

Abstract
Advances in biosensor technologies for in vitro diagnostics have the potential to transform the practice of medicine. Despite considerable work in the biosensor field, there is still no general sensing platform that can be ubiquitously applied to detect the constellation of biomolecules in diverse clinical samples (for example, serum, urine, cell lysates or saliva) with high sensitivity and large linear dynamic range. A major limitation confounding other technologies is signal distortion that occurs in various matrices due to heterogeneity in ionic strength, pH, temperature and autofluorescence. Here we present a magnetic nanosensor technology that is matrix insensitive yet still capable of rapid, multiplex protein detection with resolution down to attomolar concentrations and extensive linear dynamic range. The matrix insensitivity of our platform to various media demonstrates that our magnetic nanosensor technology can be directly applied to a variety of settings such as molecular biology, clinical diagnostics and biodefense.
Medical decision making is increasingly based on molecular testing; quantitative detection of disease-specific proteins in serum and other bodily fluids forms the foundation of many diagnostic tests to direct therapy in diverse areas of clinical medicine1–5. Current methods for protein detection, however, are limited by their sensitivity, multiplexing capacity or, most importantly, uncontrollable response to the composition of complex biological samples. Detection across varied samples is crucial; for instance, a urologist may provide urine, a neurologist cerebrospinal fluid, a cardiologist blood or an oncologist cell lysates. The diversity of such matrices has hindered the generalizability and sensitivity of the majority of protein detection platforms, thus greatly reducing their clinical utility. Here we present a magnetic nanosensing protein detection technology that overcomes the problems associated with other methodologies.
In the vast majority of protein detection platforms, the binding event of a protein to a specific recognition molecule must be detected with a signal transducer. In ELISAs, protein microarrays6,7 and quantum dot8 detection platforms, the readout is based on a fluorescent or colorimetric signal. Inherent autofluorescence or optical absorption of the matrix of many biological samples or reagents becomes a major limiting factor. Similarly, nanowires9, microcantilevers10, carbon nanotubes11 and electrochemical biosensors12 rely on charge-based interactions between the protein or tag of interest and the sensor, making each system unreliable in conditions of varying pH and ionic strength. Even a 0.14 M salt solution (similar to human serum) has sufficient Debye screening to shield nanowires from detecting protein binding events13. Accordingly, these sensors require the samples to be presented in pure water or precisely controlled salt solutions, an unrealistic requirement for practical settings. For nanowires to detect proteins in serum samples, for example, desalting steps must be performed before detection9. Therefore, making the transition from highly sensitive protein detection in an ideal salt solution in the laboratory to diverse biological matrices in the clinical realm has been challenging.
The matrices of even the most complex biological samples lack a detectable magnetic background signal and do not interfere with the magnetic transduction mechanism. Therefore, a magnetic field–based detection platform is well suited for protein detection in clinical samples. Giant magnetoresistive (GMR) sensors, which were originally developed for use as read heads in hard-disk drives, are multilayer thin-film structures that operate on the basis of a quantum mechanical effect, wherein a change in the local magnetic field induces a change in resistance of the sensor14–16. Here we show a matrix-insensitive protein detection assay in which an array of GMR sensors (Fig. 1a–c) is used to detect binding events of proteins to arrays of surface-bound antibodies with the use of magnetic nanoparticle tags17–23 and in real time24. Our technology employs a ‘sandwich’ assay in which the target antigen is sandwiched between two antibodies, one bound to the sensor and the other tagged with a superparamagnetic nanoparticle. Under an external magnetic field, the nanoparticles magnetize, and their presence or absence can be detected by the underlying GMR sensor (Fig. 1d–h). Using chips measuring 1.2 cm × 1 cm, each containing an array of 64 GMR sensors, we show rapid, multiplex protein detection with a linear dynamic range of over six orders of magnitude for a diverse range of biological fluids.
Figure 1.
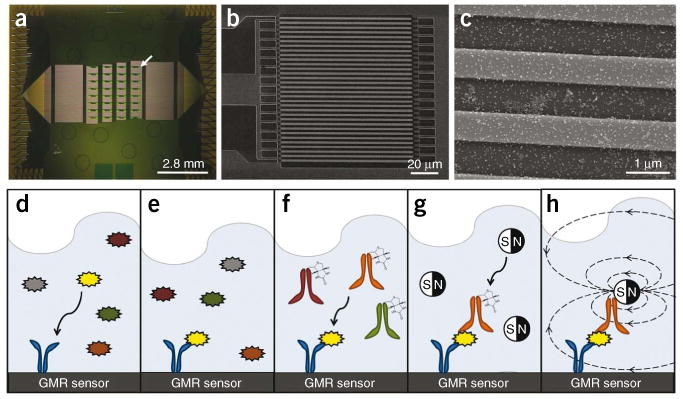
Sensor architecture and assay. (a) Image of our magnetonanosensor chip containing 64 sensors in an 8×8 array. Each green square is a uniquely addressable GMR sensor (white arrow). The horizontal lines leaving the sensors are leads connecting each sensor to a unique bond pad. (b) Scanning electron microscope (SEM) image of the sensor's serpentine architecture at 800×. (c) SEM image at 50,000× showing the sensor (light gray stripes) with magnetic nanoparticle tags (white dots). (d–h) A schematic of the sandwich assay. (d) Capture antibodies (blue) that are complementary to a chosen antigen (yellow) are immobilized onto the surface of each sensor. (e) The noncomplementary antigens are subsequently washed away. (f) After adding a cocktail of detection antibodies, the biotinylated detection antibody (orange) complementary to the antigen of interest binds in a sandwich structure, and the noncomplementary antibodies are washed away. (g) Finally, a streptavidin-labeled magnetic nanoparticle tag is added to the solution, and it binds the biotinylated detection antibody. (h) As the magnetic tags diffuse to the GMR sensor surface and bind the detection antibody, the magnetic fields from the magnetic nanoparticles can be detected by the underlying GMR sensor in real-time in the presence of a small external modulation magnetic field.
