences in the VP1-VP2 and VP3 capsid overlap regions of the cap ORF (immediately following residue 138 or 587) have produced AAV with alternative cellular receptor usage. Insertions after residue 138 (N terminus of VP2) expand the tropism of AAV (27, 45, 60), as they do not disturb the capsid residues involved in binding cellular heparan sulfate proteoglycan (23, 34). Ligands inserted after residue 587 (11, 13, 30, 33, 35, 36, 40, 41, 45, 46, 60) reside at the particle's threefold axis between critical residues involved in cell binding via heparan sulfate proteoglycan (23, 34, 63), the primary viral receptor. Thus, these insertions can simultaneously restrict viral entry and redirect it to an alternative receptor. Still, these inserted sequences have been restricted in size (∼30 amino acids), consisting of linear receptor binding epitopes. One limitation to manipulating the cap ORF in the direct insertion approach is that modification of only one capsid across its entire sequence, leaving the remaining two capsids unaltered, is not possible. Only one region of the cap ORF allows for modification of a single capsid (VP1, residues 1 to 137), and this region contains a phospholipase A motif that is critical for efficient viral infection (12). A single report (64) has shown that a significantly larger single-chain antibody-coding sequence can be incorporated into recombinant particles if it is fused to the N terminus of VP2 and coexpressed with wild-type VP1, VP2, and VP3 capsids. These particles were capable of retargeting the vector to the CD34 molecule, but recombinant titers were extremely low. Since many receptor-ligand interactions employ such larger, nonlinear binding domains, the ability to insert significantly larger polypeptides into the AAV particle was the focus of this work.
In this study, using missense mutation of cap start codons, we generated plasmids that expressed only one or two of the capsid proteins and tested their ability to produce AAV particles. We observed that AAV-like particles are produced as long as VP3 is present. Characterization of the physical titers of these AAV-like particles that lacked specific capsid proteins demonstrated that the VP2 protein is apparently redundant and is not essential for viral infectivity. Importantly, using these constructs, we designed a method of producing AAV-like particles with large peptide insertions in VP1 and VP2 or VP2 exclusively, by expressing the modified protein separately and providing the remaining wild-type capsids in trans. Finally, AAV-like particles with altered capsid composition could be produced if VP2 was significantly overexpressed.
MATERIALS AND METHODS
Plasmids.
Plasmid pIM45 contains the rep and cap coding sequences of AAV with their expression controlled by their native promoters (28). It was used as a parent template for construction of all mutant plasmids. Plasmid pXX6 (62) supplies the Ad helper gene products in trans to allow recombinant AAV (rAAV) production in an Ad-free environment and was kindly supplied by Jude Samulski. Plasmid pTR-UF5 (68) supplies the rAAV DNA to be packaged. It contains a cytomegalovirus promoter driving expression of a green fluorescent protein (GFP) reporter gene flanked by the AAV terminal repeats. Plasmid pTR-dsRed is identical to pTR-UF5 except that the GFP-coding sequence is replaced with the red fluorescent protein (RFP) coding sequence.
Construction of mutant plasmids.
Site-directed mutagenesis (Stratagene) was performed on plasmid pIM45 according to the manufacturer's instructions. For each mutant plasmid, two complementary PCR primers containing a missense mutation in the individual capsid protein start codons were used to introduce changes in the cap ORF of pIM45. The oligonucleotides used for mutagenesis are listed in Table 1. These plasmids were screened for restriction sites inserted by silent mutations, and the mutations were confirmed by DNA sequencing.
TABLE 1.
Sequences of oligonucleotides used for mutagenesis
| Oligonucleotidea | Sequence (5′ to 3′)b |
|---|---|
| VP1-M1L | gatttaaatcaggtCTGgctgccgatggttatcttccagattggctcg |
| VP2-T138A | ggaaccggttaagGCGgctccgggaaaaaagaggccggt |
| VP2-T138M | ggaaccggttaagATGgctccgggaaaaaagaggccggt |
| VP3-M203L | cccctctggcctaggaactaatacgCTGgctacaggcagtggcgc |
| VP3a-M211L | gctaccggtagtggcgcaccaCTGgcagacaataacgagggcgcc |
| VP3b-M235L | tggcattgcgattccacatggCTGggcgacagagtcatcaccacc |
| pIM45-E/M138 | aggaacctgttaagacgCGGCCGACGCGTgctccgggaaaaaagag |
| VP2A-E/M138 | aggaacctgttaagATGCGGCCGACGCGTgctccgggaaaaaagag |
| FKN insert | cgCGGCCGtctggttcaggtagcggttctggtcagcacctcggcatgacgaaatgc (+) |
| cgACGCGTaccgctgccagaacctgagccgctaccatttctagtcagggcagcggt (−) | |
| LEP insert | cgCGGCCGgtgcccatccaaaaagtccaagat (+) |
| cgACGCGTgcacccagggctgaggtccagctg (−) | |
| GFP insert | cgCGGCCGatgagcaagggcgagggaactg (+) |
| cgACGCGTcttgtacagctcgtccatgcc (−) |
Top group, complementary oligonucleotides. Bottom group, sense (+) and antisense (−) oligonucleotides.
Upper- and lowercase letters indicate mutant and wild-type sequences, respectively.
Construction of AAV capsid mutant plasmids for directional cloning of insertions at amino acid position 138.
The same site-directed mutagenesis strategy was used to insert an EagI-MluI cloning site immediately after amino acid position 138 in pIM45. The same oligonucleotide pair with an additional T138M mutation was used to introduce these sites into pVP1,2A and pVP2A. The resulting plasmids were called pIM45-E/M138, pVP1,2A-E/M138, and pVP2A-E/M138. The cDNAs for the rat fractalkine chemokine domain (FKN [CX3CL1]) (accession number NM134455), the human hormone leptin (LEP) (accession number BC060830), and GFP (accession number U50963) flanked by EagI and MluI restriction sites were generated by PCR (Table 1). The PCR products were cloned into pIM45-E/M138, pVP1,2A-E/M138, and pVP2A-E/M138.
Cell Culture.
Human embryonic kidney 293 and cervical carcinoma HeLa C12 cells (8) were grown in Dulbecco modified Eagle medium (Invitrogen) supplemented with 100 U of penicillin per ml, 100 U of streptomycin per ml, 10% bovine calf serum, sodium pyruvate, and 2 μM glutamine. Cells were incubated at 37°C in a 5% CO2 atmosphere.
Production of AAV particles.
To produce AAV virions with wild-type capsid proteins, low-passage 293 cells were transfected at ∼80% confluence by using a modification of the triple-transfection protocol (26, 62, 68). All plasmids were transfected in equivalent molar ratios by using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's suggestions. One or two pIM45-based plasmids carrying the appropriate capsid protein mutation(s) or ligand insertions, pXX6, and either pTRUF5 or pTR-dsRed (total DNA, 70 to 90 μg) were transfected into three 15-cm2 dishes, and 24 h later the transfection efficiency was determined by fluorescence microscopy. Efficiencies were consistently above 75% with this method. The three dishes were then pooled, and vector purification was carried out as previously described by using an iodixanol step gradient alone or in combination with heparin column chromatography (17, 67, 69).
Virus titer determination.
To determine the concentration of intact AAV particles, the A20 enzyme-linked immunosorbent assay (ELISA) (American Research Bioproducts) was used. The A20 antibody detects intact, fully assembled particles, both full and empty (14, 15). Iodixanol-purified stocks were serially diluted and processed by the manufacturer's recommended protocol. Only readings within the linear range of the assay were averaged.
To determine the concentration of DNA-containing particles, we performed real-time PCR (7, 54) with a Perkin-Elmer Applied Biosystems (Foster City, Calif.) Prism 7700 sequence detector system. Equal volumes of virus stocks were treated with 600 U of benzonase per ml in 50 mM Tris-Cl (pH 7.5)-10 mM MgCl2-10 mM CaCl2 at 37°C for 30 min. The reaction mixtures were adjusted to 10 mM EDTA and 5% sodium dodecyl sulfate (SDS) and incubated with 280 U of proteinase K per ml at 37°C for 30 min. The reaction mixtures were then extracted with phenol-chloroform-isoamyl alcohol (25:24:1), and the packaged DNA was precipitated overnight with ethanol and glycogen carrier. The precipitated DNA pellets were dissolved in 100 μl of water, and 5 μl was used for real-time PCR analysis in a reaction mixture that included 900 nM (each) GFP forward (5′-TTCAAAGATGACGGGAACTACAA-3′) and reverse (5′-TCAATGCCCTTCAGCTCGAT-3′) primers, 250 nM TaqMan probe (5′-6FAM-CCCGCGCTGAAGTCAAGTTCGAAG-TAMRA-3′), and 1× TaqMan universal PCR master mix in a total volume of 50 μl. The cycling parameters were 1 cycle each of 50°C for 5 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Only values within the linear portion of a standard curve having a coefficient of linearity of greater than 0.98 were accepted. The average real-time PCR titer was calculated from virus preparations assayed three times.
