

Abstract
The goals of bioengineering strategies for targeted cancer therapies are (1) to deliver a high dose of an anticancer drug directly to a cancer tumor, (2) to enhance drug uptake by malignant cells, and (3) to minimize drug uptake by nonmalignant cells. Effective cancer-targeting therapies will require both passive- and active targeting strategies and a thorough understanding of physiologic barriers to targeted drug delivery. Designing a targeted therapy includes the selection and optimization of a nanoparticle delivery vehicle for passive accumulation in tumors, a targeting moiety for active receptor-mediated uptake, and stimuli-responsive polymers for control of drug release. The future direction of cancer targeting is a combinatorial approach, in which targeting therapies are designed to use multiple targeting strategies. The combinatorial approach will enable combination therapy for delivery of multiple drugs and dual ligand targeting to improve targeting specificity. Targeted cancer treatments in development and the new combinatorial approaches show promise for improving targeted anticancer drug delivery and improving treatment outcomes.
1. INTRODUCTION
Cancer is the leading cause of death worldwide (Lovett et al., 2012) and the second leading cause of death in the U.S., accounting for one in every four deaths (Siegel et al., 2012). The American Cancer Society reported in 2012 that over one million new cancer diagnoses and half a million cancer deaths are recorded each year. The National Institutes of Health estimated that $103.8 billion was spent on direct health care costs for cancer treatment in 2007 (ACS, 2012). Advances in cancer detection and treatment have led to a decline in cancer deaths by one percent per year over the past decade; however, survival rates for several types of cancer remain low. The lowest survival rates have been recorded for cancer of the esophagus (17%), liver (14%), lung and bronchus (16%), stomach (26%), brain (35%), and pancreas (6%) (Howlader et al., 2011). Survival rates for head and neck cancer, which are currently 40-50%, have not significantly improved over the past few decades (Leemans et al., 2011). Patients diagnosed with these cancer types may benefit from new, targeted approaches to cancer therapy.
Radiation and chemotherapy are standards of care for cancer treatment; however, traditional radiation and chemotherapy have many limitations. Although radiation therapy is focused on the cancer tumor, this therapy risks severe damage to nonmalignant tissues that are in the path of the radiation beam (Shepard et al., 1999). Radiation also has limited effectiveness in treating metastasized cancers because it requires the detection and treatment of each tumor. Chemotherapy is a systemic treatment that typically targets highly proliferative cells. Systemic delivery exposes all cells to the drug. This lack of specificity also results in damage to highly proliferative non-malignant cells, such as bone marrow, gonads, gastrointestinal mucosa, and hair follicles (Corrie, 2008), resulting in acute complications and systemic toxicity (Liu et al., 2007b; Sahoo and Labhasetwar, 2003). Furthermore, non-specific uptake of the chemotherapy drug by nonmalignant cells reduces the dose delivered to the target malignant cells, and as a result, higher doses of the cytotoxic drugs must be administered systemically to achieve treatment efficacy (Yotsumoto et al., 2009). Traditional chemotherapy is also ineffective in overcoming multidrug resistance, a condition in which cancer cells become resistant to anticancer drugs. Although the traditional radiation and chemotherapy treatments can successfully fight cancer, there is an urgent need for a more targeted approach that will increase treatment efficacy and reduce treatment side effects.
The goal of targeted cancer therapy is (1) to deliver a high dose of an anticancer drug directly to the site of a tumor, (2) to enhance drug uptake by malignant cells, and (3) to minimize drug uptake by nonmalignant cells. The general approach for designing targeted cancer therapies is to design the drug delivery system to exploit the features that are unique to tumor cells and tumor tissues. Targeted delivery research has focused on unique features of the tumor microenvironment, such as leaky vasculature, overexpressed cell surface receptors, and intratumoral pH differences, as well as features of the cell uptake process, such as endosomal pH. Innovation in micro- and nano-technology has led to the development of micro- and nanoparticles, such as liposomes and micelles, that can encapsulate and deliver drugs (Egusquiaguirre et al., 2012; Gong et al., 2012; Kedar et al., 2010; Malam et al., 2009). Nanoparticles are typically defined as particles that are less than 100 nm in size. Some delivery vehicle types discussed may be fabricated at both micro- and nano-scales. Due to the inherent physiologic barriers, particles used for targeted cancer therapy are predominantly nanoscale. We have used the term nanoparticle in reference to any particle type or particle research that is at the nanoscale. These particles have been shown to enhance drug delivery to malignant cells. Advances in cancer research have identified receptors that are overexpressed in various types of cancer. Ligands that bind overexpressed receptors have been successfully used to target malignant cells (Byrne et al., 2008; Das et al., 2009; Yu et al., 2010). Stimuli responsive polymers have also been developed to control release of chemotherapy drugs in response to environmental triggers (Cabane et al., 2012).
Advances in cancer research in combination with advances in biomaterials and nanotechnology have enabled the development of targeted anticancer drug delivery and a more tailored approach to treating individual cancer types. A multidisciplinary approach that includes cancer biology, biomaterials, and nanotechnology has the potential to improve treatment outcomes while minimizing harmful side effects. The design of an effective targeted therapy will require optimization of therapeutic particles, cancer cell targeting, and drug release mechanisms. Both passive and active targeting mechanisms may be utilized to enhance targeted delivery. Physical properties of particles can be modified to decrease toxicity to nonmalignant cells and increase circulation time. Targeting moieties, ligands that bind to receptors overexpressed on malignant cells, can be conjugated to particles to increase cellular uptake, and as a result, enhance treatment efficacy. Environmentally responsive polymers can be used to achieve controlled release under defined conditions. This review discusses targeted cancer therapies currently under development and focuses on the design and optimization of individual components required to achieve effective cancer treatment.
2. CANCER-TARGETING MECHANISMS
Cancer targeting can be divided into two general categories: signaling and delivery. Traditional chemotherapy drugs can be thought of as signal transduction therapeutics because they target and inhibit signal transduction essential for tumor survival. Targets for signaling inhibitors include cell proliferation, cell survival, angiogenesis, and nuclear factors (Klein et al., 2007). These drugs target cellular processes; they do not specifically target malignant cells. A more recent therapeutic approach to cancer targeting is gene therapy, which involves the delivery of DNA or RNA to cancer cells (Ali et al., 2012; Gao et al., 2010b). There are several strategies for cancer treatment using gene therapy, such as blocking expression of oncogenes or insertion of a suicide gene or suppressor gene in tumor cells. However, similar to chemotherapy, delivery of RNA or DNA alone does not specifically target malignant cells and cell transfection is inefficient. Targeted delivery, on the other hand, is the process by which carriers specifically deliver a drug to malignant cells, while avoiding delivery to nonmalignant cells. In this review, the terms drugs and anticancer drugs are broadly defined to include chemotherapy agents, DNA or RNA for gene therapy, recombinant proteins, or any other therapeutic that can be delivered by particles for cancer therapy. Targeted delivery is essential to improve cancer treatment efficacy and reduce side effects of anticancer drugs. Particles, ligands, and controlled release mechanisms can be used in combination to design a drug-particle complex that achieves targeted delivery. To design an effective targeted cancer therapy, it is essential to understand the two primary mechanisms of cancer targeting: passive targeting and active targeting.
2.1. Passive targeting
Passive targeting, first described in 1986, takes advantage of the greater vascular permeability and poor lymphatic drainage of tumors that result in the accumulation of micro- and nano-particles in tumor tissue (Matsumura and Maeda, 1986). The particles accumulate through passive diffusion, a phenomenon known as the enhanced permeability and retention effect (EPR) (Maeda, 2001; Maeda et al., 2009; Maeda et al., 2001).
Enhanced permeability of the EPR effect is the result of the leaky vasculature in tumor tissue. Vessels in tumors are irregularly shaped, leaky, and dilated due to rapid growth and abnormal blood flow (Iyer et al., 2006). Endothelial junctions, gaps in the endothelium that mediate passage of macromolecules from the blood to tissue, vary between nonmalignant and malignant tissue. In normal vasculature, endothelial junctions between cells are narrow, ranging from 5-10 nm in width (Haley and Frenkel, 2008). However, in tumor tissue, these junctions range from 100-780 nm depending on the tumor type (Hobbs et al., 1998; Rubin and Casarett, 1966; Shubik, 1982). The larger endothelial gaps allow extravasation of particles out of circulation and into the tumor tissue (Fig. 1A).
Figure 1.
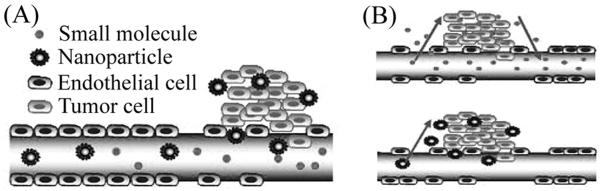
Passive targeting. (A) Nanoparticles accumulate in tumor tissue due to the enhanced permeability and retention (EPR) effect. Enhanced permeability is due to large endothelial gaps that result in leaky vasculature. Enhanced retention is due to poor lymphatic drainage. (B) Smaller particles are able to quickly enter and exit tumor tissue through large endothelial gaps, while larger particles diffuse more slowly, resulting in the accumulation of a greater number of particles and drug. Figure modified and used with permission from (Danhier et al., 2010).
Enhanced retention, the second component of the EPR effect, is the result of poor lymphatic drainage of cancer tumors. Lymphatic vessels are responsible for draining fluid from the tissue and returning it to the vascular system. However, tumor tissues either lack lymphatic vessels or the vessels they contain are non-functional, resulting in the retention of macromolecules in the tumor interstitium (Iyer et al., 2006). Poor lymphatic drainage allows particles that extravasate into tumor vasculature to be retained in the tissue. Thus, the EPR effect is a passive mechanism that enables the accumulation of particle-encapsulated drugs in tumor tissue.
Particle size must be tailored to enable permeation of particles into tumor tissue and their retention within the tissue. Very small nanoparticles (<20 nm) are able to quickly diffuse out of the vasculature through the large endothelial gaps. However, very small nanoparticles are also less likely to be retained because they can just as easily diffuse out of the tumor tissue (Perrault et al., 2009). Larger nanoparticles (50-100 nm) have longer circulation times, allowing sufficient time for the nanoparticles to travel to, and diffuse into, the tumor tissue (Liu et al., 1992; Perrault et al., 2009). Larger particles diffuse out of tumor tissue very slowly, and as a result, they accumulate in tumors (Fig. 1B).
The EPR effect has become a gold standard in the design of cancer therapeutics (Maeda, 2001) because particles can be specifically designed to take advantage of passive targeting. By using passive targeting, anticancer drugs encapsulated in particles will accumulate in tumor tissue, reaching concentrations 10-100 times higher than that of free drug alone (Sinha et al., 2006). The EPR effect also promotes prolonged drug retention (Maeda et al., 2009) due to poor lymphatic drainage.
Although passive targeting promotes drug accumulation in cancer tumors, there are also limitations to this approach. Results of passive targeting are not consistent because tumor vascularization and angiogenesis vary for different cancer types. Consequently, particle concentrations achieved will vary with tumor type and site. For example, the EPR effect is not observed in hypovascular tumors, which are common in prostate and pancreatic cancers (Maeda et al., 2009); therefore, passive targeting alone will not promote particle accumulation in these tumors. The EPR effect can be enhanced in hypovascular tumors by using chemotherapeutic agents, such as nitroglycerin (Seki et al., 2009) and S-1, a prodrug of 5-Fluorouracil (Nakamura et al., 2011), to alter the tumor microenvironment (Asai, 2012). Particle accumulation in tumors is also influenced by particle size. The EPR effect favors accumulation and retention of larger particles, greater than 100 nm in size (Danhier et al., 2010), smaller particles will be retained to a lesser degree using passive targeting, which may result in nonspecific delivery of anticancer drugs to nonmalignant cells. Passive targeting results in higher concentrations of particles in tumor tissue; however, it does not eliminate non-specific uptake of particles by nonmalignant tissue. When nonspecific uptake occurs, higher doses of particles must be administered systemically to achieve a therapeutic dose within the tumor.
2.2. Active targeting
Active targeting uses ligands to specifically target receptors that are overexpressed on malignant cells. Ligands are molecules, such as folate, transferrin, epidermal growth factor (EGF), and aptamers, which bind to receptors on the surface of a cell. Ligands are conjugated to anticancer drugs or particle-encapsulated drugs to target malignant cells or tumor endothelium. Conjugation is the physical or chemical attachment of a ligand directly to an anticancer drug or attachment to a particle encapsulating an anticancer drug. Ligand candidates for cancer treatment target receptors that are overexpressed on malignant cells. The folate receptor (FR) and the epidermal growth factor receptor (EGFR) are two examples of receptors that are overexpressed on many types of malignant cells. Therefore, conjugation of these ligands to drugs or particles will result in receptor-mediated active targeting and higher drug or particle concentration in malignant cells than in nonmalignant cells.
Active targeting promotes internalization of ligand-conjugated drug carriers into a cell via receptor-mediated endocytosis (Fig. 2). The drug may be released either at the surface of the cell or upon internalization. The ligand-conjugated particle and receptor are first internalized via invagination, and then an endosome is formed. The anticancer drug must escape the endosome before it fuses with the lysosome to avoid being damaged or destroyed by lysosomal enzymes. After release of the drug and receptor from the endosome, some receptors are recycled back to the surface of the cell where they will be available for another cycle of endocytosis (Mohanty et al., 2011).
Figure 2.

Active targeting. Active targeting of particles is achieved by conjugation of ligands that target overexpressed receptors on the surface of cancer cells. Ligands bind to overexpressed receptors on malignant cells and are endocytosed. Anticancer drugs must then escape the endosome to avoid degradation and to perform their function within the cell.
The active targeting approach addresses many of the goals for improving cancer therapies. Ligands conjugated to cancer drugs often help protect the cancer drug from degradation and enhance the physical and chemical stability of the drug (Mohanty et al., 2011). Ligand binding also increases the drug dose delivered to malignant cells, which permits systemic administration of smaller doses. Particle internalization that occurs by active targeting has been shown to enhance therapeutic effects (Iinuma et al., 2002; Kobayashi et al., 2007), an important advantage over passive targeting. However, active targeting alone will not achieve optimal results. If an anticancer drug is delivered systemically, the ligand-conjugated drug or drug-particle complex must first reach the cancer tumor before the advantages of active targeting can be realized. Passive targeting mechanisms using the EPR effect are still necessary for extravasation and drug or particle accumulation in tumor tissue. Therefore, it is necessary to use a combination of active and passive targeting in designing drug carriers to improve targeted delivery of cancer therapeutics.
3. DESIGN CRITERIA FOR CANCER TARGETING
The design goals for effective cancer targeting therapeutics include (1) selective targeting of malignant cells, (2) release of anticancer drugs at the target site, and (3) elimination of cytotoxicity to nonmalignant tissue. When cancer drugs enter the body, they face many physiological barriers that can prevent them from reaching the target site and achieving these design goals. These physiological barriers dictate the design parameters for targeted cancer treatments. A thorough understanding of these barriers is necessary for the development of effective targeting. Particles provide a flexible platform for targeted treatment development. Particle selection, size, surface modification, ligand conjugation, and controlled release mechanisms all contribute to overcoming physiological barriers to targeted delivery and to enhancing therapeutic outcomes (Table 1).
Table 1.
Bioengineering design strategies to overcome physiologic barriers to targeted delivery
| Barrier | Design Feature | Strategy |
|---|---|---|
| Opsonization and MPS | Hydrophobicity | Increase hydrophilicity. |
|
|
||
| Size | Decrease particle size (< 200 nm). | |
|
| ||
| Extravasation | Surface charge | Use cationic particles to increase extravasation and uptake. |
|
|
||
| Size | Tailor particle size to endothelial junction size of tumor tissue (<200 nm, with enhanced tissue penetration and retention at ~50-100 nm). |
|
|
| ||
| Drug uptake and release | Ligand conjugation | Conjugate ligands to particles that target overexpressed receptors on malignant cells. Use multiple ligands and oblique particles to increase binding avidity. |
|
|
||
| Stimuli-responsive element |
Use pH, temperature, or enzyme responsive polymers or linkers to trigger release. |
|
|
| ||
| Multidrug resistance | Particle encapsulation |
Encapsulate therapeutics using particles such as micelles, liposomes, polymeric particles, dendrimers, or carbon nanotubes. |
|
| ||
| Particle elimination | Biodegradable | Use biodegradable materials to avoid accumulation in the liver and spleen. |
|
|
||
| Size | Particles <20 nm are excreted, while larger, nondegradable particles accumulate in the liver and spleen. |
|
3.1. Opsonization and the mononuclear phagocytic system
When cancer drugs are introduced into the circulatory system, they first encounter the blood. Proteins in the blood called opsonins are absorbed onto the surface of the drug or particle in a process called opsonization. Opsonized drugs or particles are recognized by macrophages of the mononuclear phagocytic system (MPS), which trigger removal of drugs or particles from circulation, preventing them from reaching the cancer tissue (Owens III and Peppas, 2006). An advantage of the drug-particle complex over unconjugated free drug is that it can be designed to reduce elimination by the MPS.
Surface hydrophobicity determines the amount of opsonins absorbed onto the surface of a drug or drug-particle complex, which will ultimately determine the fate of the drug. Hydrophilic surfaces repel plasma proteins that cause opsonization, thus preventing particle elimination by the MPS (Brigger et al., 2002; Moghimi et al., 2005). “Stealth” particles, created by pegylation through covalent attachment of polyethylene glycol (PEG), a non-ionic hydrophilic biocompatible polymer, are used to improve biocompatibility and prevent elimination by the MPS (Storm et al., 1995). Hydrophilic polymers can also be used to coat the surface of cytotoxic materials to reduce their toxicity (Jokerst et al., 2011). Hydrophobic particle surfaces are not beneficial for drug delivery because they promote particle agglomeration, resulting in rapid removal by the MPS (Veiseh et al., 2010).
Particles must be small enough to avoid uptake by the MPS (Cho et al., 2008). Large particles (>200 nm) are quickly recognized by the MPS and are removed from circulation (Albanese et al., 2012). Small (<100 nm) hydrophilic nanoparticles have less interaction with the MPS and reach their cellular targets in higher numbers (Dong and Feng, 2004; Park et al., 2005). A particle will have a longer circulation time, and thus a higher probability of reaching its target, if it avoids removal by the MPS (Kumari et al., 2009).
3.2. Extravasation
After a drug-particle complex successfully avoids uptake by the MPS, it must then extravasate out of circulation into the target tissue. However, to enter the tissue, it must first traverse endothelial junctions, as described in Section 2.1. While particles must be small enough to traverse the endothelial junctions, they must also be large enough to be retained in the leaky tumor vasculature (Cho et al., 2008). There are also size limitations specific to the type of target site. Endothelial junctions in tumor vasculature range from 100 to 780 nm (Hobbs et al., 1998; Rubin and Casarett, 1966; Shubik, 1982), while nanoparticles with a hydrodynamic size (size in solution accounting for movement and viscosity) of 15 to 50 nm are able to cross the blood-brain barrier (Sonavane et al., 2008). Diffusion across the blood-brain barrier requires a low molecular weight lipid-soluble carrier (Koo et al., 2006; Pardridge, 2002).
