

Abstract
Purpose of review
The molecular mechanisms that underlie chromosome 9 open reading frame 72 (C9orf72)-associated amyotrophic lateral sclerosis and frontotemporal dementia are rapidly emerging. Two potential disease mechanisms have been postulated – gain or loss of function. We provide an overview of recent advances that support or oppose gain-of-function and loss-of-function mechanisms.
Recent findings
Since the discovery that a noncoding repeat expansion in C9orf72 was responsible for chromosome 9-linked amyotrophic lateral sclerosis and frontotemporal dementia in 2011, a plethora of studies have investigated clinical, pathological and mechanistic aspects of the disease. Loss of function is supported by reduced levels of C9orf72 in patient brain and functional work, revealing a role of the C9orf72 protein in endocytic and autophagic pathways and motor function. Gain of function is supported by the presence in patient brain of both repeat RNA and protein aggregates. Repeat RNA aggregates termed RNA foci, a hallmark of noncoding repeat expansion diseases, have been shown to sequester proteins involved in RNA splicing, editing, nuclear export and nucleolar function. Repeat-associated non-ATG dependent translation gives rise to toxic dipeptide repeat proteins that form inclusions in patient tissue. Antisense oligonucleotides targeting C9orf72 have shown promise for combating gain-of-function toxicity.
Summary
Rapid progress is being made towards understanding this common genetic cause of amyotrophic lateral sclerosis and frontotemporal dementia. Overall, the weight of data currently sits in favour of gain of function as the most important disease mechanism, which has important implications for the development of effective and targeted therapies.
Keywords: amyotrophic lateral sclerosis, C9orf72, frontotemporal dementia, gain or loss of function
INTRODUCTION
A noncoding repeat expansion in C9orf72 is a common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTD) (C9FTD/ALS) [1,2]. Disease may occur through loss of function of C9orf72, or two distinct gain-of-function mechanisms: first, the formation of repeat RNA aggregates, termed RNA foci, in neuronal nuclei that sequester important RNA-binding proteins and second, the generation of toxic, dipeptide repeat (DPR) proteins, due to the repeat RNA mediating its own translation (Fig. 1) [3]. In this review, we will cover recent findings concerning the role of each of these mechanisms in C9FTD/ALS and their relevance for developing therapeutics. Key findings relating to loss or gain of function are summarized in Table 1[4,5▪–7▪,8▪▪,9▪–11▪,12–14,15▪,16▪,17▪▪,18,19▪–26▪,27▪▪,28▪▪,29▪–31▪].
FIGURE 1.
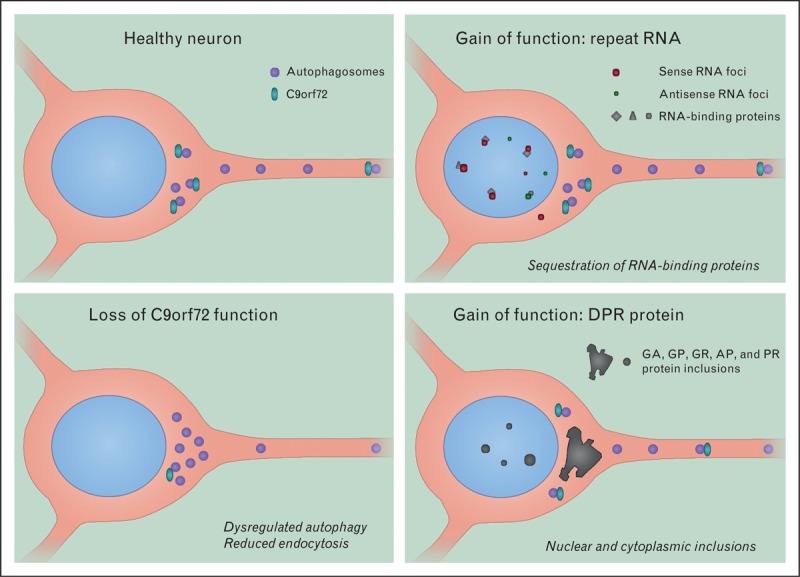
Potential mechanisms of disease in C9FTD/ALS. AP, alanine-proline; DPR, dipeptide repeat; GA, glycine-alanine; GP, glycine-proline; GR, glycine-arginine; PR, proline-arginine.
Table 1.