Results
Performance characteristics
To evaluate the sensitivity and dynamic range of our assay, we compared calibration curves generated using our magnetic nanosensor arrays to ELISA, the current gold standard in protein detection (Fig. 2). Here we decided to detect carcinoembryonic antigen (CEA), a wellknown colon and breast cancer tumor marker, spiked into a solution of 0.1% BSA in PBS. To obtain high specificity, we immobilized a monoclonal capture antibody on a sensor surface, and, to increase the signal, we used a biotinylated polyclonal detection antibody to subsequently capture streptavidin-coated magnetic nanoparticle tags. In both the ELISA and the magnetic nanosensor assay, we used the same capture and detection antibodies (for the ELISA, the tag was streptavidin–horseradish peroxidase). We found quantitative protein detection down to the single-femtomolar (10-15) level without the need for amplification (Fig. 2a). Furthermore, after only a single amplification step, where we tethered several magnetic tags to the originally captured magnetic tag, we were able to distinguish concentrations as low as 50 attomolar (10-18) above background (Fig. 2b) (P < 0.05). This places magnetonanosensors on par with the most sensitive biosensors. In addition, an examination of these data shows that our magnetic nanosensors have linear calibration curves (on a log-log plot) over a range of six orders of magnitude (Fig. 2a). In contrast, when testing the same antibody pairs by ELISA, the linear dynamic range was approximately two orders of magnitude, with a lower limit of detection around 1–5 pM (10-12), demonstrating that our sensors are over 1,000 times more sensitive than ELISA. We obtained similar performance characteristics for detection of lactoferrin, a urinary marker of urinary tract infections (Supplementary Fig. 1)25.
Figure 2.
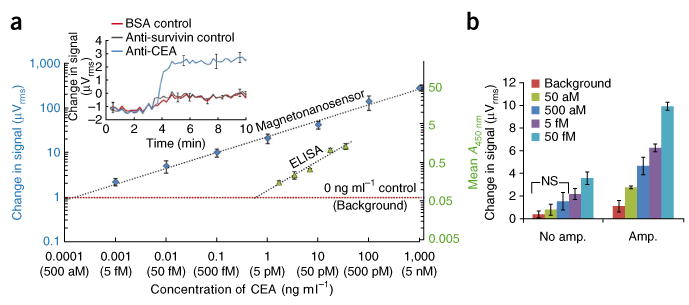
Sensitivity and linear dynamic range (on a log-log plot) of magnetonanosensors and ELISA. (a) Superimposed serial dilution curves of CEA detection on the magnetic nanosensor and ELISA comparing the linear dynamic range and the lower limit of detection in 0.1% BSA in PBS (the same antibody pairs were used for both assays). μVrms is the unit of GMR sensor signal, whereas A450nm is the unit of ELISA. The background is defined as the average signal with no (0 ng ml-1) CEA spiked into the reaction well for each technology plus 2 s.d. The error bars represent means ± s.d. Inset, real-time monitoring of change in voltage over time when 5 fM CEA is spiked into the reaction well when compared to the BSA control and a noncomplementary antibody to survivin control (anti-survivin). The error bars represent means ± s.d. (b) Demonstration of protein detection using amplification to quantifiably distinguish (P < 0.05) protein concentrations in the attomolar concentration regime. NS indicates no significant difference in signal according to Welch's t test. The error bars represent means ± s.d.
Sensor response to pH and temperature
In laboratory settings, exceptionally sensitive protein detection has been documented using a variety of nanosensing technologies, such as nanowires26, microcantilevers10, carbon nanotubes11 and biobarcode assays27. A more substantial challenge, however, is the application of such sensitive protein detection to biological samples in nonideal situations. Therefore, we investigated the performance of our magnetonanosensors for diverse media. To our knowledge, such a broad study has never been reported before for any nanosensor.
