

Abstract
Autoinflammatory diseases are characterized by seemingly unprovoked, pathological activation of the innate immune system in the absence of autoantibodies or autoreactive T cells. Discovery of the causative mutations underlying several monogenic autoinflammatory diseases has identified key regulators of innate immune responses. Recent studies have highlighted the role of misfolding, oligomerization and abnormal trafficking of pathogenic mutant proteins in triggering autoinflammation, and suggest that more common rheumatic diseases may have an autoinflammatory component. This coincides with recent discoveries of new links between endoplasmic reticulum stress and inflammatory signalling pathways, which support the emerging view that autoinflammatory diseases may be due to pathological derangement of stress sensing pathways that normally function in host defence.
Rheumatic and autoimmune diseases have traditionally been categorized according to the target of the abnormal immune response. Type I diabetes, for example, is an organ-specific autoimmune disease involving a specific immune attack on pancreatic islet β-cells, whereas systemic lupus erythematosus is the prototypical systemic autoimmune disease, in which autoimmune responses to multiple antigens can be measured and multiple organs are targeted. Recent discoveries have added a second dimension to consider, the degree to which the adaptive vs. innate immune system is involved in the disease. Diseases involving abnormal innate immune responses without the involvement of autoantibodies or autoreactive T cell are termed ‘autoinflammatory’, while diseases that depend on autoreactive B or T cells are classified as autoimmune (Figure 1) 1. Autoinflammatory diseases have emerged as a distinct entity characterized by pathological inflammation that often stems from abnormal activation of innate immune cells by endogenous or exogenous or stimuli. In the familial autoinflammatory syndromes, mutations in key genes that regulate innate immune cell function have been identified 2. The uric acid crystals that trigger gout as well as other particulate materials have been found to be potent activators of innate immune signalling pathways 3. These findings have begun to provide a molecular basis for both sporadic and familial autoinflammatory diseases. It is also becoming clear that many diseases are a mix of autoinflammatory and autoimmune components (Figure 1). In this review we will explore recent advances in the genetic and cell biological basis of inflammation triggered by mutations in mendelian autoinflammatory diseases as well as in more common rheumatic diseases such as spondylarthropathies and inflammatory muscle diseases. An emerging theme in this field has been the concept that misfolding and aggregation of protein complexes can trigger autoinflammation with or without triggering the unfolded protein response.
Figure 1. The spectrum of autoimmune and autoinflammatory diseases.

Polygeneic diseases are boxed in green and menedelian disorders boxed in red. The horizontal axis depicts the range of organ-specfic vs. systemic disease, and the vertical axis, the degree of involvement of the parts of the immune system in immunopathology: innate (autoinflammatory, bottom) vs. adaptive (autoimmune, top).
Autoinflammatory and other disease states associated with protein misfolding
Both autoimmune and autoinflammatory diseases can be episodic, but autoinflammatory diseases are unique in that flares can have a characteristic frequency and length, which prompted them to be termed periodic fever syndromes. Flares of familial Mediterranean fever and gout are particularly acute, generally lasting one week or less. Subclinical signs of autoinflammatory diseases, such as elevated levels of acute phase proteins or inflammatory cytokines, can often be detected in the absence of symptoms. Although autoreactive memory T and B cells are thought to maintain autoimmunity over the lifetime of an individual, what sustains the inflammation in autoinflammatory diseases? In environmentally triggered autoinflammatory diseases such as gout, persistence of the inciting uric acid crystals is responsible for driving recurrent attacks. Therapies that lower the levels of uric acid are highly effective in preventing recurrences of gout and progression to chronic disease. In the genetic autoinflammatory diseases, the inciting stimulus comes from within, in the form of pathogenic mutations that enhance inflammatory signalling pathways, presumably by lowering the threshold for triggering an inflammatory response, or in extreme cases, leading to continuous inflammation.
Several genetic autoinflammatory diseases are inherited in an autosomal dominant manner (Table 1), suggesting that gain of function mutations in innate immune signaling proteins may be common triggers for disease. Many mutations linked to dominant autoinflammatory diseases cause the proteins involved to misfold or form abnormal oligomers. Cellular responses to misfolded proteins can vary dramatically depending on the degree and recognition of protein misfolding (Figure 2) 4. In the endoplasmic reticulum (ER), misfolded or partially folded polypeptides can be recognized and eliminated via the ER-associated degradation (ERAD) pathway. This leads to disease due to loss of function, such as in haemophilia due to factor VIII mutations 5 or cystic fibrosis resulting from mutations in the cystic fibrosis transmembrane regulator 6. Misfolded proteins can also accumulate both inside and outside of cells, leading to a gain of function through mechanisms related to the physiological role of the protein in signal transduction, or via activation of other pathways, such as the unfolded protein response (UPR) triggered by ER stress.
Table 1. Molecular features and molecular causes of familial autoinflammatory disorders.
Diseases are listed in order of their discovery. Abbreviations and Definitions: Blau Syndrome: Granulomatous synovitis, with Uveitis and Cranial Neuropathies (also known as familial juvenile systemic arthrocutaneouveal granulomatosis); PAPA syndrome: Pyogenic Sterile Arthritis, Pyoderma Gangrenosum, and Acne; DIRA: Deficiency of IL-1 Receptor Antagonist; DITRA: Deficiency of IL-36R Receptor Antagonistm; CANDLE: Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature. JMP syndrome: Joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced childhood-onset lipodystrophy
| Disease | Gene | Protein | Key References | Inheritance pattern | Clinical Features |
|---|---|---|---|---|---|
| Familial Mediterranean Fever (FMF) | MEFV | Pyrin | 63 | autosomal recessive or Gene-dosage dependent autosomal dominant | Periodic Fevers (3-7days) serositis, arthritis |
| Tumor Necrosis Factor Associated Periodic Syndrome (TRAPS) | TNFRSF 1A | Tumor necrosis factor receptor 1 | 53 | autosomal dominant with dependence on WT allele | Periodic Fevers (1-6 weeks), serositis, rash, episcleritis |
| Hyper IgD Syndrome | MVK | Mevalonate kinase | 97 | autosomal recessive | Periodic Fevers (3-7 days), non-destructive arthritis, lymphadenopathy, vasculitic skin lesions |
| Cryopyrin-Associated Periodic Syndromes FCAS/MWS/NOMID | NLRP3 | NLRP3, Cis1 | 98,99 | autosomal dominant | Cold-induced autoinflammation>Cochl ear Inflammation> fevers & Sterile Meningitis, Bone lesion |
| Blau syndrome | NOD2 | Nod2 | 100,101 | autosomal dominant | Granulomatous Dermatitis, Uveitis. Arthritis |
| PAPA syndrome | CD2BP1 | Pstpip1 | 102 | autosomal dominant | Pyogenic arthritis, pyoderma granolosum, acne |
| DIRA | IL1RN | IL-1R antagonist | 103 | autosomal recessive | Fevers, Pustular skin rash, osteolytic bone lesions |
| DITRA (Deficiency of IL-36R antagonist) | IL36RN | IL-36R antagonist | 104 | autosomal recessive | Generalized pustular psoriasis, |
| Familial Psoriasis (PSORS2) and CAMPS (CARD14 Mediated Pustular Psoriasis) | CARD14 | caspase recruitment domain family, member 14 | 17,18 | ||
| CANDLE/ Nakajo-Nishimura syndrome/JMP syndrome | PSMB8 | b5i immunoprot easome subunit | 19-22 | autosomal recessive | Lipodystrophy associated inflammatory disease |
Figure 2. Consequences of protein misfolding and intracellular signaling complexes that activate autoinflammatory disease.