Summary of the key evidence for gain-and loss-of-function mechanisms in C9FTD/ALS
| For (+) or against (−) hypothesis | Study | |
| Loss-of-function | ||
| Reduced mRNA and protein expression | ||
| All mRNA isoforms reduced in C9FTD/ALS patient tissue and iPSC neurons | + | [1,4,5▪–7▪,8▪▪] |
| Reduced C9orf72 protein in C9FTD/ALS patient brain | + | [9▪] |
| Hypermethylation of C9orf72 promoter reduces transcript levels and correlates with shorter disease duration | + | [10▪] |
| Hypermethylation of C9orf72 promoter reduces transcript levels, increasing resistance to cellular stress and reducing RNA foci and DPR proteins | – | [11▪] |
| Models of loss of protein | ||
| Dysregulation of cellular trafficking associated with reduction of C9orf72 protein | + | [12–14] |
| C9orf72 orthologue knockdown in zebrafish has a motor phenotype | + | [15▪] |
| C9orf72 orthologue knockout in Caenorhabditis elegans has a motor phenotype | + | [16▪] |
| C9orf72 orthologue knockdown in mice has no motor phenotype | – | [17▪▪] |
| Genetics | ||
| Lack of coding mutations in C9orf72 protein in C9ALS | – | [18] |
| Homozygous C9FTD case not as severe clinically and pathologically as pure loss-of-function diseases | – | [5▪] |
| Gain-of-function: RNA | ||
| RNA foci | ||
| Sense and antisense foci identified in C9FTD/ALS patient tissue and iPSC neurons | + | [1,6▪,8▪▪,17▪▪,19▪–22▪] |
| Sense foci burden correlates with age-at-onset in C9FTD | + | [19▪] |
| Sequestration of RNA-binding proteins | ||
| ADARB2 – siRNA reduces sense RNA foci in C9ALS iPSC neurons and exacerbates glutamate-induced toxicity in control iPSC neurons | + | [8▪▪] |
| hnRNP A1 and pur-alpha – colocalization with sense RNA foci in iPSC motor neurons | + | [22▪] |
| hnRNP A3 – identified in neuronal cytoplasmic and intranuclear inclusions | + | [23▪] |
| hnRNP H – colocalization with sense RNA foci in patient brain | + | [24▪] |
| Nucleolin – colocalization with sense RNA foci in patient brain and cells, indications of nucleolar stress | + | [25▪] |
| Pur-alpha – overexpression rescues GGGGCC-dependent degeneration in Drosophila | + | [26▪] |
| Antisense oligonucleotide (ASO) treatment | ||
| ASOs reduce sense RNA foci and reverse transcriptome changes and toxicity in patient fibroblasts and iPSC neurons | + | [8▪▪,17▪▪,22▪] |
| Gain-of-function: DPR proteins | ||
| Inclusion pathology | ||
| All DPR proteins found in neuronal cytoplasmic inclusions in C9FTD/ALS patient brain and iPSC neurons | + | [6▪,20▪,21▪,27▪,28▪,29▪] |
| Poly-GA DPR protein inclusions found prior to TDP-43 inclusions in patient brain | + | [30▪] |
| Toxicity | ||
| GA DPR protein inclusion distribution does not correlate with neurodegeneration in C9FTD/ALS patient brain | – | [31▪] |
| Poly-GP and poly-PR DPR proteins made from GGGGCC repeats are toxic to HEK293 cells (other DPR proteins not assessed) | + | [21▪] |
ALS, amyotrophic lateral sclerosis; DPR, dipeptide repeat; FTD, frontotemporal dementia; GA, glycine-alanine; GP, glycine-proline; hnRNP, heterogeneous nuclear ribonucleoprotein; iPSC, induced pluripotent stem cell; PR, proline-arginine.
Box 1.
no caption available
GAIN AND LOSS OF FUNCTION IN NONCODING REPEAT EXPANSION DISORDERS
Noncoding repeat expansions have been commonly assigned into two groups: those which cause loss of function of the protein in which the mutation resides (fragile X syndrome and Friedreich's ataxia), and those in which a gain-of-function mechanism has been identified because of repeat RNA species [myotonic dystrophy, fragile X-associated tremor/ataxia syndrome (FXTAS), Huntington's disease-like 2 and spinocerebellar ataxia types 8, 10, 12 and 31]. In the fragile X mental retardation protein (FMRP) gene, CGG repeat size determines the clinical syndrome; expansions of 55–200 repeats result in the gain-of-function disease FXTAS [32,33], whereas expansions of more than 200 repeats lead to hypermethylation of the FMRP gene, which silences transcription, leading to a loss of FMRP function and fragile X syndrome [34]. There is no evidence for such a bimodal mechanism in C9FTD/ALS, indeed, a large study found no effect of repeat size on clinical presentation [35▪]. The first mechanism attributed to the gain-of-function noncoding repeat expansion diseases was toxic functions of the repeat RNA [36]. Recently, a novel mechanism was identified whereby expanded CAG repeats are translated in the absence of an ATG initiation codon, termed repeat-associated non-ATG dependent (RAN) translation [37]. RAN translation has now been found to be common to several noncoding repeat expansions, including C9FTD/ALS [38], increasing the spectrum of potential mechanisms in disease.