For AAV particles with GFP inserted in VP1 and VP2 or VP2 exclusively, the RFP gene from pTR-dsRed was packaged and particle titers were determined by dot blotting as described previously (67). Equal-volume aliquots of the vector preparations were incubated with DNase I, inactivated with EDTA, digested with proteinase K, phenol-chloroform extracted, and precipitated with ethanol. The DNA was then transferred to nitrocellulose and probed with radiolabeled RFP probe.
To determine the infectious titer of the wild-type and mutant virus stocks, we performed a fluorescent-cell assay (FCA) essentially as previously described (67). Briefly, HeLa C12 cells were seeded in a 96-well plate so that they were approximately 75% confluent at infection. Cells were infected with 10-fold serial dilutions of the vector preparations and Ad type 5 (Ad5) at a multiplicity of infection (MOI) of 10. Cells were incubated at 37°C in a 5% CO2 atmosphere for 24 h and examined by fluorescence microscopy. The average FCA titer was calculated by averaging the number of green fluorescent cells (or red fluorescent cells in the case of virus that contained a GFP insert in the particle) from preparations assayed three times. Particle-to-infectivity ratios were calculated by dividing the average DNA titer by the average FCA titer.
Confocal microscopy of AAV-like particles with GFP inserted in VP1 and VP2.
HeLa cells were seeded in eight-chamber tissue culture slides (Falcon) at 24 h prior to infection with VP1,2A-GFP particles at an MOI of 10,000 in the absence and presence of Ad5 (MOI of 20). Tissue cultures were fixed in 4% ice-cold paraformaldehyde solution for 4 h. To reduce nonspecific labeling, the slides were incubated in 1% bovine serum albumin in 0.01 M phosphate buffered saline (PBS) (pH 7.2 to 7.4) for 1 h at room temperature (RT). The primary rabbit anti-early endosomal antigen 1 antibody (Novus Biologicals, Inc., Littleton, Colo.), which was diluted at 1:1,000 with 0.1% bovine serum albumin and 0.3% Triton in PBS, was incubated for 24 h at 4 C. The secondary antibody, Cy5-conjugated donkey anti-rabbit immunoglobulin G at a 1:100 dilution in PBS (Jackson Immunoresearch Laboratories, West Grove, Pa.), was applied for 1 h at RT. Between each incubation step, slides were rinsed in PBS for 30 min at RT. For propidium iodide (PI) staining, the slides were briefly equilibrated in 2× SSC (0.3 M NaCl, 0.03 M sodium citrate [pH 7.0]) and incubated in 100 μg of DNase-free RNase per ml in 2× SSC for 20 min at 37°C. Cover slips were then applied to the slides by using Vectashield mounting medium with PI (Vector Laboratories, Inc., Burlingame, Calif.). Sections were examined with a confocal laser scanning microscope (Bio-Rad Olympus) illuminated by three lasers (argon, “green” helium-neon, and “red” helium-neon), which supply excitation lines at 458, 488, 514, 543, and 633 nm. This allowed simultaneous confocal imaging of the three fluorophores (i.e., GFP, PI, and Cy5). Cells on each slide were examined first for GFP staining. The focal plane was adjusted so that the number of detectable cell bodies was maximized, and the green GFP image was then stored in memory. The procedure was repeated for the red PI image and the blue Cy5 image. Finally, a superimposition of the three colored images was made and stored. All manipulations of contrast and illumination on color images were done by using Adobe PhotoShop 6.0 software on a PC.
RESULTS
Direct insertion of large peptides after residue 138 of the AAV capsid ORF does not yield particles.
Residue 138 was chosen because ligands inserted at this position are present on the surface of the particle and result in alternative receptor recognition by AAV vectors (27, 45, 60). Furthermore, this position does not directly interrupt the phospholipase A2 motif of VP1 (12) or interfere with the structurally critical VP3 β-barrel arrangement (63). To test the direct insertion of larger peptides into cap, we inserted the directional cloning sites EagI and MluI immediately after residue 138 of the cap ORF in plasmid pIM45, which contains the wild-type rep and cap sequences. The choice of these restriction enzymes meant that ligands inserted into the resulting plasmid (pIM45-E/M138) were flanked by Arg and Pro on the N-terminal side and by Arg and Thr on the C-terminal side. These additional four amino acids had little effect on capsid expression, particle formation, or titers (Fig. 1; Table 2). The 8-kDa FKN (76 residues)- and the 18-kDa LEP (146 residues)-coding sequences were chosen because they are approximately half of the size (FKN) or the same size (LEP) as the VP1 N-terminal extension of VP2 (137 residues). These sequences were inserted into pIM45-E/M138, and the resulting plasmids, pIM45-FKN138 and pIM45-LEP138, were transfected into 293 cells in the presence of Ad5 (MOI of 10). Western blot analysis of equivalent volumes of 293 whole-cell lysates with B1 antibody, which recognizes a linear epitope in the C-terminal regions of all three capsid proteins (59), showed a severe loss of the most abundant capsid protein, VP3 (Fig. 1A). In addition, the expression level of the modified VP2 also appears to decrease with the larger LEP insertion. Both VP1 and VP2 had the expected increased molecular weight due to the insertion of FKN and LEP.
FIG. 1.

Western blot analysis of AAV capsid proteins in 293 cell lysates (A) and iodixanol-purified virus stocks (B) following insertion of FKN or LEP peptides after residue 138 in the EagI-MluI cloning site engineered in the VP1-VP2 overlap region. Equal volumes of lysates or virus stocks were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting with the B1 antibody. The diagram illustrates the position of the insertion of the E/M cloning site and the FKN and LEP ligands.
TABLE 2.
Properties of AAV and AAV-like particles
| Category and virus | Particle titera
|
Infectious titer (IU/ml)b | Particle-to-infectivity ratioc | Empty/full ratiod | |
|---|---|---|---|---|---|
| A20/ml | Genomes/ml | ||||
| VP3 N terminus | |||||
| Wild type | 7.2 × 1012 | 3.6 × 1011 | 1.8 × 1010 | 20 | 20 |
| M203L | No virus | ||||
| M211L | No virus | ||||
| M235L | 2.9 × 1012 | 2.2 × 1011 | 9.0 × 109 | 24 | 13 |
| (−) capsid proteins | |||||
| VP1, 2 | No virus | ||||
| VP1, 2A | No virus | ||||
| VP1, 3 | 6.2 × 1012 | 1.0 × 1011 | 4.6 × 109 | 22 | 62 |
| VP2, 3 | 6.7 × 1012 | 1.4 × 1011 | 4.5 × 106 | 31,111 | 48 |
| VP2A, 3 | 2.0 × 1012 | 4.0 × 1010 | 9.0 × 104 | 444,444 | 50 |
| VP1 | No virus | ||||
| VP2 | No virus | ||||
| VP2A | No virus | ||||
| VP3 | 5.0 × 1012 | 1.3 × 1011 | 5.0 × 104 | 2,600,000 | 38 |
| Complementation | |||||
| VP0 + wild type | 5.2 × 1012 | 3.6 × 1011 | 3.5 × 109 | 103 | 14 |
| VP1 + VP2, 3 | 4.6 × 1012 | 3.4 × 1011 | 1.6 × 1010 | 21 | 14 |
| VP2 + VP1, 3 | 8.8 × 1012 | 5.8 × 1011 | 1.6 × 1010 | 36 | 15 |
| VP2A + VP1, 3 | 5.8 × 1012 | 3.4 × 1010 | 1.8 × 108 | 189 | 170 |
| VP3 + VP1, 2 | 4.6 × 1012 | 4.6 × 1011 | 1.6 × 1010 | 29 | 10 |
| pIM45-E/M138 inserts | |||||
| E/M138 | 1.8 × 1012 | 1.7 × 1011 | 2.7 × 109 | 63 | 11 |
| FKN138 | No virus | ||||
| LEP138 | No virus | ||||
| VP1-VP2 peptide inserts | |||||
| VP1, 2A-FKN + VP3 | 3.9 × 1012 | 6.0 × 1010 | 2.8 × 105 | 214,286 | 65 |
| VP2A-FKN + VP1, 3 | 6.8 × 1012 | 1.2 × 1011 | 1.4 × 109 | 86 | 57 |
| VP1, 2A-LEP + VP3 | 3.1 × 1012 | 4.4 × 1010 | 3.4 × 105 | 129,411 | 70 |
| VP2A-LEP + VP1, 3 | 5.9 × 1012 | 1.2 × 1011 | 1.8 × 109 | 66 | 49 |
| VP1, 2A-GFP + VP3 | 2.0 × 1012 | 4.0 × 109 | <1 × 104 | >400,000 | 500 |
| VP2A-GFP + VP1, 3 | 4.3 × 1012 | 1.9 × 1010 | 7.0 × 105 | 27,143 | 226 |
A20 particle titers were determined as described in Materials and Methods, using the A20 ELISA. Genomic titers were determined by real-time PCR.