We first investigated how the sensor itself (before addition of the detection antibody) responds to various reaction conditions, including pH, temperature and turbidity. In contrast to nanowires, in which a change of 0.5 pH causes considerable signal fluctuations13, our sensing technology was unaffected by changes in ionic strength and pH change between pH 4-10 (Fig. 3a). In addition, unlike microcantilevers, for which even a 0.5 °C change causes substantial cantilever deflection28, our sensors are unaffected by changes in the temperature of our sample (Fig. 3b), provided that we implement a simple temperature correction algorithm that is performed in real time without having to rely on reference sensors (details are described in Supplementary Fig. 2). Finally, optical activity or turbidity of sample solutions had no effect on our detection platform, as it does not use optical-based detection methods as do ELISAs, protein microarrays and quantum dots.
Figure 3.
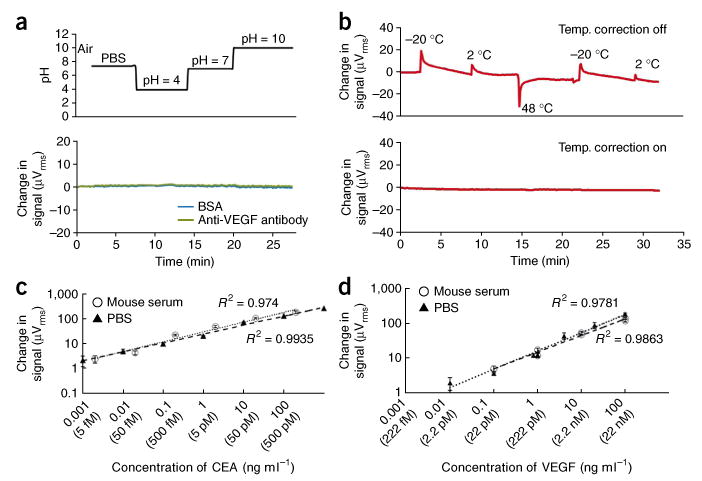
Magnetonanosensors exhibit matrix-insensitive detection. (a) Sensor response to changes in media with surface-bound BSA and antibody to VEGF. The pH of the solution is plotted above the sensor response. (b) Sensor response to temperature changes before (top) and after (bottom) background temperature correction. The numbers at the top indicate the initial temperature of the solution that was loaded into the reaction well. An exponential decay is observed in the uncorrected signal due to equilibration of the sample toward room temperature. A detailed discussion on how the temperature correction works is presented in Supplementary Figure 2.(c,d) Comparison of calibration curves detecting CEA and VEGF when spiked into 0.1% BSA in PBS and into mouse serum. The calibration curves generated in the two media are virtually identical for both proteins. The error bars represent means ± s.d.
Assay generalizability and reproducibility
We next investigated the device's generalizability for diverse media by comparing CEA and vascular endothelial growth factor (VEGF) detection in PBS to their respective detection in mouse serum, a more complex matrix. Detection signals were remarkably similar in the two media over the entire range of protein concentrations tested (Fig. 3c,d). Furthermore, the 0 ng ml-1 analyte controls in both PBS and serum yielded the same minute signal (Fig. 4), indicating that the complexity of the serum matrix did not contribute any measurable background noise to our sensors. These were the first major steps in confirming a matrix-insensitive detection platform.
Figure 4.
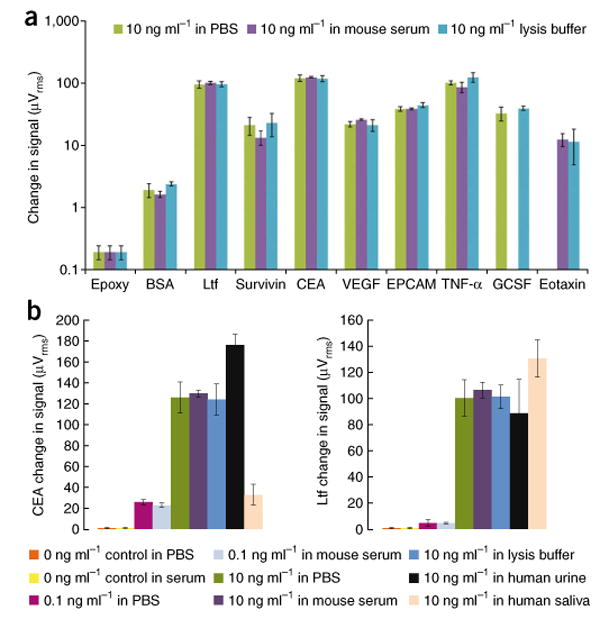
Multiplex protein detection in a diversity of media. (a) A panel of eight human tumor markers and a BSA negative control and epoxy control indicate matrix-insensitive protein detection when shifting from PBS to mouse serum to lysis buffer. No bar graph is shown for the 10 ng ml-1 GCSF spiked into mouse serum or 10 ng ml-1 of eotaxin spiked into PBS due to sensor corrosion during the experiment. The error bars represent means ± s.d. Ltf, lactoferrin. (b) Matrix-insensitive protein detection across a range of concentrations (0 ng ml-1 control, 0.1 ng ml-1 spiked samples and 10 ng ml-1 spiked samples) for CEA and Ltf in PBS, mouse serum and lysis buffer. In addition, detection of 10 ng ml-1 CEA and Ltf spiked in PBS, mouse serum, lysis buffer, human urine and human saliva is presented. The error bars represent means ± s.d.
