

Abstract
Background
Studies conducted at the whole muscle level have shown that smooth muscle can maintain tension with low ATP consumption. Whereas it is generally accepted that this property (latch-state) is a consequence of the dephosphorylation of myosin during its attachment to actin, free dephosphorylated myosin can also bind to actin and contribute to force maintenance. We investigated the role of caldesmon (CaD) in regulating the binding force of unphosphorylated tonic smooth muscle myosin to actin.
Methods
To measure the effect of CaD on the binding of unphosphorylated myosin to actin (in the presence of ATP), we used a single beam laser trap assay to quantify the average unbinding force (Funb) in the absence or presence of caldesmon, ERK-phosphorylated CaD, or CaD plus tropomyosin.
Results
Funb from unregulated actin (0.10 ± 0.01 pN) was significantly increased in the presence of CaD (0.17 ± 0.02 pN), tropomyosin (0.17 ± 0.02 pN) or both regulatory proteins (0.18 ± 0.02 pN). ERK phosphorylation of CaD significantly reduced the Funb (0.06 ± 0.01 pN). Inspection of the traces of the Funb as a function of time suggests that ERK phosphorylation of CaD decreases the binding force of myosin to actin or accelerates its detachment.
Conclusions
CaD enhances the binding force of unphosphorylated myosin to actin potentially contributing to the latch-state. ERK phosphorylation of CaD decreases this binding force to very low levels.
General Significance
This study suggests a mechanism that likely contributes to the latch-state and that explains the muscle relaxation from the latch-state.
Keywords: caldesmon, myosin, latch-state, phosphorylation, in vitro motility assay laser trap, tropomyosin
1. Introduction
Tonic smooth muscle exhibits the unique ability of maintaining force for long periods of time, with low energy consumption. A formal description of this property was elaborated by Dillon and coworkers in 1981 in which they termed this state of force maintenance, the latch-state [1]. They suggested that the deactivation (dephosphorylation) of attached cross-bridges transforms them into non- or slowly cycling latch-bridges, capable of maintaining force. Hai and Murphy [2] subsequently adapted Huxley's model [3] of cross-bridge cycling to include the regulation of shortening velocity by myosin light chain phosphorylation and the latch-state. One assumption of this model was that myosin must first be phosphorylated in order to attach to actin [2]. However, we and others have shown that unphosphorylated myosin can bind to actin [4-7] and while it may not be actively cycling, it likely contributes to force maintenance.
In their initial description of the latch-state, Murphy and co-workers implied that phosphorylation and dephosphorylation of the myosin light chain was the only regulatory mechanism needed for smooth muscle myosin function [1, 2]. However, several studies since then have suggested a role for the actin regulatory proteins in the latch-state [8-12]. In particular, caldesmon (CaD) is known to have both actin and myosin binding sites [13, 14] and could potentially act as a cross-linker and contribute to force maintenance. Its binding to actin and myosin is also regulated by various phosphorylation mechanisms [10, 13, 15] allowing fine tuning of its function at various steps of the cross-bridge cycle.
The structure and function of CaD have been reviewed before [14, 16]. Briefly, the carboxyl terminus of CaD is responsible for its binding to actin whereas the amino-terminus binds to myosin [14, 16]. CaD inhibits the actin activated myosin ATPase activity [15, 17]. This inhibition is also known to be amplified by tropomyosin [18-20]. CaD also decreases the velocity of actin propulsion in the in vitro motility assay [21, 22] and this inhibition is again facilitated by tropomyosin [22, 23]. CaD can be phosphorylated at several sites but each of their functions has yet to be identified [10, 13, 14, 22-24]. Phosphorylation by calmodulin dependant protein kinase II (PK2) in the amino-terminal region weakens the binding of CaD to myosin [25] whereas phosphorylation in the carboxyl terminus by myosin light chain kinase (MLCK) increases its binding affinity for phosphorylated myosin filaments [10]. Extracellular signal-regulated kinases (ERK) phosphorylation of CaD, however, has been shown to remove the inhibition of movement in the in vitro motility assay [26]. ERK is also known to colocalize with CaD during contraction [27].
At the whole muscle level, the role of CaD in smooth muscle relaxation has been studied by exogenous addition to permeabilized fibres [28] or more recently by studying muscle strips from CaD knockout mice [29]. Both studies reported that CaD accelerates the rate of smooth muscle relaxation and suggested that this is accomplished by inhibiting the cooperative reattachment of dephosphorylated myosin [28, 29]. In the current study, we investigated at the molecular level, the role of CaD in the binding of unphosphorylated myosin to actin. We report that CaD enhances the force of binding of unphosphorylated myosin to actin. Furthermore, this force of binding decreases to very low levels upon ERK phosphorylation of CaD, thus promoting relaxation.
2. Materials And Methods
2.1 Purification of proteins
CaD was purified from pig whole stomach [10] and myosin from pig stomach fundus [30]. Tropomyosin was purified from chicken gizzard [31] and actin from chicken pectoralis acetone powder [32]. Actin was fluorescently labeled by incubation with tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (P1951, Sigma-Aldrich Canada) [6]. Phosphorylation of proteins: For the protocols requiring myosin activation, myosin was thiophosphorylated [33]. For the protocols requiring CaD activation, CaD was phosphorylated according to Hedges and coworkers [24] with slight modifications. Briefly, CaD (0.1 mg/ml) was phosphorylated by active ERK (32.6 μg/ml; MAP kinase 2, active, phosphorylated by MEK1; Millipore Billerica, MA) in a buffer containing 25mM Tris, 10 mM magnesium acetate, 0.1 mM EGTA, 1mM DTT, pH 7.5, followed by a 60 min incubation at 30°C.
2.2.Western blot analysis of CaD phosphorylation
The phosphorylation of CaD was assessed by Western blot analysis. Proteins were run on a 4-15% ready gradient SDS-PAGE gel (Bio-Rad, Hercules, CA). 0.7 μg of CaD and phosphorylated CaD were loaded in each well. Proteins were transferred electrophoretically onto PVDF membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 1% BSA and probed with an anti-phos CaD/serine 789 antibody (SC12931, Santa Cruz, CA), followed by an anti-goat-HRP antibody (Ab6741, AbCam,Cambridge, MA). Antibody detection was done with Clarity substrate (Bio-Rad, Hercules, CA). Image acquisition was performed with ChemiDoc software (Bio-Rad, Hercules, CA). Total CaD was assessed by Coomassie blue staining of the same membranes which was possible because it is a purified protein.