Determined by FCA as described in Materials and Methods.
Calculated by dividing the average genomic titer as determined by real-time PCR by the average fluorescent-cell assay titer.
Determined by dividing the A20 particle titer by the average genomic titer.
As expected, this aberrant capsid protein expression did not result in the formation of AAV particles. Following transfection of pIM45-E/M138, pIM45-FKN138, or pIM45-LEP138 with pXX6 and pTR-UF5, particles were purified by iodixanol density gradient centrifugation. In contrast to the case for the parental plasmid pIM45-E/M138, essentially no particles were recovered from cells transfected with pIM45-FKN138 or pIM45-LEP138 (Fig. 1B; Table 2). The parental plasmid pIM45-E/M138, which had a 4-amino-acid insertion in VP1 and VP2, produced virus with approximately the same yield of particles and particle-to-infectivity ratio as pIM45, which contained wild-type capsid proteins (Table 2).
Construction of mutants that lack expression of specific capsid proteins.
The loss of VP3 following insertion of large ligands after residue 138 suggested that VP3 would have to be provided in trans to complement the ligand-extended VP1 and VP2. For this purpose we generated a complementary capsid protein expression system that would allow for a single capsid protein to be modified in a region of sequence overlap (e.g., genetic modifications of VP2 exclusively at residue 138). To generate the necessary plasmids that expressed either one or two capsid proteins, we employed missense mutations in the AAV cap ORF translational start codons as reported previously by others (31, 44).
(i) Mutants expressing two capsid proteins.
With pIM45 as our template, we mutated the VP1 start codon, M1L, generating the construct pVP2,3, which should make only VP2 and VP3 (Fig. 2A; Table 3). Similarly, we mutated the VP2 start codon, T138A, generating the construct pVP1,3, which would make only VP1 and VP3. Finally, the VP3 start codon, M203L, was mutated in an initial attempt to generate the construct pVP1,2 (Fig. 2A). Western blotting analysis of capsid protein expression in 293 cell lysates demonstrated that while the expression of VP1 and VP2 was eliminated by single point mutations (Fig. 2A), pM203L expressed a VP3-like species (VP3a) that migrated slightly faster than VP3 (Fig. 2A, lane pM203L). This had been seen previously by Ruffing et al. (44), who had used a similar strategy to eliminate VP3 expression. To evaluate the role of alternative downstream translational start codons in the production of VP3a, further point mutations in Met residues downstream of the native VP3 start codon were generated in the pM203L background. Examination of the VP3-coding region revealed that nine additional Met residues (M211, M235, M371, M402, M434, M523, M558, M604, and M634) are present. Of these, only positions M211, M235, M523, M558, and M604 were in a favorable Kozak context for translational initiation. As VP3a is only slightly smaller than VP3, we initially examined the roles of M211 and M235 in the production of VP3-like species. We mutated M211L alone and with M235L on an M203L background (Fig. 2B), generating the constructs pM203,211L and pM203,211,235L. Western blot analysis of capsid protein expression in whole-cell lysates revealed that all three Met residues had to be mutagenized to eliminate VP3 expression (Fig. 2B). The robust expression of VP3a was again seen with pM203L (Fig. 2B). Additionally, transfection of pM203,211L resulted in weaker expression of a second, smaller VP3-like species, VP3b (Fig. 2B, lane pM203,211L), while expression of all VP3-like species was eliminated in the triple mutant M203,211,235L, finally generating the plasmid pVP1,2 (pM203,211,235L), which makes only VP1 and VP2 (Fig. 2B, lane pVP1,2). (Weak doublets present at the VP3 position in the pVP1,2 lane are due to cellular proteins that cross-react with the B1 antibody [data not shown].)
FIG. 2.

Mutants that express only two capsid proteins, showing Western blot analysis of capsids in cell lysates produced from 293 cells transfected with mutants that eliminate expression of one of the three AAV capsid proteins. Equal volumes of extracts were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting with the B1 antibody. (A) The missense mutations within the start codons of the three capsid proteins (M1L, T138L, and M203L) are illustrated along with the capsid proteins expressed from each mutant on an SDS-acrylamide gel blotted with B1 antibody. (B) VP3-like proteins that result from readthrough translation. A mutation in the normal VP3 start codon produces a truncated capsid protein, VP3a; mutations in the first two methionines (pM203,211L) produce a second truncated protein, VP3b; and mutations in the first three methionines (pM203,211,235L; pVP1,2) eliminate all VP3-like proteins. (C) An alternative approach to eliminating VP3 expression while maximizing VP2 expression. pVP1,2A contains a standard ATG start codon for VP2 instead of ACG, a T138M mutation, thereby increasing VP2 expression and eliminating VP3 expression (compare pVP1,2A in panel C to pVP1,2 in panel B).
TABLE 3.
Plasmid combinations for production of AAV-like particles with genetic modifications in specific capsid proteinsa
| Modified capsid protein (mutations) | Complementing plasmid |
|---|---|
| pVP0 (M1L; T138A; M203,211,235L) | pIM45 (wild type) |
| pVP1 (T138A; M203,211,235L) | pVP2,3 (MIL) |
| pVP2 (MIL; M203,211,235L) | pVP1,3 (T138A) |
| pVP2A (T138M) | pVP1,3 (T138A) |
| pVP3 (M1L; T138A) | pVP1,2 (M203,211,235L) |
Capsid mutant complementation groups are cotransfected with pXX6 and pTRUF5 in 293 cells to produce particles.
An alternative approach to eliminating VP3 expression has been reported (31, 44), in which mutation of the VP2 start codon to the stronger ATG (T138M) results in loss of VP3 expression. As this approach minimizes the number of mutations in VP1 and VP2 while maximizing the expression of VP2, we mutated the VP2 start codon (T138M) on a pIM45 template, generating the construct pVP1,2A (Fig. 2C). Western blot analysis of capsid protein expression in lysates from cells transfected with pVP1,2A confirmed that this approach produced normal levels of VP1, significant overexpression of VP2, and loss of VP3 expression (Fig. 2C).
(ii) Mutants expressing a single capsid protein.
Generation of capsid mutants that express a single capsid protein was accomplished by sequential mutation of start codons in the mutants that express two capsid proteins (Fig. 3). The construct that expressed only VP1 (pVP1) had the VP2 start codon mutation T138A and the M203,211,235L mutations that were required to eliminate VP3-like species (Table 3). The construct pVP2 had the VP1 start codon mutation M1L and the M203,211,235L mutations, while construct pVP2A (to over express VP2 alone) had the VP1 start codon mutation M1L and the VP2 start codon mutation T138M. Finally, the construct pVP3 had the VP1 start codon mutation M1L and the VP2 start codon mutation T138A. Western blot analysis of capsid protein expression in 293 cells transfected with these plasmids showed that indeed these constructs expressed only a single capsid protein, as expected (Fig. 3). Finally, the construct pVP2A significantly increased expression of VP2 in the absence of VP1 or VP3 (Fig. 3).
FIG. 3.
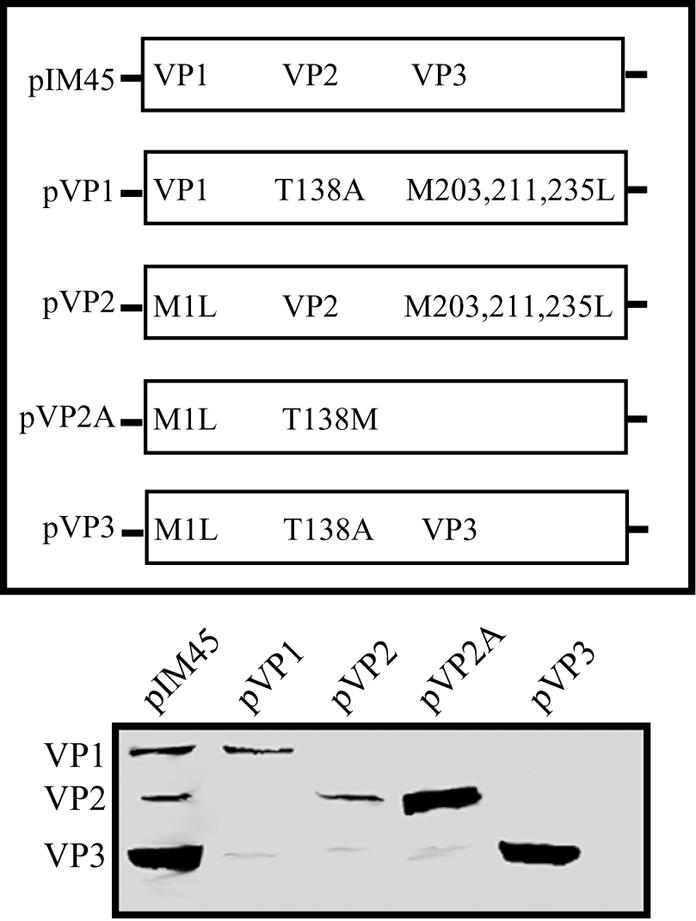
Mutants that express only a single capsid protein. Equal volumes of 293 cell extracts transfected with capsid mutants that express a single capsid protein were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting with B1 antibody. The diagram illustrates the missense mutation(s) in each construct.