LOSS OF FUNCTION OF C9ORF72 PROTEIN
The main support for a loss-of-function mechanism in C9FTD/ALS is numerous reports of decreased transcript levels of all three C9orf72 mRNA variants in patient-derived cells and tissue [1,4,5▪–7▪,8▪▪]. Reduced levels of C9orf72 transcripts may be due to hypermethylation of the C9orf72 promoter or increased histone methylation [7▪,10▪,11▪]. Unexpectedly, lower levels of C9orf72 transcripts are also identified in ALS/FTD cases without C9orf72 repeat expansion [15▪], suggesting that loss of C9orf72 could be part of a common pathway affected in these diseases. Current studies on C9orf72 protein are limited by a lack of specific antibodies. However, one study developed a new C9orf72 antibody which detects a protein of 48 kDa in human cell lines that is specifically reduced following treatment with siRNA targeting C9orf72[9▪]. This antibody was used to show reduced C9orf72 protein in frontal cortex, but not cerebellum, of C9FTD/ALS patient brain compared to ALS cases without C9orf72 repeat expansion [9▪], consistent with findings of reduced transcript levels.
When the mutation was discovered in 2011, C9orf72 protein was of unknown function. However, recent studies show that C9orf72 has a high homology to differentially expressed in normal and neoplasia proteins [13,14]. This family of proteins function as guanine nucleotide exchange factors (GEFs) that activate Rab guanosine 5’-triphosphate (GTP)ase and therefore regulate membrane trafficking [13,14]. Consistent with this, knockdown of C9orf72 leads to reduced endocytosis and dysregulated autophagy in human neuroblastoma cells [12]. Impaired autophagy and endolysosomal degradation are implicated in neurodegenerative diseases [39], thus these data provide a basis for loss of function playing a role in C9FTD/ALS. However, better antibodies are urgently needed to definitively determine the cellular distribution and function of C9orf72 protein; analysis of innate GEF activity will also provide important insight into C9orf72 function.
Knockdown of the zebrafish orthologue of C9orf72 (zC9orf72) with antisense morpholino oligonucleotides leads to axonopathy and motor deficits, which can be rescued by expression of human C9orf72[15▪]. Homozygous knockout of the worm orthologue of C9orf72 (alfa-1) also results in motor phenotypes [16▪]. Together, these data suggest that the loss of C9orf72 protein can lead to motor abnormalities, which argues for a role of loss of function in C9FTD/ALS. Conversely, intracerebroventricular delivery to adult mice of antisense oligonucleotides (ASOs) targeting C9orf72 leads to knockdown of C9orf72 throughout the central nervous system but does not result in any motor or behavioural phenotypes [17▪▪]. It is possible that knockdown of C9orf72 during development has differing effects to knockdown in adults which could explain the discrepancy between the models. Also arguing against a loss-of-function mechanism, no mutations have been found in coding regions of the C9orf72 gene [18]. Additionally, a rare homozygous C9orf72 repeat expansion case did not exhibit clinical or pathological features outside the usual disease spectrum as would be expected from reports of homozygous cases in pure loss-of-function diseases [5▪].
Reduced C9orf72 transcription via cytosine-phosphate-guanine (CpG) hypermethylation correlated with shorter disease duration suggesting C9orf72 reduction may play a role in disease [10▪]. A second study showed C9orf72 promoter CpG hypermethylation reduced C9orf72 mRNA levels, but also decreased RNA foci and RAN protein formation in C9FTD/ALS lymphoblasts and brains. Furthermore, treatment with a demethylating agent increased the vulnerability of C9FTD/ALS lymphoblasts to external stressors, suggesting reduction of C9orf72 levels may be a protective mechanism [11▪].
On the basis of evidence described above, haploinsufficiency of C9orf72 may cause defects in endosomal and autophagic processes and motor function. Therefore, the degree of reduction of C9orf72 may well modulate the disease phenotype. However, clinical data suggest that loss of function is unlikely to be the predominant causative mechanism for neurodegeneration in C9FTD/ALS.
