

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV), like all herpesviruses, encodes a protease (KSHV Pr), which is necessary for the viral lytic cycle. Herpesvirus proteases function as obligate dimers; however, each monomer has an intact, complete active site which does not interact directly with the other monomer across the dimer interface. Protein grafting of an interfacial KSHV Pr α-helix onto a small stable protein, avian pancreatic polypeptide, generated a helical 30-amino-acid peptide designed to disrupt the dimerization of KSHV Pr. The chimeric peptide was optimized through protein modeling of the KSHV Pr-peptide complex. Circular dichroism analysis and gel filtration chromatography revealed that the rationally designed peptide adopts a helical conformation and is capable of disrupting KSHV Pr dimerization, respectively. Additionally, the optimized peptide inhibits KSHV Pr activity by 50% at a ∼200-fold molar excess of peptide to KSHV Pr, and the dissociation constant was estimated to be 300 μM. Mutagenesis of the interfacial residue M197 to a leucine resulted in an inhibitory concentration which was twofold higher for KSHV Pr M197L than for KSHV Pr, in agreement with the model that the dimer interface is involved in peptide binding. These results indicate that the dimer interface, as well as the active sites, of herpesvirus proteases is a viable target for inhibiting enzyme activity.
Herpesviruses are enveloped double-stranded DNA viruses that are the causative agents of many diseases in humans and other animals (35). The nine known human herpesviruses have been classified into three subfamilies: alpha, beta, and gamma. All herpesviruses express a serine protease late in the viral lytic cycle; this serine protease processes the assembly protein (AP) scaffold upon which the immature viral capsid is initially formed. Processing of the roughly 103 AP molecules contained in each immature capsid, at a conserved maturation site near the AP carboxyl terminus, leads to release of the AP scaffold from the capsid, capsid angularization, and DNA encapsidation (8, 30). A temperature-sensitive mutant of the herpes simplex virus type 1 protease prevents the virus from forming mature infectious virions at the nonpermissive temperature, revealing the necessity of herpesvirus proteases for completion of the viral lytic cycle and spread of the virus (7, 26).
Herpesvirus proteases exist in a monomer-dimer equilibrium in solution; the dimeric protease is the only active species (5, 6, 12, 17, 32). All crystal structures of herpesvirus proteases show the same overall structure. Each monomer contains a complete active site with a catalytic triad consisting of a serine and two histidines instead of the typical Ser-His-Asp triad found in other serine proteases. Additionally, the active site of each monomer does not participate in any direct interactions with the active site of the other monomer across the dimer interface (3, 13, 27-29, 33, 36). However, dimerization is still required for activity. Inhibitory molecules that bind the dimer interface are expected to have high specificity for the protease due to the unique surfaces and large hydrophobic pockets which make up the dimer interface.
In addition, inhibitors disrupting dimerization are attractive agents against herpesvirus proteases because they target a second site, separate from the active site, and because this class of inhibitors will presumably be able to bind to monomeric forms of the protease. Binding to the inactive monomeric protease before the enzyme is able to dimerize and become active would allow this class of inhibitors to bind to the protease in the cytoplasm as well as in the nucleus, where it is believed to form active dimers (34). Binding to the protease before encapsidation would remove the problem of transporting the inhibitor into both the nucleus and the immature capsid independently of the target protease. This may increase the local concentration of the inhibitor at the site of action by changing the compartments where protease binding can occur and removing the necessity for the difficult task of targeting molecules to specific organelles. However, there are no published reports of dimerization-disrupting inhibitors of herpesvirus proteases, and therefore the exact effects of dimer disruption are as yet unknown.
Kaposi's sarcoma (KS), the most common neoplasm affecting end stage AIDS patients, is caused by a gammaherpesvirus, KS-associated herpesvirus (KSHV). Like all herpesviruses, KSHV expresses and requires a virally encoded protease, KSHV Pr (24, 25, 29, 37). Like those of other herpesvirus proteases, the dimer interface of KSHV Pr is composed primarily of helix α5. The helix α5 of one monomer interacts with residues in the helix α5 of the other monomer across the dimer interface as well as with helices α1 and α2 of the opposite monomer, forming the dimer interface and stabilizing the active dimeric conformation. In this study, we describe a new class of herpesvirus protease inhibitors that disrupt KSHV Pr dimerization and thereby inhibit the activity of the protease.
The peptide inhibitor was rationally designed to mimic the interfacial helix α5, based on the crystal structure of KSHV Pr. The KSHV Pr α5 helix was “grafted” onto the scaffold provided by avian pancreatic polypeptide (aPP) (38) to construct a stabilized form of the interfacial helix of KSHV Pr. aPP is a stable soluble protein consisting of an α-helix stabilized by a type II polyproline helix. Modeling of the KSHV Pr monomer-peptide complex followed the protein grafting in order to optimize and minimize the peptide sequence. The 30-amino-acid peptide designed disrupts dimerization and inhibits KSHV Pr activity, indicating that the dimer interface of herpesvirus proteases is a viable target for the inhibition of herpesvirus proteases and a potential therapeutic target for the treatment of herpesvirus diseases.
MATERIALS AND METHODS
Materials.
A 16-amino-acid peptide (PLETLMAKAIDAGFIR) was synthesized by solid-phase peptide synthesis using the 9-fluorenylmethoxy carbonyl (Fmoc) strategy and was purified with a reversed phase-C18 column. The amino terminus of the peptide was acetylated. A 30-amino-acid peptide (PSQPTYPGDDAPLEDLMAFAIDLSFYLGVV) was purchased from the Biomolecular Resource Center at the University of California San Francisco. The 16- and 30-amino-acid peptides are referred to here as the α5 peptide and the KSDD (KSHV Pr dimer disrupter) peptide. A variant of the KSDD peptide (PSQPTYPGDDAPLEDLLAFAIDLSFYLGVV) was also purchased and is referred to as KSDD M17L. The identities and purity of these peptides were verified by matrix-assisted laser desorption ionization mass spectrometry (Precision Biosystems, Framingham, Mass.) and high-preformance liquid chromatography with an analytical C4 column. The calculated and observed masses were 1,788.1 and 1,791.0 for the α5 peptide, 3,265.7 and 3,266.7 for the KSDD peptide, and 3,247.6 and 3,250.0 for the KSDD M17L peptide, respectively.
KSHV Pr variants.
Mutations encoding the amino acid substitutions S114A, M197L, M197F, A203S, A203E, and S204D were introduced into the KSHV Pr S204G gene by using the QuikChange mutagenesis kit (Stratagene). Residues S114, M197, A203, and G204 are shown in the crystal structure of KSHV Pr S204G in Fig. 2B. The integrity of the KSHV Pr open reading frame and the presence of the mutations were verified by DNA sequencing. KSHV Pr S204G (referred to here as KSHV Pr) and its variants were expressed and purified as described previously (24).