A) The outcomes of misfolding of secretory protewins in the ER are depicted at the bottom of the figure. Degradation of misfolded protein can cause a loss of function, whereas accumulation of misfolded proteins can trigger abnormal intracellular signaling, or at higher levels, induction of the unfolded protein response (UPR), which can also lead to the induction of inflammation and programmed cell death. Different foci of abnormal cellular signaling that trigger autoinflammatory diseases are depicted in the cell, clockwise from lower left. B) In PSMB8 deficiency, reduced degradation of misfolded proteins and peptides by the immunoproteasome leads to accumulation of ubiquinated proteins and cellular stress, which leads of Interferon-β production. Type I Interferon in turn upregulates the synthesis of immunoproteasome sububits, perpetuating the abnormalities. C) In TRAPS, extracellular mutations in TNFR1 lead to accumulation of the mutant receptor in the ER, which triggers an abnormal inflammatory response that is amplified by TNF or LPS signaling through cell surface receptors. D) In the Cryopyrin-Associated Perodic Syndromes (CAPS), mutations in NLRP3 enhance activation of the NLRP3 inflammasome and processing of IL-1β into its active form. E) In the spondylarthropathies, high-level expression of HLA-B27, which is enhanced in inflammation, fails to fold properly and is retained in the ER, triggering a partial ER stress response which leads to type I interferon and IL-23 production.
Unrestrained ER stress responses can lead to cell death or inflammation. In tissues that divide slowly, such as neurons, glial cells and pancreatic β cells, the primary outcome appears to be cell death, leading to neurodegenerative disease or diabetes. The C96Y mutation found in the insulin 2 (INS2) gene in the Akita mouse alters an essential disulphide bond that normally links the two insulin polypeptides. As a result of the mutation, insulin is misfolded and retained in the ER of pancreatic β cells, causing ER stress 8, which leads to loss of β cells and diabetes7. In the retinal degenerative syndrome retinitis pigmentosa, mutations in the CAIV gene cause the encoded protein carbonic anhydrase IV to aggregate in the ER, triggering the UPR and increased cell death. Dorzolamide, a carbonic anhydrase inhibitor that is thought to work as a cellular chaperone, 9 can rescue cells that have CAIV mutations from cell death. In other diseases, the mutant protein may acquire functions that are related to its normal role. An example of this is superoxide dismutase protein 1 (SOD1) in amyotrophic lateral sclerosis. SOD1 mutations provoke disease in the majority of cases of familial amyotrophic lateral sclerosis. Under normal physiological conditions, SOD1 functions as a reactive oxygen species (ROS) scavenger by converting superoxide into hydrogen peroxide. However, the mutant form of SOD1 has been shown to bind Rac1 and enhance NOX2-induced generation of ROS 10. This, in turn, leads to increased rates of cell death.
In addition to triggering cell death, protein misfolding in innate immune cells can activate inflammatory pathways that function in host defence. Although the ‘classic’ UPR is dedicated to restoring homeostasis, in part by a transient blockade of protein synthesis followed by upregulation of ER chaperone expression, recent studies have identified signalling pathways triggered by ER stress that drive inflammatory cellular response programmes. In addition to mediating the splicing of X-box-binding protein 1 (XBP1), which activates the transcriptional response to ER stress, the ER stress sensor inositol-requiring enzyme 1 (IRE1) also activates c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein (MAP) kinases, which activate signalling cascades that culminate in inflammatory response gene expression 11. In hepatocytes, the transcription factor cAMP-responsive-element-binding protein H (CREBH) can be activated by ER stress and leads to transcriptional activation of genes encoding acute phase proteins, such as serum amyloid P and C-reactive protein 12. Innate immune stimuli such as lipopolysaccharide (LPS) can trigger low level XBP1 splicing, which sustains the transcription of pro-inflammatory genes without activating other arms of the UPR that regulate chaperone expression 13. This response is independent of JNK and p38 MAP kinases but requires the adaptor protein TNFR-associated factor 6 (TRAF6). Taken together, these results indicate that the signalling pathways originally discovered in response to ER stress may also be part of the innate immune response and as such may be involved in the pathogenesis of autoinflammatory diseases.
Misfolded proteins that accumulate extracellularly can also trigger IL-1β secretion, which may be part of the inflammatory cascade that causes tissue damage in autoinflammatory diseases. Extracellular aggregates of amylin, an islet amyloidogenic polypeptide, trigger inflammasome activation and secretion of IL-1β, resulting in dysfunction of pancreatic β cells 14. In Alzheimer's disease, aggregates of extracellular β-amyloid can also lead to lysosomal damage followed by activation of the inflammasome 15. How commonly extracellular protein aggregates trigger other autoinflammatory conditions remains to be determined.
Molecular basis of familial autoinflammatory diseases
In the last decade, the genetic causes of several familial autoinflammatory syndromes have been discovered (Table 1). The genes that are responsible for these syndromes have been found to encode key sensors and transducers of inflammatory signal transduction pathways. Some of the genes involved, such as TNF receptor 1 (TNFR1), were previously well known, whereas the discovery of mutations in other others, such as NLRP3 (also known as cryopryrin and NLRP3), highlighted their importance in inflammatory signalling pathways16. The recent identification of gain of function mutations in CARD14, also known as CARMA2, associated with autosomal dominant familial or de-novo severe pustular psoriasis, has prompted renewed investigation into which receptors activate NF-kB through this adapter protein, particularly in the skin 17,18. The discovery of mutations in the PSMB8 immunoproteasome subunit associated with an autoinflammatory disease was surprising, and suggests new links between protein homeostasis and inflammation 19-22.
Cryopyrin-associated periodic syndromes (CAPS) and the inflammasome
CAPS are a spectrum of autosomal dominant autoinflammatory diseases including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal onset multisystem inflammatory disease (NOMID; also known as chronic neurologic cutaneous and articular syndrome, CINCA). All of these syndromes have been linked to mutations in the NLRP3 gene 23. FCAS is characterized by cold-induced urticaria and mild symptoms of systemic inflammation. In addition to these features, patients with MWS also develop progressive sensorineural deafness due to cochlear inflammation, arthrialgias, and recurrent fevers. In NOMID, inflammatory symptoms are nearly continuous and typically begin in infancy. NOMID patients also suffer from chronic meningitis that can affect central nervous system development, and hypertrophic bone and cartilaginous lesions in the epiphyses of the long bones that can lead to severe deformities and disability. Mutations in NLRP3 occur de novo in NOMID, while are often inherited in the other less severe syndromes.