Surface charge influences particle-cell interactions and particle cytotoxicity. The plasma membrane of cells is naturally negatively charged. Therefore, cationic carriers have more electrostatic interaction with the cell membrane, which results in increased extravasation and cell uptake (Nigavekar et al., 2004). However, this increased interaction also results in greater toxicity and nonspecific uptake by nonmalignant cells (Merdan et al., 2002). Nonspecific accumulation of cationic particles is typically found in the liver, kidney, spleen, and pancreas (Roberts et al., 1996). Electrostatic interactions of cationic particles with endothelial cells also shortens the particle circulation time (Lee et al., 2005; Malik et al., 2000; Wijagkanalan et al., 2011). Anionic and neutral particles are non-toxic to cells (Fant et al., 2010). However, anionic and neutral particles have reduced electrostatic interactions with cell membranes, which results in lower cellular uptake, decreased ability to bind to vascular walls (Boas and Heegaard, 2004), and increased circulation time (Kukowska-Latallo et al., 2005).
3.3. Drug uptake and release
After the therapeutic agent reaches the target tissue, receptor binding/uptake and drug release are required to achieve a therapeutic effect. As described in Section 2.2, cell binding and uptake are facilitated through receptors on the cell surface. The level of overexpression of individual receptors on malignant cells will determine which ligand(s) are best suited for conjugation to the particle for active targeting. Ligand conjugation also decreases toxicity by preventing binding/uptake of particles by cells in nonmalignant tissues where the cancer drug has been nonspecifically distributed through passive mechanisms (Vega-Villa et al., 2008). Ligands and other moieties are discussed in detail in Section 5.
Particle shape influences ligand binding avidity, which is the bond strength of multiple ligand-receptor interactions. Ligand-conjugated oblique particles, which have a flatter surface, have enhanced binding avidity relative to spherical particles (Decuzzi and Ferrari, 2006) due to increased multivalent interactions. A flatter particle surface is able to present a greater number of ligands to the malignant cell surface, increasing interaction and enhancing binding strength.
Drug release mechanisms of particles can be designed to decrease toxicity. If the particle is stimuli-responsive, nonmalignant cells can be protected from exposure. For example, polymers can be designed to release drug when exposed to either the acidic intratumoral environment or the acidic intracellular environment of the endosome. Thermoresponsive polymers such as poly(N-isopropyl acrylamide) (pNIPAM) can be used to trigger release when heat is applied to the tumor region (Peng et al., 2011). Drug release mechanisms are discussed in detail in Section 6.
3.4. Multidrug resistance
A major barrier to effective cancer chemotherapy is multidrug resistance, in which malignant cells become resistant to chemotherapy drugs. The mechanism of multidrug resistance is associated with at least two proteins found in the cell membrane of malignant cells, p-glycoprotein and multidrug resistance protein (Leslie et al., 2005). These proteins function as transmembrane transporters, or efflux pumps, that recognize and bind to drugs as they enter the cell membrane. Drug binding activates an adenosine triphosphate (ATP) binding domain, and hydrolysis of ATP results in a shape change of p-glycoprotein and release of the drug into the extracellular space (Gottesman, 2002). One of the key advantages of particle encapsulation of chemotherapy drugs is the ability to combat multidrug resistance. Particle-encapsulated drugs accumulate in cells because they are not recognized by the drug-expelling pumps (Cho et al., 2008), thus avoiding multidrug resistance and improving treatment efficacy. Particle-encapsulated drugs are transported into the cell in endosomal vesicles where they are released out of the reach of the drug efflux pumps, increasing the likelihood that they will reach their target (Hu and Zhang, 2009).
3.5. Particle elimination
The mechanisms of particle elimination from the body also affect particle toxicity. Biodegradable particles do not accumulate and disrupt cellular processes because they are degraded and metabolized (Haley and Frenkel, 2008). If the particle is not degradable, particle size affects the way it is eliminated. Nanoparticles with a molecular weight of <5,000 (Owens III and Peppas, 2006) and a size of <20 nm (Banerjee et al., 2002) are excreted by the renal system. Larger particles that are nondegradable accumulate in the spleen and liver where they are eliminated by the MPS (Albanese et al., 2012). Particle elimination is also affected by particle shape. Anisotropically shaped (non-spherical) particles have a lower bioelimination rate, possibly due to decreased interaction with phagocytes (Geng et al., 2007).
IV. DELIVERY VEHICLES
Particle delivery vehicles are an important and promising tool for the design of targeted cancer therapies. Particle delivery vehicles aid in drug delivery by protecting the surrounding environment from nonspecific effects. Particle features, including size, shape, surface charge, and hydrophobicity, affect drug delivery and vary among the particle types. Each of these features must be considered when selecting and optimizing a particle for targeted delivery of cancer drugs. There are already several FDA-approved cancer drugs that use particle drug encapsulation technology. Particles discussed in this section include liposomes, micelles, dendrimers, polymeric particles, and carbon nanotubes (CNTs).
4.1. Liposomes
Liposomes are composed of a bilayer structure of either natural or synthetic phospholipids. Phospholipids are a key structural component of the cell membrane. They are amphiphilic in nature, possessing both hydrophilic and hydrophobic regions. Amphiphilic phospholipids self-assemble into bilayers (Malam et al., 2009) by arranging their hydrophilic groups outward to interact with aqueous environments and arranging their lipophilic groups toward the center of the bilayer (Fig. 3). Liposomes can be unilamellar (Fig 3A) or multilamellar (Fig 3B) depending on the number of lipid bilayers formed (Haley and Frenkel, 2008).
Figure 3.

Structure of liposomes. (A) Unilamellar liposomes contain a large aqueous core for storage of water-soluble drugs. (B) Multilamellar liposomes are composed of multiple layers of phospholipid groups for storage of lipid-soluble drugs between the hydrophobic tail groups in each layer.
Liposome surface properties can be easily manipulated for drug delivery. Surface charge can be modified by adjusting the lipid composition to add either more neutral or cationic lipids, which directly influences liposome interactions with the negatively charged cell membrane. Neutral liposomes have no significant cell membrane interaction, and the neutral charge results in liposome aggregation (Sharma and Sharma, 1997). Aggregation is a key issue for drug delivery because particle aggregates are rapidly cleared by the MPS, which greatly reduces drug delivery. Anionic liposomes are internalized through clathrin-mediated endocytosis (Straubinger et al., 1983), while cationic liposomes deliver their contents by membrane fusion (Felgner et al., 1994) and by endocytosis. However, at high doses, cationic liposomes cause toxicity and nonspecific uptake, which harms nonmalignant cells.
Liposomes can be used to carry water- or lipid-soluble drugs. Unilamellar liposomes have an aqueous core that is used to carry water-soluble drugs, while multilamellar liposomes have lipophilic layers between hydrophobic tail groups that are used to carry lipid soluble drugs. There are currently four liposome-based cancer drugs available on the market, Doxil, DaunoXome, LipoDox, and Myocet, and many others are in clinical trials (Chang and Yeh, 2012). The four available drug formulations are all unilamellar liposomes containing water-soluble drugs. Doxil and its second generation drug, LipoDox, both contain doxorubicin encapsulated within a liposome with a PEG-modified surface. Doxil has been proven effective in treating drug-resistant tumors in clinical trials (Chou et al., 2006). Both Doxil and LipoDox have been used successfully to treat many cancers including Kaposi’s sarcoma, ovarian cancer, and metastatic breast cancer (Tejada-Berges et al., 2002). Myocet is an unpegylated liposomal doxorubicin approved for use in Canada and Europe to treat metastatic breast cancer (Lorusso et al., 2007). DaunoXome, which is a pegylated liposomal daunorubicin, has been approved to treat blood tumors (Chang and Yeh, 2012).
Liposomes have advantages and disadvantages to consider when developing targeted drug delivery therapies. Liposomes can be used to encapsulate hydrophilic and hydrophobic drugs. Liposomes also have the potential to treat multidrug resistant cancers. However, liposomes have poor control over drug release, often dispersing the loaded drug in a rapid burst release. Liposomes also exhibit low drug encapsulation efficiency due to poor solubility of many drugs in solution (Das et al., 2009), and as a result, the larger lipsomes are only useful for very potent drugs (Alexis et al., 2010). Liposomes are physically unstable; the bilayer structure can quickly disintegrate in response to hydrophobic, electrostatic, and van der Waals forces (Haley and Frenkel, 2008), resulting in particle aggregation, drug leakage, and a reduced shelf life (Sharma and Sharma, 1997). Liposomes measure up to 400 nm in size, and as a result, they are rapidly cleared by the MPS (Malam et al., 2009). Pegylation of the liposome surface reduces opsonization and clearance by the MPS. Liposomes also cause severe side effects due to nonspecific uptake in skin tissue.
4.2. Micelles
Micelles are composed of amphiphilic macromolecules that contain both hydrophilic and hydrophobic segments (Sutton et al., 2007). Depending on the size of these segments, micelles with various morphologies, including spheres, rods, tubules, lamellae, and vesicles can be created (Choucair and Eisenberg, 2003). A micelle consists of a core and a shell, where hydrophobic end groups form the core and hydrophilic head groups form the outer shell (Fig. 4), or vice versa (Haley and Frenkel, 2008). Amphiphilic micelles are formed through self assembly of unimers with hydrophilic and hydrophobic segments in solution. In aqueous solutions, water insoluble (hydrophobic) drugs are encapsulated during self assembly (Kedar et al., 2010). While in non-aqueous solutions, water-soluble (hydrophilic) drug molecules, including proteins, peptides, and nucleic acids, are encapsulated during assembly (Allen, 1998; Momekova et al., 2007). Micelle nanoparticles range in size from 5 to 100 nm (Oerlemans et al., 2010). Many different polymers can be used to create micelles; however, the selection is limited for drug delivery applications because the micelle must be biocompatible and biodegradable. PEG is commonly used to fabricate micelles because it is neutral, nontoxic, and water-soluble. Other hydrophilic polymers used include poly(N-vinyl pyrrolidone) (PVP) and poly(N-isopropyl acrylamide) (pNIPAM) (Chung et al., 1998; Chung et al., 2000; Chung et al., 1999). Degradable hydrophobic polyesters are commonly used to make the hydrophobic segment of the amphiphilic macromolecule (Sutton et al., 2007). Micelles are intrinsically stealth particles when formed with a hydrophilic outer shell, and they are able to avoid uptake by the MPS without further modification.
Figure 4.
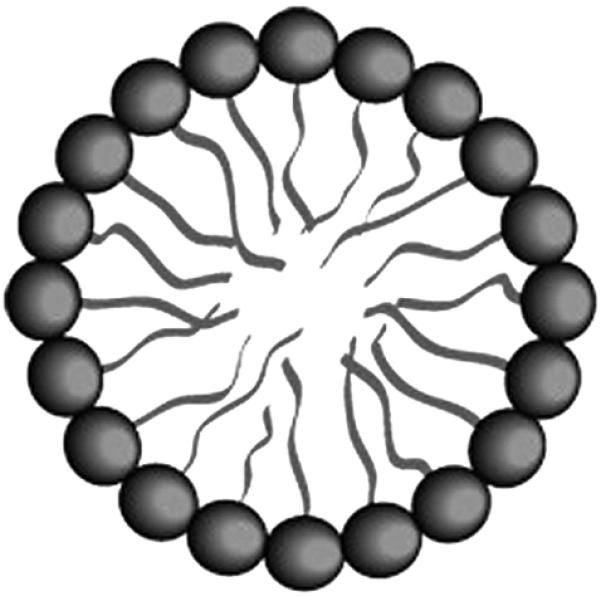
Micelle structure. Micelles are composed of phospholipids, with hydrophilic head groups forming the outer shell. Micelles encapsulate water-insoluble drugs in their hydrophobic cores. Figure modified and used with permission from (Husseini and Pitt, 2008).
Passive and active targeting can be achieved using micelles. Drug-encapsulated micelle systems increase targeting of tumors due to passive targeting via the EPR effect (Kwon et al., 1994). Polymeric micelles that are not ligand-conjugated accumulate in tumor tissue and release occurs intratumorally, in the tumor tissue outside of the malignant cells. Ligands can be attached to micelles for active targeting and drug release within the cell. Ligand-conjugated micelles are internalized via pinocytosis (Shuai et al., 2004).
Micelles can be used to deliver water-insoluble drugs in their hydrophobic core (Moghimi et al., 2005). One-third of all drugs used in cancer therapy are hydrophobic (Lee et al., 2012). Amphiphilic copolymers used to fabricate micelles are ideal for delivery of water-insoluble drugs because the outer shell creates a hydrophilic corona to stabilize and protect the hydrophobic drug. Polymeric micelles can increase water solubility of drugs by 10- to 500-fold (Savić et al., 2006), which enables the intravenous injection of micelle-encapsulated hydrophobic drugs. For example, paclitaxel is a water-insoluble drug; however, when encapsulated in a micelle, its water solubility is significantly enhanced (Soga et al., 2005).
Drug loading in micelles is achieved by physical entrapment or chemical conjugation; drug release is dependent on the loading method (Kedar et al., 2010). Drugs loaded by chemical conjugation are released by bulk degradation or surface erosion of the polymer, while drugs loaded by physical entrapment are released by diffusion. Release is also influenced by the extent of cross-linking in the core of the micelle, with cross-linked micelles resulting in slower release, which results in longer release times (Liu et al., 2007a). Three micelle-based cancer drugs have been approved for clinical trials for treatment of solid tumors; one that delivers doxorubin and two others that deliver paclitaxel (Chen et al., 2011a).
A key advantage of micelles over other particles is the core-shell structure. The hydrophobic core enables solubilization of hydrophobic drugs in water, and the hydrophilic shell, or corona, protects the drug by preventing elimination by the MPS (Kedar et al., 2010), which enables prolonged circulation (Sutton et al., 2007). Another advantage of micelles is that they generally possess low toxicity and can be eliminated through renal filtration if the molecular weight of the micelle polymer chains is below the critical value for renal filtration (Yokoyama, 2011). Disadvantages of micelles include poor drug loading efficiency, poor physical stability in vivo, and insufficient cellular interaction of neutral micelles with malignant cells for uptake (Kim et al., 2010). Additionally, micelles have a smaller drug loading capacity than liposomes.
4.3. Dendrimers
Dendrimers are the smallest of the nanoparticles (Svenson and Tomalia, 2005), with a radius ranging from 2.5 to 8 nm (Kaminskas et al., 2011). Dendrimers are spherical, with a three-dimensional branched structure, which contains a core, branching repeat units arranged radially from the core, and outer terminal functional groups (Roberts et al., 1996) (Fig.5). Branches can be created by polymerization of a monomer that diverges from the core or converges from the periphery. The size of the dendrimer is easily controlled via polymerization of repeat layers, with each layer making up a generation. Dendrimers can be made cationic, anionic, or neutral depending on their terminal groups. For example, amine-, carboxyl-, or hydroxyl-terminated dendrimers would create cationic, anionic, and neutral surface charges, respectively. Poly(amido amine) (PAMAM) dendrimers are the most widely studied. PAMAM is described as a “starburst dendrimer” because its branched structure has a star-like pattern (Labieniec et al., 2009). PAMAM dendrimers contain an initiator core of ethane-1,2-diamine with radially extending repeat branching layers. Full generation PAMAM dendrimers are amine-terminated, while half-generations are carboxylic acid-terminated.
Figure 5.

Dendrimer branch structure. Dendrimers are created by polymerization of a core monomer to create a branched polymer structure. Each polymerized layer radiates from the core and correlates to the generation of the dendrimer. Various surface functional groups may be used to modify the surface charge of the dendrimer. Figure used with permission from (Kaczorowska and Cooper, 2008).
Dendrimer size and surface charge directly affect their drug delivery properties. As discussed in Section 3, surface charge influences carrier uptake. Cationic dendrimers tend to be rapidly cleared from circulation due to electrostatic binding to tissue and vasculature, while anionic and neutral dendrimers have longer circulation times. Smaller dendrimers (<25 kDa) are also rapidly cleared from circulation through urinary excretion (Kaminskas et al., 2011). Dendrimers can be pegylated to increase their size, resulting in increased circulation time and decreased clearance by the MPS. Pegylation or acetylation of cationic dendrimers also reduces their cytotoxicity (Chen et al., 2004; Kolhatkar et al., 2007). Acetylation, which is the chemical attachment of acetyl groups, reduces the surface charge by shielding cationic groups to create a more neutral dendrimer; the resulting surface charge depends on the degree of acetylation.
Dendrimers have desirable properties for drug conjugation or encapsulation. Drugs can be attached to functional groups on branch ends or encapsulated in dendritic channels within the branches (Hughes, 2005; Moghimi et al., 2005). Incorporating degradable linkages between the drug and the dendrimer controls drug release. Drug loading in the dendritic channels can be manipulated by varying the dendrimer generation. Drug loading is performed by physical entrapment or through interactions between drug molecules and dendritic groups (D’Emanuele and Attwood, 2005). Dendrimers synthesized with hydrophobic cores are able to encapsulate hydrophobic drugs (Frechet, 2002; Patri et al., 2002), and electrostatic interactions between the dendrimer and the drug increase drug solubility at high pH (Milhem et al., 2000). Encapsulation of hydrophobic drugs is achieved through hydrophobic interactions between the drug and hydrophobic regions of the polymer when the drug and polymer are mixed. Currently, there are no FDA approved dendrimer cancer drugs; however, they are promising nanoparticles that are receiving much attention for future anticancer drug delivery.
One of the key advantages of dendrimers is that they are highly customizable. They can be uniformly reproduced with monodisperse sizes during synthesis by controlling the amount of monomer used. End groups can also be tailored to produce neutral, cationic, or anionic dendrimers. Dendrimers have a high drug loading capacity and may encapsulate both hydrophilic and hydrophobic drugs (Sampathkumar and Yarema, 2007). Further, dendrimers can be functionalized and conjugated with many different therapeutic molecules. Their small size may also help them avoid recognition by the MPS. A possible disadvantage of dendrimers is that steric crowding of the branching arms occurs at higher generation numbers, which limits growth to a larger size (Medina and El-Sayed, 2009) and results in decreased drug encapsulation. A key issue for cationic dendrimer drug delivery systems is the balance between function and surface charge. The positively charged surface promotes cellular uptake and endosome escape, but it also results in toxicity to nonmalignant cells. Surface charge must be balanced to optimize drug delivery and minimize cytotoxicity.