FIG. 2.

Strategy for the design of the KSDD peptide. (A) Sequence alignment of aPP and the α5 helix in KSHV Pr, and designed sequences of the KSaPP and KSDD peptides. Specific residue numbers are given above peptide sequences, and the corresponding residue numbers in KSHV Pr are given in parentheses. The nine residues in the α5 helix of one monomer of KSHV Pr that interact with the second monomer of KSHV Pr are shown in red. Residues in the aPP sequence that are required for stabilizing the helical structure are shown in green. Based on the model structure of the KSHV Pr-KSaPP complex (panel C), conflicting or functionally unnecessary residues in KSaPP are shown in yellow. The mutation site for attenuation of autolysis is G13 in the α5 peptide. (B) Dimer interface in the crystal structure of KSHV Pr. Residues in the dimer interface are shown in red and purple, and those in the catalytic triad are shown in orange. (C) A model structure of the KSHV Pr-KSaPP peptide complex was constructed by replacing the α5 helix in one monomer of KSHV Pr with aPP. Formation of the hydrophobic aPP core requires residues on only one face of the α-helix (shown in green in panel A). The opposite, solvent-exposed face of the α-helix is available for recognition of KSHV Pr; these residues (shown in red) interact with the other monomer. A combined sequence was generated and referred to as KSaPP. The wild-type KSHV Pr serine residue was reintroduced at the position of G13 in the KSaPP peptide because the autolysis site present in KSHV Pr and the α5 peptide no longer exists in KSaPP. The KSDD peptide was finally designed by replacing N29 with a glycine and by removing G1 and T32 through Y36.
CD analysis.
Circular dichroism (CD) spectra were collected on a JASCO spectrapolarimeter. Fifty micromolar solutions of the peptides in 25 mM potassium phosphate were placed in a 0.1-cm-pathlength cuvette, and CD spectra were measured from 185 to 250 nm. The spectra were normalized by using a buffer solution without peptide. An α-helix content was calculated by a Kohonen neural network with a 2-dimensional output layer (1).
Gel filtration.
Size exclusion chromatography was performed with a Superdex-75 HR column (Pharmacia). Samples (100 μl) were loaded in 25 mM potassium phosphate (pH 7.0), 150 mM NaCl, 0.1 mM EDTA, 1 mM β-mercaptoethanol at room temperature. The concentration of KSHV Pr was 5 μM, and that of the α5 or KSDD peptide was 500 μM, resulting in a 100-fold excess of peptide over KSHV Pr. In mixing experiments with KSHV Pr and the α5 or KSDD peptide, samples were loaded after overnight incubations at 4°C.
Protease activity.
KSHV Pr activity assays were performed by using either a synthetic oligopeptide substrate containing a fluorescent donor-quencher pair connected by the native KSHV R-site sequence as described previously (referred to below as the R-site substrate) or 7-amino-4-carbamoylmethyl coumarin (ACC) attached to a peptide with the sequence Tyr-tert-butylglycine-Gln-Ala or Tyr-Val-Glu-Ala; these were identified as substrates from a substrate library (11, 18). Only the Tyr-tert-butylglycine-Gln-Ala-ACC substrate results are shown in Fig. 1 and 5; however, the profiles obtained for the R-site and Tyr-Val-Glu-Ala-ACC substrates were identical to those determined by using the ACC substrate. The assay buffer used consisted of 25 mM potassium phosphate (pH 8.0), 150 mM KCl, 0.1 mM EDTA, and 1 mM β-mercaptoethanol. Assays were performed at 30°C in 0.2 ml of assay buffer on a Microplate spectrofluorometer (Molecular Devices) as described previously (25). The concentration of KSHV Pr was 2 μM throughout the experiments, except that 0.5 μM KSHV Pr was used in the mixing experiment with KSHV Pr S114A. The concentrations of KSHV Pr S114A and the peptides were varied as shown in Fig. 1 and 5. Activity assays of mixtures containing KSHV Pr with either KSHV Pr S114A or one of the peptides were monitored after overnight incubations at 4°C. In order to estimate the inhibitory reaction rate of the KSDD peptide, assays were performed 0.08, 0.52, 0.92, 1.4, 1.9, 2.4, 3.1, 4.4, and 6.0 h after mixing of 2 μM KSHV Pr with 800 μM KSDD peptide.
FIG. 1.

Dependence of KSHV Pr (WT) specific activity on the relative concentration of KSHV Pr S114A. Experimental concentrations of KSHV Pr are 2 (open diamonds) and 0.5 (filled circles) μM. Theoretical curves for the relative specific activities are shown at KSHV Pr concentrations of 2 (thin lines) and 0.5 (thick lines) μM. If the heterodimer of KSHV Pr and KSHV Pr S114A is active (dashed lines) or inactive (solid lines), the specific activity is expected to increase or decrease, respectively. The experimental data are in agreement with the heterodimer being active. Note that specific activity at 5.5 μM, which is the highest concentration of KSHV Pr S114A in this experiment, was not detectable, verifying that KSHV Pr S114A does not display any activity at these concentrations.
FIG. 5.

(A) Peptide concentration dependence of KSHV Pr specific activity. The KSDD peptide inhibits KSHV Pr (filled circles) or KSHV Pr M197L (filled diamonds) activity by 50% at a ∼200- or 400-fold molar excess of peptide over KSHV Pr or KSHV Pr M197L, respectively. The inhibitory concentrations of the KSDD M17L peptide for KSHV Pr (open circles) and KSHV Pr M197L (open diamonds) were shifted lower than those of the KSDD peptide with the corresponding protease. In contrast, the α5 peptide did not inhibit KSHV Pr activity at concentrations up to 400-fold higher than KSHV Pr concentrations (open triangles). (B) Time dependence of KSDD peptide inhibition, assayed 0.08, 0.52, 0.92, 1.4, 1.9, 2.4, 3.1, 4.4, and 6.0 h after the mixing of 2 μM KSHV Pr with a 400-fold excess of the KSDD peptide, the highest concentration used in panel A.
RESULTS
Increase in KSHV Pr activity in the presence of the defective mutant.