The identification of NLRP3 mutations in CAPS coincided with the realization that the NLRP3 protein is a key component of an inflammasome, a multiprotein complex that can activate caspase 1. Inflammasomes are required to process the pro-forms of the inflammatory cytokines IL-1β and IL-18 into products that can be secreted from the cell 24. There are at least four distinct inflammasome complexes in cells, which sense distinct classes of stimuli using different sensor and adaptor proteins but all converge on IL-1 activation (Figure 3). In addition, NLRP6 has recently been identified as a component of an inflammasome that activates IL-18 and negatively regulates colonic inflammation through altering the intestinal microbiota 25-27. The NLRP3 inflammasome is the most intensively studied and linked to autoinflammatory disease 28. NLRP3 is a large protein containing a C-terminal leucine-rich repeat (LRR) domain which likely acts as a sensor, a central nucleotide-binding domain (NBD) that binds ATP or dATP, and an N-terminal pyrin domain. The activation of inflammasomes is thought to be triggered by conformational changes in the NLRP3 LRR domain, which releases it from an autoinhibited basal state. Activated NLRP3 oligomerizes and interacts with the adaptor protein ASC and caspase 1, resulting in the enzymatic activation of caspase 1. Studies in NLRP3-deficient mice have shown that NLRP3 is required for IL-1β processing in response to a wide variety of molecules, including bacterial and viral RNA, Gram-positive bacteria such as Staphylococcus aureus and Listeria monocytogenes, uric acid and calcium pyrophosphate crystals, and aggregated proteins such as amyloid-β. The molecular diversity of these NLRP3 activating ligands suggests that at least some of them may not bind the LRR of NLRP3 directly, but instead trigger a common intracellular mediator that activates NLRP3. An initial priming step is also required to induce expression of inflammasome components such as IL-1β and NLRP3. This signal can be provided by Toll-like receptor (TLR) ligands, such as LPS.
Figure 3. The NLRP1, NLRP3, IPAF and AIM2 inflammasomes.

The NLRP1 inflammasome is activated by Bacillus anthracis lethal toxin and muramyl dipeptide (MDP). The NLRP3 inflammasome is activated by exposure to whole pathogens, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) and environmental irritants. The IPAF inflammasome is activated by Gram-negative bacteria with type III or IV secretion systems, and the DNA inflammasome senses double stranded DNA. Other complexes, such as NLRP6-containing complexes that regulate IL-18, also exist 24, and pyrin likely regulates an inflammasome complex containing ASC but not NLRP3 that activates IL-1β processing 69. EMCV – Encephalomyocarditis virus, ATP – Adenosine triphosphate, MSU – Monosodium urate.
Gain of function missense mutations in NLRP3 have been identified in the majority of CAPS patients, although one patient with atypical CAPS symptoms was reported to be heterozygous for an NLRP3 truncation mutation 29. Somatic missense NLRP3 mutations have also been detected in some patients who have biochemical and clinical features of CAPS but do not have germline mutations in NLRP3 23, 30.
Mechanisms of normal inflammasome activation and disruption in familial fever syndromes
Several mechanisms have been proposed to explain how diverse stimuli gain access to the cytoplasmic NLRP3 inflammasome and trigger its activation. The first model ascribes inflammasome activation to the formation of a pannexin 1 hemichannel, through which pathogen-associated molecular patterns (PAMPs), such as muramyl dipeptides, and endogenous damage-associated molecular patterns (DAMPs) may enter the cell 31,32. Although studies have shown a crucial role for pannexin 1 in phagocytosis of bacterial muramyl dipeptide 33, it is unlikely that all NLRP3 activating stimuli enter the cell through the pannexin 1 pore, particularly crystals or particulates. For these stimuli, ‘frustrated phagocytosis’, the process of phagocytosis followed by rupture of phagolysosomes, has been suggested to trigger inflammasome activation. In this second model, lysosomal perhaps triggered by mechanical disruption of lysosomes by crystalline material, may allow diffusion of phagocytosed particles into the cytoplasm where they may directly interact with the NLRP3 inflammasome. Alternatively, lysosomal enzymes, such as cathepsins, may trigger inflammasome activation in the cytoplasm through proteolytic cleavage 34,35. However, cathepsin B-deficient mice do not consistently show diminished caspase 1 cleavage and IL-1β secretion, suggesting that leakage of other lysosomal components may be required as well 36.
A third model proposes a role for reactive oxygen species (ROS), generated as a result of various types of cellular stress, in inflammasome activation. A direct effect of ROS on inflammasome activation was suggested by studies on the protein TXNIP, a protein that was previously found to be involved in glucose homeostasis 37. Under resting conditions, TXNIP associates with the oxidoreductase thioredoxin at two redox-sensitive cysteine residues. With increasing ROS, TXNIP was found to dissociate from thioredoxin and associate with NLRP3 through the LRR and NACHT domains. However, TXNIP-deficient macrophages exhibited reduced, but not absent, IL-1β secretion in response to NLRP3 agonists 38, suggesting that other mechanisms of IL-1β processing also exist. Chemically induced ER stress has recently been shown to activate the NLRP3 inflammasome via ROS production, but independent of classical UPR activation 39.
The source of ROS that activate the inflammasome has been of considerable interest. Initially, the NOX family of NADPH oxidases was implicated in generating the ROS that are essential for inflammasome activation, as these enzymes are known to have a role in innate immunity through the generation of the bacteriocidal respiratory burst. Pharmacological inhibitors of NADPH oxidases such as diphenyleneiodonium (DPI) or apocynin, or small interfering RNA-mediated knockdown of expression of p22 phox, an essential component of NOX1–NOX4, reduced NLRP3 inflammasome activation 36,40,41. Antioxidants thought to act on NADPH oxidases reduced IL-1β secretion in both CAPS patients and healthy donors 42. However, DPI and apopcynin can impair other flavin-containing enzyme complexes that generate ROS such as those in the electron transport pathway in mitochondria, calling into question its role as a specific inhibitor of NADPH oxidases 43. Furthermore, macrophages from patients with chronic granulomatous disease who are deficient in subunits of the NOX2 NADPH oxidase produce IL-1β normally in response to LPS, and NOX2 deficient mice exhibit increased pro-inflammatory responses in response to a number of sterile inflammatory stimuli 44-46. For these reasons, it seems likely that ROS generated through other pathways besides NADPH oxidase may enable NLRP3 inflammasome activation.