4.4. Polymeric particles
Polymeric particles can be fabricated as nanospheres or nanocapsules (Fig. 6). Nanospheres are solid spherical structures composed of a polymer matrix. Drugs can be conjugated to the nanosphere surface or absorbed within the bulk of the polymer matrix. Nanocapsules are vesicular hollow polymers that encapsulate a drug solution within a polymeric membrane. Polymeric particles consist of a polymeric backbone that is typically composed of a biodegradable monomer (Malam et al., 2009). Polymer particles provide flexibility in design because they can either be biodegradable or non-biodegradable, natural or synthetic (Alexis et al., 2010). Typical natural polymers used to make polymer particles are alginate, albumin, dextran, and chitosan. Commonly used synthetic polymers include PLA/PLGA and PEG block copolymers, which create stealth particles with longer particle circulation times (Gref et al., 1994) because they inhibit opsonization and elimination by the MPS.
Figure 6.
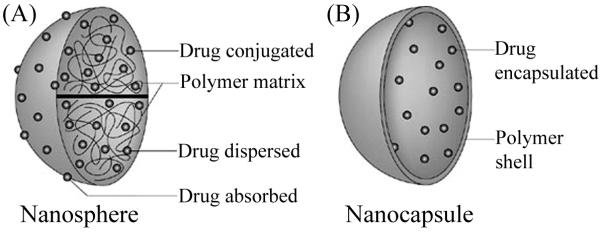
Structure of polymeric nanoparticles. (A) Polymer nanospheres are solid structures composed of a polymer matrix. Drugs can be loaded into the matrix by absorption or chemically conjugated to the surface. (B) Polymer nanocapsules are hollow structures containing an outer polymer shell. Drug can be encapsulated in the core of nanocapsules during formation or absorbed into the polymer shell. Figure modified and used with permission from (Griffiths et al., 2010).
Conjugation, functionalization, and targeted delivery can be achieved using polymeric particles. The surface of polymeric particles is typically sterically stabilized by using grafting, conjugation, or pegylation to increase repulsion between particles, which prevents their aggregation (Gref et al., 2000). Steric stabilization results in reduced elimination by the MPS and longer circulation times (Gref et al., 2000; Peracchia et al., 1999). Ligands can be conjugated to polymeric particles for active targeting. Polymeric particles can also be coated with other polymers to enhance drug delivery. Hydrophobic particles can be coated with PEG to create stealth particles. Polymeric nanoparticles may also be tailored for delivery across the blood-brain barrier by coating them with polysorbate 80 (Kreuter, 2001). Absorption of apolipoprotein E onto the surface produces a coating that mimics low-density lipoprotein (LDL) and results in transport, via LDL receptors, across the blood-brain barrier (Hans and Lowman, 2002).
Drugs can be incorporated into polymeric particles via conjugation, absorption, and encapsulation. Anticancer drugs can be chemically conjugated to the surface of both nanocapsules and nanospheres using ester or amide bonds (Malam et al., 2009). The drug is released in vivo by the hydrolysis of the ester or amide bonds (Malam et al., 2009). The anticancer drug does not become active until the bond has been hydrolyzed. Drugs can also be absorbed into solid nanospheres and entrapped in the inner polymer matrix. Nanocapsules containing a core and shell can either have drug encapsulated in the core or absorbed into the polymer matrix of the outer shell (Panyam and Labhasetwar, 2003; Parveen et al., 2012). Encapsulation is performed either during particle formation (for nanocapsules) or via adsorption following particle synthesis (for both nanocapsules and nanospheres).
The mechanism of encapsulation affects the rate of drug release. Drugs loaded using adsorption have a burst effect, in which the bulk of the drug is quickly released, while drugs that are chemically conjugated have a slower controlled release (Fresta et al., 1995). There are a few polymeric nano particle anticancer formulations in clinical trials for cancer treatment. A cyclodextrin-PEG nanoparticle conjugated with camptothecin, a broad-spectrum anticancer drug, has exhibited significant anti-tumor activity (Svenson et al., 2011). A PEG-PLGA nanoparticle encapsulating docetaxel, BIND-014, has exhibited antitumor activity in mouse models (Farokhzad et al., 2006), and it has been approved for clinical trials in patients with advanced or metastatic cancer (Service, 2010).
Polymeric particles have some unique advantages for drug delivery. Particles composed of natural polymers have greater biocompatibility than synthetic polymers and low recognition by the MPS. Like dendrimers, polymeric particles are highly customizable; polymer composition, conjugation, and drug release can each be modified to optimize targeted delivery for a specific cancer type. Biodegradable particles can be used to control drug release and avoid issues with elimination. Polymeric particles also have potential for large-scale production for drug delivery applications using several fabrication techniques (Fang and Zhang, 2011). Maintaining purity and sterility during manufacturing will be imperative for commercialization of polymeric particles for cancer drug delivery (Fang and Zhang, 2011).
4.5. Carbon nanotubes
CNTs are hollow tubes formed from rolled sheets of graphene rings, which are composed of sp2 hybridized carbons (Prakash et al., 2011) (Fig. 7). There are two types of CNTs: single-walled carbon nanotubes (SWNTs) and multi-walled carbon nanotubes (MWNTs). SWNTs are made of one layer of graphene and have a diameter of 1 to 2 nm, while MWNTs are made from multiple coaxially arranged layers of graphene and have diameters from 5 to 100 nm (Liang and Chen, 2010).
Figure 7.

Carbon nanotube structure. Carbon nanotubes (CNTs) are hollow tubes composed of graphene sheets. CNTs can be either (A) single-walled or (B) multi-walled.
CNTs are naturally hydrophobic; however, they can be functionalized via adsorption, electrostatic interaction, or covalent bonding to create a hydrophilic surface (Prakash et al., 2011). CNTs are functionalized through linkages created with the sidewall carbons or ends of the nanotubes. The tips or ends of the CNTs are typically more reactive than the sidewalls (Tasis et al., 2006). CNTs can be reacted with acids to purify them and create carboxylic acid groups for further functionalization (Bianco et al., 2005). Hydrophilically functionalized CNTs exhibit increased tumor tissue accumulation via passive targeting compared to non-functionalized CNTs (Pantarotto et al., 2004). CNTs can be conjugated with ligands for active targeting. Water-soluble CNTs can be conjugated with peptides, proteins, nucleic acids, and other water-soluble therapeutic agents for active targeting and drug delivery (Bianco et al., 2005).
CNTs exhibit effective drug loading and release. Binding and release of anticancer drugs from SWNTs can be regulated by pH. CNTs have a high drug loading capacity, a CNT with a diameter of 80 nm has been shown to contain approximately five million drug molecules (Kam and Dai, 2005). Drug loading capacity decreases as pH decreases, and drug release is triggered in acidic conditions; both effects are due to increased hydrophilicity and solubility of the CNT (Liu et al., 2007c). CNTs have not yet been approved for use in clinical trials, but they have shown promising results in drug delivery studies. SWNTs functionalized with platinum(IV)-prodrug conjugates increased internalization and reduced toxicity of anticancer drugs in testicular carcinoma cells vitro compared to free platinum(IV) (Feazell et al., 2007). Paclitaxel-conjugated SWNTs exhibited enhanced uptake by malignant cells compared to free drug in a mouse model (Liu et al., 2008).
CNTs have properties that are advantageous for cancer targeting. CNTs have a high drug loading capacity. Additionally, CNTs are triggered to release their drug load in acidic environments. However, one of the main disadvantages of CNTs is that they may cause inflammation and toxicity even after functionalization (Roda et al., 2011). The CNT purification process is also laborious, and commercially available CNTs are contaminated with particles that make them nonbiocompatible. Noncovalent bonding of drugs can result in inefficient attachment and release of the drug before it reaches its therapeutic target (Gottesman et al., 2002; Shi Kam et al., 2004). These limitations must be addressed before CNT-based cancer therapies can advance to clinical trials.
5. TARGETING MOIETIES
Targeting moieties are ligands that bind to receptors that are overexpressed on cancer cells. Conjugating targeting moieties to the surface of particles promotes uptake and intracellular retention of particles by malignant cells, both of which enhance therapeutic efficacy (Sahoo and Labhasetwar, 2005). Active targeting of malignant cells using ligands promotes receptor-mediated endocytosis. After a ligand binds to its corresponding cell surface receptor, the receptor-ligand-particle complex is endocytosed. Active targeting promotes direct cell kill and enhances cytotoxicity of anticancer drugs against malignant cells, while passive targeting promotes accumulation of particles in tumor tissue (Pastorino et al., 2006). Targeting moieties discussed here include folate, transferrin, monoclonal antibodies (MAbs), peptides, EGF, and aptamers.
Ideally, unique cell-surface antigens would be expressed exclusively on, and homogeneously among, cancer cells (Danhier et al., 2010). However, receptors overexpressed on cancer cells are also expressed on nonmalignant cells in lower numbers. Therefore, ligands should have high affinity and specificity for overexpressed cell surface receptors (Brumlik et al., 2008). Additional considerations for ligand selection include whether or not the ligand promotes internalization of the anticancer drug or drug-particle complex into malignant cells and the ease of ligand conjugation to the anticancer drug or particle.
5.1. Folate
Folate is the most extensively researched receptor for cancer targeting. Folate, or folic acid, is an essential vitamin necessary for cell function and synthesis of purine and pyrimidine for DNA. FR is typically overexpressed on malignant cells, and may be upregulated on cancer cells by up to two orders of magnitude (Low and Antony, 2004). FR is overexpressed in lung, head, neck, brain, ovarian, uterine, bone, breast, and renal cancer (Elnakat and Ratnam, 2004; Shmeeda et al., 2006; Vasir and Labhasetwar, 2005). FR is present in a reduced form on most nonmalignant cells as a low affinity carrier that selectively transports reduced forms of folate into the cell (Hilgenbrink and Low, 2005). Folate uptake on most cells is mediated by either the reduced folate carrier or a proton-coupled folate transporter, neither of which display affinity for folate conjugates (Low and Kularatne, 2009). In contrast, a high affinity form of FR is overexpressed on many cancer cells and has limited expression on nonmalignant cells. There are two FR isoforms known to be overexpressed in cancer: FR-α is found in epithelial cancer and FR-β is found in myeloid leukemia and macrophages of chronic inflammatory disease. FR-α is found in low levels in nonmalignant epithelia (Lu and Low, 2002), with the exception of the placenta and the choroid plexus (Sudimack and Lee, 2000). However, FR-α is found on the apical surface of the nonmalignant epithelia, and it is therefore not accessible to folate conjugates on the surface of cancer targeted delivery vehicles (Low and Kularatne, 2009). FR-β is generally found in low levels in nonepithelial and hematopoietic cells (Lu and Low, 2002).
Folate is easily conjugated to particles, and it has a high binding affinity for the FR (Low and Antony, 2004; Shmeeda et al., 2006; Stella et al., 2000). PAMAM-folate-methotrexate dendrimers have been shown to decrease toxicity and enhance efficacy 10-fold relative to free methotrexate in mice with human KB tumors (Kukowska-Latallo et al., 2005). KB cells are a keratin-producing cell line established by HeLa cell contamination (Lacroix, 2008). Additionally, liposomes encapsulating doxorubicin and coated with a folate-conjugated poly(L-lysine) polymer exhibited increased toxicity in both human lung and nasopharyngeal cancer cells in vitro compared to non-targeted doxorubicin encapsulated liposomes (Watanabe et al., 2012). Folate-conjugated particles are also used to target various malignancies for tumor imaging in mouse models (Chen et al., 2011b; Hou et al., 2011).
Folate has many advantages as a targeting ligand. Folate is inexpensive, non-immunogenic, available in large quantities, and easily conjugated to particles and anticancer drugs (Lee and Low, 1994; Sudimack and Lee, 2000). Folate is small in size and has a high affinity for its receptor. Some folate conjugates are released from the FR in the endosome during acidification (Lee et al., 1996; Wileman et al., 1985). Folate-conjugated particles have demonstrated endosomal escape through an unknown mechanism (Hilgenbrink and Low, 2005), preventing damage to the anticancer drug by lysosomal enzymes. Further research on this phenomenon is necessary to elucidate its mechanism. It has also been demonstrated that monovalent FR-containing endosomes are only mildly acidic (pH 6.8), whereas endosomes of multivalent FR-conjugates are more acidic (pH 5) (Lee et al., 1996; Yang et al., 2007). The pathway of multivalent folate conjugates can be exploited to trigger drug release using pH-responsive linkers (Low and Kularatne, 2009). FRs also exhibit limited expression on nonmalignant cells. Cells that do express folate are inaccessible to folate-drug conjugates in circulation because they are localized to apical surfaces of polarized epithelia (Hilgenbrink and Low, 2005).
5.2. Transferrin
Transferrin is a glycoprotein that binds iron and transports it into cells via transferrin receptor-mediated endocytosis. Because cancer cells have a high rate of proliferation, they have a greatly increased need for iron to carry out cellular functions, which results in an over expression of the transferrin receptor (Gatter et al., 1983). Transferrin receptors are particularly overexpressed on metastasizing and drug resistant cells. In fact, tumor cells typically overexpress transferrin receptors by 2- to 10-fold compared to nonmalignant cells (Anabousi et al., 2006).
Several transferrin-conjugated drugs have been tested and have shown improved outcomes in clinical trials, including adriamycin, cisplatin, and diphtheria toxin (Faulk et al., 1990; Head et al., 1997; Rainov and Soling, 2005). Transferrin-polymer-drug conjugation has been used to treat several types of malignant cells in mouse models (Hu-Lieskovan et al., 2005). Transferrin-conjugated particles loaded with paclitaxel have exhibited enhanced antiproliferative activity compared to free drug and nontargeted particles due to increased cellular uptake and reduced exocytosis (Sahoo and Labhasetwar, 2005). These particles have proven effective against breast cancer cells in vitro (Sahoo and Labhasetwar, 2005) and prostate cancer in mouse models (Sahoo et al., 2004). Lipid particles (blend of solid and oil lipid) conjugated with transferrin also exhibited increased uptake in leukemia cells in vitro (Khajavinia et al., 2012). Transferrin-conjugated particles can cross the blood-brain barrier and target rat brain tumor glioma cells in in vitro models (Pulkkinen et al., 2008).
Transferrin has several advantages for targeted delivery to cancer cells. Human transferrin is non-immunogenic; therefore, it can be safely delivered without causing toxicity. Transferrin-conjugated drugs have been shown to prevent cardiotoxicity and drug resistance (Singh, 1999). Transferrin has been used to successfully deliver many cancer drugs. Additionally, the likelihood of transferrin binding to tumor cells is higher compared to nonmalignant cells because transferrin receptors on tumor cells recycle back to the surface more quickly (Singh, 1999). Transferrin can also be easily obtained from human sources. However, there are some disadvantages to using transferrin as a targeting ligand. Delivery of transferrin-conjugated drugs in high doses may result in damage to other cells that express low levels of transferrin receptor (Laske et al., 1997). Transferrin ligands may target liver cells and highly proliferative nonmalignant cells (Daniels et al., 2006). Further, free transferrin in the blood may competitively bind to malignant cell receptors, preventing uptake and limiting efficacy (Xu et al., 1997; Xu et al., 2001).
5.3. Epidermal growth factor
EGFR is a transmembrane receptor that contains an extracellular binding domain (Tai et al., 2010). EGFR has been found to play a role in the progression of many malignancies (Danhier et al., 2010), and it is indicative of the metastatic capability of a malignant tumor (Radinsky et al., 1995; Verbeek et al., 1998). EGFR is overexpressed in many cancer types including colorectal, lung, head and neck, ovarian, kidney, prostate, and pancreatic cancer (Lurje and Lenz, 2009). EGFR is overexpressed by 100-fold in some cancer cell types (Salomon et al., 1995). EGFR is actually a family of receptors that includes EGFR, human epidermal growth factor receptor-2 (HER-2), HER-3, and HER-4 (Zwick et al., 2001). HER-2 is overexpressed in some invasive breast cancers and correlates with a poor prognosis (Sakai et al., 1986). EGFR and HER-2 have been the most extensively studied for cancer-targeting applications because their activation leads to rapid growth, differentiation, migration, and survival of cancer cells (Kaptain et al., 2001; Laskin and Sandler, 2004).
Epidermal growth factor (EGF) and transforming growth factor-α are both ligands for EGFR (Byrne et al., 2008). MAbs can also be used as ligands to target EGFR, and they are discussed in Section 5.6. EGF-conjugated particles with encapsulated gemcitabine exhibited greater uptake and anti-proliferative activity in human breast cancer cells compared to nontargeted particles in mouse tumor models (Sandoval et al., 2012). EGF-conjugated liposomes have been shown to overcome low specificity and inefficient drug accumulation in an in vitro model using human epithelial cancer cells and neuronal glioblastoma cells (Carlsson et al., 2003; Kullberg et al., 2003). Additionally, EGF-conjugated gold nanoparticles have been shown to target glioma brain tumors, increasing uptake by 10-fold over untargeted nanoparticles in mice (Cheng et al., 2011).
There are several advantages to using EGF as a targeting ligand. EGF binds to EGFR with high affinity. A key advantage of EGF relative to other ligands is that it enables drug delivery to the cell nucleus. EGFR is able to translocate from the cell surface into the nucleus through an endocytic pathway in highly proliferative cells (Lin et al., 2001; Torrisi et al., 1999; Wang et al., 2001). Internalization of EGF also occurs more quickly than antibody-conjugated anticancer drugs (Fan et al., 1994). A disadvantage of EGF is that it is mitogenic (Kitchens et al., 1994; Norman et al., 1987) and therefore may promote cell division.
5.4. Peptides
Peptides are molecules consisting of 2 to 50 amino acids linked by peptide bonds (Reubi, 2003). Some peptide receptors overexpressed in tumor cells include somatostatin, vasoactive intestinal peptide (VIP), luteinizing hormone-releasing hormone (LHRH), cholecystokinin (CCK), gastrin-releasing peptide, and bombesin receptor. Somatostatin peptides regulate the endocrine system and bind to G-coupled peptide receptors with high affinity (Graff et al., 2005). Somatostatin receptors are overexpressed on many tumor cells and in tumor blood vessels (Honer et al., 2011). Somatostatin is the most commonly used radiopeptide, a radioactively labeled peptide that can be used for diagnosis and treatment of malignances (Graham and Menda, 2011). VIP is a vasodilator that is secreted from some tumors of the pancreas and nervous system (Das et al., 2009). LHRH is a key regulator of reproduction, and it is overexpressed in many cancer types including breast, prostate, endometrial, and ovarian (Kakar et al., 2008). CCK is a digestive hormone, and CCK receptors bind CCK or gastrin peptides. CCK subtype two receptor (CCK-2R) is overexpressed in medullary thyroid carcinomas, neuroendocrine tumors, small cell lung carcinomas, and colorectal cancers (Sosabowski et al., 2009). Bombesin peptides stimulate gastrin release and are overexpressed in small cell carcinoma of the lung, gastric cancer, and neuroblastoma (Ohlsson et al., 1999).