The ability of a catalytically inactive protease variant to stabilize or destabilize wild-type KSHV Pr is shown in Fig. 1. To allow comparisons on the same scale at the different enzyme concentrations, the activities in Fig. 1 are plotted as the relative values divided by those of 2 and 0.5 μM KSHV Pr without KSHV Pr S114A, respectively. The competition of the variant for binding to the substrate is negligible, because the substrate concentration (160 μM) is >29-fold higher than the protease concentration. At the highest total protease concentration used in this experiment, no activity was observed for KSHV Pr S114A alone, which lacks the nucleophilic serine residue (data not shown). However, the activity of KSHV Pr increased in the presence of increasing amounts of KSHV Pr S114A. At concentrations of 2 and 0.5 μM KSHV Pr in assay buffer, the protease is in equilibrium between the monomer and the dimer, with only the dimer having activity. The increase in KSHV Pr specific activity in the presence of increasing amounts of KSHV Pr S114A indicates that the KSHV Pr monomer is capable of forming an active heterodimer with the inactive KSHV Pr S114A when the two proteins are mixed. The increase in the specific activity at 0.5 μM KSHV Pr was higher than that at 2 μM, since the dimer fraction, without KSHV Pr S114A, at 0.5 μM KSHV Pr is less than that at 2 μM KSHV Pr. Heterodimer formation increases the concentration of the active form of KSHV Pr, leading to the increased specific activity of KSHV Pr in the presence of the inactive variant. This result is consistent with the results of a related experiment on the human cytomegalovirus protease, a member of the family of herpesvirus proteases (16).
Theoretical curves were generated from equations 7 and 8 (see Appendix) for the KSHV Pr specific activity depending on the KSHV Pr S114A concentration (Fig. 1). If the activity of the KSHV Pr-KSHV Pr S114A heterodimer is exactly half that of the KSHV Pr dimer, the specific activity is expected to increase, which is in agreement with the experimental data at both 2 and 0.5 μM KSHV Pr. In contrast, if the heterodimer is inactive, decreased specific activity should be observed. Comparison of experimental data with the theoretical curves for 0.5 and 2 μM KSHV Pr suggests that KSHV Pr and the defective protease form an active heterodimer, and one active site is sufficient for protease activity. The defective monomer is presumably able to stabilize the active conformation of KSHV Pr.
Design of the helical inhibitory peptide.
Both homodimeric and heterodimeric formations stabilize the active conformation of KSHV Pr. A minimal interface containing the major structural element for dimerization, α5, but lacking the α1 and α2 helices was constructed (Fig. 2B). A 16-amino-acid peptide was designed to disrupt dimerization and inhibit KSHV Pr activity by interfering with interactions across the dimer interface. The KSHV Pr interfacial α5 helix amino acid sequence used for the peptide includes a naturally occurring autolysis site. Autolysis of the protease was attenuated through the S204G mutation (24), and the corresponding S13G mutation was included in the peptide to provide stability against proteolysis by KSHV Pr, resulting in the α5 peptide (Fig. 2A). Since the α5 peptide lacks the other elements of the KSHV Pr interface which may contribute to helix formation, we considered a strategy that would stabilize the helix. Formation of the α5 helical structure present in the KSHV Pr dimer, free in solution, would remove the necessity for helix folding upon binding to KSHV Pr and should improve the binding affinity of the peptide.
Protein grafting was used as a method of designing a stabilized α-helix that would mimic the interfacial helix of KSHV Pr. aPP was chosen as the scaffold for protein grafting because it is a small, well-folded protein consisting of an α-helix stabilized by hydrophobic interactions with a type II polyproline helix (9). Only residues on one face of the aPP α-helix are required for stabilizing the helical structure (38). Solvent-exposed residues on the opposite face of the α-helix can be replaced with amino acids from the KSHV Pr α5 helix that are critical for the recognition of the complementary face of the dimer interface. Inspection of the crystal structure of KSHV Pr reveals nine residues in the α5 helix of one monomer of KSHV Pr that interact with the second monomer of KSHV Pr. Alignment of the aPP and α5 helix sequences shows that any amino acid position that is needed for stabilization of the α-helix in the aPP structure is independent of the amino acids used for recognition of the KSHV Pr dimer interface. It is therefore possible to design a peptide incorporating all amino acids necessary for both helix stability and KSHV Pr interface recognition without any conflicts or loss of functionally important residues. This initial fusion peptide is referred to as KSaPP.
The fusion peptide was further refined through protein modeling of the KSHV Pr-KSaPP complex, formed by one monomer from the X-ray crystal structure of the KSHV Pr dimer and a second KSHV Pr monomer in which helix α5 had been replaced with aPP. Because the main chain trace of the α-helix in KSaPP was virtually superimposable on the α5 helix in KSHV Pr, the docked model was considered to be a good representation of the desired interaction between KSaPP and KSHV Pr. The model of the KSaPP-KSHV Pr complex revealed steric clashes between the side chains of N29, T32, and the C-terminal residues in KSaPP and residues in KSHV Pr. In addition, it has been reported that the C-terminal residues in the aPP structure are disordered (9). We hypothesized that neither G1 in the N terminus nor the C-terminal residues of KSaPP are necessary either for interaction with KSHV Pr or for formation of the α-helix. Therefore, because of potential steric clashes between KSaPP and KSHV Pr, N29 in the KSaPP sequence was replaced with a glycine, G1 in the N terminus was removed, and T32 through Y36 in the C terminus were removed. These additional modifications to KSaPP resulted in a 30-amino-acid peptide (the KSDD peptide) designed to disrupt the KSHV Pr dimer.
Peptide secondary-structure determination.
CD experiments were performed to characterize the secondary structure of the α5 and KSDD peptides (Fig. 3). The CD spectrum of the α5 peptide did not possess characteristics representative of α-helical structure, suggesting that this peptide sequence in itself does not form a stable α-helix. In contrast, CD analysis of the KSDD peptide revealed that it has a helical conformation, showing the expected spectral minimums at 208 and 222 nm, characteristic of α-helices. Although the KSDD peptide contains 10 substitutions and truncations at the N and C termini relative to the aPP sequence, the retained residues that stabilize the helical conformation appear to be sufficient to preserve its scaffold.
FIG. 3.

CD spectra of the KSDD and α5 peptides. Spectra were acquired by scanning 10 times at 20°C and were normalized by using a buffer solution without peptide.
The secondary-structure analysis program calculated an α-helical content of 40% for the CD spectrum of the KSDD peptide. The helicity calculated for aPP from the CD spectra was previously determined to be 40% (22). For neuropeptide Y and peptide YY, which are in the same PP-fold family, the helicities were determined to be 28% (23) and 42% (15), respectively. The helicity of the KSDD peptide, which includes truncation at the disordered C-terminal end, was expected to be slightly higher than that of aPP. However, our result is consistent with the range of helicity. The helicity of the truncated peptide is expected to be between 34 and 51%, based on the previously reported helicities for the PP-fold family. This result suggests that the aPP structure is maintained in the KSDD peptide.