AAV-like particle formation from capsid mutant constructs.
The construction of plasmids that made only one or two of the capsid proteins allowed us to reexamine the ability of various combinations of VP1, -2, and -3 to make viable AAV particles.
(i) VP3 N-terminal mutations.
Since the mutation of the N- and C-terminal regions of VP3 has been reported to abolish AAV particle formation (60), we examined the effects of the VP3 N-terminal M203L, M211L, and M235L mutations on particle formation (Fig. 4A; Table 2). These mutations, individually and combined in a pIM45 background (pM203L, pM211L, pM235L, and pM203,211,235L), were transfected into 293 cells with pXX6 and pTR-UF5. Particles were purified from 293 cell lysates at 72 h posttransfection by use of iodixanol step gradients, and equal volumes of the virus-containing fraction were Western blotted and probed with the B1 antibody. While AAV particles were obtained from pM235L, the importance of the VP3 N-terminal region in particle assembly is illustrated by the fact that both the pM203L and pM211L mutant plasmids produced no particles (Fig. 4A). It was not clear whether this defect was due solely to mutation of the VP3 N terminus or was because the M203L and M211L mutations were also present in the VP1 and VP2 proteins expressed from the pM203L and pM211L mutant plasmids.
FIG. 4.

Which capsid mutants can make a virus particle? Western blot analysis of AAV virus purified by use of iodixanol step gradients as described in Materials and Methods following transfection of the indicated capsid mutants into 293 cells is shown. Equal volumes of the iodixanol fraction were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting with B1 antibody. (A) Effect of the M203L, M211L, and M235L mutations on particle formation. (B) Particle formation from mutants that lack a specific capsid protein. (C) Particle formation from mutants that express a single capsid protein.
(ii) Mutants expressing two capsids.
To determine whether any of the capsid proteins were nonessential for particle formation, we examined the recovery of AAV-like particles lacking a specific capsid protein. Constructs pVP2,3, pVP1,3, pVP1,2, and pVP1,2A were transfected individually into 293 cells in combination with pXX6 and pTRUF5 at equivalent molar ratios. Particles were purified from 293 cell lysates at 72 h posttransfection by use of iodixanol step gradients, and equivalent volumes of the vector preparations were Western blotted and probed with B1 antibody (Fig. 4B). We determined the titers of the particles as described in Materials and Methods (Table 2; see Fig. 7B).
FIG. 7.

Capsid protein stoichiometry and infectivity of AAV virus stocks missing a capsid protein or containing a ligand insertion. (A) Western blot of virus stocks purified by use of iodixanol gradients and heparin sulfate column chromatography. Approximately 1011 AAV-like particles were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting with the B1 antibody. WT, wild type. (B) Particle-to-infectivity ratios of AAV-like particles relative to that of pIM45. The particle-to-infectivity ratio for each particle was calculated by dividing the average genomic titer by the average FCA titer (see Material and Methods and Table 2). The particle-to-infectivity ratio for each type of virus was then normalized to that of wild-type virus (pIM45) by dividing the particle-to-infectivity of each AAV-like particle by the particle-to-infectivity of pIM45, and the log10 value of the ratio was plotted. The wild-type pIM45 ratio equals zero and is indicated by the dashed line. Grey bars, particles with infectivity comparable to that of pIM45 (within 1 log unit); white bars, particles with significantly reduced infectivity (1- to 4-log-unit-lower infectivity); black bars, particles that were essentially noninfectious (>4-log-unit-lower infectivity). (C) Western blot of approximately 1011 AAV-like particles with GFP inserted in the capsid. Virus samples were purified as for panel A above, fractionated by SDS-10% polyacrylamide gel electrophoresis, and analyzed by Western blotting with the B1 antibody.
As expected, AAV-like particles composed of VP2 and VP3 were obtained following transfection of pVP2,3. Due to the lack of the capsid sequences unique to VP1, these particles displayed the lip phenotype with a particle-to-infectivity ratio approximately 3 log units lower than wild type (Table 2). This has been shown previously by us and others (12, 18, 52, 60) and is presumably due to the absence of the VP1 phospholipase A activity. Surprisingly, an AAV-like particle formed in the absence of the previously reported critical VP2 capsid protein (20, 31, 44) when VP1 and VP3 were present (Fig. 4B, lane pVP1,3). Furthermore, these VP2-negative particles had virtually the same properties and yield as wild-type particles (Table 2). Finally, the constructs that made only VP1 and VP2 (pVP1,2 and pVP1,2A) were unable to assemble a particle in the absence of VP3, irrespective of the level of VP2 expression (Fig. 4B; Table 2).
(iii) Mutants expressing a single capsid protein.
We next tested the ability of a single capsid protein to form an AAV-like particle. Constructs pVP1, pVP2, pVP2A, and pVP3 were transfected individually into 293 cells in combination with pXX6 and pTR-UF5 in at equivalent molar ratios. As before, particles were purified from 293 cell lysates at 72 h posttransfection and equivalent volumes of the vector preparations were Western blotted and probed with B1 antibody. Since the expression of VP1 and VP2 together did not form particles (see above), we did not anticipate the formation of particles from them individually. While no particles formed in the presence of the two less abundant capsid proteins, an AAV-like particle composed of VP3 alone was readily obtained (Fig. 4C and Table 2). This result was in agreement with a previous insertional mutagenesis study, which also suggested that particles could form with VP3 alone (40).
rAAV production system using complementary capsid protein mutants.
Since direct insertion of larger peptides after residue 138 leads to loss of VP3 expression, we hypothesized that significant modification of VP1 and VP2 at residue 138 would require that wild-type VP3 be provided in trans for efficient AAV production. We therefore tested the ability to complement a missing capsid protein by using the combinations of plasmids described above and summarized in Table 3, which express one capsid protein and various combinations of two capsid proteins. To control for twice the Rep expression resulting from combining the two pIM45-based plasmids that are used in this approach, we generated a construct, pVP0, that eliminates expression of all of the capsid proteins with the mutations M1L, T138A, M203L, M211L, and M235L (Table 3). The capsid protein complementation groups include pIM45 plus pVP0, which makes wild-type capsid proteins; construct pVP1 plus pVP2,3, which allows for exclusive modification of VP1; construct pVP2 plus pVP1,3, which allows for exclusive modification of VP2; pVP2A plus pVP1,3, which allows for exclusive modification of and significant overexpression of VP2; and construct pVP3 plus pVP1,2 which allows for exclusive modification of VP3. As before, these groups were transfected into 293 cells (in combination with pXX6 and pTR-UF5 at equivalent molar ratios), and particles were purified from 293 cell lysates at 72 h posttransfection by use of iodixanol step gradients and heparin column chromatography. Equivalent volumes of the vector preparations were Western blotted and probed with B1 antibody (Fig. 5A), and titers were determined as described in Materials and Methods (Table 2).
FIG. 5.

Complementation of mutants that make a single capsid protein. (A) Western blot analysis of AAV particles purified by use of iodixanol step gradients and heparin column chromatography following transfection of 293 cells with the complementation groups described in Table 3. (B) Western blot analysis of iodixanol fractions of particles obtained from transfection with pVP2A, pVP3, or both plasmids. Equal volumes of purified virus stocks were separated by SDS-10% acrylamide gel electrophoresis and analyzed by Western blotting with the B1 antibody.