Peptides have been used in many tumor targeting studies. In vitro, LHRH peptide-conjugated particles enhanced internalization in cancer cells overexpressing LHRH receptors when compared to nontargeted particles (Taheri et al., 2011). Divalent gastrin peptides, which are composed of two peptides, have been used to target CCK-2Rs. Contrast agent-conjugated gastrin peptide increased binding affinity and tumor uptake of the peptide in mice (Sosabowski et al., 2009). VIP-conjugated micelles loaded with anticancer drug had significantly higher toxicity than nontargeted micelles when targeting VIP receptor-expressing breast cancer cells in vivo (Onyuksel et al., 2009). VIP-conjugated liposomes and micelles exhibited increased accumulation in malignant cells compared to nontargeted particles in breast cancer in situ tumor models in rats (Dagar et al., 2003; Onyuksel et al., 2009). Somatostatin has been conjugated to many anticancer drugs, and it has displayed significant somatostatin receptor-selective antitumor ability in mouse models (Sun and Coy, 2011). The bombesin analog, a man-made peptide comparable to bombesin, also exhibits specific targeting in vivo. The bombesin analog has been used to target gastrin-releasing peptide receptors to treat prostate carcinoma in a mouse model (Honer et al., 2011). A peptide that has been used to treat hypovascular tumors is the Ala-Pro-Arg-Pro-Gly (APRPG) peptide. APRPG targets angiogenic vessels; it is not specific to tumor cells. However, these peptides are beneficial for treating hypovascular tumors because they interact with endothelial cells facing the circulating blood, and APRPG peptides damage angiogenic vessels without the need for particle extravasation (Asai, 2012). Anticancer drug-loaded liposomes (Yonezawa et al., 2007) and micelles (Saito et al., 2007) exhibited increased tumor regression relative to nontargeted particles in a mouse model of a hypovascular orthotopic pancreatic tumor.
In addition to targeting peptide receptors, peptides are commonly used as ligands to target integrin. Integrin, a transmembrane receptor that mediates adhesion between cells and surrounding tissue, is typically overexpressed in tumor neovasculature (Arias, 2011). There are many types of integrin; however, αvβ3 is the most commonly targeted integrin receptor for cancer drug delivery. αvβ3 integrin is a receptor on endothelial cells that binds extracellular matrix proteins that contain the arginine-glycine-aspartic acid (RGD) sequence (Byrne et al., 2008). The RGD peptide has high binding affinity for αvβ3, which is expressed on angiogenic endothelium in malignant tissue (Brooks et al., 1994). RGD peptides are commonly used to target αvβ3 integrin (Byrne et al., 2008). The αvβ3 ligand has been used for in vivo gene delivery to cancer cells using cationic polymerized liposomes (Hood et al., 2002). RGD10, a high affinity peptide conjugated to doxorubicin-loaded liposomes, resulted in improved uptake compared to free doxorubicin and untargeted liposomes in a mouse colon cancer model (Hölig et al., 2004). Additionally, cyclic RGD peptide targeted αvβ3 positive cells in ovarian carcinoma tumors in a xenograft mouse model (Dijkgraaf et al., 2007). Doxorubicin encapsulated in RGD-conjugated particles resulted in selective apoptosis of αvβ3-positive cells and a 15-fold increase in anticancer activity in a mouse melanoma model (Murphy et al., 2008).
There are several advantages to using peptides as ligands for cancer targeting. Generally, peptides have low immunogenicity, are small in size, and are excreted via the liver or kidney (Dijkgraaf et al., 2007). Peptides have high binding affinity for their receptors and are designed to duplicate the target region of the physiological receptor, resulting in high specificity. RGD peptides have high chemical stability in vivo, and they are easy to fabricate (Gu et al., 2007). Additionally, RGD peptides for integrin targeting have strong avidity, due to the formation of multiple ligand-receptor bonds, and extended blood circulation half-lives compared to other peptides (Montet et al., 2006). Diagnostic imaging has been achieved in clinical trials using radiopeptides to target malignant cells and also to administer treatment using radiotherapy (Graham and Menda, 2011). However, peptides are rapidly degraded by peptidases, enzymes that catalyze hydrolysis of peptide linkages (Reubi, 2003); therefore, stable analog peptides will be required for clinical applications.
5.5. Aptamers
Aptamers are DNA or RNA oligonucleotide sequences that are folded into unique 3D conformations and bind to target antigens (Pestourie et al., 2005). Aptamers have potential as cancer-targeting ligands because they can facilitate delivery of particles to tumor antigens on the surface of malignant cells (Smith et al., 2007). Nucleic acid aptamers can be engineered using in vitro selection or SELEX (systemic evolution of ligands by exponential enrichment) (Huang et al., 2009). SELEX is used to select aptamers that bind to their targets with high affinity (Ellington and Szostak, 1990; Tuerk and Gold, 1990). Aptamers from a DNA or RNA pool can be selected and enriched via repetitive binding to their target molecule. Aptamers can target proteins, small molecules, nucleic acids, cells, and tissues (Huang et al., 2009).
Aptamers can be conjugated directly to anticancer drugs or particles, and they can be used to target receptors on the cell surface or ECM molecules expressed by tumors (Das et al., 2009). Covalent conjugation of aptamers to particles can be achieved using succinimidyl ester amine chemistry that produces stable amide linkages or through maleimide thiol chemistry (Farokhzad et al., 2004) to form thioether bonds. Noncovalent conjugation involves affinity interactions and metal coordination (Farokhzad et al., 2006; Farokhzad et al., 2005). PLGA-PEG particles conjugated with prostate-specific membrane antigen aptamer have been engineered to deliver an increased drug dose to prostate cancer cells compared to nontargeted particles in xenograft mouse models (Dhar et al., 2011).
There are several advantages to using aptamers as ligands for cancer drug delivery. The advantages include high specificity and binding affinity for the target antigen, minimal immunogenicity, and easy modification (Drolet et al., 2000; Torchilin, 2005). Aptamers are stable over wide temperature and pH ranges, and they are stable in organic solvents (Nimjee et al., 2005; Wilson and Szostak, 1998). Additionally, aptamers are able to bind to proteins, molecules, nucleic acids, cells, or tissues (Burgstaller et al., 2002). Aptamers also exhibit effective tissue penetration (Hicke and Stephens, 2000). A key advantage of aptamers is their ease of synthesis and capability for large scale production. However, aptamers are susceptible to nuclease degradation, have very short circulation half-lives, and can be quickly cleared by the kidneys (Das et al., 2009). The aptamer circulation half-life can be extended by using pyrimidine or PEG linkages between the aptamer and particle (Ulrich et al., 2005) to prevent opsonization.
5.6. Monoclonal antibodies
MAbs are produced by clones of a single B lymphocyte and bind to a single epitope of a target antigen. MAbs are used to target specific receptors and interfere with signal transduction, which disrupts cancer cell proliferation (Danhier et al., 2010). Examples of MAbs include trastuzumab, bevacizumab, and etaracizumab, which are antibodies for ERBB2, vascular endothelial growth factor (VEGF), and αvβ3, respectively.
MAbs can be conjugated to liposomes for cancer targeting. MAb-conjugated liposomes are called immunoliposomes. Immunoliposomes can be prepared through conjugation of the MAb to the phospholipid head or PEG tail group (Sapra and Allen, 2003). Immunoliposomes have been used to target HER-2. Anti-HER-2 immunoliposomes encapsulating doxorubicin exhibit enhanced anticancer activity compared to untargeted liposomes in HER-2-overexpressing cancer cells in rat xenograft tumor models (Park et al., 2002; Park et al., 2001). MAbs have been used in targeted therapy for breast cancer. Cytokeratin is a MAb used to target invasive breast cancer epithelial cells. Drug-loaded PLGA particles conjugated with cytokeratin showed improved efficacy in treating breast cancer cells in vitro (Kos et al., 2009). Additionally, HER-2 trastuzumab antibody-conjugated PLGA particles exhibited improved cellular uptake and anticancer activity compared to nontargeted polymers in breast cancer cells in vitro (Sun et al., 2008).
The use of MAbs has also been explored for targeting cancer stem cells (CSCs). CD44, a cell-surface ECM receptor, is an established marker of CSCs (Deonarain et al., 2009). CD44 may exhibit various signaling functions depending on its protein interaction, including proliferation, apoptosis, survival, migration, and differentiation (Deonarain et al., 2009). Anti-CD44 antibodies have been used for targeted anti-CSC therapy. H90, a MAb specific to CD44, mediated eradication of acute myeloid leukemic stem cells in non-obese diabetic-severe immune deficient mice (Jin et al., 2006). A patented humanized version of H460-16-2, an anti-CD44 MAb, reduced tumor growth in pancreatic cancer xenografts in mice (Young et al., 2007).
There are advantages and disadvantages to using MAbs as targeting ligands. MAbs have high affinity and specificity for their target receptor, and they are able to achieve targeted delivery across the blood-brain barrier. OX26 is a monoclonal antibody that targets the transferrin receptor. Drug-OX26 conjugates have been shown to cross the blood-brain barrier using receptor-mediated transcytosis via brain capillary endothelial transferrin receptors (Bickel et al., 2001). However, MAbs are also immunogenic and have short half-lives. The high molecular weight of monoclonal antibodies also hinders extravasation, resulting in slow diffusion of MAb-conjugated particles into the target tissue (Dijkgraaf et al., 2007).
5.7. Vascular endothelial growth factor
VEGF mediates tumor angiogenesis, a common target for cancer drugs. Angiogenesis enables growth of new blood vessels, which are necessary to provide nutrients to a rapidly growing malignant tumor. Inhibiting angiogenesis inhibits tumor growth. VEGF is upregulated in neoplastic tumor cells due to hypoxia in the tumor tissue and oncogene expression, which results in overexpression of VEGF receptors-1 and -2 on tumor endothelial cells (Kremer et al., 1997; Shweiki et al., 1992). VEGF expression correlates to the degree of vascularization and to the stage of the cancer (Brown et al., 1993; Jain et al., 1998). VEGF is upregulated in bone marrow of patients with hematological tumors (Malik and Gerber, 2003). VEGF receptors (VEGFRs) are commonly expressed on endothelial cells of tumor vasculature, in solid tumors, leukemias, and other hematological tumors (Bozec et al., 2008). VEGFR-2 is typically targeted in therapeutic application because it binds VEGF and is overexpressed on malignant cells and endothelial cells of tumor vasculature.
VEGF-targeting strategies are different than those of other receptors. VEGF and VEGFR ligands are used therapeutically for their inhibitory functions, and not simply for targeting a receptor or antigen. Targeting angiogenesis is typically executed using two approaches: targeting VEGFRs to decrease binding of VEGF or targeting VEGF to inhibit its binding to VEGFRs (Backer et al., 2005; Veikkola et al., 2000). The most studied approaches include inhibition of VEGF and VEGFRs by MAbs to inhibit receptor signaling (Malik and Gerber, 2003). Bevacizumab, an antibody that binds to VEGF and inhibits tumor angiogenesis, has been FDA approved to treat colorectal cancer (Ferrara, 2005). Bevacizumab-conjugated polymer microspheres loaded with paclitaxel have been demonstrated to target angiogenesis resulting in growth inhibition of prostate tumors in mice (Lu et al., 2008). Further, boronated dendrimers conjugated with human recombinant VEGF121, a splice variant of VEGF containing 121 amino acids, have the ability to selectively target VEGFR-2 receptors on breast cancer tumor cells and inhibit tumor growth through receptor binding and endocytosis of anticancer drugs in a mouse model (Backer et al., 2005). Additionally, pegylated liposomal doxorubicin effectively reduced breast tumor growth when conjugated with anti-VEGFR-2 MAbs in various cancer types using a mouse model (Wicki et al., 2012).
One of the advantages of targeting VEGF and VEGFR is that tumor angiogenesis is directly targeted to inhibit tumor growth. VEGFRs overexpressed both in the tumor endothelium and tumor cells may be targeted. However, ligands that target VEGFR may also bind to other VEGF-related receptors and cause harmful side effects (Ferrara, 2005). Using recombinant VEGF to target VEGFRs is also very costly due to difficulty of manufacture and their short half-lives (Cao, 2001).
6. DRUG RELEASE
Methods of controlled drug release can be categorized as either temporal control or distribution control (Ulrich et al., 2005). The aim of temporal control is to enable release at a specific time and/or to extend duration of release. The goal of distribution control is to release drug at a target site. Distribution control provides additional strategies to enhance cancer targeting by maximizing the drug concentration at the cancer site, thus minimizing side effects to nonmalignant cells. This section will focus on methods for distribution control. The simplest method for achieving distribution control is to inject or implant anticancer drugs at the target site; however, direct delivery is only feasible if the target site is safely and easily accessible and the drug remains at the target site (Ulrich et al., 2005). An engineering approach for enhancing cancer targeting is to use stimuli-responsive particles to trigger drug release. Particles can be designed to interact with their physiological environment to promote drug release. Stimuli for release may be intrinsic, exploiting factors such as physiological pH or enzymes present, or stimuli may be extrinsic, using application of heat or other applied stimuli to trigger release.
The temporal and spatial drug release profiles that will be required for therapeutic efficacy are fundamental considerations when designing stimuli-responsive particles for targeted cancer therapy. Stimuli-responsive particles should enhance uptake by malignant cells and stimulate intracellular release in order to maximize the drug dose delivered to malignant cells. The particles should also be designed to promote endosomal escape after endocytosis for delivery to intracellular compartments. After a particle has been endocytosed, endosomal trafficking from the endosome to the lysosome occurs within 2 to 3 minutes (Liechty and Peppas, 2012). Particles must be designed to escape the endosome to avoid damage to the drug by the lysosomal enzymes. To avoid harming nonmalignant cells, particles must also be strictly designed to release their payloads at the targeted cancer sites and not in nonmalignant cells or tissues. If the pH- or thermo-response of a particle is not tightly controlled, it may result in drug release to non-targeted cells. The fate of the particle polymer component after drug release is also an important consideration. As reviewed in Section 3.5, polymer particles that can be degraded or excreted by the kidneys will have lower toxicity. Stimuli-responsive particles should also enable attachment of ligands to promote active targeting of the treatment site before the drug release is triggered.
6.1. pH-responsive drug release
Drug release can be triggered by taking advantage of the pH of the intratumoral environment or the intracellular environment. In nonmalignant body tissue, pH is maintained near 7.4. The pH of solid tumors varies in the range of approximately 5.7 to 7.6 due to accumulation of acidic metabolites that are the result of low blood pressure and hypoxia caused by abnormal tumor vessels (Dellian et al., 1996; Stubbs et al., 1999; Tannock and Rotin, 1989). The pH variation is even greater within the cell. Once drug-particle complexes are endocytosed, endocytotic vesicles convert to early endosomes, where the pH range is 5.9 to 6.8, and then to late endosomes, where the pH range is 5 to 6 (Liechty and Peppas, 2012). Late endosomes then fuse with lysosomes, in which the protein concentration is 100-fold higher than physiological conditions, and the pH decreases to an average of 5 (Bae et al., 2003).
pH sensitivity can be achieved by conjugating the drug to particles using acid-cleavable linkers or by conjugating a drug to, or encapsulating it within a pH sensitive polymer. Acid-cleavable linkages can be used to conjugate the drug to the polymer to form a prodrug that will remain inactive until the polymer has been cleaved. Prodrug particles are easily hydrolyzed through cleavage of their chemical bonds with water molecules at lower pH. Common pH-responsive linkages include acetals, anhydrides, cis-aconityl, and hydrazones (Duncan, 2003). Dimethylmaleic anhydride has been used as a pH-sensitive linker for polymeric particles (Kamada et al., 2004). The linker binds to amine groups at pH greater than 8 and reverts to anhydride to release the drug at pH less than 7. Hydrazone is another pH-sensitive linker that can be used to create an environmentally reactive cancer drug. Doxorubicin-conjugated polymer particles linked with hydrazone are stable at physiological pH; however, at pH 5, they release almost 50% of the drug (Etrych et al., 2001). Experiments in a mouse model demonstrated that these pH-sensitive particles slowed T cell lymphoma tumor growth when compared to free doxorubicin (Ulbrich et al., 2003) due to a triggered and controlled release of DOX at the tumor site.
In addition to pH-sensitive linkers, pH-responsive polymers can be used to trigger drug release under acidic conditions. pH sensitivity is found in ionizable groups, such as amine, azo, carboxylic acid, imidazole, pyridine, and thiol groups (Kim et al., 2009). pH-responsive polymers are triggered to escape the endosome by endosomal acidification and release their payload. Under low pH, pH-responsive particles absorb protons in the endosome, which increases endosome osmotic pressure, resulting in disruption of the endosomal membrane and particle release into the cytoplasm (Gao et al., 2010a). This mechanism is called the ‘proton sponge’ effect (Behr, 1997). The most common pH-sensitive liposomes contain phosphatidylethanolamine (PE) in the membrane bilayer (Simoes et al., 2004). Poly(ethylene oxide)-poly(propylene oxide) (PEO-PPO) copolymer modified with poly(β-amino ester) (PβEA) creates a pH-sensitive particle. This modified PEO-PPO-PβEA particle is stable at physiological pH and exhibits burst release at pH less than 6.5 (Shenoy et al., 2005). When loaded with paclitaxel, these pH-sensitive particles exhibited increased accumulation and anti-tumor activity in mouse xenograft tumor models of ovarian cancer (Devalapally et al., 2007). Acetal polymers can also be used for controlled release in pH-sensitive micelles. The acetal groups, which form the hydrophobic core to encapsulate the drug, are hydrolyzed under acidic conditions (Gillies and Frechet, 2005), resulting in micelle disruption and drug release.
One of the key advantages of pH-responsive particles is that they are able to exploit pH variances to promote endosomal escape. pH-sensitive particles enhance endosomal escape using the proton sponge effect. However, pH-sensitive particles can be toxic to nonmalignant cells if they are not designed to release their drug payload only within a precise pH range (Kim et al., 2009). Few pH-responsive delivery systems for cancer therapy have progressed to clinical trials due to concerns of toxicity to nonmalignant tissues, regulatory issues, and inefficient translation of in vitro efficacy to in vivo efficacy (Liechty and Peppas, 2012).
6.2. Enzyme-responsive drug release
Enzyme-responsive materials can be used to trigger drug release. After a particle has been endocytosed, it is exposed to pH changes and enzymatic degradation in the lysosome. When an endosome fuses with a lysosome, the particle is exposed to lysosomal enzymes such as glycosidases, proteases, and sulfatases that digest cellular waste. Enzymatically degradable spacers can be conjugated to polymeric particles to produce enzyme-mediated drug release (Hoste et al., 2004). Cancer drugs can be attached to the spacers, and upon lysosomal fusion, hydrolases degrade the spacer and release the drug. Lysosomal enzymes are not only expressed intracellularly, they are often overexpressed intratumorally, making them a promising target for enzyme controlled drug release. Enzymes including cathepsin B and D and metalloproteinase are overexpressed in tumor tissue and play a role in cancer progression (Gialeli et al., 2010; Masson et al., 2011; Podgorski and Sloane, 2003).