Disruption of KSHV Pr dimerization.
To examine the disruption of KSHV Pr dimerization by the peptides, the monomer-dimer equilibrium of KSHV Pr was analyzed in the presence of the α5 or KSDD peptide. At a KSHV Pr concentration of 5 μM, gel filtration chromatography shows that the protease is in equilibrium between the monomer and the dimer, with virtually equal amounts of the monomeric and dimeric species (Fig. 4A). In the presence of a 100-fold molar excess of the α5 peptide (500 μM), the gel filtration profile was quite similar to that without the peptide, except that the α5 peptide was in the later fraction (data not shown). This result shows that KSHV Pr dimerization was not disrupted by the α5 peptide. In contrast, in the presence of a 100-fold molar excess of the KSDD peptide (500 μM), the ratio of the KSHV Pr dimer to the KSHV Pr monomer is dramatically reduced, indicating that protease dimerization is disrupted by the KSDD peptide. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis verified the lack of dimeric protease in the presence of the KSDD peptide compared to the uninhibited protease (Fig. 4B). The results showed that the peaks of interest are KSHV Pr and that addition of the KSDD peptide perturbs the dimerization of KSHV Pr. A high-molecular-weight species also eluted from the gel filtration column at 8 ml (Fig. 4A). SDS-PAGE revealed only small amounts of KSHV Pr in this fraction, suggesting that the main part of the peak is from the aggregated KSDD peptide.
FIG. 4.

(A) Perturbation of the dimerization of KSHV Pr by the KSDD peptide. The thick black line represents 5 μM KSHV Pr, showing the equilibrium between monomer and dimer. The amounts of the two fractions are almost equal at this concentration. The thin black line represents 5 μM KSHV Pr in the presence of 500 μM KSDD peptide, showing that dimerization is disrupted by the KSDD peptide. (B) SDS-PAGE of each fraction in the gel filtration chromatography experiments. KSHV Pr was observed in the dimer fraction (d) in the absence of the KSDD peptide but was almost completely absent in the presence of the KSDD peptide.
In Fig. 4A, KSHV Pr was observed as a monomer-dimer equilibrium in the absence of the peptide. The equal amounts of the monomeric and dimeric species observed in the chromatogram under these experimental conditions are in agreement with the Kd of KSHV Pr dimerization. The Kd was previously reported to be 1.7 ± 0.9 μM by sedimentation equilibrium analysis in an assay buffer containing 25% glycerol (24). The presence of glycerol has been shown to decrease the Kd of this class of enzymes in all human herpesviruses studied (10). Additional experiments monitoring the specific activity of KSHV Pr in a high-salt assay buffer as the concentration of protease was varied resulted in a Kd of 585 ± 135 nM (25). The 5 μM concentration of the sample used in the gel filtration experiment is slightly higher than these previously reported Kd's. However, the results of the size exclusion chromatography are consistent with these values, considering the protein concentration, the presence of glycerol, and the salt conditions. This is because the sample used for gel filtration chromatography is diluted in the column, and the dimerization affinity of KSHV Pr is reduced due to the lower salt and glycerol buffer concentrations.
The KSDD peptide contains two tyrosine residues whose absorbance can be readily monitored at 280 nm. The peptide was observed at two retention volumes (14 and 15.5 ml) by gel filtration (Fig. 4A). These two peaks were also observed when the identical gel filtration chromatography experiment was conducted in the absence of KSHV Pr. The intensity ratio of the two KSDD peptide peaks varies with the concentration of the KSDD peptide, indicating that the two peaks are due to dimeric and monomeric fractions of the KSDD peptide (data not shown). This result is consistent with those of previous studies showing that members of the family of peptides including aPP, neuropeptide Y, and peptide YY form dimers in the micro- and millimolar concentration ranges (14, 20).
Inhibition of KSHV Pr activity.
Inhibition of KSHV Pr activity by the α5 and KSDD peptides was investigated by monitoring the hydrolysis of protease substrates. Inhibitory curves were generated using relative specific activities at constant protease and substrate concentrations, while the peptide concentration was varied. Traditional inhibition analysis was not attempted, since binding of the peptide to the KSHV Pr monomer leads to inhibition by actually changing the concentration of the dimeric enzyme. Peptide binding to a KSHV Pr monomer is expected to be a competition between the peptide and other monomers in solution. The concentration of KSHV Pr used in all experiments was 2 μM, and that of the substrate was 160 μM, while the concentration of the peptides was varied as shown in Fig. 5A. The α5 peptide was incapable of inhibiting KSHV Pr activity even at concentrations 400-fold higher than the KSHV Pr concentration. In contrast, the specific activity of KSHV Pr was reduced as the concentration of the KSDD peptide increased, indicating that the KSDD peptide inhibits KSHV Pr activity by 50% at approximately a 200-fold molar excess of peptide over KSHV Pr under this experimental condition. Based on the inhibitory concentration and equation 10 (see Appendix), the dissociation constant was estimated to be 300 μM. This result shows that stabilization of the α-helical conformation of the KSDD peptide by the aPP scaffold is crucial to its inhibitory capability.
Estimation of the KSDD peptide inhibitory rate was conducted by measuring protease activity 0.08, 0.52, 0.92, 1.4, 1.9, 2.4, 3.1, 4.4, and 6.0 h after mixing of 2 μM KSHV Pr with a 400-fold excess of KSDD peptide (Fig. 5B). The KSDD peptide concentration was the highest ratio of peptide to enzyme used in specific activity measurements (Fig. 5A). The observed activities decayed exponentially, reaching a plateau at approximately 15% of full activity. This reduction in activity is consistent with the inhibition in the steady state experiments, where protease activity was monitored after overnight incubation at 4°C. The KSDD peptide inhibition rate at 2 μM KSHV Pr was 0.13 ± 0.012 min−1 under these conditions, and approximately 3 h of preincubation was necessary for maximum inhibition.
KSDD peptide inhibition of a dimer interface mutant.
To verify the binding site of the KSDD peptide, several dimer interface variants were generated and the inhibitory capability of the peptide for these mutants was compared with wild-type inhibition. Residues M197, A203, and S204 were originally chosen for mutagenesis of the dimer interface with the intent of improving the binding affinity between the monomers for the purpose of testing the dimer disruption capabilities of the KSDD peptide against an altered dimer interface. M197, which interacts with the M197 of the other monomer across the dimer interface, was replaced with a leucine or a phenylalanine in an effort to increase the hydrophobic interactions across the dimer interface. In contrast, A203 was mutated to a serine or a glutamate, while S204 was replaced with an aspartate for the purpose of creating new hydrogen bonding interactions (Fig. 2B).