We also investigated the sensor-to-sensor and chip-to-chip reproducibility. We monitored 4-16 sensors and compared experiments performed weeks to months apart. Real-time change in voltage-versus-time measurements recorded on our sensors showed reproducible signals in virtually every condition tested (Supplementary Fig. 3). Finally, we performed a direct comparison between quantitative protein detection by ELISA and by magnetonanosensors. Our experimental results showed a remarkably similar ability of the two platforms to quantify the concentration of a given protein in an unknown sample at the relatively high concentrations needed for ELISA, further demonstrating the precision of our technology (Supplementary Fig. 4).
Multiplex tumor marker detection across many biological fluids
We next applied our magnetonanosensors to simultaneously monitor real-time binding events of multiple tumor markers in a number of biological fluids. We functionalized magnetosensors with antibodies to a representative panel of tumor markers. We analyzed a total of ten reaction conditions simultaneously in quadruplicates, making up a total of 40 sensors per reaction well. We spotted primary antibodies to lactoferrin, survivin, CEA, VEGF, epithelial cell adhesion molecule (EpCAM), granulocyte colony–stimulating factor (G-CSF), tumor necrosis factor-α (TNF-α) and eotaxin over four unique sensors per chip (we chose these markers to show the diversity and multiplex capacity of our technology, and they are not specific to any one disease process). Additionally, we functionalized four sensors in the array with BSA as negative control to measure nonspecific binding, and we deposited four sensors in the array with epoxy to monitor any systematic fluctuations in the electronics. We made replicas of this chip and tested them in PBS (pH 7.4), mouse serum, lysis buffer (pH 8.0), human urine (pH 5.15), human saliva and human serum (Supplementary Fig. 5 shows an illustration of the general experimental setup for each chip and the results from a human serum study).
We observed that when we introduced 10 ng ml-1 of each tumor marker in PBS, mouse serum or lysis buffer, we obtained virtually identical signals for each protein across the three media (Fig. 4a). Unfortunately, two signals are not shown, owing to sensor corrosion caused by inadvertent sensor contact during antibody spotting. In the majority of the experiments, the spiked serum samples produced considerably lower s.d. than both PBS and lysis buffer (Fig. 4b). In addition, the 0 ng ml-1 control, BSA negative control and noncomplementary capture antibody negative control tests in each media gave negligible signals (Fig. 4b). These results indicate that the more complex matrices of mouse serum and lysis buffer had no effect on the background signal.
We were also able to perform protein detection in human serum, human urine and human saliva. Because these samples were human, however, basal levels of human proteins were present in the matrix before the addition of 10 ng ml-1 of each marker. Therefore, we expected to see a higher signal when compared to PBS, mouse serum and lysis buffer experiments (all of which lacked human proteins before introducing 10 ng ml-1 of each protein). For example, the basal concentration of CEA in a healthy urine sample is typically around 20 ng ml-1, leading our CEA detection signal to be appropriately higher than we would expect from previously discussed samples containing only the 10 ng ml-1 spiked CEA (Fig. 4b)29. Therefore, although we used nonhuman biological fluids for demonstration purposes, the same protein detection capabilities extend to human fluids.
We were even able to quantitatively detect spiked proteins in the complex matrix of human saliva (Fig. 4b). However, the protein signals were systematically lower for all analytes, with the exception of lactoferrin. Owing to the high protease content, the higher viscosity of the sample (requiring longer diffusion times for the magnetic nanoparticles) or both, we observed lower signals. The lactoferrin signal, in contrast, was elevated because the basal concentration of lactoferrin in human saliva is approximately 5 μg ml-1, as it has a major role in the innate immune system in the human mouth30.
Multiplex detection of human colorectal cancer in mouse models
A compelling application of our magnetic nanosensing technology is for multiplex profiling over time of blood tumor markers in individuals with cancer. Rapid, sensitive and multiplex diagnostic tools for monitoring the progression of tumors will have a high impact not only in clinical diagnostics but also in biomedical research for investigating key components in signaling pathways involved in tumor growth, invasion and malignant transformation, as well as in monitoring response to therapies.