2.3 Buffers
Myosin buffer (300 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 30 mM DTT; pH adjusted to 7.4); Actin buffer (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 30 mM DTT, with an oxygen scavenger system consisting of 0.25 mg/ml glucose oxidase, 0.045 mg/ml catalase, and 5.75 mg/ml glucose; pH adjusted to 7.4); Assay buffers: The in vitro motility assay buffer consisted of actin buffer to which was added methylcellulose (0.5%), to favor binding of myosin to actin, and MgATP (2 mM). The laser trap assay buffer consisted of actin buffer to which methylcellulose (0.3%) and MgATP (200 μM) were added.
2.4 In vitro motility assay
Our in vitro motility assay to measure the actin filament propulsion velocity (νmax) has been described before [5]. Ultracentrifugation (Optima ultracentrifuge L-90K and 42.2 Ti rotor, Beckman Coulter, Fullerton, CA) of myosin (500 μg/ml) with equimolar filamentous actin and 1 mM MgATP in myosin buffer was used to eliminate the non-functional myosin molecules. The functional myosin was then perfused in a flow through chamber [6] kept at 30°C, at a concentration of 125 μg/ml for all velocity measurements and let to randomly attach to the nitrocellulose for 2 min. Next, the following solutions were perfused sequentially in the flow through chamber (all in actin buffer): BSA (0.5 mg/ml), unlabeled G-actin (1.33 μM) to bind to any remaining non-functional myosin, followed by MgATP (1 mM) to remove the unlabeled actin from the functional heads. Then two washes of actin buffer were followed by TRITC labeled actin (30 nM), with or without CaD (300 nM) or phosphorylated CaD (300 nM), or tropomyosin (300 nM) or both CaD and tropomyosin each at 300 nM, incubated for 1 min, and finally, motility assay buffer. The actin filament movement was then observed with an inverted microscope (IX70, Olympus, Melville, NY) with a high numerical aperture objective (X100 magnification Ach 1.25 numerical aperture, Olympus, Melville, NY) and equipped for rhodamine epifluorescence. The images were captured with an intensified video camera (KP-E500 CCD Camera, Hitachi Kokusai Electric, Woodbury, NY, 720 × 480 resolution, 68.6 μm × 45.7 μm real frame size, 29.94 frame/s, 8 bit grayscale) and recorded on computer (custom built by Norbec Communication, Montreal, QC) using a frame grabber (Pinnacle Studio AV/DV V.9 PCI Card) and image acquisition software (AMCap software V9.20) at 29.94 Hz. νmax was obtained by dividing the total path followed by the filaments by the elapsed time using our automated version of the National Institutes of Health tracking software (NIH macro in Scion Image 4.02, Scion) coded in Matlab (R2009b) [34]. Only filaments present for at least 20% of the recorded video time (50 s) and following a path of at least 3μm were considered.
2.5 Laser trap assay
The force measurements were performed using our single beam laser trap assay [4, 5] comprised of a Laser Tweezers Workstation (Cell Robotics, Albuquerque, NM) and the motility assay described above. Pedestals were made with 4.5 μm polystyrene microspheres (Polybead, Polysciences, Warrington, PA) as previously described [5]. The trapping microspheres (3 μm polystyrene, Polybead, Polysciences, Warrington, PA) were prepared by incubation for 30 min at room temperature with N-ethylmaleimide-modified (NEM) skeletal myosin [6]. Unphosphorylated myosin was perfused in the flow through chamber at a concentration of 16.7μg/ml for all force measurements and let to randomly attach to the nitrocellulose for 2 min. Next, BSA (0.5 mg/ml) was perfused followed by two washes of actin buffer. Then, equal volumes of microspheres (13×103 microspheres/μl) and TRITC-labeled actin or regulated actin were mixed in laser trap assay buffer to yield the following final concentrations: actin (1 nM), CaD (10-500 nM), tropomyosin (5-50 nM), phosphorylated CaD (10 nM), or CaD and tropomyosin each at 10 nM. Note that the regulatory proteins were used at saturating concentrations to ensure regulation even at this low concentration of actin. The laser trap was created with a diode pumped Nd:YAG solid-state laser (TEM00, 1.5 W, 1064nm). This trap was used to capture a microsphere which was visualized in bright field by a charge coupled device (CCD) camera (XC-75, Sony Corporation of America, New York, NY). An actin filament (with or without the actin regulatory proteins), visualized by fluorescence imaging as described above, was bound to the trapped microsphere (Fig.1A) and then brought in contact with unphosphorylated myosin molecules randomly adhered to a pedestal. Contact between myosin and actin was allowed (∼10s) during which the microsphere baseline position in the trap was recorded (Fig.1 B). The microscope stage, and thus the pedestal, was then moved away from the trap at a slow and constant velocity (∼0.5 μm/s; Fig. 1B-C). Once the filament was taut (Fig.1C), the trapped microsphere moved with the pedestal (Fig.1D) until the force exerted on it by the trap became greater than the binding force of myosin to actin. At that point, the microsphere sprang back to its unloaded baseline position (trap center, Fig.1 E).
Figure 1.
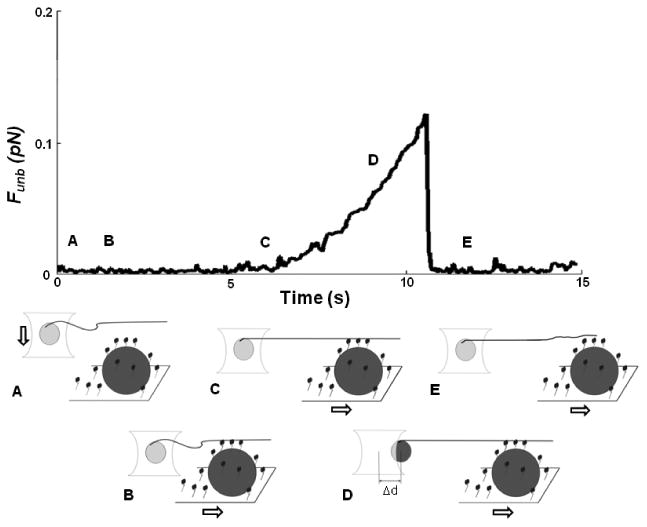
Sample trace of unbinding assay using the single beam laser trap: A) A polystyrene microsphere (light gray) to which is attached a TRITC-labeled actin filament is captured in the laser trap. B) Actin is brought in contact with unphosphorylated myosin that randomly coats a pedestal (dark grey) on a coverslip, time is allowed for protein interaction, and then the filament is made taut by moving the stage. C) The pedestal/coverslip is then moved away from the laser trap at a constant and slow velocity. D) This movement drags the trapped microsphere away from the trap center as a result of the attachment of unphosphorylated myosin to actin. E) When the force exerted by the laser trap on the microsphere exceeds the force of binding of unphosphorylated myosin to actin, the microsphere snaps back into the trap center, its unloaded position. The unbinding force (Funb) is then calculated as the maximal distance between the microsphere and the trap center (Δd) multiplied by the trap stiffness, calculated by the Stokes force method. (Figure not to scale).