GAIN OF FUNCTION: REPEAT RNA
RNA gain of function is a common mechanism in noncoding repeat expansion diseases [40]. The proposed mode of action in these diseases is via sequestration of essential RNA-binding proteins into aggregates of repeat-containing RNA foci, in the nucleus of affected cells. The most clearly defined molecular mechanism occurs in myotonic dystrophy (DM), a neuromuscular disease caused by CTG or CCTG repeat expansions in the dystrophia myotonica protein kinase (DMPK) or zinc finger protein 9 (ZNF9) genes (DM type 1 or 2, respectively) [36]. In DM type 1 or 2, RNA foci containing CUG repeat RNA sequester the splicing factor muscleblind-like protein 1, whose loss results in the mis-splicing of a muscle-specific chloride channel that is directly responsible for the myotonia observed in patients [41]. On the basis of this very clear example of RNA gain of function, great effort has focussed on investigating this mechanism in C9FTD/ALS. RNA foci composed of sense and antisense repeat RNA are present in frontal cortex, hippocampus, cerebellum and spinal cord of C9FTD/ALS patients [1,6▪,8▪▪,17▪▪,19▪–22▪]. In support of a role in disease, increased burden of RNA foci in neurons correlates with lower age-at-onset of disease in C9FTD cases, with the strongest correlation with sense foci in the frontal cortex, the region most affected in FTD [19▪]. RNA foci were present in several types of glial cells (astrocytes, microglia and oligodendrocytes), but are predominantly a neuronal phenotype [17▪▪,19▪,20▪], which is reflective of relative expression levels of C9orf72 in these cell types in mouse [42]. This raises the possibility that toxicity could arise from non-cell-autonomous routes. Indeed, astrocytes derived from familial and sporadic ALS patients, including C9ALS cases, can exert toxicity to motor neurons [43].
Although RNA foci can be found in the cytoplasm, the vast majority are localized in the nucleus [17▪▪,19▪]. Several studies have employed biochemical techniques to identify binding partners of the expanded sense repeat in vitro using differing methods and sources of protein, reviewed in [44]. Sequestration of some of these proteins into sense RNA foci has been assessed in patient-derived cells and tissue (Table 2). Some inconsistency has been observed between studies, but investigations in larger cohorts will be needed to clarify whether this is due to variation between brain regions or patients or differences due to detection protocols. Splicing factors constituted a large proportion of identified RNA-binding proteins. Of these, the splicing factors heterogeneous nuclear ribonucleoprotein (hnRNP) A1, hnRNP H and serine/arginine-rich splicing factor 2 (SRSF2; also known as SC35) are found to be sequestered into sense RNA foci [22▪,24▪,45▪]. Loss of function of these proteins would be predicted to cause downstream changes in splicing in target mRNAs. All these splicing factors affect splicing of a wide variety of targets, so a major challenge will be to determine whether specific targets are responsible for neurodegeneration and to confirm specific alteration of these targets in C9FTD/ALS patients.
Table 2.
Reported sequestration of RNA-binding proteins into sense RNA foci in patient cells and tissue
| RNA-binding protein | C9ALS or FTD | Patient-derived cells | Patient tissue | Study |
| ADARB2 | ALS | iPSC-differentiated neurons | Motor cortex | [8▪▪] |
| ALYREF | ALS | ND | Cerebellum and spinal cord | [45▪] |
| hnRNP A1 | ALS and ALS/FTD | iPSC-differentiated motor neurons | [22▪] | |
| ALS | ND | Cerebellum | [45▪] | |
| hnRNP Ha | ALS | ND | Cerebellum | [24▪] |
| ALS | Cerebellum and cerebellum and spinal cord | [45▪] | ||
| Nucleolin | ALS | ND | Motor cortex | [25▪] |
| Pur-alpha | ALS and ALS/FTD | iPSC-differentiated motor neurons | [22▪] | |
| SRSF2 (SC35) | ALS | ND | Cerebellum | [24▪] |
| ALS | ND | Cerebellum and spinal cord | [45▪] |
ADARB2, adenosine deaminase, RNA-specific, B2; ALS, amyotrophic lateral sclerosis; ALYREF, Aly/REF export factor; FTD, frontotemporal dementia; hnRNP, heterogeneous nuclear ribonucleoprotein; iPSC, induced pluripotent stem cell; ND, not determined; SRSF2 (SC35), serine/arginine-rich splicing factor 2 (also known as SC35).
aSequestration of hnRNP H into RNA foci was not observed in C9FTD iPSC-neurons [6▪].
Proteins involved in nuclear mRNA export also bind to GGGGCC repeats and are sequestered into foci containing sense RNA transcripts [22▪,45▪]. Aly/REF export factor (ALYREF) functions as an adaptor for mature RNA, transferring it through the nuclear RNA export factor 1 pathway to the nuclear pore for export [46]. Pur-alpha has also been implicated in nuclear export and trafficking in dendritic RNA granules for the regulation of local translation [47,48]. Another RNA-binding protein, hnRNP A3, which binds GGGGCC repeat RNA in vitro, but does not colocalize with RNA foci [23▪], is also proposed to function in a similar manner [49]. These proteins may contribute to export of C9orf72 repeat-containing RNA from the nucleus for C9orf72 and RAN protein translation or degradation. Sequestration of these factors into RNA foci may also prevent export of their other target mRNAs and affect downstream cellular functions.