Regardless of the complementation group employed, particles containing all three capsid proteins were recovered by using this rAAV production system. Interestingly, we also observed that overexpression of VP2 results in the recovery of a particle in which VP2 is overrepresented (Fig. 5A, lane pVP2A + pVP1,3). These particles contained smaller amounts of VP1 and VP3 and contained VP2 levels that were nearly equivalent to those of VP3. (The slightly lower infectivity of the VP2A-containing particle [Table 2] might be a reflection of the smaller amounts of VP1 in these particles, but this was not further explored.) All of the complementation groups produced virus yields and particle-to-infectivity ratios that were within 1 log unit of those for wild-type virus. We interpreted this to mean that we could now attempt to individually modify specific capsid proteins in regions of overlap (e.g., residue 138). We also noted that the mutations M203L and M211L, which are present in VP1 and VP2 when they are synthesized from pVP1,2 (Table 3), have little if any effect on the function of VP1 and VP2 in particle formation when complemented with a wild-type VP3 synthesized from pVP3 (Table 2). Thus, the effect of these mutations in the context of pIM45 (Table 2, mutants M203L and M211L) appeared to be entirely due to loss of VP3 function.
AAV-like particles with FKN or LEP inserted into VP1 and VP2.
Because direct insertion of large peptides after residue 138 resulted in the loss of VP3 expression and the complementary capsid protein groups produced viable rAAV particles, we next tested the ability to produce AAV-like particles with larger peptides inserted after residue 138 either simultaneously in VP1 and VP2 or exclusively in VP2 (Fig. 6A). Constructs that contained insertions in both VP1 and VP2 were complemented with pVP3, while those with insertions only in VP2 were complemented with pVP1,3. To make ligand insertion easier, we again inserted EagI-MluI cloning sites after amino acid position 138 in pVP1,2A and pVP2A as described earlier for pIM45 to create the plasmids pVP1,2A-E/M138 and pVP2A-E/M. The VP2-overexpressing background was chosen to increase the incorporation of VP2-ligand fusion proteins into viral particles. Both the FKN- and LEP-coding sequences were inserted into pVP1,2A-E/M138 and pVP2A-E/M138 to make pVP1,2A-FKN, pVP2A-FKN, pVP1,2A-LEP, and pVP2A-LEP (Fig. 6; Table 2). These plasmids were transfected into 293 cells in combination with pVP3 or pVP1,3 and pXX6 and pTR-UF5 at equivalent molar ratios, and the resulting virus particles were purified with iodixanol step gradients. Equivalent volumes of the various preparations were then Western blotted in duplicate and probed with B1 or ligand-specific antibodies (anti-FKN or anti-LEP) (Fig. 6B and C). In all cases we obtained novel AAV-like particles in which the inserted sequences were present in VP1 and VP2 or just in VP2. This was illustrated by an increase in the size of the VP1 and VP2 capsid proteins in blots probed with B1 antibody and was confirmed with the ligand-specific (FKN or LEP) antibodies. Titers in these iodixanol fractions were then determined as described in Materials and Methods (Table 2; see Fig. 7B).
FIG. 6.

Capsid complementation strategy for creating particles with large peptide insertions in the VP1-VP2 overlap region, showing Western blotting of equal volumes of iodixanol stocks of AAV-like particles containing FKN or LEP insertions at position 138. (A) Diagram of constructs used to complement insertions at amino acid 138 in both VP1 and VP2A or just VP2A. (B) Particles with the FKN insertion were purified by use of iodixanol gradients and probed on SDS-10% polyacrylamide gels with anticapsid (B1) antibody or anti-FKN antibody. (C) Particles with the LEP insertion were purified by use of iodixanol gradients and probed on SDS-10% polyacrylamide gels with anticapsid (B1) antibody or anti-LEP antibody.
Characterization of AAV-like particles.
To characterize the novel particles described in this study further, a portion of all of the virus stocks described above that were either missing a capsid protein or contained a modified capsid were purified by heparin column chromatography. Subsequently, approximately 1011 particles were Western blotted and probed with B1 antibody (Fig. 7A) to compare the stoichiometries of the capsid proteins in the various particles. Generally, the levels of individual capsid proteins were similar to those of the wild type, with the following exceptions. First, as shown above (Fig. 2, 3, and 5), overexpression of VP2 (VP2A) leads to an altered capsid ratio in a particle composed of VP2 and VP3 (Fig. 7A, lane VP2A + VP3). This was true even when peptides of 76 (FKN) or 146 (LEP) amino acids were inserted after amino acid 138 of VP2A (compare Fig. 7A, lanes WT and VP2,3 with FKN or LEP inserted particles). Additionally, the relative amount of VP1-ligand fusion protein (and often wild-type VP3) was reduced in these particles. Finally, the fact that the particles with FKN and LEP inserted in VP1 and VP2 could be purified by heparin chromatography suggested that ligands of up to 18 kDa may not affect binding to heparan sulfate proteoglycan when inserted after residue 138.
To determine the relative abilities of the novel particles to assemble, package DNA, and infect cells, we determined the titers of the particles by use of the A20 ELISA (to estimate the total particles, empty and full), the real-time PCR assay (to determine the titer of genome-containing DNase-resistant full particles), and the FCA (to determine the infectious particle titer). These assays were all performed on the iodixanol purified stocks (Table 2), and then the log relative infectivity was calculated (Fig. 7B).
With the exception of the mutants discussed above, all of the virus stocks contained A20 particle titers that were similar to wild type (Table 2) (approximately 2 × 1012 to 8 × 1012/ml). This was also true of the particles that contained an FKN or LEP insertion in VP1 and VP2 or in VP2 alone. Thus, the FKN and LEP insertions, and even a larger GFP insertion (discussed below), did not seem to affect viral assembly as judged by use of the conformation-dependent A20 antibody (Table 2). When we examined the relative packaging efficiency of the rAAV-like particles containing FKN or LEP ligands (Table 2), the analysis revealed that these particles package DNA nearly as well as the wild type, within 1 log unit (Table 2). A striking difference, however, was noticed when the FKN and LEP particles were tested for infectivity. Particles that contained FKN and LEP insertions only in VP2 had particle-to-infectivity ratios that were essentially the same as wild type (Table 2 and Fig. 7B, compare E/M138, WT, and VP1,3 with VP2A-FKN + VP1,3 and VP2A-LEP + VP1,3). However, particles that had an FKN or LEP insertion in both VP1 and VP2 were 4 to 5 log units less infectious. The loss in infectivity was comparable to that seen with all particles that had wild-type AAV capsid proteins but were missing VP1 (Table 2; Fig. 7B, lanes pVP2,3, pVP2A,3, and VP3). Thus, it appeared that if the foreign ligand was inserted exclusively into the N terminus of the nonessential VP2 capsid, a ligand as large as 138 amino acids could be tolerated with minimal loss of packaging efficiency or infectivity.
AAV-like particles with GFP inserted into VP1 and VP2.
Since FKN and LEP had little effect on overall vector yields, we wanted to determine whether insertions significantly larger than the VP1 unique region (137 residues) are still able to form particles. Therefore, we inserted the coding sequence for the 30-kDa GFP protein (238 residues) into pVP1,2A-E/M138 and pVP2A-E/M138 for complementation with pVP3 and pVP1,3, respectively. These particles were purified by using iodixanol step gradients followed by heparin chromatography, and titers were determined as described in Materials and Methods (Fig. 7B; Table 2). Western blot analysis of equal volumes revealed that both VP1 and VP2 had the expected increased molecular weight due to the insertion of GFP (Fig. 7C). While this experiment was primarily meant to be a test of the size limit for insertions after residue 138, the development of a fluorescently tagged vector was also a potentially interesting tool for studying the cellular entry and trafficking of rAAV particles. As with the FKN and LEP insertions, insertion of the GFP sequence into both VP1 and VP2 was much less successful than insertion into VP2 alone. While the yield of particles obtained with GFP inserted into both VP1 and VP2 appeared to be similar to wild type (Fig. 7C and Table 2), these vectors had a more severe defect in packaging (Table 2) (almost 2 log units lower) and were severely defective for infectivity (Table 2 and Fig. 7B) (approximately 5 log units). In contrast, GFP insertions into VP2A alone produced stocks that were 3 to 4 log units lower in infectivity (Table 2 and Fig. 7B).
To determine whether the particles that contained GFP inserts in both VP1 and VP2 (VP1,2A-GFP + VP3) behaved normally with respect to entry and trafficking, we used confocal microscopy. Confocal microscopic analysis of these particles in the absence (Fig. 8, top panels) and presence (Fig. 8, bottom panels) of helper Ad5 infection revealed that in the absence of helper virus, these AAV-like particles slowly accumulated in endosomes and/or cytoplasm perinuclearly over a 24-h period. However, dramatic changes were observed when helper virus was present, with the appearance of the viral GFP signal within the nucleus as early as 1 h. These results were in agreement with a previous report from our laboratory on the facilitation of AAV trafficking by Ad (61). Thus, the particles containing a 30-kDa GFP insertion in VP1 and VP2 behaved essentially like wild-type virus with respect to infection and trafficking in response to Ad coinfection.
FIG. 8.