The most common type of spacer for enzyme-mediated drug delivery is an oligopeptide, a peptide composed of 2 to 20 amino acids. The rate of cleavage or enzymatic degradation of the peptide depends on the amino acid composition. Drug release can be modified by adjusting the length and composition of the spacer. Oligopeptide spacers with a C-terminal glycine are degraded more quickly, whereas hydrophobic terminal amino acids, such as leucine and phenylalanine, are degraded slowly (De Marre et al., 1994a; De Marre et al., 1994b). In an in vitro study, polymer-coated gold nanoparticles with glycine-phenylalanine-leucine-glycine spacers were designed to effectively control release of cancer drugs upon enzymatic degradation of the spacer (Schneider et al., 2009).
Controlled release and delivery of cytotoxic drugs can also be achieved by using antibody-directed enzyme prodrug therapy (ADEPT) (Sharma et al., 2010). In ADEPT, enzymes are first delivered to tumors using antibody-enzyme conjugates to target tumor cells. After enzymes bind to receptors on tumor cells and residual enzymes are cleared from circulation, prodrug is administered that is then enzymatically degraded within the tumor by the bound enzymes to create an active drug. ADEPT has been successful in clinical trials for the treatment of malignant tumors (Napier et al., 2000).
Enzyme-responsive drug carriers have advantages for cancer targeting because they minimize toxicity to nonmalignant cells. Through the use of prodrugs, the anticancer drug only becomes active at the target cancer site, limiting exposure of nonmalignant cells to the harmful effects of the drug. The prodrug-enzyme therapy enables delivery of large amounts of drug to the tumor site. However, one of the disadvantages of this therapy is the poor solubility of prodrugs (Hoste et al., 2004). Furthermore, even though ADEPT therapy results in local delivery, some active drug may escape the tumor site and cause damage to nonmalignant cells (Springer et al., 1993).
6.3. Thermoresponsive drug release
Hyperthermia can be used to trigger drug release from particles composed of thermoresponsive polymers. Local hyperthermia can be generated using infrared light, microwaves, hot water bath, ultrasound, or radiofrequency, depending on the location of the tumor (Hosokawa et al., 2003). Magnetic particles, in conjunction with an alternating magnetic field, can also be used to generate localized hyperthermia (Kobayashi, 2011). Regional hyperthermia is typically generated via perfusion by heating the blood. The temperature range for hyperthermia is between 37 and 42°C; at 42°C, five degrees above physiologic temperature, cells begin to show signs of thermal damage (Crile, 1962). Thermosensitive particles can be delivered intratumorally through passive accumulation due to the EPR effect. After particle accumulation, heat can be applied locally to stimulate drug release from the thermosensitive particles. Hyperthermia also increases the permeability of the heated tissue and cells, promoting intracellular uptake of drug or particle-drug conjugates. Electroporation, a technique in which an electric field is generated around the target tissue, also increases permeability, and it can be used to ablate tissue through irreversible electroporation (Davalos et al., 2005).
Poly(N-isopropyl acrylamide) (PNIPAAm) is a commonly used thermoresponsive polymer. The thermosensitivity of PNIPAAm results from a balance between hydrophobic and hydrophilic polymer segments; interactions between these segments causes chain aggregation and cross-linking (Kim et al., 2009). PNIPAAm is soluble at room temperature and gels above its lower critical solution temperature (LCST) (Kaneko et al., 1999; Nakayama et al., 2006), the temperature below which the polymer is miscible. The LCST of PNIPAAm is close to body temperature; therefore, the polymer will gel and trap drug molecules upon injection. However, PNIPAAm is commonly blended with other polymers to create particles or particle coatings. LCST of PNIPAAm is tunable via copolymerization of hydrophobic monomers or adjusting the molecular weight of the polymer. Hydrophobic monomers and high molecular weight decrease LCST, while hydrophilic monomers increase LCST (Kim et al., 2009). Doxorubicin-encapsulated magnetic particles coated with PNIPAAm exhibit drug release when heated at 42°C in vitro (Purushotham et al., 2009). A PNIPAAm copolymer loaded with camptothecin anticancer drug in conjunction with hyperthermia suppressed tumor growth in a mouse model of colon cancer (Peng et al., 2011).
Thermosensitive polymers that have been used to encapsulate cancer drugs include block copolymers of PEG-PLGA and PEO-PPO (Jeong et al., 2000; Kabanov et al., 2002). A multiblock polymer composed of poly(ε-caprolactone), PEG, and poly(propylene glycol) segments was synthesized to promote controlled release of paclitaxel (Loh et al., 2012). The copolymer exhibited sustained drug release for more than two weeks, and it arrested growth of cervical cancer-derived HeLa cells in vitro compared to paclitaxel encapsulated in non-thermosensitive particles. Adriamycin encapsulated in a thermosensitive liposome had greater delivery across the blood-brain barrier and greater anti-tumor activity in hyperthermia-treated mice with glioma than treatment with free drug or Adriamycin encapsulated in non-thermosensitive liposomes (Gong et al., 2011). Preliminary research has shown that permeability of the liposome across an in vitro model of the blood-brain barrier increased significantly with elevated temperature. Another liposomal formulation termed ‘Hyperthermia-activated cytoToxic’ (HaT), resulted in greater cell membrane permeability and drug release for mammary cancer cells in mice at elevated temperature, 40 to 41°C (Tagami et al., 2011). The liposomal HaT increased uptake of doxorubicin into heated tumor tissue by 5.2-fold and reduced delivery to the heart by 15-fold compared to free doxorubicin. Using hyperthermia, administration of a single intravenous treatment of the doxorubicin-encapsulated thermosensitive liposome resulted in greater mammary carcinoma tumor regression compared to free doxorubicin and temperature-sensitive lysolipid (phospholipid containing one carbon chain) liposomes in a mouse model (Tagami et al., 2011).
Thermosensitive drug carriers have several advantages for cancer targeting. Hyperthermia increases accumulation of drug in malignant tissue and cells (Gaber et al., 1996; Kong et al., 2001) by increasing the permeability of tumor vessels and tumor cells. Hyperthermia also increases cell sensitivity to thermally-induced injury (Fajardo and Prionas, 1994), which increases the death rate of malignant cells. Additionally, thermoresponsive polymers can solubilize hydrophobic drugs, and they can be used with poorly soluble drugs (Vukelja et al., 2007; Zentner et al., 2001).
7. FUTURE DIRECTIONS IN CANCER TARGETING
The successes of current research in targeted anticancer drug delivery demonstrate that the future of cancer-targeting therapy is promising. Particle drug encapsulation, passive and active targeting, and controlled release each enhance the targeting capabilities of anticancer drugs. Targeted cancer treatments such as particle encapsulation, targeting moiety conjugation, and stimuli-responsive drug release have already demonstrated improved patient outcomes relative to untargeted systemic drug administration in clinical trials. Research is ongoing to further improve targeted drug delivery for a variety of cancers. However, the tumor microenvironment is very complex, and optimizing targeted delivery requires a sophisticated drug delivery system that utilizes more than one targeting approach, for example, combining targeting moiety conjugation with stimuli-responsive drug release. Recent studies have shown that particle systems designed to have multiple functions further enhance the specificity of targeted drug delivery that is required to improve treatment efficacy and reduce treatment side effects. These systems combine design elements, such as particle encapsulation of the anticancer drug, ligand conjugation, controlled release, and imaging, to improve targeted delivery and to allow greater control over cancer therapy. Multifunctional systems have explored the delivery of therapeutics beyond chemotherapy, including DNA and RNA for cancer gene targeting and silencing. Multidrug therapy has also proven successful in many particle systems (Parhi et al., 2012). Additionally, dual ligand targeting has been employed to enhance cancer targeting specificity to receptors on malignant cells. The goal of the combinatorial approach is to further enhance targeted delivery.
7.1. Multifunctional delivery systems
Recently, multifunctional delivery systems have been designed to provide a comprehensive approach to targeting cancer. Multifunctional delivery systems are particles that have multiple functionalities, including diagnostic imaging, targeted drug delivery, and controlled drug release. These particles enable active monitoring of the cancer during administration of chemotherapy, which allows further control over the treatment process. Multifunctional particles combine multiple-targeting methods in an attempt to achieve optimal therapeutic results. These carriers may also support combination drug therapy by loading the particle with multiple drugs to achieve a better therapeutic outcome. Single drug chemotherapy may sometimes result in poor prognosis due to multidrug resistance, toxicity, and undesirable side effects (Parhi et al., 2012). Particle-based combination therapy provides a synergistic effect that enhances treatment effectiveness and overcomes limitations of single drug therapy. Multifunctional delivery systems can be used to deliver and monitor combination drug therapy to further improve cancer treatment.
There are many design options for multifunctional delivery systems. For example, a poly(propylene imine) dendrimer was designed for multifunctional drug delivery by loading it with both small interfering RNA (siRNA) and superparamagnetic iron oxide (SPIO) nanoparticless followed by conjugation with LHRH peptide, (Taratula et al., 2009). SPIOs are used as magnetic resonance imaging (MRI) contrast agents. The combination of the LHRH peptide and the SPIO contrast agent enabled successful targeting and monitoring of siRNA nanoparticles to LHRH receptor-positive ovarian and lung cancer cells, which resulted in tumor suppression in a mouse model. In another recent example, magnetic particles loaded with an anticancer superparamagnetic manganese complex were functionalized with PEG and RGD peptides and doped with fluorescent dye (Chen et al., 2012). The magnetic particles enabled in vitro imaging and differentiation between malignant and nonmalignant liver cells using MRI. RGD peptides enabled effective targeting and internalization of particles into HeLa cells in vitro. Multifunctional degradable polymeric particles have been developed for cancer “theranostics,” which combines diagnostic and therapeutic capabilities (Wang et al., 2012). Photosensitive and fluorescent imaging agents were incorporated into a polymer particle matrix with PEG and peptide targeting ligand conjugated to the surface. The degradable particles efficiently targeted breast cancer cells in vitro and enabled fluorescent imaging. The photosensitive drug also caused damage when tumor cells were irradiated with light at the appropriate wavelength.
Microbubbles (MBs), another type of multifunctional theranostic agent, have recently been studied for drug and gene delivery applications. MBs contain a gas core and an outer shell composed of lipids, proteins, or polymers (Sirsi and Borden, 2009). MBs have decreased solubility and diffusivity due to the high molecular weight gases contained in their core, making the bubble robust and able to recirculate in the blood stream several times (Greco et al., 2010). MBs exhibit useful properties for diagnostic imaging and drug delivery when subjected to ultrasound. MBs produce a backscattered echo under ultrasound that can be used to detect the bubble, making it useful as an imaging contrast agent. A MB also functions as a vehicle for gene and drug delivery. The MB is triggered for local release of its cargo by an ultrasound ‘destruction’ pulse in a technique referred to as ultrasound-targeted MB-destruction (UTMD) (Sirsi and Borden, 2009). The UTMD approach has been used to enhance therapeutic outcomes by delivering cancer terminator viruses (Das et al., 2012). MBs and UTMD have been used for delivery of a combination of adenoviruses that resulted in tumor growth inhibition in a mouse model of prostate cancer (Dash et al., 2011). In another study, ultrasound-guided MBs, complexed with a cancer terminator virus, eradicated targeted therapy-resistant tumors and nontargeted distant tumors in xenograft mouse models of prostate cancer (Greco et al., 2010).
7.2. Dual ligand targeting
The dual ligand targeting approach involves conjugating more than one type of ligand to the surface of a particle. Dual ligand targeting is based on the idea that cancer cells tend to overexpress multiple types of surface receptors (Li et al., 2011). For example, both HER-2 and EGFR are often overexpressed in breast cancer, while both EGFR and bombesin peptide are often overexpressed in prostate cancer. A key challenge for targeted cancer treatment is that many receptors that are overexpressed on malignant cells are also expressed on nonmalignant cells at lower, normal expression levels. Conjugation of two different ligands to particles to target overexpressed receptors has been shown to result in a higher rate of malignant cell recognition than that of either of the individual ligands (Li et al., 2011) by increasing specificity. Dual ligands increase the binding avidity of the particle complex to the malignant cell by binding two different ligands to two different receptors on the surface of the same cell (Meng et al., 2010). Dual ligands can further improve targeted delivery by enhancing the specificity of malignant cell targeting, thereby reducing toxicity to other cells.
Dual ligand targeting was demonstrated in vitro with human KB cells that overexpress both FR and EGFR (Saul et al., 2006). Both folic acid and MAbs for EGFR were conjugated to liposomal particles. When KB cells expressing both receptors were tested against cells in which one or both of the receptors were blocked, the results showed that the dual ligand improved detection of the KB cells.
Dual ligands have been shown to enhance cancer targeting in several other studies. Dual ligand gold nanoparticles (GNPs) have been designed to promote multivalent interactions between GNPs and FR-overexpressing cancer cells (Li et al., 2011). GNPs with folate and glucose conjugation enhanced cellular recognition of FR-overexpressing cells. Cellular uptake was increased by 3.9- and 12.7-fold compared to GNP-folate and GNP-glucose. In another study, αv integrins and neuropilin-1 (NRP-1), a receptor for VEGF, were chosen as simultaneous tumor targets (Meng et al., 2010). RGD was used to target αv integrins and a heptapeptide was used to target NRP-1. Targeting was tested in vitro in lung carcinoma cells and human umbilical vein endothelial cells, both of which express αv integrins and NRP-1. The dual targeting ligands increased uptake of paclitaxel in both cell lines. Dual targeting of brain glioma was achieved using the TGN peptide, a 12 amino acid peptide that targets the blood-brain barrier, and AS1411 aptamer, a DNA aptamer that binds to nucleolin protein in glioma cells (Gao et al., 2012). The dual ligand polymeric particles not only targeted endothelial and glioma cells, they also penetrated the endothelial monolayers and tumor cells to reach the core of the tumor spheroids in vitro (Gao et al., 2012). A similar strategy was used by another group to also successfully target brain glioma using p-aminophenyl-α-D-mannopyranoside (MAN) and transferrin-conjugated liposomes to enhance penetration of the blood-brain barrier and targeted delivery to glioma, respectively, in a rat model (Ying et al., 2010).
8. CONCLUDING REMARKS
This review has focused on bioengineering approaches to improve targeted delivery of cancer therapeutics, which include particles, targeting moieties, and stimuli-responsive drug release mechanisms. Design criteria must be understood in order to achieve the goals of cancer targeting, which include (1) selective targeting of cancer cells, (2) efficient release of anticancer drugs at the target site, and (3) elimination of cytotoxicity to non-cancerous tissue. To achieve these goals, barriers to targeted delivery, including opsonization, nonspecific uptake by nonmalignant cells, and multidrug resistance, must be addressed when designing targeted anticancer drugs. By combining particles, targeting ligands, and triggered release mechanisms, cancer targeting drugs can be improved and customized to achieve targeted cancer treatment.
Advances in nanotechnology and biomaterials have enabled the development of enhanced drug carriers for cancer targeting. Particles have been designed to encapsulate drugs to reduce toxicity to nonmalignant cells and to take advantage of passive targeting mechanisms using the EPR effect. Additionally, the surface properties of particles can be customized using techniques such as pegylation to enhance biocompatibility and to reduce cytotoxicity. Micelle and liposomal particles can be used to encapsulate hydrophobic drugs for delivery; however, there is a need for improved drug-loading capacity. CNTs have high drug-loading capacity but efficient purification methods are needed before CNTs can be used commercially as nanocarriers for cancer drugs. Dendrimers and other polymeric particles show promise for many cancer drug delivery applications because they are both highly customizable, and they can be designed with minimal immunogenicity and high drug loading capacity.
Targeting ligands can be attached to particles to promote active targeting and endocytosis of the drug by the targeted cancer cell. Ligands are selected to target receptors that are overexpressed on the surface of malignant cells. Folate is a widely used targeting ligand because its receptor is overexpressed in many cancer cells, but not on most nonmalignant tissues. Transferrin is non-immunogenic and can be easily obtained from human sources, but it can also cause toxicity to nontarget tissue due to nonspecific uptake in nonmalignant cells that have low-level expression of transferrin receptors. MAbs have high affinity and specificity for their target receptors, but are also immunogenic and diffuse more slowly due to a high MW. Peptides have potential for many cancer targeting applications due to the large selection of targeting ligands, high binding affinity and specificity, and low immunogenicity.
Controlled release of the anticancer drug from a particle carrier can be achieved using pH, enzyme, or thermo-responsive particles or conjugates. Stimuli-responsive particles promote release of the drug under the physiological conditions of the cancer tissue or cell. pH-sensitive particles take advantage of the intratumoral or intracellular pH differences to promote intratumoral or intracellular drug release. Enzyme-responsive spacers create prodrugs that are nontoxic until enzymes in the cancer tissue degrade the spacer. Thermosensitive release is achieved by applying external heat to enhance permeability of tumor tissue and cells, which increases the accumulation of particle encapsulated anticancer drugs in malignant cells. Stimuli-responsive polymers or spacers must be tightly controlled to avoid toxicity to noncancerous tissues.
The future of bioengineering strategies for cancer targeting will be to use a combinatorial approach to customize delivery of anticancer drugs and improve treatment outcomes. Multifunctional drug delivery systems are being designed that achieve imaging, targeted delivery, and controlled release from each particle. In addition, dual ligand targeting to multiple overexpressed receptors on malignant cells enhances specificity of particles for malignant cells while further reducing nonspecific uptake by nonmalignant cells. This combinatorial and customized approach to the design of particles shows promise for optimizing targeted cancer drug delivery to improve efficacy and to minimize side effects associated with drug delivery to nonmalignant cells.
ACKNOWLEDGMENTS
This work was made possible by the MUSC Hollings Cancer Center Translational Research Pilot Funds and the William H. Goodwin Endowment Funds (X.W.). This work was also supported by the AO Foundation (S0955V: W.V.F., X.W.) and the National Institutes of Health (T32DE017551: W.V.F, A.A.B; K99DE023123: W.V.F.). The authors thank Drs. Andrew G. Jakymiw and Frank Alexis for their critical reviews of this work.