Other proteins found to interact with GGGGCC repeats were adenosine deaminase, RNA-specific, B2 (ADARB2) [8▪▪] and nucleolin [25▪]. ADARB2 colocalizes with sense RNA foci in C9ALS induced pluripotent stem cell (iPSC)-differentiated motor neurons and patient motor cortex, leading to significant nuclear accumulation of the protein compared with controls. Unexpectedly, knockdown of ADARB2 results in reduced RNA foci in iPSC motor neurons, suggesting a role in foci formation or stabilization. ADARB2 (also known as ADAR3 or RED2) is a member of the ADAR (adenosine deaminase, RNA specific) RNA editing family, but unlike the other members, lacks editing activity [50]. Decrease in another member of this family, ADAR2, is thought to underlie excitotoxic loss of motor neurons in ALS via reduced editing of AMPA receptors subunits [51].
GGGGCC RNA and DNA repeats exhibit stable G-quadruplex formation in vitro[25▪,52▪,53]. These structures are involved in telomere stability, RNA splicing, transport and degradation and regulation of translation [54–56]. A recent study investigated binding of proteins to GGGGCC RNA hairpins, GGGGCC G-quadruplexes and antisense GGCCCC hairpins [25▪]. The nucleolar protein nucleolin was shown to have specificity for sense G-quadruplex structures, and dispersal of nucleolin staining in the nucleus was observed in C9ALS patient-derived cells and tissue, with sequestration into sense RNA foci visible in the motor cortex. Consequences of impaired nucleolar function were also observed, including decreased RNA processing and an increase in the number of P bodies, which are ribonucleoprotein complexes involved in the degradation of untranslated RNAs. These changes could be recapitulated when GGGGCC repeats were expressed in a cell line, implicating nucleolar stress as a gain-of-function RNA mechanism in C9FTD/ALS.
It is intriguing to speculate whether the heterogeneity seen in C9FTD/ALS could be attributed to differential sequestration of RNA-binding proteins between brain regions and patients, because of protein abundance or availability or RNA foci burden. No protein sequestration into antisense RNA foci has yet been identified, but this could be important as ASOs targeting sense RNA did not reverse all transcriptome changes in C9FTD/ALS-derived fibroblasts [17▪▪]. This suggests antisense transcripts may also cause transcriptional changes, potentially through sequestration of RNA-binding proteins. Repeat RNA may also exert neurotoxic effects through processes other than RNA foci generation. In a fly model of myotonic dystrophy and a cell model of Huntington's disease, expression of expanded repeats led to double stranded RNAs that were processed by the dicer complex into short siRNAs that exert toxicity by silencing complementary CAG or CTG repeat-containing transcripts, respectively [57,58]. It will be interesting to determine whether this pathway contributes to C9FTD/ALS. In a similar manner, antisense transcripts may regulate the levels of sense transcripts, and vice versa, via antisense-mediated RNA degradation. This may explain why sense and antisense RNA foci are rarely found within the same cell [19▪,21▪]. In Huntington's disease, antisense transcripts reduce huntingtin protein expression, partially via dicer [59]. This mechanism could also contribute to the reduced C9orf72 transcript levels observed in C9FTD/ALS patients.
In summary, the greatest evidence in support for a role of RNA gain of function in C9FTD/ALS are the abundant sense and antisense RNA foci in patient tissue that correlate with clinical phenotypes and can sequester RNA-binding proteins. However, a clear mechanism definitively linking sequestration of specific RNA-binding proteins to disease pathogenesis (as is the case for myotonic dystrophy) is currently lacking.
GAIN OF FUNCTION: DIPEPTIDE REPEAT PROTEINS
The other novel and potentially toxic species in C9FTD/ALS are the DPR proteins formed by RAN translation of the expanded repeat [20▪,21▪,27▪▪,28▪▪,29▪]. DPR proteins are translated from all frames of the GGGGCC repeat resulting in polymers of glycine-alanine (GA), glycine-proline (GP) and glycine-arginine (GR) in the sense frames, and glycine-proline (GP), alanine-proline (AP) and proline-arginine (PR) in the antisense frames. Although poly-GP is translated from both sense and antisense RNA, translation has been found to continue after the repeat expansion (using antibodies against downstream regions), and thus these poly-GP proteins have different carboxy terminal tails that may affect their function [21▪]. All DPR proteins form widespread neuronal cytoplasmic inclusions in patient brain [20▪,21▪,27▪▪,28▪▪,29▪] that frequently colocalize with p62-positive [but not TAR DNA-binding protein 43 (TDP-43)-positive] inclusions [28▪▪,29▪,31▪,60]. Poly-GP, and poly-GA DPR proteins additionally display dot-like neuronal intranuclear inclusions [27▪▪,28▪▪,31▪]. Unlike RNA foci, DPR inclusions appear to be an exclusively neuronal phenotype [27▪▪,31▪,60], possibly reflective of the clearance ability of mitotic cells.