Mitochondrial damage and consequent ROS production have been recently implicated in the activation of the NLRP3 inflammasome. Blocking mitophagy, the autophagic removal of damaged mitochondria, resulted in increased mitochondrial ROS production and NLRP3 inflammasome activation, whereas inhibition of ROS reversed the increased IL-1β production 47,48. Oxidative damage to mitochondrial DNA may provide a link between ROS generation and inflammasome activation as oxidized mitochondrial DNA can directly trigger NLRP3-dependent IL-1 processing 49. However, kinetic studies have shown that mitochondria-targeted antioxidants also block the transcription of inflammatory genes, including NLRP3, prior to inflammasome activation, suggesting a role for mitochondrial ROS in in the priming phase of inflammasome activation 45,50.
How do NLRP3 mutations in CAPS enhance inflammasome activation? Most of the 110 known disease-associated mutations are concentrated in exon 3, encoding the nucleotide binding domain (NBD), suggesting that this domain has functional importance in disease process 51. The NLRP3 NBD binds ATP and acts as an ATPase, and this ability is required for IL-1β processing 52. Intact nucleotide binding is also required for the oligomerization of NLRP3 itself. Importantly, mutant NLRP3 proteins from CAPS patients still bind ATP, and this binding is required for IL-1β processing 52. Modeling has suggested that CAPS-associated NLRP3 mutant proteins oligomerize, potentially having an open form to favour nucleotide binding, resulting in inflammasome activation in the presence of reduced or absent exposure to activating stimuli. Cells harboring CAPS-associated NLRP3 mutations release IL-1β spontaneously and are hyper-responsive to pro-inflammatory stimuli such as TLR ligands. Notably, cells harboring NLRP3 mutations lose the requirement for extracellular ATP to activate IL-1β processing. These effects are seen in cells from CAPS patients who are heterozygous for NLRP3 mutations, suggesting that these mutations function as dominant gain of function alleles.
TNFR-associated periodic syndrome (TRAPS)
TRAPS is an autosomal dominant autoinflammatory disease associated with mutations in TNFRSF1A, encoding TNFR1 53. Patients with TRAPS experience recurrent prolonged episodes of fever lasting for up to six weeks, rash, abdominal pain, and internal inflammatory manifestations, such as serositis, fasciitis and episcleritis 54. Systemic amyloidosis occurs in approximately 10% of TRAPS patients. Seventy-nine distinct mutations in TNFRSF1A have been found to be associated with TRAPS 51 in the hetrozygous state55. Strikingly, almost all TRAPS-associated mutations in TNFR1 are missense mutations that result in amino acid substitutions or small deletions and insertions in the extracellular domain of TNFR1, which is responsible for receptor pre-association and TNF binding. About half of these mutations occur at highly conserved cysteines and other conserved residues that are important for maintaining the secondary structure of the TNFR1 extracellular domain. Another class of mutations associated with TRAPS occur in 1-3% of asymptomatic subjects, including mutations that lead to R92Q and P46L amino acid substitutions, suggesting that they may be more akin to functional polymorphisms. The autoinflammatory phenotype of TRAPS was originally thought to result from reduced proteolytic cleavage of the soluble extracellular domain of TNFR1, but only one mutation, V173D, has been shown to be in or near the metalloprotease cleavage site in TNFR1 56. Reductions in cleavage of cell surface TNFR1 in cells harboring wild-type and mutant TNFR1 have been variable 54. In addition, treatment of TRAPS patients with TNF blocking agents, such as etanercept (a TNFR2-Fc fusion protein), improves symptoms but does not fully suppress disease or prevent amyloidosis, indicating that there may be alternative mechanisms by which mutant TNFR1 causes inflammation in TRAPS 57,58. TRAPS-associated TNFR1 mutant proteins have several abnormalities that probably stem from aberrant folding of the extracellular domain of the protein. Mutant TNFR1 receptors that fail to bind TNF, did not physically interact with wild-type receptor and have reduced or absent expression on the cell surface. The bulk of mutant TNFR1 molecules are retained intracellularly, primarily in the ER, probably because of abnormal protein folding through non-physiological disulphide binding, and accumulate to levels more than 10-fold higher than wild-type TNFR1 59-62.
These findings raise the question of whether TNFR1 mutant proteins might trigger ER stress and the UPR. Cells harboring heterozygous TRAPS-associated TNFR1 do not have altered baseline levels of the ER chaperone protein BiP or the ER stress responsive protein CHOP or aberrant increases in these proteins following induction of the UPR 59,62. However, JNK and p38 MAP kinases are spontaneously activated in cells harboring TRAPS-associated TNFR1 mutations, with further increases occurring after LPS treatment 62. These findings suggest that mutant TNFR1, either through spontaneous TNF-independent signalling due to intracellular protein accumulation, or induction of a low level of ER stress, alters the balance of MAP kinase activation. These alterations in kinase activation have inflammatory consequences, as cells from TRAPS patients and mice engineered to express heterozygous TNFR1 mutations homologous to those linked to TRAPS produce excess inflammatory cytokines in response to LPS in a JNK- and p38 MAP kinase-dependent manner 62. Interestingly, full expression of the inflammatory phenotype in TRAPS seems to require the wild-type as well as the mutant TNFR1 protein, because cells from mice homozygous for TRAPS-associated mutant TNFR1 mimic TNFR1-deficient mice in their resistance to LPS-induced septic shock 62. Thus, the mutant TNFR1 in TRAPS seems to function as an unusual gain of function protein, signalling from within the cell to enhance inflammatory responses, but requiring cooperation with the wild-type receptor to produce the clinical manifestations of TRAPS. These results may explain the partial efficacy of TNF blockade in this syndrome 58, as TNF blockade would only affect extracellular TNF binding to the wild-type TNFR1, not the intracellular effects of the mutant receptor.
The study of TNFR1 mutations in TRAPS has also revealed new insights into mitochondrial biology and the origin of pro-inflammatory ROS. The enhanced MAP kinase activation seen in cells with TRAPS-associated TNFR1 mutations depends on ROS, probably because MAP kinase phosphatases are inactivated by ROS. As was found in recent studies of inflammasome activation, enhanced inflammation in TRAPS was dependent on mitochondrial respiration rather than on NADPH oxidases. Elevated levels of mitochondrial ROS were seen in cells from TRAPS patients and mice with engineered TRAPS-associated TNFR1 mutations 43. MitoQ, a mitochondrially targeted coenzyme Q antioxidant, could reduce inflammatory responses in these cells. Unlike cells in which mitophagy is blocked and ROS leak from a damaged respiratory chain, TRAPS cells have increased oxygen consumption and respiratory capacity. Whether other diseases display this type of increased ROS production due to enhanced respiration would be an important subject for future investigation.