The total unbinding force (Total Funb) of the myosin molecules was:
| (1) |
where k is the trap stiffness and Δd is the maximal displacement of the trapped microsphere from baseline. k was calibrated using the Stokes force (Ff) approach [4, 5], i.e. a viscous drag was applied to a trapped microsphere by moving it at a constant velocity (ν) in 0.3% methylcellulose while measuring Δd. Stokes' law states that the frictional force exerted on spherical objects with very small Reynolds numbers is calculated as:
| (2) |
where η is the dynamic viscosity and r is the microsphere radius. The viscosity of 0.3% methylcellulose was measured with a viscometer (DV-I at 60 rpm, Brookfield, Middleboro, MA), using a UL (ultra-low viscosity) adapter and was equal to 10.4 cP, at 30°C. Thus,
| (3) |
k (0.013 pN/nm, R2=0.95) was averaged from several measurements done at different ν and then used to obtain the force values.
The average binding force per myosin head (Funb) was calculated as we previously reported [5]. First, the length of actin filament in contact with the pedestal (ℓ) was measured by fluorescence imaging. That is, the actin filament was brought in focus and the bound portion (ℓ) was measured using NIH macro in Scion Image 4.02, (Scion Corp. Frederick, MD). Unbound actin moved in and out of focus due to Brownian motion so was discarded from the length measurements. We then used estimates of the number of active myosin heads on the motility surface, for a given concentration, previously reported by several groups [35-38]. The density of active myosin heads was calculated by dividing this number by the surface area and the number of active myosin head per actin filament length was calculated by assuming that all myosins could interact with actin within a 26 nm wide band [35-37].
2.6 Microsphere displacement analysis software
The movement of the trapped microsphere was tracked using Matlab customized software for optimal fitting of a reference image, as previously described in detail [5]. Briefly, a section of the first frame of the video (720×480 resolution, 68.6×40.3 μm real frame size, 29.94 frame/s, 8 bit grayscale) containing the microsphere image was compared to a section of the same size of each subsequent frame. This microsphere reference image is centered on the center of mass of the largest area of connected pixels above a threshold gray value and was visually confirmed to always contain the microsphere. The microsphere location in the analyzed frame is the location at which the summed absolute difference in pixel grey values between the current frame and the reference image was minimized. Sub-pixel resolution of this position was achieved by interpolation of the absolute difference values for the surrounding 8 pixels using a cubic interpolation algorithm. The microsphere baseline location, i.e. the trap center, was computed as the average microsphere position in the frames where no pedestal movement occurred and displacements were computed relative to this baseline. The local displacement maxima, which denote the detachment of myosin from actin, were found by searching in user defined regions. These maxima, corresponding to the maximal force exerted by the laser trap, were used to calculate the associated total Funb.
2.7 Statistical analysis
Differences in νmax and Funb between multiple conditions were tested using one way ANOVAs. When the within group variances differed between the groups, a Satterthwaite T-test, accounting for unequal variances, was performed [39]. Post-hoc comparisons between conditions were adjusted with a Bonferroni correction. Differences in νmax and Funb with only two conditions were tested using the Student's t-test, with t-tests for unequal variance when it applied. A value of p < 0.05 was considered significant. The Systat Software Inc., (San Jose, CA) was used to compute the exact p values of ANOVAs or Student's t-tests. For the motility assay, N represents the number of flow-through chambers studied. A minimum of three locations in each flow through chambers were analyzed; each location contained at least 10 filaments. Thus, a minimum of 30 filaments were analyzed per chamber to make up an N of 1. For the laser trap assay, N represents the number of actin filaments analyzed, with 1 to 3 events per filament. All data are presented as mean±SE.
3. Results
To assess whether CaD alters the average binding force of unphosphorylated myosin to actin filaments, we measured the Funb using the laser trap assay. A significant increase in the Funb was observed in the presence of 10 nM CaD (0.17± 0.02 pN) as compared to control, i.e. unregulated actin (0.1± 0.01 pN; p<0.001, Fig.2). Raising the CaD concentration further did not significantly increase the Funb above the value obtained at 10 nM (0.14± 0.02 pN, p = 0.120 at 50nM; 0.17± 0.02 pN, p = 0.714 at 250nM; 0.14± 0.03 pN, p = 0.160 at 500nM). Because tropomyosin has been reported to mechanically potentiate the effect of CaD [22, 23], we tested their combined concentration of tropomyosin was also chosen to be within physiological ratio range with actin and its effect on the Funb was tested independently of CaD. A significant increase in the Funb was observed with a tropomyosin concentration as low as 5nM (0.17± 0.02 pN; p=0.005), as compared to unregulated actin. Raising the tropomyosin concentration further did not significantly increase the Funb above the value obtained at 5nM (0.16± 0.02 pN; p= 0.632, at 25nM; 0.17± 0.05 pN; p= 0.928, at 50nM; average value shown in Fig.2). Thus, both CaD and tropomyosin enhance the Funb of unphosphorylated myosin to actin but their effects are not synergistic.
Figure 2.
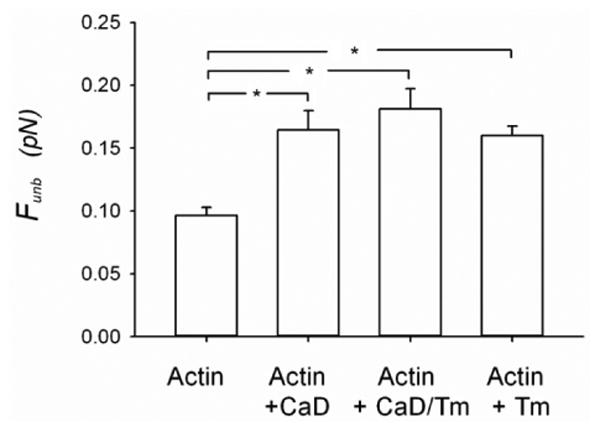
Unbinding force (Funb) of unphosphorylated myosin from actin measured using the laser trap assay. The measurements were performed with unregulated actin filaments (1nM) (N=20) and actin filaments (1nM) regulated by CaD (10nM) (N=14), or by CaD (10nM) + tropomyosin (10nM) (N=14) or tropomyosin (5nM to 50nM) (N=14). The results are reported as average force per myosin head. CaD: Caldesmon, Tm: tropomyosin and *: p < 0.05.