A detailed pathological analysis of C9FTD/ALS cases found that TDP-43, but not poly-GA pathology, correlates with neurodegeneration [31▪], suggesting a lack of pathogenicity of poly-GA inclusions. However, this does not rule out toxicity of soluble GA polymers or other DPR proteins. Interestingly, rare C9orf72 cases have been reported with DPR but not TDP-43 pathology [28▪▪,30▪]. This suggests DPR proteins can be toxic without invoking TDP-43 dysfunction.
DISSECTION OF GAIN-OF-FUNCTION MECHANISMS
We have reviewed evidence for gain-of-function and loss-of-function mechanisms in C9FTD/ALS. However, it still remains to be determined which species and pathways are responsible for neurodegeneration and clinical phenotypes in disease. Co-occurrence of sense and antisense RNA foci with DPR, p62 or TDP-43 protein inclusions is only as frequent as expected by chance [8▪▪,19▪–21▪,45▪], suggesting a lack of interdependence between species and distinct toxic mechanisms. Overexpression of expanded GGGGCC repeats (outside the context of the C9orf72 gene) can induce RNA foci formation [20▪,24▪] and DPR protein production [20▪,21▪,28▪▪]. Repeats can also exert toxicity in cell lines [21▪,24▪,25▪], flies [26▪] and zebrafish [24▪], suggesting that gain-of-function mechanisms are sufficient for neurodegeneration. Studies have yet to clearly attribute observed toxicity to repeat RNA or DPR protein species, but one study showed that increasing expression of C9orf72 repeats specifically in the GP (sense) and PR (antisense) frames can exacerbate toxicity in human HEK293T cells [21▪], showing that these DPR proteins can affect cell viability. Impaired degradation through the autophagic system is consistent with the accumulation of p62 and ubiquitin pathology that is abundant in C9FTD/ALS cases [61], and the sensitivity of C9FTD iPSCs specifically to autophagic stressors [6▪]. However, it is not clear whether these effects are due to loss of the normal cellular function of C9orf72 or gain of function due to the accumulation of protein aggregates.
THERPAEUTIC TARGETTING OF GAIN-OF-FUNCTION MECHANISMS
There is considerable excitement about the possibility of ASOs for the treatment of C9FTD/ALS. One reason for this is that ASOs should ameliorate both repeat RNA and DPR protein toxicity, and therefore do not need to wait for a better understanding of the contribution of each of these mechanisms to disease pathogenesis. ASOs will not alleviate loss of C9orf72 function, but as discussed above, the weight of evidence currently suggests gain of function is likely to be the primary mechanism to address therapeutically. ASOs targeting sense transcripts reduce sense RNA foci and ameliorate transcriptome changes and toxicity in C9FTD/ALS-derived cells [8▪▪,17▪▪,22▪]. ASOs targeting antisense transcripts may also be required as it was proposed that antisense RNA-mediated mechanisms were responsible for the proportion of transcriptional changes that remain dysregulated after treatment of patient cells with ASOs targeting sense repeats [17▪▪]. In addition, antisense DPR proteins have been shown to be toxic to cells [21▪]. Given the precedent of a clinical trial for ASOs targeting superoxide dismutase 1 (SOD1) for ALS patients with SOD1 mutations [62], ASO treatment is currently the most promising prospect for treating C9FTD/ALS. The development of small molecules that specifically bind the secondary structure formed by the GGGGCC repeats is another promising area for therapeutic intervention [52▪,63▪]. Such small molecules would also be predicted to prevent both repeat RNA and DPR protein mechanisms, with the potential advantage of simpler delivery.
CONCLUSION
In the 3 years since the discovery of the C9orf72 mutation in ALS and FTD, intense investigations have begun to unfold the mechanisms at play in these diseases. Haploinsufficiency of C9orf72 may cause defects in endosomal and autophagic processes that lead to dysfunction of the motor system, but clinical data do not support causation of disease. Cellular and animal models inform us that gain-of-function mechanisms from expanded C9orf72 repeats are sufficient to cause neurodegeneration. As has been found in other diseases classically thought of as either loss-of-function or gain-of-function diseases, it is likely that both mechanisms contribute to different aspects of the heterogeneous phenotypes in these diseases. As the same C9orf72 mutation appears to result in a spectrum of disease phenotypes, differential regulation of toxic species or the degree of loss of function may play a role in differing disease presentations. Finally, current evidence suggests gain-of-function-based therapies such as ASOs hold promise for C9FTD/ALS.