All KSHV Pr variants were expressed and purified by the same protocol used for the purification of KSHV Pr. KSHV Pr M197L behaved similarly to KSHV Pr during the purification process, and its Kd was approximated by using gel filtration chromatography. At a protease concentration of 5 μM, KSHV Pr M197L formed a significantly larger fraction of dimers than KSHV Pr, demonstrating that the binding affinity between the monomers of KSHV Pr M197L had increased (Fig. 6A). In contrast, the dimer fractions of the M197F, A203S, A203E, and S204D KSHV Pr variants were dramatically smaller than that of KSHV Pr (data not shown). Under identical size exclusion conditions, the monomers of these mutants were the dominant species, and the fractions of dimers were less than 5%, indicating that their binding affinities were more than 36-fold lower than that of KSHV Pr. These results suggest that the dimer interface is finely developed and sensitive to perturbations.
FIG. 6.

(A) Comparison of the gel filtration profiles of 5 μM KSHV Pr (thick line) and KSHV Pr M197L (thin line). The dimerization affinity of KSHV Pr M197L is higher than that of KSHV Pr. (B) Stimulation of the specific activities of KSHV Pr (filled circles) and KSHV Pr M197L (open diamonds) upon dimerization (25). Assays were performed with 500 μM total substrate and 0.24 to 8 μM total enzyme. The best-fit Kd's for KSHV Pr and KSHV Pr M197L are 1.3 ± 0.24 and 0.40 ± 0.12 μM, respectively, as shown by the dashed lines.
The dependence of the specific activity of wild-type and mutant KSHV Pr on the protease concentration is shown in Fig. 6B. Specific activities were measured between 0.24 and 8 μM total enzyme with a large excess (500 μM) of the substrate. This analysis yielded a Kd of 1.3 ± 0.24 μM for KSHV Pr, which is consistent with previous measurements (24, 25), and a Kd of 0.40 ± 0.12 μM for KSHV Pr M197L, showing that the binding affinity between the monomers of KSHV Pr M197L is 3.3-fold higher than that for the wild type. This is consistent with the presence of a larger population of KSHV Pr M197L active dimers, as shown by the gel filtration experiments.
The inhibition by the KSDD peptide of KSHV Pr M197L was compared with that of KSHV Pr by monitoring protease substrate hydrolysis (Fig. 5A). The specific activity of KSHV Pr M197L was reduced in the presence of the KSDD peptide in a concentration-dependent manner. However, the inhibitory concentration for the variant was shifted approximately twofold higher than that for KSHV Pr, indicating that the dimer interface is involved in peptide binding. This result suggests that the KSDD peptide binds in the dimer interface and that it inhibits protease activity through dimer disruption.
KSDD M17L peptide.
The KSDD M17L peptide, in which residue 17 corresponds to M197 of KSHV Pr (Fig. 2), was designed to allow assessment of the effect of the M17L mutation on inhibitory activity. KSHV Pr M197L has a higher binding affinity between monomers, and M17 of the KSDD peptide is expected to interact with M197 of KSHV Pr. Therefore, the KSDD M17L peptide should have an altered binding affinity. The inhibition of KSHV Pr and KSHV Pr M197L by the KSDD M17L peptide was monitored as described above (Fig. 5A). The inhibitory concentrations of the KSDD M17L peptide were shifted lower than those of the KSDD peptide for the corresponding protease. These profiles show that the binding affinity of the KSDD M17L peptide is higher than that of the original peptide and that M17 of the KSDD peptide is involved in binding to KSHV Pr. This result suggests that the binding site in the KSDD peptide is located in the α-helix, which is consistent with the model structure of the complex in Fig. 2.
DISCUSSION
Although enzyme active sites are attractive targets for inhibition, a shallow substrate cleft such as that of KSHV Pr is particularly challenging. The possibility of cross-reactivity with other enzymes, particularly proteases, becomes a concern in developing selective inhibitors. To compound the problem, viral enzyme targets are prone to the rise of resistant mutations. Identification of a unique site other than the substrate binding pocket could provide exquisite inhibitor selectivity. Furthermore, new and complementary inhibitor binding sites on the same enzyme would allow simultaneous two-site inhibition of the enzyme of interest, reducing the probability of resistance development. Herpesvirus proteases are strictly regulated by their monomer-dimer equilibrium, which relies upon the interaction of a large, unique dimer interface comprised of many diverse interactions, including large hydrophobic pockets. Molecules which bind to this surface, disrupting dimerization and inactivating the enzyme, are expected to possess high specificity due to the unique nature of the interfacial surface. All mutations to the dimer interface, with the exception of KSHV Pr M197L, but including KSHV Pr L196A, M197D, and M197K, which have been reported previously (25), result in a reduction of at least 1 order of magnitude in the binding affinity between monomers. This suggests that the dimer interface is finely developed and sensitive to perturbations, which in turn greatly affect the activity of the enzyme. These mutagenesis studies show that the dimer interface is a highly effective site through which to alter the enzyme's catalytic activity by using specific inhibitors designed against herpesvirus proteases.
A helical peptide was designed, as a dimerization disruptor of KSHV Pr, to mimic the α5 helix in KSHV Pr and was grafted onto the aPP scaffold. The PP-fold has been shown previously to maintain integrity in protein-grafting experiments (4, 21, 38). In these previous studies, peptides were designed purely by sequence alignment with aPP and the domain of interest. In this study, the main chain traces of aPP and the α5 helix of KSHV Pr are superimposable, suggesting that the exposed residues in aPP can be replaced with KSHV Pr α5 helix residues in order to form an interfacial helix mimic. After protein grafting was conducted based on sequence alignment, the model structure of the complex was constructed in order to optimize the peptide sequence. The binding affinity of the inhibitory peptide was expected to be equivalent to or less than that of another KSHV Pr molecule because the peptide lacks other areas of the dimer interface which may be important for binding. Since the binding of the peptide to a KSHV Pr monomer is a competition between the KSDD peptide and other monomers in solution, this optimization of the peptide is crucial for the effectiveness of the KSDD peptide. The optimized KSDD peptide, containing 10 substitutions derived from aPP, has a helical conformation revealed by CD analysis, suggesting that the aPP structure is maintained in the KSDD peptide.