Familial mediterranean fever
Mutations in the MEFV gene, which encodes the protein pyrin, were found to underlie most cases of the autoinflammatory disease FMF, which is characterized by shorter episodes of fevers and inflammation involving the joints and serosal surfaces 63. FMF was previously accompanied by high rates of amyloidosis until the introduction of colchicine therapy in the 1970s. Pyrin is an adaptor protein that contains an N-terminal eponymous pyrin domain and a domain in the C-terminal end termed the B30.2 or PRY-SPRY domain. Interestingly, the B30.2 domain is present in primate and human pyrin but not rodent pyrin 64, and most missense mutations associated with FMF in humans are in this domain. Pyrin can assemble with ASC and caspase 1 in human cells to activate caspase 1 cleavage 65. However, pyrin has also been reported to function as a negative regulator of inflammasome function, through interactions between the B30.2 domain of pyrin and caspase 1 66-68. FMF-associated mutations in the B30.2 domain reduce interactions with caspase 1 and potentiate caspase 1 activation 67,68. Gain of function mutations in pyrin exert their effects independent of NLRP3, suggesting that a distinct inflammasome may be the target of regulation by pyrin 69.
Although FMF has been traditionally thought of as an autosomal recessive disease, sequencing of the MEFV locus in FMF patients revealed a significant fraction of patients with only one mutated MEFV allele, which raises the possibility that FMF could also result from MEFV haploinsufficiency, a dominant negative mechanism or a dose-dependent gain of function 70. The fact that nearly all MEFV mutations are missense point mutations rather than nonsense mutations or deletions suggests that the mutant protein may have pro-inflammatory functions 70. Indeed, mice engineered to express a human-mouse chimeric pyrin with B30.2 domain of human pyrin harboring FMF-associated mutations developed spontaneous systemic inflammation similar to, but more severe than human FMF. This phenotype only resulted when the mutant B30.2 domain was present in both pyrin alleles, suggesting a gain of function mechanism with a gene dosage effect. Inflammation in these mice was dependent on bone marrow-derived cells expressing IL-1β and ASC, but interestingly not NLRP3, suggesting that human mutant pyrin activates an NRLP3-independent inflammasome complex 69. These data indicate that mutant pyrin enhances inflammation and the symptoms of FMF in a dominant manner that is highly dependent on gene dosage, although halploinsufficiency or a dominant-negative mechanism that inhibits an unknown inflammatory suppressive function remains conceptually possible.
A role for the UPR in polygenic autoinflammatory diseases
HLA-B27 misfolding and spondyloarthritis
Spondyloarthropathies are a family of immune-mediated inflammatory diseases that are strongly associated with the MHC class I allele HLA-B27 71. Although genetic predisposition is complex, HLAB27 constitutes up to 30-40% of overall risk. In ankylosing spondylitis, the prototypic spondyloarthropathy, there is spinal inflammation with new bone formation, resulting in fusion of sacroiliac and vertebral facet joints, along with syndesmophyte formation that links adjacent vertebral bodies. Despite years of intense investigation, the role of HLA-B27 in these disorders remains enigmatic, and a definitive role for particular peptides presented by HLA-B27 to CD8+ T cells in the pathogenesis of these diseases has not been identified. There is increasing evidence that these diseases may be more autoinflammatory rather than autoimmune in nature 72.
Recent evidence has suggested an alternative role for HLA-B27 in the pathogenesis of spondylarthropathies independent of its role in antigen presentation. A tendency of the HLA-B27 heavy chain to misfold, with prolonged ER retention, BiP binding and formation of disulphide-linked heavy chain complexes, may provide clues to its role in disease 73. In cells from transgenic rats, HLA-B27 misfolding results in ER stress and UPR activation, particularly after upregulation of expression 74,75. Occurrence of the UPR together with macrophage activation leads to synergistic induction of certain cytokines, most notably IFNβ and IL-23 after exposure to TLR agonists such as LPS. 76,77. XBP1 is required for the UPR effect on IFNβ, whereas CHOP mediates the IL-23 superinduction 78. HLA-B27 may trigger the UPR more easily than other misfolded proteins, such as TNFR1 in TRAPS, because it is a relatively abundant cellular protein that can be substantially upregulated. The correlation between upregulation of HLA-B27 by stimuli such as interferons (IFNs), and induction of the UPR supports this hypothesis 74,75.
In transgenic rats, macrophages that exhibit UPR activation tend to produce more IL-23 relative to IL-12, suggesting that HLA-B27 misfolding could be a stimulus for the activation of T helper 17 (Th17) cells 77. Consistent with this, myeloid cells from the colon of these rats exhibit UPR activation and IL-23 upregulation, and CD4+ T cells overexpress IL-17 coincident with the development of inflammation 77. These results provide a compelling model that links HLA-B27 misfolding to the pathogenesis of inflammation independent of its role in antigen presentation to CD8+ T cells. Thus, although T cells have a role in pathogenesis, there are likely to be upstream autoinflammatory signals that may converge to activate cells expressing the IL-23 receptor including Th17 cells. It is of interest that polymorphisms in the IL23R gene influence susceptibility to ankylosing spondylitis 79.
UPR activation in inflammatory myositis
Idiopathic inflammatory myopathies (IIM) are a group of inflammatory muscle diseases including juvenile and adult dermatomyositis, polymyositis and inclusion body myositis. Chronic skeletal muscle inflammation and muscle fiber damage are characteristic features of these diseases. While the adaptive immune response is involved in pathogenesis, it has recently become clear that the innate immune system also plays a key role. Notably, type I IFNs may be particularly important in dermatomyositis, where there is strong evidence for a distinct IFN-induced gene signature in muscle tissue 80,81. The cellular source of the type I IFNs has been difficult to elucidate. However, recent evidence suggests that in addition to plasmacytoid dendritic cells, immature myocytes expressing TLR3 are a local source of IFNβ 82. The IFNβ may help drive the inappropriate high-level MHC class I expression seen in these cells. Interestingly, UPR activation is prominent in certain forms of IIM in association with increased MHC class I expression. In a mouse model, enforced conditional expression of MHC class I (H-2Kb) in muscle cells initiates the UPR and causes muscle inflammation that is thought to be sustained through UPR-induced apoptosis of myocytes 83. However, the ability of spliced XBP1 to potentiate IFNβ induction by TLR3/4 agonists 76 suggests that myocytes affected by UPR activation could be a significant source of type I IFN. These studies demonstrate another potential link between UPR activation and a disease that is associated with autoantibodies, but may nevertheless have a strong component of autoinflammation.
Dysregulation of the immunoproteasome and the UPR in autoinflammation
In spondylarthropathies and inflammatory myositis, inflammation may arise in the context of activation of the UPR. However, the UPR also restores homeostasis in cells, in part by mediating a transient block in protein synthesis. When the UPR is dysfunctional, inflammation can arise as a consequence of the inability of cells to respond to unfolded proteins 84. Conditional knockouts of XBP1 in intestinal epithelial cells make mice hypersensitive to colitis induced by dextran sodium sulphate (DSS), and also cause spontaneous ileal inflammation that is probably due to the loss of secretory Paneth cells and goblet cells in the intestinal epithelia 85. Paneth cells secrete antimicrobial peptides and IL-1 in response to bacteria, and the loss of these cells may impair host defence against commensal organisms that results in inflammatory bowel disease (IBD). XBP1 polymorphisms were found to be a genetic risk factor for ulcerative colitis and Crohn's disease, suggesting that these findings may be relevant for human IBD 85. In contrast, deletion of XBP1 in macrophages reduces their ability to produce sustained levels of pro-inflammatory cytokines in response to TLR agonists, resulting in an inability to clear bacterial infection 13. Similarly, in B cells, an intact UPR is required for plasma cell differentiation, and XBP1-deficient B cells die when stimulated to become plasma cells 86.