A series of control experiments were then performed to ascertain that the enhancement of the Funb obtained in the presence of the actin regulatory proteins was not due to unspecific binding to the pedestal (Fig. 3 A-C). This was accomplished by skipping the myosin incubation on the coverslip and incubating directly with BSA (see methods of the in vitro motility assay). These results were reported in terms of force per ℓ because there was no myosin to normalize to, as for the results presented in figure 2 In the presence of CaD, the Funb was negligible (0.44± 0.16 pN/μm) in the absence of myosin as compared to its value in the presence of myosin (4.42± 0.43 pN/μm; p<0.001; Fig.3A). Similarly, in the presence of tropomyosin, the Funb was negligible (0.31± 0.08 pN/μm) in the absence of myosin as compared to its value (3.7± 0.34 pN/μm; p<0.001; Fig.3 B) in the presence of myosin. Finally, the Funb was also found to be negligible in the presence of both CaD and tropomyosin (0.37± 0.13 pN/μm) in the absence of myosin as compared to its value in the presence of myosin (3.47± 0.3 pN/μm; p<0.001; Fig.3C). Thus, the enhancement effect of CaD and tropomyosin on the Funb is not due to unspecific binding to the pedestal or coverslip, but to a direct action on the acto-myosin interaction.
Figure 3.
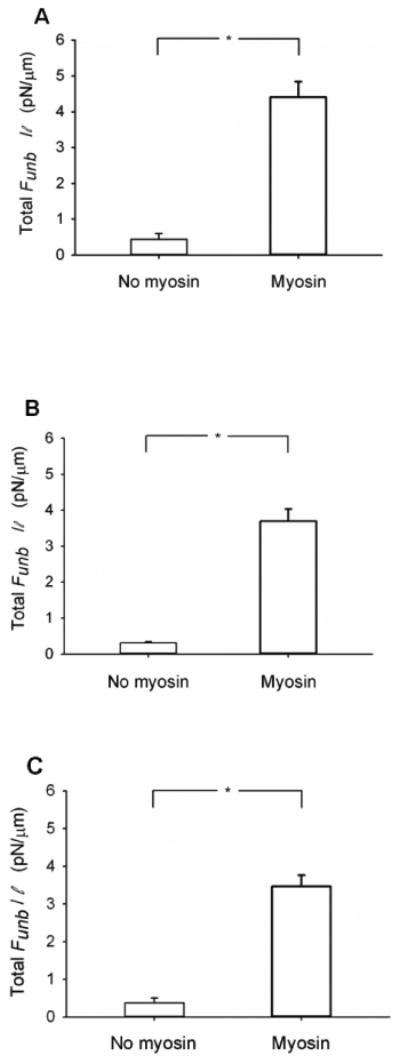
Total unbinding force normalized per actin filament length (Total Funb/ℓ) in the absence or presence of unphosphorylated myosin. The measurements were performed in the presence of 1nM actin and (A) CaD (500nM) (N=5 without and N=5 with unphosphorylated myosin), (B) tropomyosin (50nM) (N=4 without and N=6 with unphosphorylated myosin) or (C) both CaD (10nM) and tropomyosin (10nM) (N=5 without and N=5 with unphosphorylated myosin). *: p < 0.05.
Because CaD and tropomyosin have been more extensively studied for their effects on phosphorylated than on unphosphorylated myosin, we verified using the in vitro motility assay that the concentrations that we used for these actin regulatory proteins were producing results expected from the literature (Fig.4). Indeed, as previously reported [21, 23], νmax for unregulated actin (0.42±0.01 μm/s) was significantly increased in the presence of tropomyosin (0.53±0.03 μm/s, p < 0.001, Fig.4). Also, as expected from the literature [21, 22], νmax was significantly decreased in the presence of CaD (0.19±0.01μm/s; p<0.001, Fig.4). Furthermore, the combination of tropomyosin and CaD had a slight synergistic effect on νmax and significantly decreased it further (0.16±0.01 μm/s) from its value with CaD alone (p=0.003, Fig.4). Interestingly, this synergistic effect was not observed for the Funb of unphosphorylated myosin (Fig.2).
Figure 4.
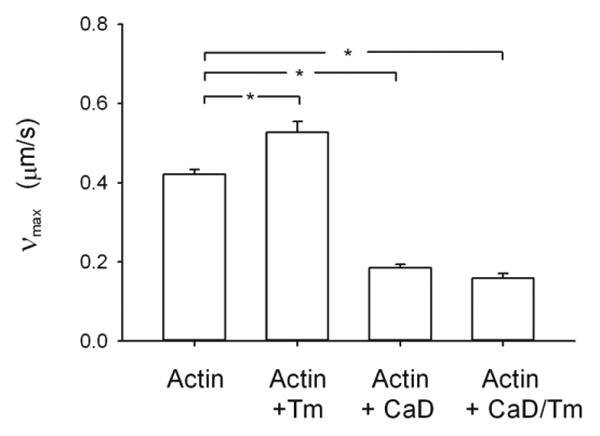
Functional assay (in vitro motility) to verify the regulation of actin by caldesmon and tropomyosin. The velocity of actin filaments (νmax) when propelled by phosphorylated myosin was measured with unregulated actin (30nM) filaments (N = 14) and actin (30nM) filaments regulated by tropomyosin (300nM) (N = 8),, actin (30nM) filaments regulated by CaD (300nM) (N = 10) and actin (30nM) filaments regulated by CaD (300nM) and tropomyosin (300nM) (N = 8). CaD: caldesmon, *: p < 0.05.
To assess the role of the phosphorylation of CaD on the average binding force of unphosphorylated myosin to actin filaments, we measured the Funb in the presence of ERK-phosphorylated CaD. The phosphorylation of CaD by ERK significantly decreased the Funb (0.06± 0.01 pN) as compared to non-phosphorylated CaD (0.2± 0.02 pN; p<0.001) and to unregulated actin (0.12± 0.0.01 pN; p<0.001; Fig.5A). CaD phosphorylation was confirmed by Western blotting (Fig.5B) and a control for the effect of the CaD phosphorylation reagents on the Funb of unphosphorylated myosin from actin but in the absence of caldesmon was also performed (Fig.5C). There was no effect of the CaD phosphorylation reagents on the Funb of unphosphorylated myosin from actin in the absence of caldesmon (p=0.725).
Figure 5.