The ability of the KSDD peptide to disrupt KSHV Pr dimerization was shown in gel filtration experiments where the dimeric fraction of KSHV Pr was greatly reduced in the presence of the inhibitory peptide. Size-exclusion chromatography also revealed that the KSDD peptide itself preferentially dimerizes at the concentrations used in the experiment and elutes mainly as a 7-kDa species. In agreement with the dimer formation of the KSDD peptide, members of the family of small proteins such as pancreatic polypeptide, neuropeptide Y, and peptide YY have a propensity to form dimers at the micro- and millimolar concentrations. The nuclear magnetic resonance structure of neuropeptide Y revealed that the monomeric and dimeric forms of the oligopeptide expose the same side of the α-helix to the solvent (20). The solvent-exposed face of both the monomer and dimer of neuropeptide Y is the side of the α-helix which was mutated to mimic the interfacial α5 helix of KSHV Pr. Therefore, both the monomer and the dimer of the KSDD peptide are expected to bind to the KSHV Pr interface. A high-molecular-weight species also eluted in the void volume of the gel filtration column (Fig. 4A). SDS-PAGE revealed only small amounts of KSHV Pr in this fraction, indicating that the main constituent of this fraction is aggregated KSDD peptide. The KSDD peptide alone does not form this high-molecular-weight species (data not shown); therefore, perhaps the presence of KSHV Pr causes the peptide to partially aggregate. The KSDD peptide inhibits KSHV Pr activity by 50% at approximately a 200-fold molar excess of peptide over KSHV Pr, and the dissociation constant was estimated to be 300 μM. In addition, binding of the KSDD peptide to the KSHV Pr dimer interface is indicated by an increase in the inhibitory concentration for the interface mutant M197L.
The crystal structure of KSHV Pr shows the two M197 residues in the dimer interacting with each other across the dimer interface. The M197L protease variant, which has a higher binding affinity between monomers, was successfully produced for the purpose of identifying dimer disruptors. This variant, which increases the binding affinity between the monomers of a herpesvirus protease, will be useful for studying the structure and activity of the dimer. The higher-affinity dimer variant may facilitate studies of the dimeric protease, given the difficulties in studying the wild-type dimeric enzyme, which is equilibrated with the monomer in solution even at micromolar concentrations (18). In addition, this variant has proved useful in determining the mechanism of inhibition of the KSDD peptide. The interaction of the KSDD peptide with KSHV Pr M197L will use a leucine-methionine interaction rather than the wild-type dimer interaction of two methionines. The inhibition of KSHV Pr and KSHV Pr M197L by the KSDD and KSDD M17L peptides was monitored by hydrolysis of the protease substrate (Fig. 5A). The inhibitory concentration for KSHV Pr M197L was shifted approximately twofold higher than that for KSHV Pr, and the concentrations of the KSDD M17L peptide necessary for equivalent inhibition were lower than those of the KSDD peptide. These results suggest that the binding site in the KSDD peptide is located in the α-helix, which includes M17, and that a higher concentration of the KSDD peptide may be necessary for binding to KSHV Pr M197L. This may be because KSHV Pr M197L forms a more stable dimer than the wild-type enzyme.
Human immunodeficiency virus type 1 protease (HIV Pr) is also a viral protease which functions only as a dimer; however, HIV Pr is an obligate dimer because each monomer donates one catalytic aspartate residue to the shared active site of the enzyme. Inhibitors that target the active site of HIV Pr have been developed to destabilize or disrupt dimerization by binding at the dimer interface. Dominant-negative HIV Pr variants that form inactive heterodimers with wild-type HIV Pr and thus inhibit protease activity have also been reported (2, 19, 31). The active sites in herpesvirus proteases are not present in the dimer interface. This class of enzymes still requires dimerization for activation even though both monomers have separate, complete active sites. Mixing experiments of KSHV Pr with inactive KSHV Pr S114A resulted in an increase in the specific activity of KSHV Pr. This result shows that only one active site is sufficient for protease activity. Additionally, the retention and increase in the specific activity of KSHV Pr in the presence of KSHV Pr S114A, but not when KSHV Pr is mixed with the KSDD peptide, suggests the necessity of other components of the dimer interface for the stabilization of the active conformation. The KSDD peptide, which contains the α5 helix but does not include the pocket formed by helices α1 and α2 (Fig. 2B), inhibits protease activity instead of activating the monomer. This shows that the interaction between the two α5 helices at the dimer interface is not sufficient for protease activation.
In conclusion, an inhibitor has been designed which is capable of disrupting dimerization through mimicry of the natural interactions present in the dimeric protease without activating the resulting bound monomeric protease. By using this inhibitor, it has been shown that the dimer interfaces as well as the active sites of KSHV Pr and potentially other herpesvirus proteases are attractive targets for the inhibition of protease activity. Disrupting the dimerization of herpesvirus proteases has the potential to inhibit the viral lytic cycle in a highly selective fashion, providing a new avenue for blocking the spread of the virus.
Acknowledgments
We thank Volker Dötsch and R. Kiplin Guy for useful discussions. We also thank Allart Stoop, Jeonghoon Sun, Timothy Geistlinger, and Sarah Galicia for helpful suggestions and technical assistance.
This work was supported by grant GM-56531 from NIH (to C.S.C.). N.S. was supported by Ajinomoto Co., Inc.
APPENDIX
Theoretical curves.The measured specific activity, Aspec, at experimental concentrations is proportional to the concentration of the KSHV Pr dimer, [Pr2], which is related to the Kd and the total experimental KSHV Pr concentration, Epr (i.e., Epr = [Pr] + 2[Pr2]). [Pr2] can be calculated from Epr and Kd as shown in equation 1 (25).
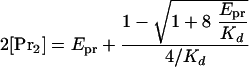 |
(1) |
In the presence of KSHV Pr S114A, five species are expected to exist in solution. Their concentrations are shown as follows: [Pr]SA, KSHV Pr monomer; [Pr2]SA, KSHV Pr dimer; [SA]SA, KSHV Pr S114A monomer; [SA2]SA, KSHV Pr S114A dimer; [Pr-SA]SA, KSHV Pr-KSHV Pr S114A heterodimer. The concentrations of the KSHV Pr dimer, 2[Pr2]SA or 2[Pr2]SA + [Pr-SA]SA, can be calculated from the total experimental KSHV Pr S114A concentration, αEpr, which is expressed as the relative concentration of KSHV Pr. This is because the ratio of the dimers (the [Pr2]SA:[Pr-SA]SA:[SA2]SA ratio) is expected to be 1:2α:α2. Note that the Kd's of the KSHV Pr dimer, the KSHV Pr S114A dimer, and the heterodimer are hypothesized to be equal. Etot is the total KSHV Pr plus KSHV Pr S114A concentration.