Time course of VP1,2A-GFP plus VP3 particle trafficking following infection in the absence (top panels) and presence (bottom panels) of Ad5. HeLa cells were infected with AAV containing a GFP insertion at an MOI of 10,000 with or without Ad5 at an MOI of 20. Vectors remained on the cells for the duration of the time course. The input capsids appear green from the native GFP fluorescence of the capsid, the nuclei are stained red with PI, and early endosomal antigen 1 is stained blue.
DISCUSSION
The AAV particle is capable of transducing a wide range of dividing and nondividing cell types. The promiscuity of this gene therapy vector is due in part to the widespread distribution of its primary receptors (23, 34, 38, 50, 51) and the strong electrostatic interaction between cell surface heparan sulfate and the spike protrusion at the particle's threefold axes (23, 34, 51). To date, most of the strategies for retargeting AAV have involved inserting short, linear targeting sequences directly into the capsid genes, normally that for VP3, which is the most abundant capsid protein (5). The major goal of the present study was to see whether it was possible to incorporate significantly larger peptides into the AAV particle as a first step in retargeting the vector to alternative receptors requiring conformation-dependent ligands. Based on the symmetry of the particle and capsid protein molecular weight estimates (63), it has been proposed that of the 60 capsid proteins that make up a given particle, approximately 3 are VP1, 3 are VP2, and 54 are VP3. Thus, depending on the position within the cap ORF, retargeting sequences can result in the incorporation of differing numbers of ligands per particle. For instance, insertions immediately after residue 138 in the VP1-VP2 region have been shown to expand the tropism of the virus (45, 60) following the incorporation of approximately six modified capsid proteins (three VP1 and three VP2).
Theoretically, the insertion of a single full-length ligand could retarget the particle to a receptor, binding its ligand with 1:1 stoichiometry. Therefore, we confined our insertions to residue 138 to minimize disruption of the overall structural features of the particle (as 60 large ligands seemed excessive and more likely to sterically hinder assembly than 6 ligands). However, direct insertion of the coding sequence for FKN and LEP at this position led to the loss of VP3 expression (Fig. 1A) and did not result in particle formation (Fig. 1B). This was seen as well by others (40) and was presumably due to disruption of the readthrough translational initiation required for production of the critical VP3 protein (1). We were thus forced to consider the alternative of using insertions in only one capsid protein at a time, with the other two being functionally wild type. To test this possibility we constructed a series of complementing plasmids (Table 3) that would allow insertions into only one of the three capsid proteins at a time.
VP3-like proteins can be translated from three different methionine codons, and the first eight amino acids of VP3 appear to be essential for VP3 capsid assembly.
While VP1 and VP2 synthesis was easily eliminated by mutation of their respective start codons (Fig. 4B), the elimination of VP3 per se was interesting, requiring multiple mutations to generate the construct pVP1,2 (Fig. 2B). Ruffing et al. (44) had also previously seen alternative VP3-like proteins when the start codon was changed to Leu. We have demonstrated here that the alternative VP3 species are due to the use of alternative start codons downstream of the normal ATG for VP3 (M203). Readthrough translational initiation on the 2.3-kb mRNA continued for an additional 32 amino acids after M203 to positions M211 and M235. Since the M203L or M211L mutation prevented particle recovery (Fig. 4A), it appears that these residues play critical roles in particle assembly and/or stability. M203L results in an N-terminal truncation of VP3 (VP3a), while M211L is a point mutation in full-length VP3. These mutations are present in all three capsid proteins but appear to be critical to VP3, as the combination of pVP1,2 and pVP3 produced essentially wild-type recombinant particles. The formation of particles from the complementation groups are examples of positional rescue of mutations at the VP3 N terminus, as the M203L and M211L mutations that are required to eliminate VP3 expression (Fig. 2B) abolish particle formation (Fig. 3A) when present in all three capsid proteins yet yield particles that are essentially wild type when these mutations are present only in VP1 and/or VP2 (Fig. 5). The design of our production system results in the VP3 protein never having the M203,211,235L mutations (Table 3). In contrast, manipulation of the common C terminus of the cap ORF is apparently different (43, 60). A recent example of positional rescue was reported for the insertion of a His6 tag (for recombinant vector purification purposes) at the extreme C terminus of the cap ORF (66). In that report, the VP1 and VP2 capsid proteins were shown to be responsible for the defects in particle formation when the insertion was present in all three capsid proteins, and this position was rescued when the tag was present only in VP3.
VP2 appears to be redundant and nonessential for viral infectivity.
Surprisingly, the AAV-like particle composed of only VP1 and VP3 had infectious titers within a factor of 4 of the wild-type value (Fig. 4B and 7B; Table 2) and particle-to-infectivity ratios which were identical to that of the wild type. Thus, VP2 appeared to be a redundant capsid that is not essential for infectivity. This made it an ideal candidate for the insertion of large peptides for the purpose of retargeting the particle.
Earlier work had found the identical cap mutant to be defective for production of infectious virus (31). We have no good explanation for this discrepancy. We can only propose that improvement in AAV production and purification may have allowed us to now characterize this particle or that there might have been additional cryptic mutations in the earlier constructs. Similarly, expression of the three capsid proteins in a baculovirus system also suggested that VP2 may play a role in particle assembly (44, 49). Thus, the isolation of AAV-like particles from pVP1,3 was unexpected, since critical aspects of nuclear localization (20, 44) and particle formation (44) have been attributed to VP2. In contrast to that work, our attempts to make particles containing VP3 only or VP2-negative particles have been consistently in the presence of AAV replication proteins, rAAV DNA, and Ad helper functions. This may partly explain the discrepancy with the baculovirus systems and earlier experiments with Cos cells. Alternatively, this may reflect a property of AAV assembly in these cell types.
Curiously, while VP2-negative particles (VP1,3) appear to be functionally wild type, the VP2A plus VP3 group or VP3 alone produces particles that are more defective than those that are missing only VP1 (VP2,3) (Fig. 7B). Thus, in the absence of VP1, VP2 may perform some function in AAV infection. A comparison of the characteristics of the VP3 particle with those of the VP2,3 particle (Fig. 7B; Table 2) suggests that the additional VP2 residues may facilitate transduction in the absence of VP1. Possibly the basic residues that cluster in the VP2 N-terminal extension of VP3, which are capable of being nuclear localization signals (20), play a role. However, the VP2A,3 particle is less infectious than VP2,3, showing that the inclusion of more VP2 unique sequence into the particle is detrimental (Fig. 7B; Table 2).
VP3 is the only capsid protein required to form genome-containing particles.
AAV-like particles were obtained from any combination of capsid proteins or capsid mutants as long as VP3 was present (Fig. 7A and C; Table 3). Furthermore, VP3 alone was sufficient to make viral particles. We obtained viral particles composed of VP2,3, VP1,3, and VP3 only at wild-type particle titers (both full and empty) (Fig. 4B and C, 5, and 7A and Table 2). As expected, particles that were missing VP1 (VP2,3, VP2A,3, and VP3) were severely defective for infectivity (Fig. 7B; Table 2). This defect is presumably due to the absence of the phospholipase activity in the N-terminal region of VP1, as previously described (12, 18, 52, 60).
The recovery of the particle containing VP3 only (Fig. 4C and 7A; Table 2) agrees with a previous insertional mutagenesis study in which a particle that appeared to be composed exclusively of VP3 was isolated (40). Taken together, these results show that neither VP1 nor VP2 is absolutely required for nuclear localization of VP3 (44, 57) and beg the question as to which nuclear localization signals are employed by the three capsid proteins.
Complementary capsid protein expression allows formation of particles with large insertions exclusively in VP2 that have only modest defects in viral infectivity.
The key finding in this study is that it is possible to insert substantially larger peptides into AAV capsid proteins than previously shown, provided that the foreign peptide is fused to only one of the three capsid proteins. In our studies thus far, we have focused primarily on insertions into the minor capsid proteins. The insertion of FKN and LEP simultaneously into VP1 and VP2 had little effect on packaging efficiency but resulted in particles with low infectious titers (Fig. 7B; Table 2). This may be partly explained by spatial distortion of the phospholipase A2 motifs, but defects in viral uncoating cannot be ruled out. To rescue position 138 for insertion of large peptides with respect to infectivity, the inserted peptide had to be confined to VP2 exclusively. These AAV-like particles had titers within 1 log unit of wild-type particle and had infectious titers and particle-to-infectivity ratios virtually identical to those of wild-type virus (Fig. 7B; Table 2). Thus, ligands as large as 146 amino acids (LEP) appear to be readily accommodated by this method. In contrast, when the 238-amino-acid GFP was inserted into VP2, there was a significant drop in the particle-to-infectivity ratio (Fig. 7B; Table 2). It may be possible to correct this by increasing the intracellular expression of VP1, which was severely underrepresented in the VP2A-GFP plus VP3 particles, or by decreasing the VP2A-ligand concentration. This is currently being explored.