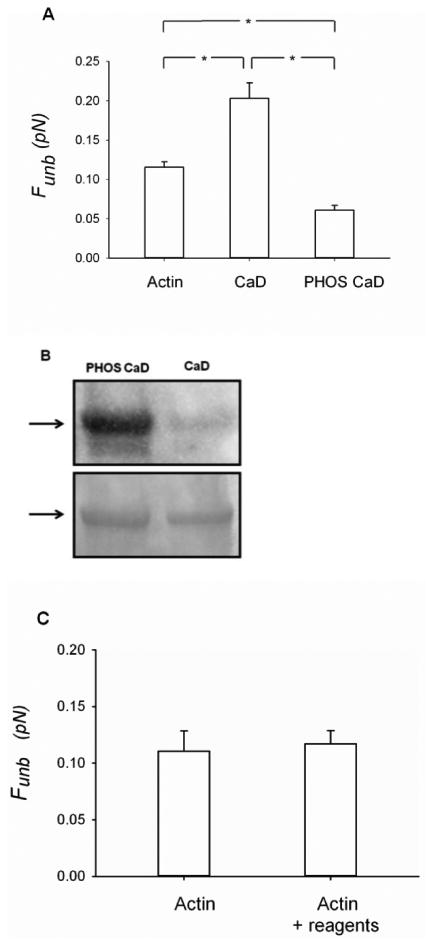
Effects of phosphorylated CaD on the unbinding force (Funb) of unphosphorylated myosin from actin. A) The measurements were performed with unregulated actin (1nM) filaments (N = 10), actin (1nM) filaments regulated by CaD (10nM) (N = 14) or actin (1nM) filaments regulated by ERK-phosphorylated CaD (10nM) (N = 10). The results are reported as average force per myosin head. CaD: caldesmon, PHOS CaD: phosphorylated caldesmon. *: p < 0.05. B) The phosphorylation of CaD was assessed by Western blotting (top panel). The total protein loading was assessed by Coomasie blue staining of the membrane (bottom panel). C) Effects of the CaD phosphorylating reagents on the unbinding force (Funb) of unphosphorylated myosin from actin, in the absence of caldesmon. The measurements were performed with unregulated actin (1nM) filaments (N=4) and with unregulated actin (1nM) filaments in the presence of the reagents used to phosphorylate CaD (see methods for details) (N=3).
Sample traces of the unbinding maneuvers are shown in figure 6. The Funb observed in the presence of actin only (Fig.6A) is seen to be increased in amplitude in the presence of CaD (Fig.6B). Interestingly, the phosphorylation of CaD decreases the Funb to very low values (Fig. 6C). Figure 6D shows again that the Funb is insignificant in the absence of myosin.
Figure 6.
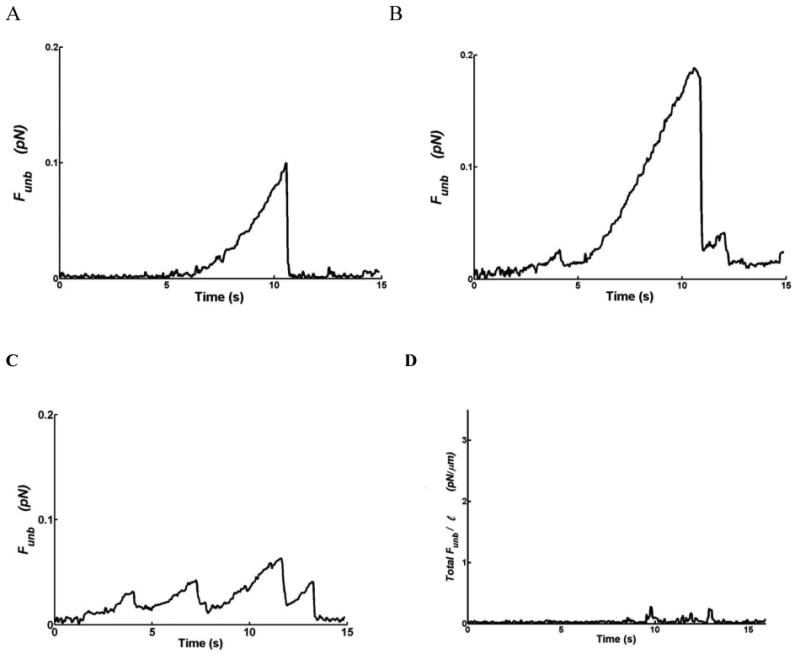
Sample traces of the unbinding force (Funb) from: A) unregulated actin (1nM) filaments, B) actin (1nM) filaments regulated by CaD (10nM), C) actin filaments regulated by phosphorylated CaD (10nM). These forces are reported as average per myosin head. D) Total Funb normalized per actin filament length (Total Funb/l) in the absence of unphosphorylated myosin but in the presence of unregulated actin (1nM) filaments. The maxima correspond to filament detachment from myosin.
The actomyosin regulation by ERK phosphorylation of CaD was verified functionally by measuring νmax of phosphorylated myosin in the in vitro motility assay. We found that the phosphorylation of CaD significantly increased νmax (0.36±0.02 μm/s) as compared to unphosphorylated CaD (0.27±0.02 μm/s; p<0.001) but that this νmax was still significantly slower than in the absence of CaD (0.48±0.01 μm/s; p<0.001; Fig.7).
Figure 7.
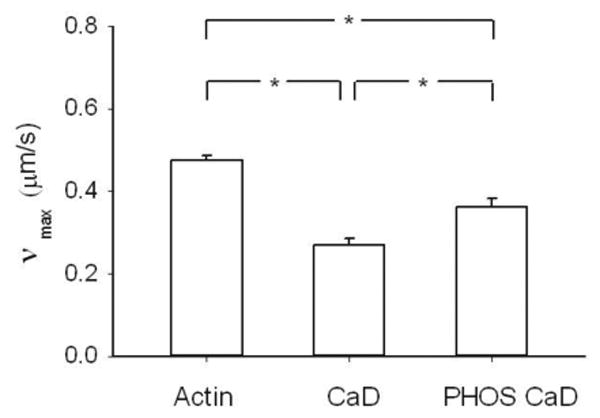
Functional assay (in vitro motility) to verify the regulation of actin by caldemon and phosphorylated caldesmon. The velocity of actin filaments (νmax) when propelled by phosphorylated myosin was measured with unregulated actin (30nM) filaments (N = 21), actin filaments regulated by CaD (300nM) (N = 17) and actin (30nM) filaments regulated by phosphorylated CaD (300nM) (N = 15). CaD: caldesmon, PHOS CaD: phosphorylated caldesmon. *: p < 0.05.
4. Discussion
The major findings of this study are: 1) CaD increases the average force of binding of unphosphorylated myosin to actin thereby promoting force maintenance; 2) upon ERK phosphorylation of CaD, the average force of binding of unphosphorylated myosin to actin decreases to very low levels, thus promoting relaxation.