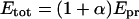 |
(2) |
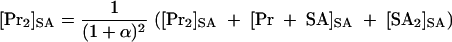 |
(3) |
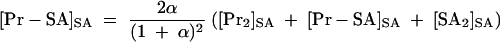 |
(4) |
Combining equations 2 through 4 with equation 1 yields equations 5 and 6.
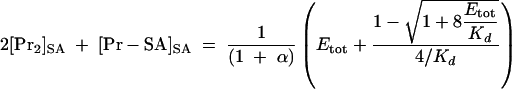 |
(5) |
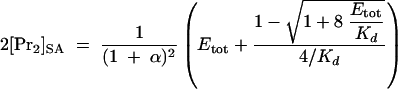 |
(6) |
If the heterodimer is active or inactive, the relative specific activities, Aactive/Aspec, or Ainactive/Aspec, are given by equations 7 and 8.
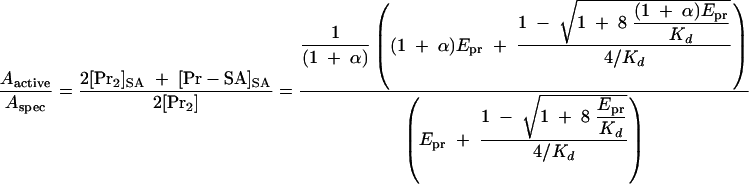 |
(7) |
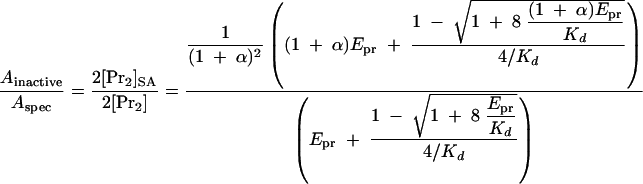 |
(8) |
Based on these equations, the theoretical curves for the relative specific activities were generated (Fig. 1) for a Kd of 1.3 μM and an Epr of 2 or 0.5 μM; Kd was the value calculated in Fig. 6B.
Estimation of the dissociation constant, Ki.If a molecule, [I], binds in the dimer interface of KSHV Pr, the dissociation constant (Ki) of the molecule is shown in the scheme below, where [Pr-I] is the complex of the KSHV Pr monomer and the KSDD peptide.
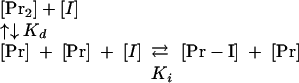 |
If Kd is much smaller than Ki, the total concentration of the inhibitor, [Itot], becomes almost equal to that of the free inhibitor. Under this condition, the concentration of the inhibited KSHV dimer, 2[Pr2]inhib, is given by equation 9.
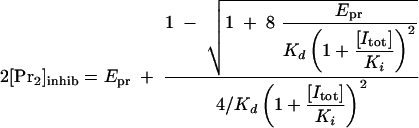 |
(9) |
Using the experimentally determined percentage of inhibited dimer, the Ki can be calculated as shown in equation 10. In this equation, the [IC50] is the concentration of inhibitor necessary to inhibit KSHV Pr activity by 50%.
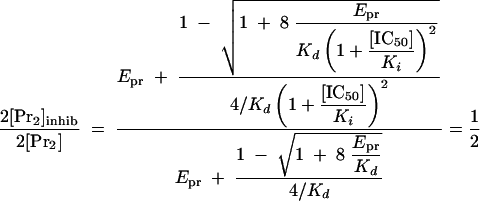 |
(10) |
REFERENCES
- 1.Andrade, M. A., P. Chacon, J. J. Merelo, and F. Moran. 1993. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6:383-390. [DOI] [PubMed] [Google Scholar]
- 2.Babé, L. M., J. Rosé, and C. S. Craik. 1995. Trans-dominant inhibitory human immunodeficiency virus type 1 protease monomers prevent protease activation and virion maturation. Proc. Natl. Acad. Sci. USA 92:10069-10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, P., H. Tsuge, R. J. Almassy, C. L. Gribskov, S. Katoh, D. L. Vanderpool, S. A. Margosiak, C. Pinko, D. A. Matthews, and C. C. Kan. 1996. Structure of the human cytomegalovirus protease catalytic domain reveals a novel serine protease fold and catalytic triad. Cell 86:835-843. [DOI] [PubMed] [Google Scholar]
- 4.Chin, J. W., and A. Schepartz. 2001. Concerted evolution of structure and function in a miniature protein. J. Am. Chem. Soc. 123:2929-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole, J. L. 1996. Characterization of human cytomegalovirus protease dimerization by analytical centrifugation. Biochemistry 35:15601-15610. [DOI] [PubMed] [Google Scholar]
- 6.Darke, P. L., J. L. Cole, L. Waxman, D. L. Hall, M. K. Sardana, and L. C. Kuo. 1996. Active human cytomegalovirus protease is a dimer. J. Biol. Chem. 271:7445-7449. [DOI] [PubMed] [Google Scholar]
- 7.Gao, M., L. Matusick-Kumar, W. Hurlburt, S. F. DiTusa, W. W. Newcomb, J. C. Brown, P. J. McCann III, I. Deckman, and R. J. Colonno. 1994. The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J. Virol. 68:3702-3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibson, W. 1996. Structure and assembly of the virion. Intervirology 39:389-400. [DOI] [PubMed] [Google Scholar]
- 9.Glover, I., I. Haneef, J. Pitts, S. Wood, D. Moss, I. Tickle, and T. Blundell. 1983. Conformational flexibility in a small globular hormone: X-ray analysis of avian pancreatic polypeptide at 0.98-Å resolution. Biopolymers 22:293-304. [DOI] [PubMed] [Google Scholar]
- 10.Hall, D. L., and P. L. Darke. 1995. Activation of the herpes simplex virus type 1 protease. J. Biol. Chem. 270:22697-22700. [DOI] [PubMed] [Google Scholar]
- 11.Harris, J. L., B. J. Backes, F. Leonetti, S. Mahrus, J. A. Ellman, and C. S. Craik. 2000. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. USA 97:7754-7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holwerda, B. 1999. Activity in monomers of human cytomegalovirus protease. Biochem. Biophys. Res. Commun. 259:370-373. [DOI] [PubMed] [Google Scholar]
- 13.Hoog, S. S., W. W. Smith, X. Qiu, C. A. Janson, B. Hellmig, M. S. McQueney, K. O'Donnell, D. O'Shannessy, A. G. DiLella, C. Debouck, and S. S. Abdel-Meguid. 1997. Active site cavity of herpesvirus proteases revealed by the crystal structure of herpes simplex virus protease/inhibitor complex. Biochemistry 36:14023-14029. [DOI] [PubMed] [Google Scholar]
- 14.Keire, D. A., M. Kobayashi, T. E. Solomon, and J. R. Reeve, Jr. 2000. Solution structure of monomeric peptide YY supports the functional significance of the PP-fold. Biochemistry 39:9935-9942. [DOI] [PubMed] [Google Scholar]
- 15.Keire, D. A., P. Mannon, M. Kobayashi, J. H. Walsh, T. E. Solomon, and J. R. Reeve, Jr. 2000. Primary structures of PYY, [Pro(34)]PYY, and PYY-(3-36) confer different conformations and receptor selectivity. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G126-G131. [DOI] [PubMed] [Google Scholar]