Although complete deficiency of the proteasome or UPR-associated genes are likely to be lethal in man, loss of function mutations in PSMB8, which encodes the β5i subunit of the immunoproteasome, have been recently identified as the cause of a spectrum of systemic autoinflammatory conditions that are associated with lipodystrophy 19-22. In addition to its role in antigen presentation, the immunoproteasome has recently been shown to mediate decay of short-lived proteins in the secretory pathway 87. The mechanism by which this occurs is not clear, although incomplete clearance of proteins by the immunoproteasome in PSMB8 deficiency results in intracellular accumulation of ubiquitylated proteins, which enhances inflammatory responses.
Applying insights from genetic disease to novel therapies for more common inflammatory disorders
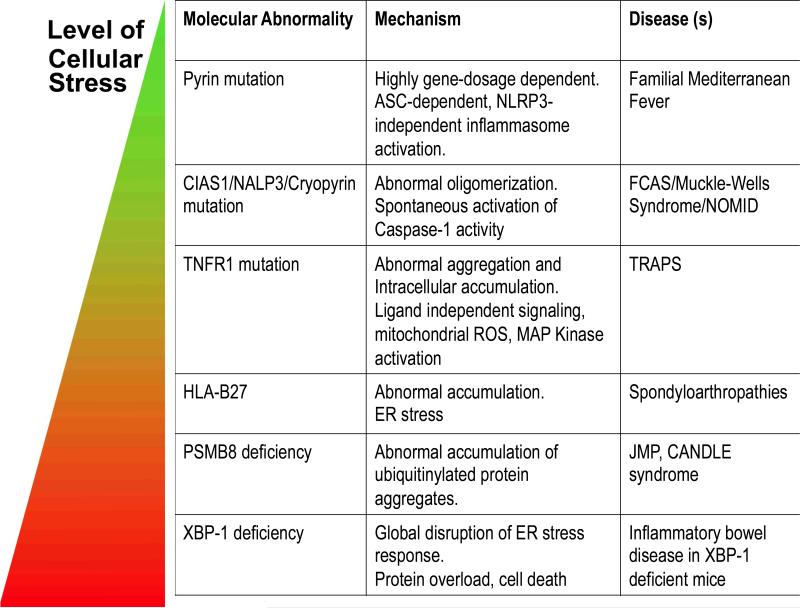
Taken together, the molecular abnormalities that connect protein misfolding to autoinflammatory disease can be seen as a spectrum of disorders, in which increasing levels cellular stress trigger commensurate pathological consequences (Table 2). In CAPS, NLRP3 mutations lead to hyperactivation of the inflammasome and production of IL-1, probably because of enhanced oligomerization of NLRP3. In TRAPS, mutations more severely affect the TNFR1 protein, causing trafficking abnormalities and abrogating the normal signalling function of TNFR1 as a TNF receptor. In diseases linked to the abundantly expressed HLA class I genes, misfolded proteins may accumulate over the threshold to trigger the UPR and induction of chaperone gene expression. In spondyloarthropathies, the UPR can cause dysregulation of cytokine production. In the extreme case of XBP1 deficiency, pathology results from the consequences of a lack of appropriate cellular responses to ER stress.
Table 2. Pathogenic mechanisms in autoinflammatory diseases.
The abnormal folding and accumulation of proteins regulating inflammation can lead to different degrees of cellular stress. The effect of mutations in pyrin and NLRP3 is primarily local, HLA-B27 and XBP1 deficiency can activate the unfolded protein response (UPR) associated with toxicity and enhanced inflammatory responses.
|
Therapeutic strategies aimed at the problems of protein misfolding and its consequences for disease are being actively pursued. Chemical chaperones and drugs that stimulate autophagy may be of some benefit in this regard. One of the consequences of misfolding of certain ER-synthesized proteins is activation of the UPR. Pharmacological targeting of the signaling molecules that mediate this response is possible, and enhancing UPR activation might be a viable strategy to improve protein homeostasis. However, the UPR is a proverbial double-edged sword in that its activation in innate immune cells may exacerbate inflammation and disease manifestations. There is a clear need to better understand the mechanisms and consequences of protein misfolding in specific autoinflammatory disorders if we are to develop safe and effective targeted therapies for these diseases.
Targeting cytokines produced in genetic autoinflammatory syndromes has been a very fruitful area of clinical investigation. The degree of success of these therapies has shed light on importance of the targeted cytokine to the pathological process. For example, therapies targeting Interleukin 1 have been highly effective in suppressing most symptoms and halting progression of organ damage in CAPS 88,89, whereas TNF blockade is only partially effective in TRAPS 58. These successes have inspired therapeutic trials in more common diseases, particularly for interleukin 1 blockade 90. In diseases where there is a clear role for the NLRP3 inflammasome in disease pathogenesis, such as gout, therapeutic trials of IL-1β inhibition have been successful in reducing symptoms in refractory disease and preventing flares during uric acid lowering therapy 91,92. In type II diabetes, the efficacy of interleukin-1 blockade has sparked interest of the role of the inflammasome in pancreatic beta cell dysfunction 93,94. Remarkably, in both gout and type II diabetes, the anti-inflammatory effects of IL-1 blockade appeared to persist far after withdrawal of therapy. The mechanism for these sustained effects needs further study. In other diseases, such systemic-onset Juvenille Inflammatory Arthritis, analysis of gene expression and cytokine production profiles has suggested a role for IL-1β, and pilot trials of IL-1 blockade been moderately successful in this pediatric inflammatory syndrome 95,96. Identification of particular inflammatory cytokines driving other more common diseases through gene expression profiling and the analysis of cytokines produced by peripheral blood cells in other disease states will likely lead to more therapeutic trials of cytokine blockade which will shed light on the pathogenesis of other inflammatory diseases and hopefully lead to better treatments. With the large number of anti-cytokine biologic therapies and small molecules targeting the JAK kinases approved for clinical use and in late-stage trials, testing the role of specific cytokines in human disease through clinical investigational trials will become an increasingly common way of determining the relevance of autoinflammation to human disease.