It is generally believed that CaD promotes relaxation [28, 29]. Albrecht et al [28], demonstrated that exogenous addition of CaD to permeabilized tissues accelerates their rate of relaxation. They suggested that CaD inhibits cooperative attachment of dephosphorylated crossbridges in the latch-state phase [28]. More recently, Guo and coworkers [29] knocked out h-CaD in arterial muscle and reported a slower rate of relaxation. They suggested that h-CaD facilitates the detachment of dephosphorylated cross-bridges, in the presence of ATP, rather than preventing phosphorylated cross-bridge formation. From our observations that CaD increases Funb of unphosphorylated myosin to actin promoting force maintenance, we suggest a different molecular mechanism by which CaD accomplishes this relaxation. Our data actually demonstrate that one additional step is required for muscle relaxation. That is, CaD must be phosphorylated to lead to smooth muscle relaxation. Indeed, the data that we present in figures 2, 5 and 6 demonstrate that CaD enhances the average force of binding of unphosphorylated myosin to actin which is contrary to what both Albrecht et al. [28] and Guo et al. [29] suggested from data obtained at the whole tissue level. However, once CaD gets phosphorylated by ERK, its Funb becomes lower than in the presence of unphosphorylated CaD, and even lower than in the absence of CaD. Thus, ERK-phosphorylated CaD favors relaxation.
The mechanism that we propose for the contribution of ERK phosphorylation of CaD to smooth muscle relaxation becomes even more appealing when we consider the published data on the timing of ERK activation. Ratz [40] showed that upon stimulation of rabbit arterial smooth muscle with 10μM phenylephrine, the maximum active force was reached within 1 min whereas the phosphorylation of ERK reached its peak only at 5 min. Thus, this time course in conjunction with our data suggest that before its activation, CaD favors force maintenance (latch state/cooperative reattachment) by increasing unphosphorylated/dephosphorylated myosin average binding force to actin (Figs. 2, 5 and 6). Later in the contraction, once CaD becomes phosphorylated by ERK, this binding force is decreased to very low levels (Figs.5 & 6), thereby allowing the muscle to relax.
How does CaD accomplish these functions becomes the next question. To address this point, it is informative to look more closely at CaD's binding sites. CaD binds via its N-terminus to the S2 region of myosin [41-44]. The C-terminal region of CaD [45-48] has two strong actin-binding clusters whereas the N-terminal region has only weak actin binding sites [48]. The cluster closest to the C-terminus has been reported to be responsible for the inhibitory action of CaD [46, 49]. It also contains the site that gets phosphorylated by ERK [50]. The ERK phosphorylation of CaD has been reported to “alleviate” the inhibitory effect of CaD on the actomyosin ATPase activity [13] and on the in vitro actin propulsion velocity [26] by detaching CaD from actin [13]. Only the phosphorylated cluster detaches while the other one remains attached [13]. It has been shown that it is kinetically advantageous that CaD stays close to actin and does not detach completely [13, 51]. Our results demonstrate that indeed, when attached to actin via its 2 carboxyl sites, CaD enhances the average force of binding of unphosphorylated myosin to actin, presumably by the cross-linking of myosin to actin by CaD. Thus, CaD promotes force maintenance. Upon ERK phosphorylation of CaD, and thus detachment of CaD's ERK-phosphorylatable site from actin, the average force of binding becomes lower than in the absence of CaD. Thus, ERK-phosphorylated CaD promotes relaxation. The exact mechanism by which this occurs will require more structure/function studies involving CaD and ERK-phosphorylated CaD, in the presence of actin and phosphorylated and unphosphorylated myosin.
Other regulatory mechanisms of CaD have also previously been studied. For instance, phosphorylation at the carboxyl terminus of CaD by MLCK increases its binding affinity for phosphorylated myosin filaments [10]. Phosphorylation of CaD by MLCK must take place early in the contraction favoring binding of myosin to actin and promoting contraction. Indeed, the actin activated ATPase rate is increased in the presence of MLCK phosphorylated CaD [10]. This mechanism is not believed to be active once myosin is dephosphorylated because MLCK phosphorylated CaD does not bind with high affinity to unphosphorylated myosin [10]. Thus, a series of mechanisms must be orchestrated for the smooth muscle cell to sequentially favor cross-bridge cycling, followed by force maintenance and finally followed by relaxation. Fine tuning seems to be possible due to the involvement of several factors including the contractile proteins expressed, the signaling involved and its exact timing.
A closer look at the maneuvers used to produce the Funb traces, i.e. the pulling at constant velocity of the pedestal/myosin away from the laser trap center, allows us to speculate on the mechanisms by which CaD alters the binding of unphosphorylated myosin to actin. CaD increases the Funb (Figs. 6A & B) whereas ERK-phosphorylated CaD decreases it to very low values (Figs.6A-C). The binding of unphosphorylated myosin to actin clearly occurs in the presence of CaD and ERK-phosphorylated CaD but in the latter case, detachment occurs before significant displacement can occur (Fig.6C). Average forces are the product of unitary forces produced by single myosin molecules and their duty cycle (the percentage of time spent bound to actin and maintaining force) [52]. Thus, CaD and phosphorylated CaD might either alter the unitary force of binding of unphosphorylated myosin to actin or its time of attachment or the time between individual myosin molecule attachments. Understanding this further will require single myosin molecular mechanics studies. Furthermore, figures 3A-C and 6D demonstrate that these behaviors are true functions of myosin because they are abolished in its absence.
Another point that is worth noting is the fact that tropomyosin is usually believed to potentiate the effects of CaD. We (Fig. 4) and others [21, 23, 48] have reported an increase in νmax as measured in the in vitro motility assay in the presence of tropomyosin, a decrease in the presence of CaD and a further decrease in the presence of both tropomyosin and CaD. As others have reported [21, 23, 48], the potentiation of the effect of CaD by tropomyosin on νmax is very small but statistically significant. However, this potentiating effect of tropomyosin on CaD does not seem to play a role in the presence of unphosphorylated myosin as can be seen in figure 2. Taking that into consideration, we chose not to pursue the mechanics measurements on unphosphorylated myosin in the presence of tropomyosin.
A limitation of our study is that unphosphorylated and not dephosphorylated myosin was used in our measurements. Although there is no information in the literature to support this idea, there is a possibility that dephosphorylated myosin is functionally different from unphosphorylated myosin. Unfortunately, it is not possible for us to work with dephosphorylated myosin because of the high probability that there would be some remaining phosphorylated myosin that would greatly overestimate our force measurements due to the sensitivity of our equipment.
It is interesting to note that there is more CaD in phasic than in tonic muscle. Furthermore, Guo and co-workers reported that, in their CaD knock-out animal, the non-muscle isoform of CaD was over-expressed in phasic but not in tonic muscle [29]. Taken together these observations suggest that CaD should play an essential role in phasic muscle. As suggested by Guo et al. [29], CaD might be important for the rapid relaxation observed in the phasic muscle but which is not seen in tonic smooth muscle.
5. Conclusions
In conclusion, CaD enhances the force of binding of unphosphorylated, and presumably dephosphorylated, myosin to actin. Furthermore, ERK phosphorylation of CaD decreases this force of binding to very low levels, thus promoting relaxation.