- 16.Khayat, R., R. Batra, G. A. Bebernitz, M. W. Olson, and L. Tong. 2004. Characterization of the monomer-dimer equilibrium of human cytomegalovirus protease by kinetic methods. Biochemistry 43:316-322. [DOI] [PubMed] [Google Scholar]
- 17.Margosiak, S. A., D. L. Vanderpool, W. Sisson, C. Pinko, and C. C. Kan. 1996. Dimerization of the human cytomegalovirus protease: kinetic and biochemical characterization of the catalytic homodimer. Biochemistry 35:5300-5307. [DOI] [PubMed] [Google Scholar]
- 18.Marnett, A. B., A. M. Nomura, N. Shimba, S. Mahrus, P. R. Ortiz de Montellano, and C. S. Craik. Communication between the spatially separate active site and dimer interface of KSHV protease revealed by small molecule inhibition. Proc. Natl. Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed]
- 19.McPhee, F., A. C. Good, I. D. Kuntz, and C. S. Craik. 1996. Engineering human immunodeficiency virus 1 protease heterodimers as macromolecular inhibitors of viral maturation. Proc. Natl. Acad. Sci. USA 93:11477-11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monks, S. A., G. Karagianis, G. J. Howlett, and R. S. Norton. 1996. Solution structure of human neuropeptide Y. J. Biomol. NMR 8:379-390. [DOI] [PubMed] [Google Scholar]
- 21.Montclare, J. K., and A. Schepartz. 2003. Miniature homeodomains: high specificity without an N-terminal arm. J. Am. Chem. Soc. 125:3416-3417. [DOI] [PubMed] [Google Scholar]
- 22.Noelken, M. E., P. J. Chang, and J. R. Kimmel. 1980. Conformation and association of pancreatic polypeptide from three species. Biochemistry 19:1838-1843. [DOI] [PubMed] [Google Scholar]
- 23.Nordmann, A., M. J. Blommers, H. Fretz, T. Arvinte, and A. F. Drake. 1999. Aspects of the molecular structure and dynamics of neuropeptide Y. Eur. J. Biochem. 261:216-226. [DOI] [PubMed] [Google Scholar]
- 24.Pray, T. R., A. M. Nomura, M. W. Pennington, and C. S. Craik. 1999. Auto-inactivation by cleavage within the dimer interface of Kaposi's sarcoma-associated herpesvirus protease. J. Mol. Biol. 289:197-203. [DOI] [PubMed] [Google Scholar]
- 25.Pray, T. R., K. K. Reiling, B. G. Demirjian, and C. S. Craik. 2002. Conformational change coupling the dimerization and activation of KSHV protease. Biochemistry 41:1474-1482. [DOI] [PubMed] [Google Scholar]
- 26.Preston, V. G., J. A. Coates, and F. J. Rixon. 1983. Identification and characterization of a herpes simplex virus gene product required for encapsidation of virus DNA. J. Virol. 45:1056-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu, X., J. S. Culp, A. G. DiLella, B. Hellmig, S. S. Hoog, C. A. Janson, W. W. Smith, and S. S. Abdel-Meguid. 1996. Unique fold and active site in cytomegalovirus protease. Nature 383:275-279. [DOI] [PubMed] [Google Scholar]
- 28.Qiu, X., C. A. Janson, J. S. Culp, S. B. Richardson, C. Debouck, W. W. Smith, and S. S. Abdel-Meguid. 1997. Crystal structure of varicella-zoster virus protease. Proc. Natl. Acad. Sci. USA 94:2874-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiling, K. K., T. R. Pray, C. S. Craik, and R. M. Stroud. 2000. Functional consequences of the Kaposi's sarcoma-associated herpesvirus protease structure: regulation of activity and dimerization by conserved structural elements. Biochemistry 39:12796-12803. [DOI] [PubMed] [Google Scholar]
- 30.Roizman, B., and A. E. Sears. 1996. Herpes simplex viruses and their replication, p. 2231-2295. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 31.Rozzelle, J. E., D. S. Dauber, S. Todd, R. Kelley, and C. S. Craik. 2000. Macromolecular inhibitors of HIV-1 protease. Characterization of designed heterodimers. J. Biol. Chem. 275:7080-7086. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt, U., and P. L. Darke. 1997. Dimerization and activation of the herpes simplex virus type 1 protease. J. Biol. Chem. 272:7732-7735. [DOI] [PubMed] [Google Scholar]
- 33.Shieh, H. S., R. G. Kurumbail, A. M. Stevens, R. A. Stegeman, E. J. Sturman, J. Y. Pak, A. J. Wittwer, M. O. Palmier, R. C. Wiegand, B. C. Holwerda, and W. C. Stallings. 1996. Three-dimensional structure of human cytomegalovirus protease. Nature 383:279-282. (Erratum, 384:288.) [DOI] [PubMed] [Google Scholar]
- 34.Stevenson, A. J., E. E. Morrison, R. Chaudhari, C. C. Yang, and D. M. Meredith. 1997. Processing and intracellular localization of the herpes simplex virus type 1 proteinase. J. Gen. Virol. 78:671-675. [DOI] [PubMed] [Google Scholar]
- 35.Tong, L. 2002. Viral proteases. Chem. Rev. 102:4609-4626. [DOI] [PubMed] [Google Scholar]
- 36.Tong, L., C. Qian, M. J. Massariol, P. R. Bonneau, M. G. Cordingley, and L. Lagacé. 1996. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 383:272-275. [DOI] [PubMed] [Google Scholar]
- 37.Ünal, A., T. R. Pray, M. Lagunoff, M. W. Pennington, D. Ganem, and C. S. Craik. 1997. The protease and the assembly protein of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J. Virol. 71:7030-7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zondlo, N. J., and A. Schepartz. 1999. Highly specific DNA recognition by a designed miniature protein. J. Am. Chem. Soc. 121:6938-6939. [Google Scholar]