Acknowledgements
AMI receives funding from Alzheimer's Research UK, The Motor Neurone Disease Association, the MHMS General Charitable Trust and the UK Medical Research Council.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lashley T, Hardy J, Isaacs AM. RANTing about C9orf72. Neuron 2013; 77:597–598 [DOI] [PubMed] [Google Scholar]
- 4.Gijselinck I, Van Langenhove T, van der ZJ, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2012; 11:54–65 [DOI] [PubMed] [Google Scholar]
- 5▪.Fratta P, Poulter M, Lashley T, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 2013; 126:401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of homozygous C9orf72 repeat expansion carrier.
- 6▪.Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 2013; 126:385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of RNA foci and DPR protein in C9FTD patient-derived iPSC neurons.
- 7▪.Belzil VV, Bauer PO, Prudencio M, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 2013; 126:895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article identifies histone methylation as a mechanism for reduction of C9orf72 transcription in C9FTD/ALS.
- 8▪▪.Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013; 80:415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of three articles showing therapeutic potential of ASOs in C9ALS/FTD patient-derived cells.
- 9▪.Waite AJ, Baumer D, East S, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 2014; 35:e5–131779 [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of reduced C9orf72 protein in C9ALS/FTD patient tissue.
- 10▪.Xi Z, Zinman L, Moreno D, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet 2013; 92:981–989 [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of CpG island methylation as a mechanism for reduced C9orf72 transcription.
- 11▪.Liu EY, Russ J, Wu K, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 2014; Epub 2014 May 8 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found a correlation between methylation status and clinical phenotype, suggesting that hypermethylation and thus decreased C9orf72 is protective.
- 12.Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 2014; 23:3579–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013; 29:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang D, Iyer LM, He F, et al. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet 2012; 3:1–10 Article 283 10.3389/fgene.2012.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15▪.Ciura S, Lattante S, Le B I. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 2013; 74:180–187 [DOI] [PubMed] [Google Scholar]; First loss of C9orf72 function in zebrafish model.
- 16▪.Therrien M, Rouleau GA, Dion PA, et al. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 2013; 8:e83450. [DOI] [PMC free article] [PubMed] [Google Scholar]; First loss of C9orf72 in Caenorhabditis elegans model.
- 17▪▪.Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013; 110:E4530–E4539 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of three articles showing therapeutic potential of ASOs in C9FTD/ALS patient-derived cells, and one of the first descriptions of antisense RNA foci in C9FTD/ALS patient tissue.
- 18.Harms MB, Cady J, Zaidman C, et al. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 2013; 34:2234–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19▪.Mizielinska S, Lashley T, Norona FE, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 2013; 126:845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first descriptions of antisense RNA foci in C9FTD/ALS patient tissue and also reports a correlation between RNA foci burden and clinical phenotypes, suggesting a role for RNA foci in C9FTD/ALS.
- 20▪.Gendron TF, Bieniek KF, Zhang YJ, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013; 126:829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first descriptions of antisense RNA foci and DPR protein in C9FTD/ALS patient tissue.
- 21▪.Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 2013; 110:E4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first descriptions of antisense RNA foci and DPR protein in C9FTD/ALS patient tissue and provides evidence for cellular toxicity of DPR proteins.
- 22▪.Sareen D, O’Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013; 5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]; One of three articles showing therapeutic potential of ASOs in C9FTD/ALS patient-derived cells, and also reports sequestration of RNA-binding proteins into RNA foci.
- 23▪.Mori K, Lammich S, Mackenzie IR, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 2013; 125:413–423 [DOI] [PubMed] [Google Scholar]; Identification of hnRNP A3 as a binding protein of expanded C9orf72 repeat RNA and its presence in a subset of neuronal inclusions.
- 24▪.Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 2013; 5:1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of hnRNP H as a protein sequestered by expanded C9orf72 repeat RNA.
- 25▪.Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014; 507:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of nucleolin as a binding protein of G-quadruplexes formed by expanded C9orf72 repeat RNA and evidence suggesting a role for nucleolar stress in C9FTD/ALS.
- 26▪.Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 2013; 110:7778–7783 [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of C9orf72 repeats causing neurodegeneration in Drosophila and identification of repeat binding proteins.
- 27▪▪.Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013; 77:639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]; The first description, with reference [28▪▪], of RAN translation of the C9orf72 repeat in C9FTD/ALS.
- 28▪▪.Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013; 339:1335–1338 [DOI] [PubMed] [Google Scholar]; The first description, with reference [27▪▪], of RAN translation of the C9orf72 repeat in C9FTD/ALS.