Highlights.
Molecular level myosin force measurements.
Molecular mechanical mechanism to explain relaxation from the latch-state.
Physiological significance of caldesmon phosphorylation sites.
Acknowledgments
We thank Professor John M. Dealy, from the department of Chemical Engineering, McGill University, for performing the methylcellulose viscosity measurements and Michel Alveis from Marvid Poultry for the procurement of the chicken breasts for the purification of skeletal muscle actin and myosin for NEM preparation.
Grants: This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) grant, National Heart, Lung, and Blood Institute grant RO1-HL 103405-02, and the Costello Fund. The Meakins-Christie Laboratories (McGill University Health Centre Research Institute) are supported in part by a center grant from Le Fonds de la Recherche en Santé du Québec (FRSQ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495–497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- 2.Hai CM, Murphy RA. Regulation of shortening velocity by cross-bridge phosphorylation in smooth muscle. Am J Physiol. 1988;255:C86–94. doi: 10.1152/ajpcell.1988.255.1.C86. [DOI] [PubMed] [Google Scholar]
- 3.Huxley AF. Muscle structure and theories of contraction. Prog Biophys. 1957;7:255–317. [PubMed] [Google Scholar]
- 4.Leguillette R, Zitouni NB, Govindaraju K, Fong LM, Lauzon AM. Affinity for MgADP and force of unbinding from actin of myosin purified from tonic and phasic smooth muscle, American journal of physiology. Cell physiology. 2008;295:C653–660. doi: 10.1152/ajpcell.00100.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roman HN, Zitouni NB, Kachmar L, Ijpma G, Hilbert L, Matusovskiy O, Benedetti A, Sobieszek A, Lauzon AM. Unphosphorylated calponin enhances the binding force of unphosphorylated myosin to actin. Biochim Biophys Acta. 2013;1830:4634–4641. doi: 10.1016/j.bbagen.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J Cell Biol. 1990;111:453–463. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haeberle JR. Thin-filament linked regulation of smooth muscle myosin. J Muscle Res Cell Motil. 1999;20:363–370. doi: 10.1023/a:1005408402323. see comments. [DOI] [PubMed] [Google Scholar]
- 8.Hemric ME, Chalovich JM. Effect of caldesmon on the ATPase activity and the binding of smooth and skeletal myosin subfragments to actin. J Biol Chem. 1988;263:1878–1885. [PubMed] [Google Scholar]
- 9.Hai CM, Kim HR. An expanded latch-bridge model of protein kinase C-mediated smooth muscle contraction. J Appl Physiol. 2005;98:1356–1365. doi: 10.1152/japplphysiol.00834.2004. [DOI] [PubMed] [Google Scholar]
- 10.Sobieszek A, Sarg B, Lindner H, Seow CY. Phosphorylation of caldesmon by myosin light chain kinase increases its binding affinity for phosphorylated myosin filaments. Biol Chem. 2010;391:1091–1104. doi: 10.1515/BC.2010.105. [DOI] [PubMed] [Google Scholar]
- 11.Horiuchi KY, Chacko S. Effect of unphosphorylated smooth muscle myosin on caldesmon-mediated regulation of actin filament velocity. J Muscle Res Cell Motil. 1995;16:11–19. doi: 10.1007/BF00125306. [DOI] [PubMed] [Google Scholar]
- 12.Sutherland C, Walsh MP. Phosphorylation of caldesmon prevents its interaction with smooth muscle myosin. J Biol Chem. 1989;264:578–583. [PubMed] [Google Scholar]
- 13.Huang R, Li L, Guo H, Wang CL. Caldesmon binding to actin is regulated by calmodulin and phosphorylation via different mechanisms. Biochemistry (Mosc) 2003;42:2513–2523. doi: 10.1021/bi0268605. [DOI] [PubMed] [Google Scholar]
- 14.Morgan KG, Gangopadhyay SS. Invited review: cross-bridge regulation by thin filament-associated proteins. J Appl Physiol. 2001;91:953–962. doi: 10.1152/jappl.2001.91.2.953. [DOI] [PubMed] [Google Scholar]
- 15.Ngai PK, Walsh MP. Inhibition of smooth muscle actin-activated myosin Mg2+-ATPase activity by caldesmon. J Biol Chem. 1984;259:13656–13659. [PubMed] [Google Scholar]
- 16.Wang CL. Caldesmon and the regulation of cytoskeletal functions. Adv Exp Med Biol. 2008;644:250–272. doi: 10.1007/978-0-387-85766-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngai PK, Walsh MP. The effects of phosphorylation of smooth-muscle caldesmon. Biochem J. 1987;244:417–425. doi: 10.1042/bj2440417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dabrowska R, Goch A, Galazkiewicz B, Osinska H. The influence of caldesmon on ATPase activity of the skeletal muscle actomyosin and bundling of actin filaments. Biochim Biophys Acta. 1985;842:70–75. doi: 10.1016/0304-4165(85)90295-8. [DOI] [PubMed] [Google Scholar]
- 19.Horiuchi KY, Miyata H, Chacko S. Modulation of smooth muscle actomyosin ATPase by thin filament associated proteins. Biochem Biophys Res Commun. 1986;136:962–968. doi: 10.1016/0006-291x(86)90426-2. [DOI] [PubMed] [Google Scholar]
- 20.Sobue K, Takahashi K, Wakabayashi I. Caldesmon150 regulates the tropomyosin-enhanced actin-myosin interaction in gizzard smooth muscle. Biochem Biophys Res Commun. 1985;132:645–651. doi: 10.1016/0006-291x(85)91181-7. [DOI] [PubMed] [Google Scholar]
- 21.Okagaki T, Higashi-Fujime S, Ishikawa R, Takano-Ohmuro H, Kohama K. In vitro movement of actin filaments on gizzard smooth muscle myosin: requirement of phosphorylation of myosin light chain and effects of tropomyosin and caldesmon. J Biochem. 1991;109:858–866. doi: 10.1093/oxfordjournals.jbchem.a123471. [DOI] [PubMed] [Google Scholar]
- 22.Shirinsky VP, Biryukov KG, Hettasch JM, Sellers JR. Inhibition of the relative movement of actin and myosin by caldesmon and calponin. J Biol Chem. 1992;267:15886–15892. [PubMed] [Google Scholar]
- 23.Fraser ID, Marston SB. In vitro motility analysis of smooth muscle caldesmon control of actin-tropomyosin filament movement. J Biol Chem. 1995;270:19688–19693. doi: 10.1074/jbc.270.34.19688. [DOI] [PubMed] [Google Scholar]
- 24.Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle, American journal of physiology. Cell physiology. 2000;278:C718–726. doi: 10.1152/ajpcell.2000.278.4.C718. [DOI] [PubMed] [Google Scholar]