Nevertheless, it was possible to obtain and visualize particles with GFP inserted into both VP1 and VP2 (Figs. 7C and 8), and these VP1,2A-GFP plus VP3 particles appeared to traffic in a fashion similar to that described previously for wild-type virus (61), suggesting that insertions as large as the 30-kDa GFP could be tolerated. We have not yet reported ligand insertions that were exclusively in VP1 or VP3 at any surface positions previously shown to accommodate shorter peptides (12, 45, 60). However, we believe that these positions also may be useful for insertion of larger ligands with the use of the separate capsid expression plasmids described here.
In summary, while VP3 alone is sufficient to form a particle capable of protecting the viral genome and VP1 is required for efficient viral infectivity, VP2 is nonessential and tolerates large peptide insertions at its N terminus. The stoichiometry of the particle can be altered if VP2 is significantly overexpressed in the presence of native levels of VP1 and VP3. While the inserted sequences studied here are themselves potential targeting ligands, this system could also be applied to the insertion of large conjugate-based linker sequences (36, 41) or for the presentation of large immunogenic peptides for vaccine development by using empty particles formed with this system as the platform for epitope presentation. Future work with the described FKN and LEP particles will involve testing their ability to bind their respective receptors. The GFP-containing particles may have potential use in real-time in vivo fluorescent monitoring of events that occur during infection. It is evident that optimal retargeting of these particles with insertions at the N terminus of VP2 may require manipulation of linker sequences between the inserted ligand and VP2 to optimize presentation of the ligand binding domain. Furthermore, mutation of the recently identified residues involved in binding heparan sulfate proteoglycan (23, 34) will also be required to restrict these vectors to cellular entry via the targeting ligand-receptor interaction. Importantly, the system described here for modifying capsid proteins with larger peptide insertions in specific capsid proteins should facilitate development of retargeted AAV vectors for clinically relevant cell types and be applicable to all AAV serotypes and chimeric-type particles (3, 10, 16, 19, 39).
Acknowledgments
We thank Eric Kolbrenner for excellent technical assistance and Jurgen Kleinschmidt for helpful discussions during the course of this work.
This work has been supported by NIH grants PO1 HL51811 and P50 HL59412 and the Edward R. Koger Chair to N.M. K.H.W is supported by NIH training grant T32 AI 7110.
N.M. is an inventor on patents related to recombinant AAV technology and owns equity in a gene therapy company that is commercializing AAV for gene therapy applications.
Footnotes
This paper is dedicated to the memory of Howard N. Martindale.
REFERENCES
- 1.Becerra, S. P., F. Koczot, P. Fabisch, and J. A. Rose. 1988. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J. Virol. 62:2745-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becerra, S. P., J. A. Rose, M. Hardy, B. M. Baroudy, and C. W. Anderson. 1985. Direct mapping of adeno-associated virus capsid proteins B and C: a possible ACG initiation codon. Proc. Natl. Acad. Sci. USA 82:7919-7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowles, D. E., J. E. Rabinowitz, and R. J. Samulski. 2003. Marker rescue of adeno-associated virus (AAV) capsid mutants: a novel approach for chimeric AAV production. J. Virol. 77:423-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buller, R. M., and J. A. Rose. 1978. Characterization of adenovirus-associated virus-induced polypeptides in KB cells. J. Virol. 25:331-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buning, H., M. U. Ried, L. Perabo, F. M. Gerner, N. A. Huttner, J. Enssle, and M. Hallek. 2003. Receptor targeting of adeno-associated virus vectors. Gene Ther. 10:1142-1151. [DOI] [PubMed] [Google Scholar]
- 6.Cassinotti, P., M. Weitz, and J. D. Tratschin. 1988. Organization of the adeno-associated virus (AAV) capsid gene: mapping of a minor spliced mRNA coding for virus capsid protein 1. Virology 167:176-184. [PubMed] [Google Scholar]
- 7.Clark, K. R., X. Liu, J. P. McGrath, and P. R. Johnson. 1999. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum. Gene Ther. 10:1031-1039. [DOI] [PubMed] [Google Scholar]
- 8.Clark, K. R., F. Voulgaropoulou, and P. R. Johnson. 1996. A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity titration of adeno-associated virus vectors. Gene Ther. 3:1124-1132. [PubMed] [Google Scholar]
- 9.Dubielzig, R., J. A. King, S. Weger, A. Kern, and J. A. Kleinschmidt. 1999. Adeno-associated virus type 2 protein interactions: formation of pre-encapsidation complexes. J. Virol. 73:8989-8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao, G., M. R. Alvira, S. Somanathan, Y. Lu, L. H. Vandenberghe, J. J. Rux, R. Calcedo, J. Sanmiguel, Z. Abbas, and J. M. Wilson. 2003. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. USA 100:6081-6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girod, A., M. Ried, C. Wobus, H. Lahm, K. Leike, J. Kleinschmidt, G. Deleage, and M. Hallek. 1999. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 5:1052-1056. (Erratum, 5:1438.) [DOI] [PubMed] [Google Scholar]
- 12.Girod, A., C. E. Wobus, Z. Zadori, M. Ried, K. Leike, P. Tijssen, J. A. Kleinschmidt, and M. Hallek. 2002. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 83:973-978. [DOI] [PubMed] [Google Scholar]
- 13.Grifman, M., M. Trepel, P. Speece, L. B. Gilbert, W. Arap, R. Pasqualini, and M. D. Weitzman. 2001. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 3:964-975. [DOI] [PubMed] [Google Scholar]
- 14.Grimm, D., A. Kern, M. Pawlita, F. Ferrari, R. Samulski, and J. Kleinschmidt. 1999. Titration of AAV-2 particles via a novel capsid ELISA: packaging of genomes can limit production of recombinant AAV-2. Gene Ther. 6:1322-1330. [DOI] [PubMed] [Google Scholar]
- 15.Grimm, D., A. Kern, K. Rittner, and J. A. Kleinschmidt. 1998. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 9:2745-2760. [DOI] [PubMed] [Google Scholar]
- 16.Hauck, B., L. Chen, and W. Xiao. 2003. Generation and characterization of chimeric recombinant AAV vectors. Mol. Ther. 7:419-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hermens, W. T., O. ter Brake, P. A. Dijkhuizen, M. A. Sonnemans, D. Grimm, J. A. Kleinschmidt, and J. Verhaagen. 1999. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 10:1885-1891. [DOI] [PubMed] [Google Scholar]
- 18.Hermonat, P. L., M. A. Labow, R. Wright, K. I. Berns, and N. Muzyczka. 1984. Genetics of adeno-associated virus: isolation and preliminary characterization of adeno-associated virus type 2 mutants. J. Virol. 51:329-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hildinger, M., A. Auricchio, G. Gao, L. Wang, N. Chirmule, and J. M. Wilson. 2001. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J. Virol. 75:6199-6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoque, M., K. Ishizu, A. Matsumoto, S. I. Han, F. Arisaka, M. Takayama, K. Suzuki, K. Kato, T. Kanda, H. Watanabe, and H. Handa. 1999. Nuclear transport of the major capsid protein is essential for adeno-associated virus capsid formation. J. Virol. 73:7912-7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter, L. A., and R. J. Samulski. 1992. Colocalization of adeno-associated virus Rep and capsid proteins in the nuclei of infected cells. J. Virol. 66:317-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janik, J. E., M. M. Huston, and J. A. Rose. 1984. Adeno-associated virus proteins: origin of the capsid components. J. Virol. 52:591-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kern, A., K. Schmidt, C. Leder, O. J. Muller, C. E. Wobus, K. Bettinger, C. W. Von der Lieth, J. A. King, and J. A. Kleinschmidt. 2003. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 77:11072-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kronenberg, S., J. A. Kleinschmidt, and B. Bottcher. 2001. Electron cryo-microscopy and image reconstruction of adeno-associated virus type 2 empty capsids. EMBO Rep. 2:997-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kube, D. M., S. Ponnazhagan, and A. Srivastava. 1997. Encapsidation of adeno-associated virus type 2 Rep proteins in wild-type and recombinant progeny virions: Rep-mediated growth inhibition of primary human cells. J. Virol. 71:7361-7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, J., R. J. Samulski, and X. Xiao. 1997. Role for highly regulated rep gene expression in adeno-associated virus vector production. J. Virol. 71:5236-5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loiler, S. A., T. J. Conlon, S. Song, Q. Tang, K. H. Warrington, A. Agarwal, M. Kapturczak, C. Li, C. Ricordi, M. A. Atkinson, N. Muzyczka, and T. R. Flotte. 2003. Targeting recombinant adeno-associated virus vectors to enhance gene transfer to pancreatic islets and liver. Gene Ther. 10:1551-1558. [DOI] [PubMed] [Google Scholar]
- 28.McCarty, D. M., M. Christensen, and N. Muzyczka. 1991. Sequences required for coordinate induction of adeno-associated virus p19 and p40 promoters by Rep protein. J. Virol. 65:2936-2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McPherson, R. A., and J. A. Rose. 1983. Structural proteins of adenovirus-associated virus: subspecies and their relatedness. J. Virol. 46:523-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller, O. J., F. Kaul, M. D. Weitzman, R. Pasqualini, W. Arap, J. A. Kleinschmidt, and M. Trepel. 2003. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 21:1040-1046. [DOI] [PubMed] [Google Scholar]
- 31.Muralidhar, S., S. P. Becerra, and J. A. Rose. 1994. Site-directed mutagenesis of adeno-associated virus type 2 structural protein initiation codons: effects on regulation of synthesis and biological activity. J. Virol. 68:170-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muzyczka, N., and K. I. Berns. 2001. Parvoviridae: the viruses and their replication, p. 2327-2360. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams and Wilkins, New York, N.Y.