REFERENCES
- ACS . Cancer Facts & Figures 2012. American Cancer Society; Atlanta: 2012. [Google Scholar]
- Albanese A, Tang PS, Chan WC. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- Alexis F, Pridgen EM, Langer R, Farokhzad OC. Nanoparticle Technologies for Cancer Therapy Drug Delivery. In: Schäfer-Korting M, editor. Handbook of Experimental Pharmacology. Vol. 197. Springer; Berlin Heidelberg: 2010. pp. 55–86. [DOI] [PubMed] [Google Scholar]
- Ali HM, Urbinati G, Raouane M, Massaad-Massade L. Significance and applications of nanoparticles in siRNA delivery for cancer therapy. Expert Rev Clin Pharmacol. 2012;5:403–412. doi: 10.1586/ecp.12.33. [DOI] [PubMed] [Google Scholar]
- Allen TM. Liposomal drug formulations. Rationale for development and what we can expect for the future. Drugs. 1998;56:747–756. doi: 10.2165/00003495-199856050-00001. [DOI] [PubMed] [Google Scholar]
- Anabousi S, Bakowsky U, Schneider M, Huwer H, Lehr CM, Ehrhardt C. In vitro assessment of transferrin-conjugated liposomes as drug delivery systems for inhalation therapy of lung cancer. Eur J Pharm Sci. 2006;29:367–374. doi: 10.1016/j.ejps.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Arias JL. Drug Targeting Strategies in Cancer Treatment: An Overview. Mini Rev Med Chem. 2011;11:1–17. doi: 10.2174/138955711793564024. [DOI] [PubMed] [Google Scholar]
- Asai T. Nanoparticle-mediated delivery of anticancer agents to tumor angiogenic vessels. Biol Pharm Bull. 2012;35:1855–1861. doi: 10.1248/bpb.b212013. [DOI] [PubMed] [Google Scholar]
- Backer MV, Gaynutdinov TI, Patel V, Bandyopadhyaya AK, Thirumamagal BT, Tjarks W, Barth RF, Claffey K, Backer JM. Vascular endothelial growth factor selectively targets boronated dendrimers to tumor vasculature. Mol Cancer Ther. 2005;4:1423–1429. doi: 10.1158/1535-7163.MCT-05-0161. [DOI] [PubMed] [Google Scholar]
- Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Ed Engl. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- Banerjee T, Mitra S, Kumar Singh A, Kumar Sharma R, Maitra A. Preparation, characterization and biodistribution of ultrafine chitosan nanoparticles. Int J Pharm. 2002;243:93–105. doi: 10.1016/s0378-5173(02)00267-3. [DOI] [PubMed] [Google Scholar]
- Behr J-P. The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit. Chimia (Aarau) 1997;51:34–36. [Google Scholar]
- Bianco A, Kostarelos K, Prato M. Applications of carbon nanotubes in drug delivery. Curr Opin Chem Biol. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Bickel U, Yoshikawa T, Pardridge WM. Delivery of peptides and proteins through the blood–brain barrier. Adv Drug Deliv Rev. 2001;46:247–279. doi: 10.1016/s0169-409x(00)00139-3. [DOI] [PubMed] [Google Scholar]
- Boas U, Heegaard PM. Dendrimers in drug research. Chem Soc Rev. 2004;33:43–63. doi: 10.1039/b309043b. [DOI] [PubMed] [Google Scholar]
- Bozec A, Gros FX, Penault-Llorca F, Formento P, Cayre A, Dental C, Etienne-Grimaldi MC, Fischel JL, Milano G. Vertical VEGF targeting: A combination of ligand blockade with receptor tyrosine kinase inhibition. Eur J Cancer. 2008;44:1922–1930. doi: 10.1016/j.ejca.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Senger DR, Dvorak HF. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in adenocarcinomas of the gastrointestinal tract. Cancer Res. 1993;53:4727–4735. [PubMed] [Google Scholar]
- Brumlik MJ, Daniel BJ, Waehler R, Curiel DT, Giles FJ, Curiel TJ. Trends in immunoconjugate and ligand-receptor based targeting development for cancer therapy. Expert Opin Drug Deliv. 2008;5:87–103. doi: 10.1517/17425247.5.1.87. [DOI] [PubMed] [Google Scholar]
- Burgstaller P, Jenne A, Blind M. Aptamers and aptazymes: accelerating small molecule drug discovery. Curr Opin Drug Discov Devel. 2002;5:690–700. [PubMed] [Google Scholar]
- Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Cabane E, Zhang X, Langowska K, Palivan C, Meier W. Stimuli-Responsive Polymers and Their Applications in Nanomedicine. Biointerphases. 2012;7:1–27. doi: 10.1007/s13758-011-0009-3. [DOI] [PubMed] [Google Scholar]
- Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33:357–369. doi: 10.1016/s1357-2725(01)00023-1. [DOI] [PubMed] [Google Scholar]
- Carlsson J, Kullberg EB, Capala J, Sjoberg S, Edwards K, Gedda L. Ligand liposomes and boron neutron capture therapy. J Neurooncol. 2003;62:47–59. doi: 10.1007/BF02699933. [DOI] [PubMed] [Google Scholar]
- Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011a;16:354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Chen H, Li L, Cui S, Mahounga D, Zhang J, Gu Y. Folate conjugated CdHgTe quantum dots with high targeting affinity and sensitivity for in vivo early tumor diagnosis. J Fluoresc. 2011b;21:793–801. doi: 10.1007/s10895-010-0772-4. [DOI] [PubMed] [Google Scholar]
- Chen HT, Neerman MF, Parrish AR, Simanek EE. Cytotoxicity, hemolysis, and acute in vivo toxicity of dendrimers based on melamine, candidate vehicles for drug delivery. J Am Chem Soc. 2004;126:10044–10048. doi: 10.1021/ja048548j. [DOI] [PubMed] [Google Scholar]
- Chen Q-Y, Tao G-P, Liu Y-Q, Yang X. Synthesis, characterization, cell imaging and anti-tumor activity of multifunctional nanoparticles. Spectrochim Acta A Mol and Biomol Spectrosc. 2012;96:284–288. doi: 10.1016/j.saa.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome AM, Burda C, Basilion JP. Addressing Brain Tumors with Targeted Gold Nanoparticles: A New Gold Standard for Hydrophobic Drug Delivery? Small. 2011 doi: 10.1002/smll.201100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic Nanoparticles for Drug Delivery in Cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- Chou H-H, Wang K-L, Chen C-A, Wei L-H, Lai C-H, Hsieh C-Y, Yang Y-C, Twu N-F, Chang T-C, Yen M-S. Pegylated liposomal doxorubicin (Lipo-Dox®) for platinum-resistant or refractory epithelial ovarian carcinoma: A Taiwanese gynecologic oncology group study with long-term follow-up. J Gynecol Oncol. 2006;101:423–428. doi: 10.1016/j.ygyno.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Choucair A, Eisenberg A. Control of amphiphilic block copolymer morphologies using solution conditions. Eur Phys J E Soft Matter. 2003;10:37–44. doi: 10.1140/epje/e2003-00002-5. [DOI] [PubMed] [Google Scholar]
- Chung JE, Yokoyama M, Aoyagi T, Sakurai Y, Okano T. Effect of molecular architecture of hydrophobically modified poly(N-isopropylacrylamide) on the formation of thermoresponsive core-shell micellar drug carriers. J Control Release. 1998;53:119–130. doi: 10.1016/s0168-3659(97)00244-7. [DOI] [PubMed] [Google Scholar]
- Chung JE, Yokoyama M, Okano T. Inner core segment design for drug delivery control of thermo-responsive polymeric micelles. J Control Release. 2000;65:93–103. doi: 10.1016/s0168-3659(99)00242-4. [DOI] [PubMed] [Google Scholar]
- Chung JE, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T. Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly(N-isopropylacrylamide) and poly(butylmethacrylate) J Control Release. 1999;62:115–127. doi: 10.1016/s0168-3659(99)00029-2. [DOI] [PubMed] [Google Scholar]
- Corrie PG. Cytotoxic chemotherapy: clinical aspects. Medicine. 2008;36:24–28. [Google Scholar]
- Crile G., Jr. Selective destruction of cancers after exposure to heat. Ann Surg. 1962;156:404–407. doi: 10.1097/00000658-196209000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Emanuele A, Attwood D. Dendrimer-drug interactions. Adv Drug Deliv Rev. 2005;57:2147–2162. doi: 10.1016/j.addr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Dagar S, Krishnadas A, Rubinstein I, Blend MJ, Önyüksel H. VIP grafted sterically stabilized liposomes for targeted imaging of breast cancer: in vivo studies. J Control Release. 2003;91:123–133. doi: 10.1016/s0168-3659(03)00242-6. [DOI] [PubMed] [Google Scholar]
- Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Das M, Mohanty C, Sahoo SK. Ligand-based targeted therapy for cancer tissue. Expert Opin Drug Deliv. 2009;6:285–304. doi: 10.1517/17425240902780166. [DOI] [PubMed] [Google Scholar]
- Das SK, Sarkar S, Dash R, Dent P, Wang X-Y, Sarkar D, Fisher PB. Adv Cancer Res. Vol. 115. Academic Press; 2012. Chapter One - Cancer Terminator Viruses and Approaches for Enhancing Therapeutic Outcomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK, Rahmani M, Wei J, Hedvat M, Dent P, Dmitriev IP, Curiel DT, Grant S, Wu B, Stebbins JL, Pellecchia M, Reed JC, Sarkar D, Fisher PB. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci U S A. 2011;108:8785–8790. doi: 10.1073/pnas.1100769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos RV, Mir IL, Rubinsky B. Tissue ablation with irreversible electroporation. Ann Biomed Eng. 2005;33:223–231. doi: 10.1007/s10439-005-8981-8. [DOI] [PubMed] [Google Scholar]
- De Marre A, Seymour LW, Schacht E. Evaluation of the hydrolytic and enzymatic stability of macromolecular Mitomycin C derivatives. J Control Release. 1994a;31:89–97. [Google Scholar]
- De Marre A, Soyez H, Schacht E. Synthesis of macromolecular Mitomycin C derivatives. J Control Release. 1994b;32:129–137. [Google Scholar]
- Decuzzi P, Ferrari M. The adhesive strength of non-spherical particles mediated by specific interactions. Biomaterials. 2006;27:5307–5314. doi: 10.1016/j.biomaterials.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Dellian M, Helmlinger G, Yuan F, Jain RK. Fluorescence ratio imaging of interstitial pH in solid tumours: effect of glucose on spatial and temporal gradients. Br J Cancer. 1996;74:1206–1215. doi: 10.1038/bjc.1996.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1:12–25. doi: 10.4161/mabs.1.1.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalapally H, Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: part 3. Therapeutic efficacy and safety studies in ovarian cancer xenograft model. Cancer Chemother Pharmacol. 2007;59:477–484. doi: 10.1007/s00280-006-0287-5. [DOI] [PubMed] [Google Scholar]
- Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf I, Kruijtzer JAW, Frielink C, Corstens FHM, Oyen WJG, Liskamp RMJ, Boerman OC. αvβ3 Integrin-targeting of intraperitoneally growing tumors with a radiolabeled RGD peptide. Int J Cancer. 2007;120:605–610. doi: 10.1002/ijc.22297. [DOI] [PubMed] [Google Scholar]
- Dong Y, Feng S-S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25:2843–2849. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- Drolet DW, Nelson J, Tucker CE, Zack PM, Nixon K, Bolin R, Judkins MB, Farmer JA, Wolf JL, Gill SC, Bendele RA. Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm Res. 2000;17:1503–1510. doi: 10.1023/a:1007657109012. [DOI] [PubMed] [Google Scholar]
- Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- Egusquiaguirre S, Igartua M, Hernández R, Pedraz J. Nanoparticle delivery systems for cancer therapy: advances in clinical and preclinical research. Clin Transl Oncol. 2012;14:83–93. doi: 10.1007/s12094-012-0766-6. [DOI] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Etrych T, Jelinkova M, Rihova B, Ulbrich K. New HPMA copolymers containing doxorubicin bound via pH-sensitive linkage: synthesis and preliminary in vitro and in vivo biological properties. J Control Release. 2001;73:89–102. doi: 10.1016/s0168-3659(01)00281-4. [DOI] [PubMed] [Google Scholar]
- Fajardo LF, Prionas SD. Endothelial cells and hyperthermia. Int J Hyperthermia. 1994;10:347–353. doi: 10.3109/02656739409010278. [DOI] [PubMed] [Google Scholar]
- Fan Z, Lu Y, Wu X, Mendelsohn J. Antibody-induced epidermal growth factor receptor dimerization mediates inhibition of autocrine proliferation of A431 squamous carcinoma cells. J Biol Chem. 1994;269:27595–27602. [PubMed] [Google Scholar]
- Fang RH, Zhang L. Dispersion-Based Methods for the Engineering and Manufacture of Polymeric Nanoparticles for Drug Delivery Applications. J Nanoeng Nanomanuf. 2011;1:106–112. [Google Scholar]
- Fant K, Esbjorner EK, Jenkins A, Grossel MC, Lincoln P, Norden B. Effects of PEGylation and Acetylation of PAMAM Dendrimers on DNA Binding, Cytotoxicity and in Vitro Transfection Efficiency. Mol Pharm. 2010 doi: 10.1021/mp1001312. [DOI] [PubMed] [Google Scholar]
- Farokhzad OC, Jon S, Khademhosseini A, Tran T-NT, LaVan DA, Langer R. Nanoparticle-Aptamer Bioconjugates. Cancer Res. 2004;64:7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- Farokhzad OC, Karp JM, Langer R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv. 2006;3:311–324. doi: 10.1517/17425247.3.3.311. [DOI] [PubMed] [Google Scholar]
- Farokhzad OC, Khademhosseini A, Jon S, Hermmann A, Cheng J, Chin C, Kiselyuk A, Teply B, Eng G, Langer R. Microfluidic system for studying the interaction of nanoparticles and microparticles with cells. Anal Chem. 2005;77:5453–5459. doi: 10.1021/ac050312q. [DOI] [PubMed] [Google Scholar]
- Faulk WP, Taylor CG, Yeh CJ, McIntyre JA. Preliminary clinical study of transferrin-adriamycin conjugate for drug delivery to acute leukemia patients. Mol Biother. 1990;2:57–60. [PubMed] [Google Scholar]
- Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. Soluble single-walled carbon nanotubes as longboat delivery systems for platinum(IV) anticancer drug design. J Am Chem Soc. 2007;129:8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgner JH, Kumar R, Sridhar CN, Wheeler CJ, Tsai YJ, Border R, Ramsey P, Martin M, Felgner PL. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J Biol Chem. 1994;269:2550–2561. [PubMed] [Google Scholar]
- Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69(Suppl 3):11–16. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- Frechet JM. Dendrimers and supramolecular chemistry. Proc Natl Acad Sci U S A. 2002;99:4782–4787. doi: 10.1073/pnas.082013899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresta M, Puglisi G, Giammona G, Cavallaro G, Micali N, Furneri PM. Pefloxacine mesilate- and ofloxacin-loaded polyethylcyanoacrylate nanoparticles: characterization of the colloidal drug carrier formulation. J Pharm Sci. 1995;84:895–902. doi: 10.1002/jps.2600840721. [DOI] [PubMed] [Google Scholar]
- Gaber MH, Wu NZ, Hong K, Huang SK, Dewhirst MW, Papahadjopoulos D. Thermosensitive liposomes: extravasation and release of contents in tumor microvascular networks. Int J Radiat Oncol Biol Phys. 1996;36:1177–1187. doi: 10.1016/s0360-3016(96)00389-6. [DOI] [PubMed] [Google Scholar]
- Gao H, Qian J, Cao S, Yang Z, Pang Z, Pan S, Fan L, Xi Z, Jiang X, Zhang Q. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials. 2012;33:5115–5123. doi: 10.1016/j.biomaterials.2012.03.058. [DOI] [PubMed] [Google Scholar]
- Gao W, Chan JM, Farokhzad OC. pH-Responsive nanoparticles for drug delivery. Mol Pharm. 2010a;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Xiao Z, Radovic-Moreno A, Shi J, Langer R, Farokhzad OC. Progress in siRNA delivery using multifunctional nanoparticles. Methods Mol Biol. 2010b;629:53–67. doi: 10.1007/978-1-60761-657-3_4. [DOI] [PubMed] [Google Scholar]
- Gatter KC, Brown G, Trowbridge IS, Woolston RE, Mason DY. Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol. 1983;36:539–545. doi: 10.1136/jcp.36.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS Journal. 2010;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- Gillies ER, Frechet JM. pH-Responsive copolymer assemblies for controlled release of doxorubicin. Bioconjug Chem. 2005;16:361–368. doi: 10.1021/bc049851c. [DOI] [PubMed] [Google Scholar]
- Gong J, Chen M, Zheng Y, Wang S, Wang Y. Polymeric micelles drug delivery system in oncology. J Control Release. 2012;159:312–323. doi: 10.1016/j.jconrel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Gong W, Wang Z, Liu N, Lin W, Wang X, Xu D, Liu H, Zeng C, Xie X, Mei X, Lü Wanliang. Improving Efficiency of Adriamycin Crossing Blood Brain Barrier by Combination of Thermosensitive Liposomes and Hyperthermia. Biol Pharm Bull. 2011;34:1058–1064. doi: 10.1248/bpb.34.1058. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Graff A, Tropel D, Raman SK, Machaidze G, Aebi U, Burkhard P. Peptidic nanoparticles: for cancer diagnosis and therapy. NanoBioTechnology. 2005;1:293–294. [Google Scholar]
- Graham MM, Menda Y. Radiopeptide imaging and therapy in the United States. J Nucl Med. 2011;52(Suppl 2):56S–63S. doi: 10.2967/jnumed.110.085746. [DOI] [PubMed] [Google Scholar]
- Greco A, Di Benedetto A, Howard CM, Kelly S, Nande R, Dementieva Y, Miranda M, Brunetti A, Salvatore M, Claudio L, Sarkar D, Dent P, Curiel DT, Fisher PB, Claudio PP. Eradication of therapy-resistant human prostate tumors using an ultrasound-guided site-specific cancer terminator virus delivery approach. Mol Ther. 2010;18:295–306. doi: 10.1038/mt.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Muller RH. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Nystrom B, Sable SB, Khuller GK. Nanobead-based interventions for the treatment and prevention of tuberculosis. Nat Rev Microbiol. 2010;8:827–834. doi: 10.1038/nrmicro2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, Langer RS, Farokhzad OC. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2:14–21. [Google Scholar]
- Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci. 2002;6:319–327. [Google Scholar]
- Head JF, Wang F, Elliott RL. Antineoplastic drugs that interfere with iron metabolism in cancer cells. Adv Enzyme Regul. 1997;37:147–169. doi: 10.1016/s0065-2571(96)00010-6. [DOI] [PubMed] [Google Scholar]
- Hicke BJ, Stephens AW. Escort aptamers: a delivery service for diagnosis and therapy. J Clin Invest. 2000;106:923–928. doi: 10.1172/JCI11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: From therapeutics to diagnostics. J Pharm Sci. 2005;94:2135–2146. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölig P, Bach M, Völkel T, Nahde T, Hoffmann S, Müller R, Kontermann RE. Novel RGD lipopeptides for the targeting of liposomes to integrin-expressing endothelial and melanoma cells. Protein Eng Des Sel. 2004;17:433–441. doi: 10.1093/protein/gzh055. [DOI] [PubMed] [Google Scholar]
- Honer M, Mu L, Stellfeld T, Graham K, Martic M, Fischer CR, Lehmann L, Schubiger PA, Ametamey SM, Dinkelborg L, Srinivasan A, Borkowski S. 18F-Labeled Bombesin Analog for Specific and Effective Targeting of Prostate Tumors Expressing Gastrin-Releasing Peptide Receptors. J Nucl Med. 2011;52:270–278. doi: 10.2967/jnumed.110.081620. [DOI] [PubMed] [Google Scholar]
- Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA. Tumor Regression by Targeted Gene Delivery to the Neovasculature. Science. 2002;296:2404–2407. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Sami M, Kato Y, Hayakawa E. Alteration in the temperature-dependent content release property of thermosensitive liposomes in plasma. Chem Pharm Bull (Tokyo) 2003;51:1227–1232. doi: 10.1248/cpb.51.1227. [DOI] [PubMed] [Google Scholar]
- Hoste K, De Winne K, Schacht E. Polymeric prodrugs. Int J Pharm. 2004;277:119–131. doi: 10.1016/j.ijpharm.2003.07.016. [DOI] [PubMed] [Google Scholar]
- Hou J, Zhang Q, Li X, Tang Y, Cao MR, Bai F, Shi Q, Yang CH, Kong DL, Bai G. Synthesis of novel folate conjugated fluorescent nanoparticles for tumor imaging. J Biomed Mater Res A. 2011;99:684–689. doi: 10.1002/jbm.a.33187. [DOI] [PubMed] [Google Scholar]
- Howlader N, Krapcho M, Neyman N, et al. SEER Cancer Statistics Review, 1975-2008. National Cancer Institute; Bethesda, MD: 2011. [Google Scholar]
- Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–8992. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10:836–841. doi: 10.2174/138920009790274540. [DOI] [PubMed] [Google Scholar]
- Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, Tan W. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009;10:862–868. doi: 10.1002/cbic.200800805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes GA. Nanostructure-mediated drug delivery. Nanomedicine. 2005;1:22–30. doi: 10.1016/j.nano.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Husseini GA, Pitt WG. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60:1137–1152. doi: 10.1016/j.addr.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iinuma H, Maruyama K, Okinaga K, Sasaki K, Sekine T, Ishida O, Ogiwara N, Johkura K, Yonemura Y. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int J Cancer. 2002;99:130–137. doi: 10.1002/ijc.10242. [DOI] [PubMed] [Google Scholar]
- Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Jain RK, Safabakhsh N, Sckell A, Chen Y, Jiang P, Benjamin L, Yuan F, Keshet E. Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: role of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 1998;95:10820–10825. doi: 10.1073/pnas.95.18.10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectable thermosensitive hydrogel of PEG-PLGA-PEG triblock copolymers. J Control Release. 2000;63:155–163. doi: 10.1016/s0168-3659(99)00194-7. [DOI] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) 2011;6:715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82:189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- Kaczorowska MA, Cooper HJ. Electron capture dissociation, electron detachment dissociation, and collision-induced dissociation of polyamidoamine (PAMAM) dendrimer ions with amino, amidoethanol, and sodium carboxylate surface groups. J Am Soc Mass Spectrom. 2008;19:1312–1319. doi: 10.1016/j.jasms.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar SS, Jin H, Hong B, Eaton JW, Kang KA, Kang KA, Harrison DK, Bruley DF. LHRH Receptor Targeted Therapy for Breast Cancer Oxygen Transport to Tissue XXIX. Vol. 614. Springer; US: 2008. pp. 285–296. [DOI] [PubMed] [Google Scholar]
- Kam NW, Dai H. Carbon nanotubes as intracellular protein transporters: generality and biological functionality. J Am Chem Soc. 2005;127:6021–6026. doi: 10.1021/ja050062v. [DOI] [PubMed] [Google Scholar]
- Kamada H, Tsutsumi Y, Yoshioka Y, Yamamoto Y, Kodaira H, Tsunoda S, Okamoto T, Mukai Y, Shibata H, Nakagawa S, Mayumi T. Design of a pH-sensitive polymeric carrier for drug release and its application in cancer therapy. Clin Cancer Res. 2004;10:2545–2550. doi: 10.1158/1078-0432.ccr-03-0544. [DOI] [PubMed] [Google Scholar]
- Kaminskas LM, Boyd BJ, Porter CJ. Dendrimer pharmacokinetics: the effect of size, structure and surface characteristics on ADME properties. Nanomedicine (Lond) 2011;6:1063–1084. doi: 10.2217/nnm.11.67. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Nakamura S, Sakai K, Kikuchi A, Aoyagi T, Sakurai Y, Okano T. Synthesis and swelling-deswelling kinetics of poly(N-isopropylacrylamide) hydrogels grafted with LCST modulated polymers. J Biomater Sci Polym Ed. 1999;10:1079–1091. doi: 10.1163/156856299x00757. [DOI] [PubMed] [Google Scholar]
- Kaptain S, Tan LK, Chen B. Her-2/neu and breast cancer. Diagn Mol Pathol. 2001;10:139–152. doi: 10.1097/00019606-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010;6:714–729. doi: 10.1016/j.nano.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Khajavinia A, Varshosaz J, Dehkordi AJ. Targeting etoposide to acute myelogenous leukaemia cells using nanostructured lipid carriers coated with transferrin. Nanotechnology. 2012;23:405101. doi: 10.1088/0957-4484/23/40/405101. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim J-H, Jeon O, Kwon IC, Park K. Engineered polymers for advanced drug delivery. Eur J Pharm Biopharm. 2009;71:420–430. doi: 10.1016/j.ejpb.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Shi Y, Kim JY, Park K, Cheng J-X. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- Kitchens DL, Snyder EY, Gottlieb DI. FGF and EGF are mitogens for immortalized neural progenitors. J Neurobiol. 1994;25:797–807. doi: 10.1002/neu.480250705. [DOI] [PubMed] [Google Scholar]
- Klein S, Levitzki A, George F.V.W.a.G.K. Targeted Cancer Therapy: Promise and Reality. In: David T.C.a.P.B.F., editor. Advances in Cancer Research. Vol. 97. Academic Press; 2007. pp. 295–319. [DOI] [PubMed] [Google Scholar]
- Kobayashi T. Cancer hyperthermia using magnetic nanoparticles. Biotechnol J. 2011;6:1342–1347. doi: 10.1002/biot.201100045. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int J Pharm. 2007;329:94–102. doi: 10.1016/j.ijpharm.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Surface acetylation of polyamidoamine (PAMAM) dendrimers decreases cytotoxicity while maintaining membrane permeability. Bioconjug Chem. 2007;18:2054–2060. doi: 10.1021/bc0603889. [DOI] [PubMed] [Google Scholar]
- Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res. 2001;61:3027–3032. [PubMed] [Google Scholar]
- Koo Y-EL, Reddy GR, Bhojani M, Schneider R, Philbert MA, Rehemtulla A, Ross BD, Kopelman R. Brain cancer diagnosis and therapy with nanoplatforms. Adv Drug Deliv Rev. 2006;58:1556–1577. doi: 10.1016/j.addr.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Kos J, Obermajer N, Doljak B, Kocbek P, Kristl J. Inactivation of harmful tumour-associated proteolysis by nanoparticulate system. Int J Pharm. 2009;381:106–112. doi: 10.1016/j.ijpharm.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Kremer C, Breier G, Risau W, Plate KH. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997;57:3852–3859. [PubMed] [Google Scholar]
- Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47:65–81. doi: 10.1016/s0169-409x(00)00122-8. [DOI] [PubMed] [Google Scholar]
- Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR. Nanoparticle Targeting of Anticancer Drug Improves Therapeutic Response in Animal Model of Human Epithelial Cancer. Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- Kullberg EB, Nestor M, Gedda L. Tumor-cell targeted epiderimal growth factor liposomes loaded with boronated acridine: uptake and processing. Pharm Res. 2003;20:229–236. doi: 10.1023/a:1022223204460. [DOI] [PubMed] [Google Scholar]
- Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2009;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Kwon G, Suwa S, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly (ethylene oxide-aspartate) block copolymer-adriamycin conjugates. J Control Release. 1994;29:17–23. [Google Scholar]
- labieniec M, Ulicna O, Vancova O, Kucharska J, Gabryelak T, Watala C. Effect of poly(amido)amine (PAMAM) G4 dendrimer on heart and liver mitochondria in an animal model of diabetes. Cell Biol Int. 2009;34:89–97. doi: 10.1042/CBI20090010. [DOI] [PubMed] [Google Scholar]
- Lacroix M. Persistent use of “false” cell lines. Int J Cancer. 2008;122:1–4. doi: 10.1002/ijc.23233. [DOI] [PubMed] [Google Scholar]
- Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med. 1997;3:1362–1368. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- Laskin JJ, Sandler AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat Rev. 2004;30:1–17. doi: 10.1016/j.ctrv.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- Lee P, Zhang R, Li V, Liu X, Sun RW, Che CM, Wong KK. Enhancement of anticancer efficacy using modified lipophilic nanoparticle drug encapsulation. Int J Nanomedicine. 2012;7:731–737. doi: 10.2147/IJN.S28783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RJ, Low PS. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J Biol Chem. 1994;269:3198–3204. [PubMed] [Google Scholar]
- Lee RJ, Wang S, Low PS. Measurement of endosome pH following folate receptor-mediated endocytosis. Biochim Biophys Acta. 1996;1312:237–242. doi: 10.1016/0167-4889(96)00041-9. [DOI] [PubMed] [Google Scholar]
- Leemans CR, Braakhuis BJM, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Li X, Zhou H, Yang L, Du G, Pai-Panandiker AS, Huang X, Yan B. Enhancement of cell recognition in vitro by dual-ligand cancer targeting gold nanoparticles. Biomaterials. 2011;32:2540–2545. doi: 10.1016/j.biomaterials.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Chen B. A review on biomedical applications of single-walled carbon nanotubes. Curr Med Chem. 2010;17:10–24. doi: 10.2174/092986710789957742. [DOI] [PubMed] [Google Scholar]
- Liechty WB, Peppas NA. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur J Pharm Biopharm. 2012;80:241–246. doi: 10.1016/j.ejpb.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- Liu D-Z, Hsieh J-H, Fan X-C, Yang J-D, Chung T-W. Synthesis, characterization and drug delivery behaviors of new PCP polymeric micelles. Carbohydr Polym. 2007a;68:544–554. [Google Scholar]
- Liu D, Mori A, Huang L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta. 1992;1104:95–101. doi: 10.1016/0005-2736(92)90136-a. [DOI] [PubMed] [Google Scholar]
- Liu Y, Miyoshi H, Nakamura M. Nanomedicine for drug delivery and imaging: a promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int J Cancer. 2007b;120:2527–2537. doi: 10.1002/ijc.22709. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Drug Delivery with Carbon Nanotubes for In vivo Cancer Treatment. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sun X, Nakayama-Ratchford N, Dai H. Supramolecular Chemistry on Water-Soluble Carbon Nanotubes for Drug Loading and Delivery. ACS Nano. 2007c;1:50–56. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- Loh XJ, Yee BJH, Chia FS. Sustained delivery of paclitaxel using thermogelling poly(PEG/PPG/PCL urethane)s for enhanced toxicity against cancer cells. J Biomed Mater Res A. 2012 doi: 10.1002/jbm.a.34198. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- Lorusso V, Manzione L, Silvestris N. Role of liposomal anthracyclines in breast cancer. Annals of Oncology. 2007;18:vi70–vi73. doi: 10.1093/annonc/mdm229. [DOI] [PubMed] [Google Scholar]
- Lovett KM, Liang BA, Mackey TK. Risks of online direct-to-consumer tumor markers for cancer screening. J Clin Oncol. 2012;30:1411–1414. doi: 10.1200/JCO.2011.37.8984. [DOI] [PubMed] [Google Scholar]
- Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56:1055–1058. doi: 10.1016/j.addr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256–262. doi: 10.1016/j.cbpa.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Lu J, Jackson JK, Gleave ME, Burt HM. The preparation and characterization of anti-VEGFR2 conjugated, paclitaxel-loaded PLLA or PLGA microspheres for the systemic targeting of human prostate tumors. Cancer Chemother Pharmacol. 2008;61:997–1005. doi: 10.1007/s00280-007-0557-x. [DOI] [PubMed] [Google Scholar]
- Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54:675–693. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77:400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Malik AK, Gerber H-P. Targeting VEGF ligands and receptors in cancer. TARGETS. 2003;2:48–57. [Google Scholar]
- Malik N, Wiwattanapatapee R, Klopsch R, Lorenz K, Frey H, Weener JW, Meijer EW, Paulus W, Duncan R. Dendrimers:: Relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release. 2000;65:133–148. doi: 10.1016/s0168-3659(99)00246-1. [DOI] [PubMed] [Google Scholar]
- Masson O, Prebois C, Derocq D, Meulle A, Dray C, Daviaud D, Quilliot D, Valet P, Muller C, Liaudet-Coopman E. Cathepsin-D, a Key Protease in Breast Cancer, Is Up-Regulated in Obese Mouse and Human Adipose Tissue, and Controls Adipogenesis. PLoS ONE. 2011;6:e16452. doi: 10.1371/journal.pone.0016452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- Medina SH, El-Sayed MEH. Dendrimers as Carriers for Delivery of Chemotherapeutic Agents. Chem Rev. 2009;109:3141–3157. doi: 10.1021/cr900174j. [DOI] [PubMed] [Google Scholar]
- Meng S, Su B, Li W, Ding Y, Tang L, Zhou W, Song Y, Li H, Zhou C. Enhanced antitumor effect of novel dual-targeted paclitaxel liposomes. Nanotechnology. 2010;21:415103. doi: 10.1088/0957-4484/21/41/415103. [DOI] [PubMed] [Google Scholar]
- Merdan T, Kopecek J, Kissel T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv Drug Deliv Rev. 2002;54:715–758. doi: 10.1016/s0169-409x(02)00046-7. [DOI] [PubMed] [Google Scholar]
- Milhem OM, Myles C, McKeown NB, Attwood D, D’Emanuele A. Polyamidoamine Starburst dendrimers as solubility enhancers. Int J Pharm. 2000;197:239–241. doi: 10.1016/s0378-5173(99)00463-9. [DOI] [PubMed] [Google Scholar]
- Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- Mohanty C, Das M, Kanwar JR, Sahoo SK. Receptor Mediated Tumor Targeting: An Emerging Approach for Cancer Therapy. Curr Drug Deliv. 2011;8:45–58. doi: 10.2174/156720111793663606. [DOI] [PubMed] [Google Scholar]
- Momekova D, Rangelov S, Yanev S, Nikolova E, Konstantinov S, Romberg B, Storm G, Lambov N. Long-circulating, pH-sensitive liposomes sterically stabilized by copolymers bearing short blocks of lipid-mimetic units. Eur J Pharm Sci. 2007;32:308–317. doi: 10.1016/j.ejps.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalent Effects of RGD Peptides Obtained by Nanoparticle Display. J Med Chem. 2006;49:6087–6093. doi: 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, Wrasidlo W, Cheresh DA. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A. 2008;105:9343–9348. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Abu Lila AS, Matsunaga M, Doi Y, Ishida T, Kiwada H. A double-modulation strategy in cancer treatment with a chemotherapeutic agent and siRNA. Mol Ther. 2011;19:2040–2047. doi: 10.1038/mt.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Okano T, Miyazaki T, Kohori F, Sakai K, Yokoyama M. Molecular design of biodegradable polymeric micelles for temperature-responsive drug release. J Control Release. 2006;115:46–56. doi: 10.1016/j.jconrel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Napier MP, Sharma SK, Springer CJ, Bagshawe KD, Green AJ, Martin J, Stribbling SM, Cushen N, O’Malley D, Begent RH. Antibody-directed enzyme prodrug therapy: efficacy and mechanism of action in colorectal carcinoma. Clin Cancer Res. 2000;6:765–772. [PubMed] [Google Scholar]
- Nigavekar SS, Sung LY, Llanes M, El-Jawahri A, Lawrence TS, Becker CW, Balogh L, Khan MK. 3H dendrimer nanoparticle organ/tumor distribution. Pharm Res. 2004;21:476–483. doi: 10.1023/B:PHAM.0000019302.26097.cc. [DOI] [PubMed] [Google Scholar]
- Nimjee SM, Rusconi CP, Harrington RA, Sullenger BA. The potential of aptamers as anticoagulants. Trends Cardiovasc Med. 2005;15:41–45. doi: 10.1016/j.tcm.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Norman J, Badie-Dezfooly B, Nord EP, Kurtz I, Schlosser J, Chaudhari A, Fine LG. EGF-induced mitogenesis in proximal tubular cells: potentiation by angiotensin II. Am J Physiol Renal Physiol. 1987;253:F299–F309. doi: 10.1152/ajprenal.1987.253.2.F299. [DOI] [PubMed] [Google Scholar]
- Oerlemans C, Bult W, Bos M, Storm G, Nijsen J, Hennink W. Polymeric Micelles in Anticancer Therapy: Targeting, Imaging and Triggered Release. Pharm Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson B, Rehfeld JF, Axelson J. CCK stimulates growth of both the pancreas and the liver. Int J Surg Investig. 1999;1:47–54. [PubMed] [Google Scholar]
- Onyuksel H, Mohanty PS, Rubinstein I. VIP-grafted sterically stabilized phospholipid nanomicellar 17-allylamino-17-demethoxy geldanamycin: a novel targeted nanomedicine for breast cancer. Int J Pharm. 2009;365:157–161. doi: 10.1016/j.ijpharm.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DE, III, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Pantarotto D, Briand JP, Prato M, Bianco A. Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem Commun (Camb) 2004:16–17. doi: 10.1039/b311254c. [DOI] [PubMed] [Google Scholar]
- Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Drug and Gene Delivery to the Brain: The Vascular Route. Neuron. 2002;36:555–558. doi: 10.1016/s0896-6273(02)01054-1. [DOI] [PubMed] [Google Scholar]
- Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012 doi: 10.1016/j.drudis.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Park EK, Lee SB, Lee YM. Preparation and characterization of methoxy poly(ethylene glycol)/poly(Îμ-caprolactone) amphiphilic block copolymeric nanospheres for tumor-specific folate-mediated targeting of anticancer drugs. Biomaterials. 2005;26:1053–1061. doi: 10.1016/j.biomaterials.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8:1172–1181. [PubMed] [Google Scholar]
- Park JW, Kirpotin DB, Hong K, Shalaby R, Shao Y, Nielsen UB, Marks JD, Papahadjopoulos D, Benz CC. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74:95–113. doi: 10.1016/s0168-3659(01)00315-7. [DOI] [PubMed] [Google Scholar]
- Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8:147–166. doi: 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Pastorino F, Brignole C, Di Paolo D, Nico B, Pezzolo A, Marimpietri D, Pagnan G, Piccardi F, Cilli M, Longhi R, Ribatti D, Corti A, Allen TM, Ponzoni M. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006;66:10073–10082. doi: 10.1158/0008-5472.CAN-06-2117. [DOI] [PubMed] [Google Scholar]
- Patri AK, Majoros IJ, Baker JR. Dendritic polymer macromolecular carriers for drug delivery. Curr Opin Chem Biol. 2002;6:466–471. doi: 10.1016/s1367-5931(02)00347-2. [DOI] [PubMed] [Google Scholar]