- 25.Hemric ME, Lu FW, Shrager R, Carey J, Chalovich JM. Reversal of caldesmon binding to myosin with calcium-calmodulin or by phosphorylating caldesmon. J Biol Chem. 1993;268:15305–15311. [PMC free article] [PubMed] [Google Scholar]
- 26.Gerthoffer WT, Yamboliev IA, Shearer M, Pohl J, Haynes R, Dang S, Sato K, Sellers JR. Activation of MAP kinases and phosphorylation of caldesmon in canine colonic smooth muscle. The Journal of physiology. 1996;495(Pt 3):597–609. doi: 10.1113/jphysiol.1996.sp021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil RA, Menice CB, Wang CL, Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ Res. 1995;76:1101–1108. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- 28.Albrecht K, Schneider A, Liebetrau C, Ruegg JC, Pfitzer G. Exogenous caldesmon promotes relaxation of guinea-pig skinned taenia coli smooth muscles: inhibition of cooperative reattachment of latch bridges? Pflugers Arch. 1997;434:534–542. doi: 10.1007/s004240050433. [DOI] [PubMed] [Google Scholar]
- 29.Guo H, Huang R, Semba S, Kordowska J, Huh YH, Khalina-Stackpole Y, Mabuchi K, Kitazawa T, Wang CL. Ablation of smooth muscle caldesmon affects the relaxation kinetics of arterial muscle. Pflugers Arch. 2013;465:283–294. doi: 10.1007/s00424-012-1178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobieszek A. Smooth muscle myosin. Molecule conformation, filament assembly and association of regulatory enzymes. In: Giembycz aMA., DR, editor. Airways smooth muscle: Biochemical Control of Contraction and Relaxation. Birkhauser; Bassel: 1994. pp. 1–29. [Google Scholar]
- 31.Sobieszek A, Small JV. Regulation of the actin-myosin interaction in vertebrate smooth muscle: activation via a myosin light-chain kinase and the effect of tropomyosin. J Mol Biol. 1977;112:559–576. doi: 10.1016/s0022-2836(77)80164-2. [DOI] [PubMed] [Google Scholar]
- 32.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 33.Trybus KM, Lowey S. Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J Biol Chem. 1984;259:8564–8571. [PubMed] [Google Scholar]
- 34.Hilbert L, Cumarasamy S, Zitouni NB, Mackey MC, Lauzon AM. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophys J. 2013;105:1466–1474. doi: 10.1016/j.bpj.2013.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem. 1993;268:14764–14768. [PubMed] [Google Scholar]
- 36.Kishino A, Yanagida T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 1988;334:74–76. doi: 10.1038/334074a0. [DOI] [PubMed] [Google Scholar]
- 37.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J Mol Biol. 1990;214:699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 38.VanBuren P, Work SS, Warshaw DM. Enhanced force generation by smooth muscle myosin in vitro. Proc Natl Acad Sci U S A. 1994;91:202–205. doi: 10.1073/pnas.91.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steel RGD, Torrie JH. In: Principles and Procedures of Statistics. McGraw-Hill, editor. New York: 1980. [Google Scholar]
- 40.Ratz PH. Regulation of ERK phosphorylation in differentiated arterial muscle of rabbits, American journal of physiology. Heart and circulatory physiology. 2001;281:H114–123. doi: 10.1152/ajpheart.2001.281.1.H114. [DOI] [PubMed] [Google Scholar]
- 41.Hemric ME, Chalovich JM. Characterization of caldesmon binding to myosin. J Biol Chem. 1990;265:19672–19678. [PMC free article] [PubMed] [Google Scholar]
- 42.Ikebe M, Reardon S. Binding of caldesmon to smooth muscle myosin. J Biol Chem. 1988;263:3055–3058. [PubMed] [Google Scholar]
- 43.Li Y, Zhuang S, Guo H, Mabuchi K, Lu RC, Wang CA. The major myosin-binding site of caldesmon resides near its N-terminal extreme. J Biol Chem. 2000;275:10989–10994. doi: 10.1074/jbc.275.15.10989. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Jiang H, Yang ZQ, Chacko S. Both N-terminal myosin-binding and C-terminal actin-binding sites on smooth muscle caldesmon are required for caldesmon-mediated inhibition of actin filament velocity. Proc Natl Acad Sci U S A. 1997;94:11899–11904. doi: 10.1073/pnas.94.22.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao Y, Patchell VB, Huber PA, Copeland O, El-Mezgueldi M, Fattoum A, Calas B, Thorsted PB, Marston SB, Levine BA. The interface between caldesmon domain 4b and subdomain 1 of actin studied by nuclear magnetic resonance spectroscopy. Biochemistry (Mosc) 1999;38:15459–15469. doi: 10.1021/bi991383k. [DOI] [PubMed] [Google Scholar]
- 46.Wang CL, Wang LW, Xu SA, Lu RC, Saavedra-Alanis V, Bryan J. Localization of the calmodulin- and the actin-binding sites of caldesmon. J Biol Chem. 1991;266:9166–9172. [PubMed] [Google Scholar]
- 47.Wang Z, Horiuchi KY, Chacko S. Characterization of the functional domains on the C-terminal region of caldesmon using full-length and mutant caldesmon molecules. J Biol Chem. 1996;271:2234–2242. doi: 10.1074/jbc.271.4.2234. [DOI] [PubMed] [Google Scholar]
- 48.Zhuang S, Mabuchi K, Wang CA. Heat treatment could affect the biochemical properties of caldesmon. J Biol Chem. 1996;271:30242–30248. doi: 10.1074/jbc.271.47.30242. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Yang ZQ, Chacko S. Functional and structural relationship between the calmodulin-binding, actin-binding, and actomyosin-ATPase inhibitory domains on the C terminus of smooth muscle caldesmon. J Biol Chem. 1997;272:16896–16903. doi: 10.1074/jbc.272.27.16896. [DOI] [PubMed] [Google Scholar]
- 50.Adam LP, Hathaway DR. Identification of mitogen-activated protein kinase phosphorylation sequences in mammalian h-Caldesmon. FEBS Lett. 1993;322:56–60. doi: 10.1016/0014-5793(93)81110-l. [DOI] [PubMed] [Google Scholar]
- 51.Graceffa P, Mazurkie A. Effect of caldesmon on the position and myosin-induced movement of smooth muscle tropomyosin bound to actin. J Biol Chem. 2005;280:4135–4143. doi: 10.1074/jbc.M410375200. [DOI] [PubMed] [Google Scholar]
- 52.VanBuren P, Waller GS, Harris DE, Trybus KM, Warshaw DM, Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc Natl Acad Sci U S A. 1994;91:12403–12407. doi: 10.1073/pnas.91.26.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]