Accordingly, we determined the ability of our system to monitor dynamic changes of CEA, VEGF and EpCAM in a human colorectal cancer xenograft mouse model. First, we drew blood samples from each mouse before transplantation and analyzed them to investigate antibody cross-reactivity and to establish a background signal. On days 9, 18 and 21, we measured the tumor volumes and drew blood samples. Subsequently, we generated plots of the absolute CEA concentration over time as the tumor grew (Fig. 5a). We observed that after tumor inoculation, human CEA concentrations in each mouse consistently increased over time from 2 fM to 300 fM, indicating a strong correlation between tumor progression and the amount of human CEA in mouse serum. (Not all mice were analyzed for the entire time, as some mice had to be killed before day 21 as a result of tumor burden.) When we ran aliquots from the same samples on an ELISA, however, CEA was consistently below the limit of detection (1–5 pM). The EpCAM concentration remained consistently low or undetectable throughout the experiment, whereas VEGF abundance decreased over the first few time points and then spiked on the last day (Fig. 5b,c). Therefore, as expected, we found CEA to be a useful marker for monitoring tumor growth in our mouse model. The concentration of EpCAM and VEGF, however, remained relatively flat as the tumor grew, indicating that neither marker is useful in predicting tumor burden for this colorectal cancer cell line. In this study, we were able to show that magnetonanosensors are capable not only of determining the most clinically relevant tumor markers in a given panel, but also of monitoring minute fluctuations of chosen markers over time in a complex biological fluid, which was not possible with conventional methods.
Figure 5.
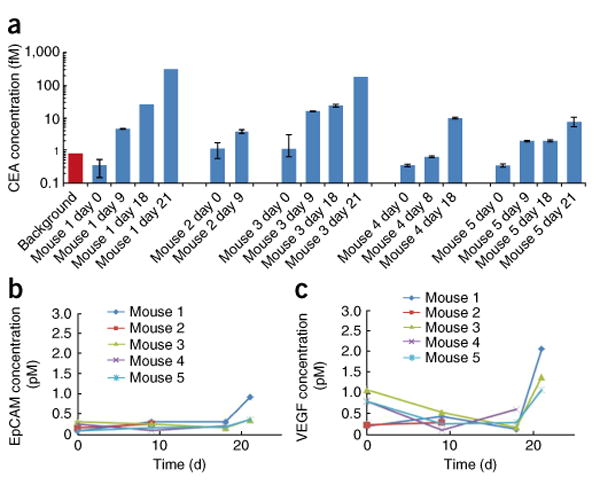
Femtomolar-level multiplex tumor marker profiling in xenograft mice. Detection of CEA, EPCAM and VEGF in xenograft tumor-bearing mice. (a) Time-course detection of CEA in each mouse. The background bar in red indicates the average background signal plus 2 s.d. The error bars represent means ± s.d. (b) Time-course detection of EpCAM in mouse serum. (c) Time-course detection of VEGF in mouse serum.
Discussion
High-sensitivity protein detection platforms often suffer from the requirement for well controlled reaction conditions and thus can only be used in a narrow range of applications. Here we have demonstrated and validated a new protein detection platform that we show to be robust and capable of sensitive and reproducible multiplex protein detection over a wide range of concentrations. Therefore, our detection strategy overcomes a considerable technological barrier to transfer benchtop application to the bedside.
When evaluating a multiplex protein detection platform, the linear dynamic range is equally as important as the sensitivity and robustness to diverse matrices. Currently, a major limitation to clinically relevant multiplex protein detection is a small linear dynamic range. Technologies such as the biobarcode assays27, carbon nanotubes31, nanowires32, protein microarrays6, quantum dots8 and ELISAs have all been reported to be limited in this way. In the biobarcode assay, for example, although exceptionally sensitive protein detection is possible, the technology relies on scanometric detection that saturates at higher signals27. Therefore, when investigating proteins of interest present at low concentrations with the biobarcode assay and others, one sacrifices the ability to simultaneously detect proteins present at concentrations only two orders of magnitude higher. For singleplex experiments, this may not be a major constraint, as the assay can be tediously rerun or performed with serial dilutions. However, when attempting to perform multiplex protein detection in biological fluids, where the protein concentrations may vary markedly depending on the marker of interest or between patients, a small linear dynamic range is a major limitation. With magnetonanosensors, however, even with sensitive protein detection, the signal does not saturate at six-log higher protein concentrations. Thus, the large linear dynamic range enables simultaneous quantitative protein detection of markers present at femtomolar concentrations and one million times higher (in the nanomolar concentration range).
The potential applications of our matrix-insensitive technology are numerous. For example, in clinical oncology, monitoring dynamic changes in tumor markers in both blood and cell lysates represents the future of cancer diagnostics. The ability to investigate an entire panel of markers, present at a wide range of concentrations, will arm physicians with the tools to make time-sensitive diagnoses of malignant diseases currently difficult to detect at a curable stage. Furthermore, the potential applications of our device in monitoring tumor response to chemotherapy are equally promising. By studying changes in tumor marker expression profiles before and after chemotherapy, physicians may be able to use our magnetonanosensors to predict tumor response to a given therapy before any detection is possible by existing imaging technologies. Such an advance could limit the undesirable side effects of an ineffective therapy and facilitate a more timely adjustment of medication to attack elusive tumors. Furthermore, the combination of the improved sensitivity, expanded linear dynamic range and matrix insensitivity of the assay will enable biomarker detection in nonserum biological fluids including urine and saliva, in which biomarker concentrations are typically orders of magnitude lower than in serum but which are less invasive to obtain.