Glossary
- Autophagy
An evolutionarily conserved process in which acidic double-membrane vacuoles sequester intracellular contents (such as damaged organelles and macromolecules) and target them for degradation through fusion to secondary lysosomes. Selective removal of mitochondria by under conditions of nutrient starvation or mitochondrial stress is termed Mitophagy
- Colchicine
An inhibitor of microtubule polymerization that has anti-inflammatory properties, possibly by blocking neutrophil chemotaxis. Colchicine is effective in treating acute attacks of gout and protecting against recurrent flares. Also effective in familial mediterranean fever, but not other genetic autoinflammatory syndromes. Damage associated molecular pattern molecules (DAMPs) are ones released following tissue injury or certain cellular pathways. DAMPs are mainly host nuclear and cellular proteins or ATP or uric acid. In contrast, Pathogen-associated molecular pattern molecules (PAMPs) are released when cells encounter pathogens. Both DAMPs and PAMPs trigger inflammatory response in innate immune cells via various pattern recognition receptors.
- ER stress
Stress caused by perturbation in ER function that is necessary for the cell homeostasis. Unfolded protein response (UPR) is the collective outcome of ER stress and must be tightly regulated otherwise directly linked to a number of disease pathogenesis.
- Gout
An autoinflammatory disease triggered by crystalline uric acid and characterized by episodic flares of arthritis which can progress to destructive joint damage.
- Immunoproteasome
The standard proteasome is composed of 14α and β subunits, of which three, β1, β2 and β5, are involved in peptide-bond cleavage. Interferon-γ induces the expression of the immunosubunits β1i, β2i and β5i that can replace the catalytic subunits of the standard proteasome to generate the immunoproteasome, which has distinct cleavage-site preferences.
- Inflammasome
A large multiprotein complex usually comprising an NLR (NOD-like receptor), the adaptor protein ASC and pro-caspase 1. The assembly of the inflammasome leads to the activation of caspase 1, which cleaves pro-interleukin-1β (pro-IL-1β) and pro-IL-18 to generate the active pro-inflammatory cytokines.
- NADPH oxidase
A plasma membrane- and phagosomal membrane-bound enzyme complex that transfers electrons from NADPH to molecular oxygen, promoting the generation of the reactive oxygen species superoxide.
- Spondylarthropathies
A group of immune-mediated inflammatory disorders that share the property of affecting the vertebral column. Spondlyarthropathies, also frequenctly affect the enthesis, areas of tendon and ligament insertion into bone.
- Sterile inflammation
Inflammation, characterized by leukocyte recruitment, that does not involve infection but is precipitated by the activation of innate immune cells by endogenous mediators (alarmins or DAMPs) that are released from host cells following tissue injury and necrotic cell death.
Footnotes
Online and other resources on Autoinflammatory Diseases.
Database of autoinflammatory disease-associated mutations: http://fmf.igh.cnrs.fr/ISSAID/infevers/.
International Society of Systemic Auto-Inflammatory Diseases: http://fmf.igh.cnrs.fr/ISSAID/
References
- 1.McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3:e297. doi: 10.1371/journal.pmed.0030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [This paper connected the pathogenesis of gout to activation of the NLRP3 inflammasome by uric acid crystals] [DOI] [PubMed] [Google Scholar]
- 4.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 5.Pipe SW, Kaufman RJ. Factor VIII C2 domain missense mutations exhibit defective trafficking of biologically functional proteins. J Biol Chem. 1996;271:25671–25676. doi: 10.1074/jbc.271.41.25671. [DOI] [PubMed] [Google Scholar]
- 6.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79:S167–173. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oyadomari S, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datta R, Waheed A, Bonapace G, Shah GN, Sly WS. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc Natl Acad Sci U S A. 2009;106:3437–3442. doi: 10.1073/pnas.0813178106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harraz MM, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urano F, et al. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 12.Zhang K, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 13.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masters SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature Immunology. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kastner D, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory Disease Reloaded: A Clinical Perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [A concise review that highlights recent advances in the pathogenesis of and treatment of autoinflammatory diseases] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jordan CT, et al. PSORS2 is due to mutations in CARD14. American journal of human genetics. 2012;90:784–795. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan CT, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. American journal of human genetics. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal AK, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. American journal of human genetics. 2010;87:866–872. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arima K, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proceedings of the National Academy of Sciences. 2011;108:14914–14919. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamura A, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. Journal of Clinical Investigation. 2011;121:4150–4160. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, et al. Mutations in PSMB8 cause CANDLE syndrome with evidence of genetic and phenotypic heterogeneity. Arthritis and rheumatism. 2011 [References 20, 21, and 22 describe recessive mutations in the PSMB8 immunoproteasome subunit that cause autoinflammatory syndromes] [Google Scholar]
- 23.Aksentijevich I, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 25.Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. JOURNAL OF IMMUNOLOGY. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisenbarth SC, Flavell RA. Innate instruction of adaptive immunity revisited: the inflammasome. EMBO molecular medicine. 2009;1:92–98. doi: 10.1002/emmm.200900014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeru I, Hayrapetyan H, Duquesnoy P, Sarkisian T, Amselem S. PYPAF1 nonsense mutation in a patient with an unusual autoinflammatory syndrome: role of PYPAF1 in inflammation. Arthritis and rheumatism. 2006;54:508–514. doi: 10.1002/art.21618. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka N, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63:3625–3632. doi: 10.1002/art.30512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marina-Garcia N, et al. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J Immunol. 2008;180:4050–4057. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- 34.Sharp FA, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [References 34-36 demonstrate activation of the NLRP inflammasome through environmental particulate stimuli and the adjuvant Aluminum Hydroxide (Alum)] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parikh H, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2009;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 39.Menu P, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012;3:e261. doi: 10.1038/cddis.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 41.Cruz CM, et al. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tassi S, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci U S A. 2010;107:9789–9794. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulua AC, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). The Journal of Experimental Medicine. 2011;208:519–533. doi: 10.1084/jem.20102049. [This study demonstrates a mitochondrial source for ROS species seen in TRAPS and suggests that enhanced inflammatory cytokine production results from ROS-triggered MAPK signaling] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollock JD, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 45.van de Veerdonk FL, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A. 2010;107:3030–3033. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meissner F, et al. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–1573. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2010 doi: 10.1038/nature09663. [This study was the first to suggest that dysfunctional mitochondria may activate the NLRP3 inflammasome, although more recent work suggest that ROS also work through increasing transcription of inflammasome component proteins (Refs 45, 50)] [DOI] [PubMed] [Google Scholar]
- 48.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1|[bgr]| production. Nature aop. 2008 doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 49.Shimada K, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauernfeind F, et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Touitou I, et al. [June, 2012];InFevers The registry of Hereditary Auto-Inflammatory Disorders Mutations. http://fmf.igh.cnrs.fr/ISSAID/infevers/42012.