- 33.Nicklin, S. A., H. Buening, K. L. Dishart, M. de Alwis, A. Girod, U. Hacker, A. J. Thrasher, R. R. Ali, M. Hallek, and A. H. Baker. 2001. Efficient and selective aav2-mediated gene transfer directed to human vascular endothelial cells. Mol. Ther. 4:174-181. [DOI] [PubMed] [Google Scholar]
- 34.Opie, S. R., K. H. Warrington, Jr., M. Agbandje-McKenna, S. Zolotukhin, and N. Muzyczka. 2003. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 77:6995-7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perabo, L., H. Buning, D. M. Kofler, M. U. Ried, A. Girod, C. M. Wendtner, J. Enssle, and M. Hallek. 2003. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol. Ther. 8:151-157. [DOI] [PubMed] [Google Scholar]
- 36.Ponnazhagan, S., G. Mahendra, S. Kumar, J. A. Thompson, and M. Castillas, Jr. 2002. Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J. Virol. 76:12900-12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prasad, K. M., and J. P. Trempe. 1995. The adeno-associated virus Rep78 protein is covalently linked to viral DNA in a preformed virion. Virology 214:360-370. [DOI] [PubMed] [Google Scholar]
- 38.Qing, K., C. Mah, J. Hansen, S. Zhou, V. Dwarki, and A. Srivastava. 1999. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 5:71-77. [DOI] [PubMed] [Google Scholar]
- 39.Rabinowitz, J. E., F. Rolling, C. Li, H. Conrath, W. Xiao, X. Xiao, and R. J. Samulski. 2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 76:791-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabinowitz, J. E., W. Xiao, and R. J. Samulski. 1999. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology 265:274-285. [DOI] [PubMed] [Google Scholar]
- 41.Ried, M. U., A. Girod, K. Leike, H. Buning, and M. Hallek. 2002. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 76:4559-4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rose, J. A., J. V. Maizel, Jr., J. K. Inman, and A. J. Shatkin. 1971. Structural proteins of adenovirus-associated viruses. J. Virol. 8:766-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruffing, M., H. Heid, and J. A. Kleinschmidt. 1994. Mutations in the carboxy terminus of adeno-associated virus 2 capsid proteins affect viral infectivity: lack of an RGD integrin-binding motif. J. Gen. Virol. 75:3385-3392. [DOI] [PubMed] [Google Scholar]
- 44.Ruffing, M., H. Zentgraf, and J. A. Kleinschmidt. 1992. Assembly of viruslike particles by recombinant structural proteins of adeno-associated virus type 2 in insect cells. J. Virol. 66:6922-6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi, W., G. S. Arnold, and J. S. Bartlett. 2001. Insertional mutagenesis of the adeno-associated virus type 2 (aav2) capsid gene and generation of aav2 vectors targeted to alternative cell-surface receptors. Hum. Gene Ther. 12:1697-1711. [DOI] [PubMed] [Google Scholar]
- 46.Shi, W., and J. S. Bartlett. 2003. RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Mol. Ther. 7:515-525. [DOI] [PubMed] [Google Scholar]
- 47.Smuda, J. W., and B. J. Carter. 1991. Adeno-associated viruses having nonsense mutations in the capsid genes: growth in mammalian cells containing an inducible amber suppressor. Virology 184:310-318. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava, A., E. W. Lusby, and K. I. Berns. 1983. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 45:555-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinbach, S., A. Wistuba, T. Bock, and J. A. Kleinschmidt. 1997. Assembly of adeno-associated virus type 2 capsids in vitro. J. Gen. Virol. 78:1453-1462. [DOI] [PubMed] [Google Scholar]
- 50.Summerford, C., J. S. Bartlett, and R. J. Samulski. 1999. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5:78-82. [DOI] [PubMed] [Google Scholar]
- 51.Summerford, C., and R. J. Samulski. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tratschin, J. D., I. L. Miller, and B. J. Carter. 1984. Genetic analysis of adeno-associated virus: properties of deletion mutants constructed in vitro and evidence for an adeno-associated virus replication function. J. Virol. 51:611-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trempe, J. P., and B. J. Carter. 1988. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J. Virol. 62:3356-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veldwijk, M. R., J. Topaly, S. Laufs, U. R. Hengge, F. Wenz, W. J. Zeller, and S. Fruehauf. 2002. Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol. Ther. 6:272-278. [DOI] [PubMed] [Google Scholar]
- 55.Vincent, K. A., S. T. Piraino, and S. C. Wadsworth. 1997. Analysis of recombinant adeno-associated virus packaging and requirements for rep and cap gene products. J. Virol. 71:1897-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weger, S., A. Wistuba, D. Grimm, and J. A. Kleinschmidt. 1997. Control of adeno-associated virus type 2 cap gene expression: relative influence of helper virus, terminal repeats, and Rep proteins. J. Virol. 71:8437-8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wistuba, A., A. Kern, S. Weger, D. Grimm, and J. A. Kleinschmidt. 1997. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J. Virol. 71:1341-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wistuba, A., S. Weger, A. Kern, and J. A. Kleinschmidt. 1995. Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing Rep and Cap proteins. J. Virol. 69:5311-5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wobus, C. E., B. Hugle-Dorr, A. Girod, G. Petersen, M. Hallek, and J. A. Kleinschmidt. 2000. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74:9281-9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu, P., W. Xiao, T. Conlon, J. Hughes, M. Agbandje-McKenna, T. Ferkol, T. Flotte, and N. Muzyczka. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 74:8635-8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao, W., K. H. Warrington, Jr., P. Hearing, J. Hughes, and N. Muzyczka. 2002. Adenovirus-facilitated nuclear translocation of adeno-associated virus type 2. J. Virol. 76:11505-11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao, X., J. Li, and R. J. Samulski. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72:2224-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xie, Q., W. Bu, S. Bhatia, J. Hare, T. Somasundaram, A. Azzi, and M. S. Chapman. 2002. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 99:10405-10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang, Q., M. Mamounas, G. Yu, S. Kennedy, B. Leaker, J. Merson, F. Wong-Staal, M. Yu, and J. R. Barber. 1998. Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum. Gene Ther. 9:1929-1937. [DOI] [PubMed] [Google Scholar]
- 65.Zadori, Z., J. Szelei, M. C. Lacoste, Y. Li, S. Gariepy, P. Raymond, M. Allaire, I. R. Nabi, and P. Tijssen. 2001. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 1:291-302. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, H. G., J. Xie, I. Dmitriev, E. Kashentseva, D. T. Curiel, H. C. Hsu, and J. D. Mountz. 2002. Addition of six-His-tagged peptide to the C terminus of adeno-associated virus VP3 does not affect viral tropism or production. J. Virol. 76:12023-12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zolotukhin, S., B. J. Byrne, E. Mason, I. Zolotukhin, M. Potter, K. Chesnut, C. Summerford, R. J. Samulski, and N. Muzyczka. 1999. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6:973-985. [DOI] [PubMed] [Google Scholar]
- 68.Zolotukhin, S., M. Potter, W. Hauswirth, J. Guy, and N. Muzyczka. 1996. A humanized green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol. 70:4646-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zolotukhin, S., M. Potter, I. Zolotukhin, Y. Sakai, S. Loiler, T. J. Fraites, Jr., V. A. Chiodo, T. Phillipsberg, N. Muzyczka, W. W. Hauswirth, T. R. Flotte, B. J. Byrne, and R. O. Snyder. 2002. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods Enzymol. 28:158-167. [DOI] [PubMed] [Google Scholar]