Many potential applications of our technology exist beyond the clinical realm. Selection of the highest affinity drugs, antibodies or aptamers can be easily performed with this assay. By simply immobilizing a unique analyte recognition molecule on a unique sensor and monitoring the sensor in real time, researchers can deduce binding events and kinetic information. As a result, the recognition molecules with the highest association constants can be easily chosen after running one simple assay. In addition, due to the high sensitivity and real-time monitoring capabilities, the array can provide researchers with the unique ability to observe protein expression and protein-protein interactions with high spatial and temporal resolution. To our knowledge, such an advance is not possible with any other biosensors and will be a focus of our future work.
In conclusion, we have shown highly sensitive and specific multiplexed detection of protein tumor markers in a matrix-insensitive assay. Using up to 64 individually addressable magnet-nanosensors, we have shown real-time measurements of protein concentrations down to the attomolar level in a variety of clinically relevant media with a linear dynamic range of over six orders of magnitude. Thus, arrays of magnetoresistive sensors offer great promise in diverse applications such as medical diagnostics, therapy, clinical research and basic science.
Online Methods
Magnetonanosensor arrays fabrication
We deposited a multilayer thin film similar to the spin valves in hard-disk drive read heads and patterned them via ion milling into unique sensors (Supplementary Figs. 6-8)24. Each sensor covers a 100 × 100 μm2 area, and the nominal resistance of each sensor is approximately 2.5 kΩ with a magnetoresistance of 12%. We radially wire-bonded the chip with gold wire to a ceramic 84-pin chip carrier (LCC08423, Spectrum Semiconductor Materials). We attached a reaction well (Tygon tubing, 0.25-inch inner diameter and 0.75-inch outer diameter, 5 mm in length) to the chip using a two-component epoxy (EP5340, Eager Plastics). Finally, we chemically modified the oxide surface as described previously 24. In addition, the magnetic amplification protocol is similar to that described previously 24.
Protein detection assay
After surface functionalization, we manually deposited 100-nl droplets of each of the desired capture probes (antibody to lactoferrin (ab10110 from Abcam), CEA (5910 from BiosPacific), GCSF (551342 from BD Biosciences), eotaxin (555035 from BD Biosciences), VEGF (ab69479 from Abcam), EpCAM (ab20160 from Abcam), survivin (monoclonal-clone 32.1; 905-627 from Assay Designs, Inc.) and/or TNF-α (Mab210 from R&D Systems)) over at least four unique sensors. Typically, the concentration of antibodies ranged from 500 μg ml-1 to 1 mg ml-1. We also spotted 1% BSA in PBS onto at least four sensors as a negative control, and we spotted epoxy resin onto four sensors to monitor systematic fluctuations in the electronics. After 1 h incubation at 20 °C at 90% relative humidity, we rinsed the chips in a rinsing buffer comprising 0.1% BSA and 0.2% Tween-20 in PBS. Next, we blocked the chips with 50 μl of 1% BSA in PBS for 1 h. We then spiked the protein standard samples (lactoferrin (L0520-5MG from Sigma Aldrich), CEA (4128-CM-050 from R&D Systems), GCSF (214-CS-025 from R&D Systems), eotaxin (555102 from BD Biosciences), VEGF (293-VE165 from R&D Systems), EpCAM (960-EP-050 from R&D Systems), survivin (H00000332-P01 from Novus Biologicals, LLC) and/or TNF-α (210-TA-010 from R&D Systems) into PBS, mouse serum (015-000-120, Jackson ImmunoResearch), lysis buffer (R&D Systems protocol for Lysis Buffer #11: 50 mM Tris, 300 mM NaCl, 10% (wt/vol) glycerol, 3 mM EDTA, 1 mM MgCl2, 20 mM β-glycerophosphate, 25 mM NaF, 1% Triton X-100, 25 μg ml-1 leupeptin, 25 μg ml-1 pepstatin, and 3 μg ml-1 aprotinin), human urine, human saliva (IR100000, Innovative Research) or human serum (009-000-121, Jackson ImmunoResearch). After 1 h incubation of 50 μl-samples containing spiked proteins, we rinsed the chips with the rinsing buffer five times and then introduced biotinylated polyclonal antibodies to the proteins of interest at a concentration of 2 μg ml-1, each diluted in the rinsing buffer. During the next hour incubation with the biotinylated detection antibodies, we transferred the chips to the measuring station. After turning on the measuring station and helmholtz coil, we rinsed the chips five times with the rinsing buffer and added 50 μl of a solution of streptavidin-coated magnetic nanoparticles (MACS 130-048-102, Miltenyi Biotec; Supplementary Fig. 9). We monitored real-time binding of the streptavidin-coated magnetic nanoparticles to the bound biotinylated antibodies until the signal reached saturation, which took up to 20 min for the highest concentration of proteins tested.