- 52.Duncan JA, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A. 2007;104:8041–8046. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDermott MF, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 54.Hull KM, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002;81:349–368. doi: 10.1097/00005792-200209000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Kimberley FC, Lobito AA, Siegel RM, Screaton GR. Falling into TRAPS – receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res Ther. 2007;9:217. doi: 10.1186/ar2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stojanov S, et al. Clinical and functional characterisation of a novel TNFRSF1A c.605T>A/V173D cleavage site mutation associated with tumour necrosis factor receptor-associated periodic fever syndrome (TRAPS), cardiovascular complications and excellent response to etanercept treatment. Ann Rheum Dis. 2008;67:1292–1298. doi: 10.1136/ard.2007.079376. [DOI] [PubMed] [Google Scholar]
- 57.Hull Treatment of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) with the Recombinant Human Tumor Necrosis Factor Receptor TNFR2:Fc Fusion Protein, Etanercept. e.a. in preparation. [Google Scholar]
- 58.Bulua AC, et al. Efficacy of etanercept in the tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Arthritis and rheumatism. 2011 doi: 10.1002/art.33416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lobito AA, et al. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood. 2006;108:1320–1327. doi: 10.1182/blood-2005-11-006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Todd I, et al. Mutant tumor necrosis factor receptor associated with tumor necrosis factor receptor-associated periodic syndrome is altered antigenically and is retained within patients' leukocytes. Arthritis Rheum. 2007;56:2765–2773. doi: 10.1002/art.22740. [DOI] [PubMed] [Google Scholar]
- 61.Rebelo SL, et al. Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–2687. doi: 10.1002/art.21964. [DOI] [PubMed] [Google Scholar]
- 62.Simon A, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A. 2010;107:9801–9806. doi: 10.1073/pnas.0914118107. [A study describing cooperativity between hunctional wild-type TNFR1 and TRAPS-associated mutant TNFR1 that leads to enhanced inflammatory responses through MAPK activation] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The International FMF Consortium Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 64.Chae JJ, et al. Isolation, genomic organization, and expression analysis of the mouse and rat homologs of MEFV, the gene for familial mediterranean fever. Mamm Genome. 2000;11:428–435. doi: 10.1007/s003350010082. [DOI] [PubMed] [Google Scholar]
- 65.Yu J-W, et al. Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 2006;13:236–249. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
- 66.Seshadri S, Duncan MD, Hart JM, Gavrilin MA, Wewers MD. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1beta processing and release. J Immunol. 2007;179:1274–1281. doi: 10.4049/jimmunol.179.2.1274. [DOI] [PubMed] [Google Scholar]
- 67.Chae JJ, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papin S, et al. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14:1457–1466. doi: 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- 69.Chae JJ, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [Using a knock-in mouse model, this study demonstrates that the human-specific B30.2 domain of pyrin that is the focus of mutations in FMF contributes to caspase-1 and interleukin-1β dependent inflammation through a mechanism independent of NLRP3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Booty MG, et al. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009;60:1851–1861. doi: 10.1002/art.24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369:1379–1390. doi: 10.1016/S0140-6736(07)60635-7. [DOI] [PubMed] [Google Scholar]
- 72.Ambarus C, Yeremenko N, Tak PP, Baeten D. Pathogenesis of spondyloarthritis: autoimmune or autoinflammatory? CURRENT OPINION IN RHEUMATOLOGY. 2012;24:351–358. doi: 10.1097/BOR.0b013e3283534df4. [DOI] [PubMed] [Google Scholar]
- 73.Smith JA, Marker-Hermann E, Colbert RA. Pathogenesis of ankylosing spondylitis: current concepts. Best Pract Res Clin Rheumatol. 2006;20:571–591. doi: 10.1016/j.berh.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 74.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56:215–223. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 75.Turner MJ, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [This is the study was the first to show that HLA-B27 may contribute to the pathogenesis of spondylarthropathies through misfolding in the ER and induction of the unfolded protein response] [DOI] [PubMed] [Google Scholar]
- 76.Smith JA, et al. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DeLay ML, et al. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goodall JC, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107:17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burton PR, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffin TA, Reed AM. Pathogenesis of myositis in children. CURRENT OPINION IN RHEUMATOLOGY. 2007;19:487–491. doi: 10.1097/BOR.0b013e32825a6a57. [DOI] [PubMed] [Google Scholar]
- 81.Liao AP, et al. Interferon beta is associated with type 1 interferon-inducible gene expression in dermatomyositis. Annals of the Rheumatic Diseases. 2011;70:831–836. doi: 10.1136/ard.2010.139949. [DOI] [PubMed] [Google Scholar]
- 82.Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature muscle precursors are a source of interferon-beta in myositis: role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis and rheumatism. 2012;64:533–541. doi: 10.1002/art.33350. [DOI] [PubMed] [Google Scholar]
- 83.Nagaraju K, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 85.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iwakoshi NN, et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 87.Seifert U, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 88.Goldbach-Mansky R, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoffman HM, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [These two reports (88-89) describe the efficacy of using interleukin-1 blockade for the inflammatory syndromes linked to dominant NLRP3 mutations] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schumacher HR, Jr., et al. Rilonacept (Interleukin-1 Trap) for prevention of gout flares during initiation of uric acid-lowering therapy: Results of the presurge-1 trial. Arthritis Care & Research. 2012 doi: 10.1002/acr.21690. [DOI] [PubMed] [Google Scholar]
- 92.So A, et al. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis and rheumatism. 2010;62:3064–3076. doi: 10.1002/art.27600. [DOI] [PubMed] [Google Scholar]
- 93.Larsen CM, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. The New England journal of medicine. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 94.Larsen CM, et al. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–1668. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. The Journal of Experimental Medicine. 2005;201:1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lequerre T, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Annals of the Rheumatic Diseases. 2008;67:302–308. doi: 10.1136/ard.2007.076034. [DOI] [PubMed] [Google Scholar]
- 97.Drenth JP, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nature Genetics. 1999;22:178. doi: 10.1038/9696. [DOI] [PubMed] [Google Scholar]
- 98.Feldmann J, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. American journal of human genetics. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aksentijevich I, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis and rheumatism. 2007;56:1273–1285. doi: 10.1002/art.22491. [These two papers (refs 98 and 99) are the initial report and spectrum of inflammatory syndromes linked to dominant mutations in the NLRP3 gene] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rose C, Martin T, Wouters C. Blau syndrome revisited. CURRENT OPINION IN RHEUMATOLOGY. 2011;23:411–418. doi: 10.1097/BOR.0b013e328349c430. [DOI] [PubMed] [Google Scholar]
- 101.Miceli-Richard C, et al. CARD15 mutations in Blau syndrome. Nature Genetics. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 102.Shoham NG, et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci U S A. 2003;100:13501–13506. doi: 10.1073/pnas.2135380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aksentijevich I, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. The New England journal of medicine. 2009;360:2426–2437. doi: 10.1056/NEJMoa0807865. [A report describing the familial syndrome DIRA linked to deficiency of the IL-1-receptor antagonist, which leads to severe systemic autoinflammation. This study confirms a critical role IL-1-receptor antagonist in dampening hyper-production of IL-1β during the inflammatory response in human disease] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marrakchi S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. The New England journal of medicine. 2011;365:620–628. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]