- 29▪.Mori K, Arzberger T, Grasser FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 2013; 126:881–893 [DOI] [PubMed] [Google Scholar]; One of the first descriptions of antisense DPR protein in C9FTD/ALS patient tissue.
- 30▪.Proudfoot M, Gutowski NJ, Edbauer D, et al. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol 2014; 127:451–458 [DOI] [PubMed] [Google Scholar]; Description of two C9orf72 repeat expansion carriers with predominant and early poly-GA pathology and limited TDP-43 pathology.
- 31▪.Mackenzie IR, Arzberger T, Kremmer E, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 2013; 126:859–879 [DOI] [PubMed] [Google Scholar]; An in-depth clinicopathological study showing that TDP-43 rather than poly-GA inclusions correlate with neurodegeneration in C9FTD/ALS.
- 32.Hagerman P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol 2013; 126:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001; 57:127–130 [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev 2012; 22:256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35▪.van Blitterswijk M, Dejesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013; 12:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]; First large study assessing repeat size and clinicopathological correlations in ALS, FTD and ALS/FTD C9orf72 repeat expansion carriers.
- 36.Cooper TA, Wan L, Dreyfuss G. R.N.A and disease. Cell 2009; 136:777–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zu T, Gibbens B, Doty NS, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 2011; 108:260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev 2014; 26C:6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983–997 [DOI] [PubMed] [Google Scholar]
- 40.Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 2011; 20:3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel premRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 2002; 10:35–44 [DOI] [PubMed] [Google Scholar]
- 42.Suzuki N, Maroof AM, Merkle FT, et al. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci 2013; 16:1725–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer K, Ferraiuolo L, Miranda CJ, et al. Direct conversion of patient fibroblasts demonstrates noncell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A 2014; 111:829–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vatovec S, Kovanda A, Rogelj B. Unconventional features of C9ORF72 expanded repeat in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiol Aging 2014; 35:e1–122421 [DOI] [PubMed] [Google Scholar]
- 45▪.Cooper-Knock J, Walsh MJ, Higginbottom A, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014; 137:2040–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of multiple RNA-binding proteins sequestered by expanded C9orf72 repeat RNA.
- 46.Stutz F, Bachi A, Doerks T, et al. REF, an evolutionary conserved family of hnRNP-like proteins, interacts with TAP/Mex67p and participates in mRNA nuclear export. RNA 2000; 6:638–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 2004; 43:513–525 [DOI] [PubMed] [Google Scholar]
- 48.Johnson EM, Kinoshita Y, Weinreb DB, et al. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res 2006; 83:929–943 [DOI] [PubMed] [Google Scholar]
- 49.Ma AS, Moran-Jones K, Shan J, et al. Heterogeneous nuclear ribonucleoprotein A3, a novel RNA trafficking response element-binding protein. J Biol Chem 2002; 277:18010–18020 [DOI] [PubMed] [Google Scholar]
- 50.Chen CX, Cho DS, Wang Q, et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000; 6:755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwak S, Hideyama T, Yamashita T, et al. AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 2010; 30:182–188 [DOI] [PubMed] [Google Scholar]
- 52▪.Fratta P, Mizielinska S, Nicoll AJ, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2012; 2:1–6Article 1016 10.1038/srep01016 [DOI] [PMC free article] [PubMed] [Google Scholar]; The first description of the tertiary structures formed by C9orf72 repeat RNA.
- 53.Reddy K, Zamiri B, Stanley SY, et al. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 2013; 288:9860–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji X, Sun H, Zhou H, et al. Research progress of RNA quadruplex. Nucleic Acid Ther 2011; 21:185–200 [DOI] [PubMed] [Google Scholar]
- 55.Subramanian M, Rage F, Tabet R, et al. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep 2011; 12:697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collie GW, Parkinson GN. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem Soc Rev 2011; 40:5867–5892 [DOI] [PubMed] [Google Scholar]
- 57.Banez-Coronel M, Porta S, Kagerbauer B, et al. A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 2012; 8:e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Z, Teng X, Bonini NM. Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet 2011; 7:e1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung DW, Rudnicki DD, Yu L, et al. A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet 2011; 20:3467–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mann DM, Rollinson S, Robinson A, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2013; 1:1–13Article 68 10.1186/2051-5960-1-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011; 122:691–702 [DOI] [PubMed] [Google Scholar]
- 62.Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 2013; 12:435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63▪.Zamiri B, Reddy K, Macgregor RB, Jr, et al. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem 2014; 289:4653–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]; The study provides proof of principle for RNA G-quadruplex binding small molecules as a therapeutic strategy in C9FTD/ALS.