Cell culture and xenograft mouse model
We grew human colorectal adenocarcinoma (LS 174T) cells (American Type Culture Collection) known to produce CEA33 in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated FBS and penicillin (100 U ml-1) and streptomycin (100 μg ml-1). We collected cells by trypsinization at 80–90% confluence, washed them in medium and suspended them in PBS. Next, we injected female athymic nude mice (nu/nu; n = 5), obtained at 8 weeks of age (Charles River Laboratories), subcutaneously in the right flank with 5 × 106 cells suspended in 100 μl PBS. We bled the mice by submandibular bleeding at days 0, 9, 18 and 21 after tumor inoculations using a 5-mm Goldenrod Animal Lancet (Medipoint, Inc.) and micro tubes with serum-gel clotting activator (Sarstedt Inc.). At each time point, we measured tumor sizes by caliper and calculated the tumor volume by the formula 0.5(length × width2). We carried out all mouse procedures according to a protocol approved by Stanford University Administrative Panels on Laboratory Animal Care. Blood samples clotted for 1 h at 20 °C. After centrifugation at 10,000g for 10 min at 4 °C, we transferred serum to new tubes and stored it at -80 °C.
Enzyme-linked immunosorbent assay
We measured serum CEA concentrations with the commercially available ELISA kit UBI MAGIWEL CEA Quantitative Test (United Biotech,). We performed the CEA ELISA according to the provided protocol with standards run in triplicates and samples in duplicates. The absorbance was read at 450 nm with a Synergy 4 Multi-Mode Microplate Reader (BioTek Instruments).
Statistical analyses
We performed statistical analysis using a Welch's t test to obtain P values relative to the background signal with a threshold of 0.05 for all P values.
Supplementary Material
Acknowledgments
This work was supported in part by US National Cancer Institute grants 1U54CA119367 and N44CM–2009-00011, US National Science Foundation grant ECCS-0801385-000, US Defense Threat Reduction Agency grant HDTRA1-07-1-0030-P00005, the US Defense Advanced Research Projects Agency/Navy Grant N00014–02-1–0807, NCI ICMIC P50 CA114747, the US Department of Veterans Affairs Merit Review B4872, the Canary Foundation and The National Semiconductor Corporation. R.S.G. acknowledges financial support from Stanford Medical School Medical Scientist Training Program and a National Science Foundation graduate research fellowship. C.H.N. acknowledges financial support from the Denmark-American Foundation and the Lundbeck Foundation.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Author Contributions: R.S.G., D.A.H., S.S.G. and S.X.W designed research; R.S.G., D.A.H., C.H.N. and K.E.M. performed research; R.S.G., D.A.H., C.H.N., S.J.O., H.Y., K.E.M., R.J.W., B.M., J.C.L., S.S.G. and S.X.W contributed new reagents and/or analytical tools; R.S.G., D.A.H. and S.X.W analyzed data; S.J.O. and S.X.W. designed the magnetic sensors; R.S.G. and H.Y. developed the biochemistry; and R.S.G., S.S.G. and S.X.W. wrote the paper.
Competing Interests Statement: The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
References
- 1.Srinivas PR, et al. Proteomics in early detection of cancer. Clin Chem. 2001;47:1901–1911. [PubMed] [Google Scholar]
- 2.Lopez MF, et al. A novel, high-throughput workflow for discovery and identification of serum carrier protein-bound peptide biomarker candidates in ovarian cancer samples. Clin Chem. 2007;53:1067–1074. doi: 10.1373/clinchem.2006.080721. [DOI] [PubMed] [Google Scholar]
- 3.Gorelik E, et al. Multiplexed immunobead-based cytokine profiling for early detection of ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:981–987. doi: 10.1158/1055-9965.EPI-04-0404. [DOI] [PubMed] [Google Scholar]
- 4.Zheng Y, et al. A multiparametric panel for ovarian cancer diagnosis, prognosis, and response to chemotherapy. Clin Cancer Res. 2007;13:6984–6992. doi: 10.1158/1078-0432.CCR-07-1409. [DOI] [PubMed] [Google Scholar]
- 5.Heath JR, Davis ME. Nanotechnology and cancer. Annu Rev Med. 2008;59:251–265. doi: 10.1146/annurev.med.59.061506.185523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell PA. perspective on protein microarrays. Nat Biotechnol. 2002;20:225–229. doi: 10.1038/nbt0302-225. [DOI] [PubMed] [Google Scholar]
- 7.Chan SM, et al. Protein microarrays for multiplex analysis of signal transduction pathways. Nat Med. 2004;10:1390–1396. doi: 10.1038/nm1139. [DOI] [PubMed] [Google Scholar]
- 8.Shingyoji M, et al. Quantum dots–based reverse phase protein microarray. Talanta. 2005;67:472–478. doi: 10.1016/j.talanta.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 9.Zheng G, et al. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nat Biotechnol. 2005;23:1294–1301. doi: 10.1038/nbt1138. [DOI] [PubMed] [Google Scholar]