- Peng C-L, Tsai H-M, Yang S-J, Luo T-Y, Lin C-F, Lin W-J, Ming-Jium S. Development of thermosensitive poly(n -isopropylacrylamide- co -((2-dimethylamino) ethyl methacrylate))-based nanoparticles for controlled drug release. Nanotechnology. 2011;22:265608. doi: 10.1088/0957-4484/22/26/265608. [DOI] [PubMed] [Google Scholar]
- Peracchia MT, Fattala E, Desmaëleb D, Besnarda M, Noëlc JP, Gomisc JM, Appela M, d’Angelob J, Couvreura P. Stealth® PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J Control Release. 1999;60:121–128. doi: 10.1016/s0168-3659(99)00063-2. [DOI] [PubMed] [Google Scholar]
- Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Mediating Tumor Targeting Efficiency of Nanoparticles Through Design. Nano Letters. 2009;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- Pestourie C, Tavitian B, Duconge F. Aptamers against extracellular targets for in vivo applications. Biochimie. 2005;87:921–930. doi: 10.1016/j.biochi.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Podgorski I, Sloane BF. Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp. 2003:263–276. doi: 10.1042/bss0700263. [DOI] [PubMed] [Google Scholar]
- Prakash S, Malhotra M, Shao W, Tomaro-Duchesneau C, Abbasi S. Polymeric nanohybrids and functionalized carbon nanotubes as drug delivery carriers for cancer therapy. Adv Drug Deliv Rev. 2011;63:1340–1351. doi: 10.1016/j.addr.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Pulkkinen M, Pikkarainen J, Wirth T, Tarvainen T, Haapa-aho V, Korhonen H, Seppala J, Jarvinen K. Three-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin-biotin technology: Formulation development and in vitro anticancer activity. Eur J Pharm Biopharm. 2008;70:66–74. doi: 10.1016/j.ejpb.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Purushotham S, Chang P, Rumpel H, Kee I, Ng R, Chow P, Tan C, Ramanujan R. Thermoresponsive core-shell magnetic nanoparticles for combined modalities of cancer therapy. Nanotechnology. 2009;20:305101. doi: 10.1088/0957-4484/20/30/305101. [DOI] [PubMed] [Google Scholar]
- Radinsky R, Risin S, Fan D, Dong Z, Bielenberg D, Bucana CD, Fidler IJ. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin Cancer Res. 1995;1:19–31. [PubMed] [Google Scholar]
- Rainov NG, Soling A. Technology evaluation: TransMID, KS Biomedix/Nycomed/Sosei/PharmaEngine. Curr Opin Mol Ther. 2005;7:483–492. [PubMed] [Google Scholar]
- Reubi JC. Peptide Receptors as Molecular Targets for Cancer Diagnosis and Therapy. Endocr Rev. 2003;24:389–427. doi: 10.1210/er.2002-0007. [DOI] [PubMed] [Google Scholar]
- Roberts JC, Bhalgat MK, Zera RT. Preliminary biological evaluation of polyamidoamine (PAMAM) Starburst dendrimers. J Biomed Mater Res. 1996;30:53–65. doi: 10.1002/(SICI)1097-4636(199601)30:1<53::AID-JBM8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Roda E, Coccini T, Acerbi D, Barni S, Vaccarone R, Manzo L. Comparative pulmonary toxicity assessment of pristine and functionalized multi-walled carbon nanotubes intratracheally instilled in rats: morphohistochemical evaluations. Histol Histopathol. 2011;26:357–367. doi: 10.14670/HH-26.357. [DOI] [PubMed] [Google Scholar]
- Rubin P, Casarett G. Microcirculation of tumors. I. Anatomy, function, and necrosis. Clin Radiol. 1966;17:220–229. doi: 10.1016/s0009-9260(66)80027-2. [DOI] [PubMed] [Google Scholar]
- Sahoo SK, Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discov Today. 2003;8:1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–383. doi: 10.1021/mp050032z. [DOI] [PubMed] [Google Scholar]
- Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112:335–340. doi: 10.1002/ijc.20405. [DOI] [PubMed] [Google Scholar]
- Saito Y, Yasunaga M, Kuroda J.-i., Koga Y, Matsumura Y. Antitumour activity of NK012, SN-38-incorporating polymeric micelles, in hypovascular orthotopic pancreatic tumour. Eur J Cancer. 2007;46:650–658. doi: 10.1016/j.ejca.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Sakai K, Mori S, Kawamoto T, Taniguchi S, Kobori O, Morioka Y, Kuroki T, Kano K. Expression of epidermal growth factor receptors on normal human gastric epithelia and gastric carcinomas. J Natl Cancer Inst. 1986;77:1047–1052. [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Sampathkumar S-G, Yarema KJ. Nanotechnologies for the Life Sciences. Vol. 7. WILEY_VCH Verlag GmbH & Co. KGaA; Weinheim: 2007. Nanomaterials for Cancer Diagnosis. [Google Scholar]
- Sandoval MA, Sloat BR, Lansakara PD, Kumar A, Rodriguez BL, Kiguchi K, Digiovanni J, Cui Z. EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity. J Control Release. 2012;157:287–296. doi: 10.1016/j.jconrel.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42:439–462. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Saul JM, Annapragada AV, Bellamkonda RV. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J Control Release. 2006;114:277–287. doi: 10.1016/j.jconrel.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Savić R, Eisenberg A, Maysinger D. Block copolymer micelles as delivery vehicles of hydrophobic drugs: Micelle–cell interactions. J Drug Target. 2006;14:343–355. doi: 10.1080/10611860600874538. [DOI] [PubMed] [Google Scholar]
- Schneider G.g.F., Subr V, Ulbrich K, Decher G. Multifunctional Cytotoxic Stealth Nanoparticles. A Model Approach with Potential for Cancer Therapy. Nano Lett. 2009;9:636–642. doi: 10.1021/nl802990w. [DOI] [PubMed] [Google Scholar]
- Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100:2426–2430. doi: 10.1111/j.1349-7006.2009.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service RF. Nanotechnology. Nanoparticle Trojan horses gallop from the lab into the clinic. Science. 2010;330:314–315. doi: 10.1126/science.330.6002.314. [DOI] [PubMed] [Google Scholar]
- Sharma A, Sharma US. Liposomes in drug delivery: Progress and limitations. Int J Pharm. 1997;154:123–140. [Google Scholar]
- Sharma SK, Bagshawe KD, Reddy LH, Couvreur P. Antibody-Directed Enzyme Prodrug Therapy (ADEPT) for Cancer Macromolecular Anticancer Therapeutics. Springer New York; 2010. pp. 393–406. [Google Scholar]
- Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-Modified Poly(Î2-amino ester) Nanoparticles as a pH-Sensitive System for Tumor-Targeted Delivery of Hydrophobic Drugs. 1. In Vitro Evaluations. Mol Pharm. 2005;2:357–366. doi: 10.1021/mp0500420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard DM, Ferris MC, Olivera GH, Mackie TR. Optimizing the Delivery of Radiation Therapy to Cancer Patients. SIAM Review. 1999;41:721–744. [Google Scholar]
- Shi Kam NW, Jessop TC, Wender PA, Dai H. Nanotube molecular transporters: internalization of carbon nanotube-protein conjugates into Mammalian cells. J Am Chem Soc. 2004;126:6850–6851. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]
- Shmeeda H, Mak L, Tzemach D, Astrahan P, Tarshish M, Gabizon A. Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol Cancer Ther. 2006;5:818–824. doi: 10.1158/1535-7163.MCT-05-0543. [DOI] [PubMed] [Google Scholar]
- Shuai X, Merdan T, Schaper AK, Xi F, Kissel T. Core-cross-linked polymeric micelles as paclitaxel carriers. Bioconjug Chem. 2004;15:441–448. doi: 10.1021/bc034113u. [DOI] [PubMed] [Google Scholar]
- Shubik P. Vascularization of tumors: a review. J Cancer Res Clin Oncol. 1982;103:211–226. doi: 10.1007/BF00409698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Simoes S, Moreira JN, Fonseca C, Duzgunes N, de Lima MC. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56:947–965. doi: 10.1016/j.addr.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Singh M. Transferrin As A targeting ligand for liposomes and anticancer drugs. Curr Pharm Des. 1999;5:443–451. [PubMed] [Google Scholar]
- Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5:1909–1917. doi: 10.1158/1535-7163.MCT-06-0141. [DOI] [PubMed] [Google Scholar]
- Sirsi S, Borden M. Microbubble Compositions, Properties and Biomedical Applications. Bubble Sci Eng Technol. 2009;1:3–17. doi: 10.1179/175889709X446507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C, Tan W. Aptamer-conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal Chem. 2007;79:3075–3082. doi: 10.1021/ac062151b. [DOI] [PubMed] [Google Scholar]
- Soga O, van Nostrum CF, Fens M, Rijcken CJ, Schiffelers RM, Storm G, Hennink WE. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J Control Release. 2005;103:341–353. doi: 10.1016/j.jconrel.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Sonavane G, Tomoda K, Makino K. Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B Biointerfaces. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Sosabowski JK, Matzow T, Foster JM, Finucane C, Ellison D, Watson SA, Mather SJ. Targeting of CCK-2 Receptor-Expressing Tumors Using a Radiolabeled Divalent Gastrin Peptide. J Nucl Med. 2009;50:2082–2089. doi: 10.2967/jnumed.109.064808. [DOI] [PubMed] [Google Scholar]
- Springer CJ, Poon GK, Sharma SK, Bagshawe KD. Identification of prodrug, active drug, and metabolites in an ADEPT clinical study. Cell Biophys. 1993;22:9–26. doi: 10.1007/BF03033864. [DOI] [PubMed] [Google Scholar]
- Stella B, Arpicco S, Peracchia MT, Desmaele D, Hoebeke J, Renoir M, D’Angelo J, Cattel L, Couvreur P. Design of folic acid-conjugated nanoparticles for drug targeting. J Pharm Sci. 2000;89:1452–1464. doi: 10.1002/1520-6017(200011)89:11<1452::aid-jps8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv Drug Deliv Rev. 1995;17:31–48. [Google Scholar]
- Straubinger RM, Hong K, Friend DS, Papahadjopoulos D. Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles. Cell. 1983;32:1069–1079. doi: 10.1016/0092-8674(83)90291-x. [DOI] [PubMed] [Google Scholar]
- Stubbs M, McSheehy PM, Griffiths JR. Causes and consequences of acidic pH in tumors: a magnetic resonance study. Adv Enzyme Regul. 1999;39:13–30. doi: 10.1016/s0065-2571(98)00018-1. [DOI] [PubMed] [Google Scholar]
- Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41:147–162. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Sun B, Ranganathan B, Feng SS. Multifunctional poly(D,L-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials. 2008;29:475–486. doi: 10.1016/j.biomaterials.2007.09.038. [DOI] [PubMed] [Google Scholar]
- Sun LC, Coy DH. Somatostatin receptor-targeted anti-cancer therapy. Curr Drug Deliv. 2011;8:2–10. doi: 10.2174/156720111793663633. [DOI] [PubMed] [Google Scholar]
- Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized Micellar Systems for Cancer Targeted Drug Delivery. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- Svenson S, Tomalia DA. Dendrimers in biomedical applications--reflections on the field. Adv Drug Deliv Rev. 2005;57:2106–2129. doi: 10.1016/j.addr.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Svenson S, Wolfgang M, Hwang J, Ryan J, Eliasof S. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J Control Release. 2011;153:49–55. doi: 10.1016/j.jconrel.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Tagami T, Ernsting MJ, Li S-D. Efficient tumor regression by a single and low dose treatment with a novel and enhanced formulation of thermosensitive liposomal doxorubicin. J Control Release. 2011;152:303–309. doi: 10.1016/j.jconrel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Taheri A, Dinarvand R, Atyabi F, Ahadi F, Nouri FS, Ghahremani MH, Ostad SN, Borougeni AT, Mansoori P. Enhanced Anti-Tumoral Activity of Methotrexate-Human Serum Albumin Conjugated Nanoparticles by Targeting with Luteinizing Hormone-Releasing Hormone (LHRH) Peptide. Int J Mol Sci. 2011;12:4591–4608. doi: 10.3390/ijms12074591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146:264–275. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock IF, Rotin D. Acid pH in Tumors and Its Potential for Therapeutic Exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- Taratula O, Garbuzenko OB, Kirkpatrick P, Pandya I, Savla R, Pozharov VP, He H, Minko T. Surface-engineered targeted PPI dendrimer for efficient intracellular and intratumoral siRNA delivery. J Control Release. 2009;140:284–293. doi: 10.1016/j.jconrel.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasis D, Tagmatarchis N, Bianco A, Prato M. Chemistry of carbon nanotubes. Chem Rev. 2006;106:1105–1136. doi: 10.1021/cr050569o. [DOI] [PubMed] [Google Scholar]
- Tejada-Berges T, Granai CO, Gordinier M, Gajewski W. Caelyx/Doxil for the treatment of metastatic ovarian and breast cancer. Expert Rev Anticancer Ther. 2002;2:143–150. doi: 10.1586/14737140.2.2.143. [DOI] [PubMed] [Google Scholar]
- Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- Torrisi MR, Lotti LV, Belleudi F, Gradini R, Salcini AE, Confalonieri S, Pelicci PG, Di Fiore PP. Eps15 is recruited to the plasma membrane upon epidermal growth factor receptor activation and localizes to components of the endocytic pathway during receptor internalization. Mol Biol Cell. 1999;10:417–434. doi: 10.1091/mbc.10.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Ulbrich K, Etrych T, Chytil P, Jelinkova M, Rihova B. HPMA copolymers with pH-controlled release of doxorubicin: in vitro cytotoxicity and in vivo antitumor activity. J Control Release. 2003;87:33–47. doi: 10.1016/s0168-3659(02)00348-6. [DOI] [PubMed] [Google Scholar]
- Ulrich H, Martins AH, Pesquero JB. RNA and DNA aptamers in cytomics analysis. Curr Protoc Cytom. 2005 doi: 10.1002/0471142956.cy0728s33. Chapter 7, Unit 7 28. [DOI] [PubMed] [Google Scholar]
- Vasir JK, Labhasetwar V. Targeted drug delivery in cancer therapy. Technol Cancer Res Treat. 2005;4:363–374. doi: 10.1177/153303460500400405. [DOI] [PubMed] [Google Scholar]
- Vega-Villa KR, Takemoto JK, Yanez JA, Remsberg CM, Forrest ML, Davies NM. Clinical toxicities of nanocarrier systems. Adv Drug Deliv Rev. 2008;60:929–938. doi: 10.1016/j.addr.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 2000;60:203–212. [PubMed] [Google Scholar]
- Veiseh O, Gunn JW, Zhang M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv Drug Deliv Rev. 2010;62:284–304. doi: 10.1016/j.addr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett. 1998;425:145–150. doi: 10.1016/s0014-5793(98)00224-5. [DOI] [PubMed] [Google Scholar]
- Vukelja SJ, Anthony SP, Arseneau JC, Berman BS, Cunningham CC, Nemunaitis JJ, Samlowski WE, Fowers KD. Phase 1 study of escalating-dose OncoGel (ReGel/paclitaxel) depot injection, a controlled-release formulation of paclitaxel, for local management of superficial solid tumor lesions. Anticancer Drugs. 2007;18:283–289. doi: 10.1097/CAD.0b013e328011a51d. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen P, Su Z-F, Vallis K, Sandhu J, Cameron R, Hendler A, Reilly RM. Amplified delivery of indium-111 to EGFR-positive human breast cancer cells. Nucl Med Biol. 2001;28:895–902. doi: 10.1016/s0969-8051(01)00262-1. [DOI] [PubMed] [Google Scholar]
- Wang S, Kim G, Koo Lee Y-E, Hah HJ, Ethirajan M, Pandey RK, Kopelman R. Multifunctional Biodegradable Polyacrylamide Nanocarriers for Cancer Theranostics - A “See and Treat” Strategy. ACS Nano. 2012 doi: 10.1021/nn301633m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kaneko M, Maitani Y. Functional coating of liposomes using a folate-polymer conjugate to target folate receptors. Int J Nanomedicine. 2012;7:3679–3688. doi: 10.2147/IJN.S32853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki A, Rochlitz C, Orleth A, Ritschard R, Albrecht I, Herrmann R, Christofori G, Mamot C. Targeting Tumor-Associated Endothelial Cells: Anti-VEGFR2 Immunoliposomes Mediate Tumor Vessel Disruption and Inhibit Tumor Growth. Clin Cancer Res. 2012;18:454–464. doi: 10.1158/1078-0432.CCR-11-1102. [DOI] [PubMed] [Google Scholar]
- Wijagkanalan W, Kawakami S, Hashida M. Designing dendrimers for drug delivery and imaging: pharmacokinetic considerations. Pharm Res. 2011;28:1500–1519. doi: 10.1007/s11095-010-0339-8. [DOI] [PubMed] [Google Scholar]
- Wileman T, Harding C, Stahl P. Receptor-mediated endocytosis. Biochem J. 1985;232:1–14. doi: 10.1042/bj2320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Szostak JW. Isolation of a fluorophore-specific DNA aptamer with weak redox activity. Chem Biol. 1998;5:609–617. doi: 10.1016/s1074-5521(98)90289-7. [DOI] [PubMed] [Google Scholar]
- Xu L, Pirollo KF, Chang EH. Transferrin-liposome-mediated p53 sensitization of squamous cell carcinoma of the head and neck to radiation in vitro. Human Gene Therapy. 1997;8:467–475. doi: 10.1089/hum.1997.8.4-467. [DOI] [PubMed] [Google Scholar]
- Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, Rait A, Chang EH. Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med. 2001;7:723–734. [PMC free article] [PubMed] [Google Scholar]
- Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Characterization of the pH of folate receptor-containing endosomes and the rate of hydrolysis of internalized acid-labile folate-drug conjugates. J Pharmacol Exp Ther. 2007;321:462–468. doi: 10.1124/jpet.106.117648. [DOI] [PubMed] [Google Scholar]
- Ying X, Wen H, Lu W-L, Du J, Guo J, Tian W, Men Y, Zhang Y, Li R-J, Yang T-Y, Shang D-W, Lou J-N, Zhang L-R, Zhang Q. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release. 2010;141:183–192. doi: 10.1016/j.jconrel.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Yokoyama M. Clinical Applications of Polymeric Micelle Carrier Systems in Chemotherapy and Image Diagnosis of Solid Tumors. J Exp Clin Med. 2011;3:151–158. [Google Scholar]
- Yonezawa S, Asai T, Oku N. Effective tumor regression by anti-neovascular therapy in hypovascular orthotopic pancreatic tumor model. J Control Release. 2007;118:303–309. doi: 10.1016/j.jconrel.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Yotsumoto F, Sanui A, Fukami T, Shirota K, Horiuchi S, Tsujioka H, Yoshizato T, Kuroki M, Miyamoto S. Efficacy of ligand-based targeting for the EGF system in cancer. Anticancer Res. 2009;29:4879–4885. [PubMed] [Google Scholar]
- Young D, Findlay H,P, Hahn S,E, Cechetto L,M, McConkey F, Arius Research Inc. Cytotoxicity Mediation of Cells Evidencing Surface Expression of CD44. United States patent WO 2007098575. 2007 Jul 9; inventors. assignee.
- Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27:286–298. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Control Release. 2001;72:203–215. doi: 10.1016/s0168-3659(01)00276-0. [DOI] [PubMed] [Google Scholar]
- Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001;8:161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]