- 10.Ji H, et al. Microcantilever biosensors based on conformational change of proteins. Analyst. 2008;133:434–443. doi: 10.1039/b713330h. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, Sood AK, Kumar N. Carbon nanotube flow sensors. Science. 2003;299:1042–1044. doi: 10.1126/science.1079080. [DOI] [PubMed] [Google Scholar]
- 12.Drummond TG, Hill MG, Barton JK. Electrochemical DNA sensors. Nat Biotechnol. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 13.Cheng MM, et al. Nanotechnologies for biomolecular detection and medical diagnostics. Curr Opin Chem Biol. 2006;10:11–19. doi: 10.1016/j.cbpa.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Barnas J, et al. Novel magnetoresistance effect in layered magnetic structures: Theory and experiment. Phys Rev B Condens Matter. 1990;42:8110–8120. doi: 10.1103/physrevb.42.8110. [DOI] [PubMed] [Google Scholar]
- 15.Prinz GA. Magnetoelectronics. Science. 1998;282:1660–1663. doi: 10.1126/science.282.5394.1660. [DOI] [PubMed] [Google Scholar]
- 16.Wolf SA, et al. Spintronics: A spin-based electronics vision for the future. Science. 2001;294:1488–1495. doi: 10.1126/science.1065389. [DOI] [PubMed] [Google Scholar]
- 17.Baselt DR, et al. A biosensor based on magnetoresistance technology. Biosens Bioelectron. 1998;13:731–739. doi: 10.1016/s0956-5663(98)00037-2. [DOI] [PubMed] [Google Scholar]
- 18.Li G, et al. Detection of single micron-sized magnetic bead and magnetic nanoparticles using spin valve sensors for biological applications. J Appl Phys. 2003;93:7557–7559. [Google Scholar]
- 19.Graham DL, et al. Single magnetic microsphere placement and detection on-chip using current line designs with integrated spin valve sensors: biotechnological applications. J Appl Phys. 2002;91:7786–7788. [Google Scholar]
- 20.Schotter J, et al. Comparison of a prototype magnetoresistive biosensor to standard fluorescent DNA detection. Biosens Bioelectron. 2004;19:1149–1156. doi: 10.1016/j.bios.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Millen RL, et al. Giant magnetoresistive sensors and superparamagnetic nanoparticles: a chip-scale detection strategy for immunosorbent assays. Anal Chem. 2005;77:6581–6587. doi: 10.1021/ac0509049. [DOI] [PubMed] [Google Scholar]
- 22.Li G, et al. Spin valve sensors for ultrasensitive detection of superparamagnetic nanoparticles for biological applications. Sens Actuators A Phys. 2006;126:98–106. doi: 10.1016/j.sna.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, et al. Giant magnetoresistive biochip for DNA detection and HPV genotyping. Biosens Bioelectron. 2008;24:99–103. doi: 10.1016/j.bios.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osterfeld SJ, et al. Multiplex protein assays based on real-time magnetic nanotag sensing. Proc Natl Acad Sci USA. 2008;105:20637–20640. doi: 10.1073/pnas.0810822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arao S, et al. Measurement of urinary lactoferrin as a marker of urinary tract infection. J Clin Microbiol. 1999;37:553–557. doi: 10.1128/jcm.37.3.553-557.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern E, et al. Label-free immunodetection with CMOS-compatible semiconducting nanowires. Nature. 2007;445:519–522. doi: 10.1038/nature05498. [DOI] [PubMed] [Google Scholar]
- 27.Georganopoulou DG, et al. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc Natl Acad Sci USA. 2005;102:2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu G, et al. Bioassay of prostate-specific antigen (PSA) using microcantilevers. Nat Biotechnol. 2001;19:856–860. doi: 10.1038/nbt0901-856. [DOI] [PubMed] [Google Scholar]
- 29.Zimmerman R, Wahren B, Edsmyr F. Assessment of serial CEA determinations in urine of patients with bladder carcinoma. Cancer. 1980;46:1802–1809. doi: 10.1002/1097-0142(19801015)46:8<1802::aid-cncr2820460818>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee S, et al. A longitudinal study of unsaturated iron-binding capacity and lactoferrin in unstimulated parotid saliva. Biol Trace Elem Res. 1997;57:1–8. doi: 10.1007/BF02803864. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, et al. Protein microarrays with carbon nanotubes as multicolor Raman labels. Nat Biotechnol. 2008;26:1285–1292. doi: 10.1038/nbt.1501. [DOI] [PubMed] [Google Scholar]
- 32.Cui Y, et al. Nanowire nanosensors for highly sensitive and selective detection of biological and chemical species. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 33.Tom BH, et al. Human colonic adenocarcinoma cells. I. Establishment and description of a new line. In Vitro. 1976;12:180–191. doi: 10.1007/BF02796440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.