

Summary
Germline determinants of gene expression in tumors are less studied due to the complexity of transcript regulation caused by somatically acquired alterations. We performed expression quantitative trait locus (eQTL) based analyses using the multi-level information provided in The Cancer Genome Atlas (TCGA). Of the factors we measured, cis-acting eQTL saccounted for 1.2% of the total variation of tumor gene expression, while somatic copy number alteration and CpG methylation accounted for 7.3% and 3.3%, respectively. eQTL analyses of 15 previously reported breast cancer risk loci resulted in discovery of three variants that are significantly associated with transcript levels (FDR<0.1). In a novel trans- based analysis, an additional three risk loci were identified to act through ESR1, MYC, and KLF4. These findings provide a more comprehensive picture of gene expression determinants in breast cancer as well as insights into the underlying biology of breast cancer risk loci.
Introduction
Prior studies unambiguously demonstrate that inherited variation is a determinant of gene expression (Cheung et al., 2003; Montgomery et al., 2010; Pickrell et al., 2010; Schadt et al., 2003; Stranger et al., 2005). Polymorphisms associated with mRNA levels are typically referred to as expression quantitative trait loci (eQTLs). eQTL studies have shed light on the genetic architecture of gene expression. eQTLs have also provided key insights into genes and pathways underlying the associations by providing an intermediate phenotype between non-protein coding genetic variants and complex traits (Chen et al., 2008; Cookson et al., 2009; Emilsson et al., 2008; Grisanzio et al., 2012; Musunuru et al., 2010; Pomerantz et al., 2009). In fact, trait-associated loci are enriched for eQTLs (Nicolae et al., 2010). Most of our knowledge, however, is based on data derived from cell lines and normal tissues (Dimas et al., 2009; Myers et al., 2007; Nica et al., 2010). By contrast, cancer studies typically generate more expression data on tumors than in normal tissues.
Mapping eQTL-target gene associations in tumor tissue presents additional analytical challenges. Tumors acquire frequent genetic and epigenetic alterations, which can substantially affect gene expression. For example, somatic copy number changes and DNA methylation status are known to strongly influence transcript abundance in tumors (Curtis et al., 2012; Portela and Esteller, 2010). Consequently, the effect of these somatic alterations may obscure the association between germline genetic polymorphisms and gene expression. The creation of publicly available large-scale datasets, such as the Cancer Genome Atlas (TCGA), and Encyclopedia of DNA Elements (ENCODE) provide comprehensive catalogs of multiple data types performed on the same set of samples. In this study, we use these resources to develop a general method that models transcript levels as having inputs from germline and somatic factors.
eQTL-based strategies provide a straightforward method to link non-protein coding risk alleles discovered through genome wide association studies (GWAS). Understanding the genes and pathways underlying common risk alleles remains a formidable challenge because the majority of trait-associated polymorphisms are outside of known protein coding regions (Hindorff et al., 2009). Many of these loci are thought to be involved in transcriptional regulation (Freedman et al., 2011; Nicolae et al., 2010). Therefore, eQTL-based approaches are well suited to identifying candidate genes acting through these loci. We apply our method to known breast cancer risk alleles discovered through GWAS to link the risk variants with their target genes.
Results
The results are presented in two main sections. First, a general approach is described to identify cis- acting eQTL-target gene pairs in tumor genomes while adjusting for other factors that can also influence transcript abundance. Using this method, we conducted a study to identify determinants of transcript variation in the TCGA breast cancer samples.
Next, to gain biologic insights into known breast cancer risk polymorphisms, we hypothesized that the breast cancer risk alleles previously discovered through GWAS are eQTLs. We performed separate cis- and trans- based eQTL analyses for 15 previously identified risk loci in the TCGA breast cancer datasets.
Breast cancer data set
We selected 407 patients with both tumor samples and matched normal blood samples from the TCGA breast cancer dataset. For each of these samples, we obtained the germline genotypes from the normal blood sample and somatic copy number, methylation, mRNA gene expression measures from the matched tumor sample (Table S1, related to Figure 1). Ancestry was inferred using genotype data resulting in 273 cases of European ancestry. In the analyses below, 171 ER-positive cases and 48 ER-negative cases (as determined by ESR1 expression levels) were used (Methods).
Figure 1.

Effects of three determinants on gene expression in ER-positive breast cancer:cis-acting SNP loci, somatic copy number and CpG methylation. (a) Venn diagram shows the number of genes that are under regulation of one or multiple factors. (b) Pie chart shows the relative and absolute fraction of variance of gene expression explained by three factors. Please see also Figure S1, Figure S2, Table S1, Table S2, Table S3, Table S5.
eQTL analysis of ER-positive breast cancer
For a given gene i and a SNP locus j, we consider three factors of transcript abundance (T), the germline genotypes as the genetic determinants (G), the somatic copy number alterations (Sc) and the CpG methylation levels in the promoter region (M), we first used multivariate linear regression (1) to compute the residual expression εi of Sci and Mi:
| (1) |
Then we regress on the residual expression εi to the germline genotypes Gi where ω ωi is the random error.
| (2) |
Thus, the effects of the genetic determinants, the somatic copy number changes, and methylation levels on the transcript abundance are estimated separately.
Using this model, we evaluated the association between the germline genotypes of 816,362 SNP loci and genes within 1 Mb on either side of each SNP locus. In order to further control the false positive rate, we excluded 396 genes with low expression levels (less 10% present call), 85 genes with probes having a known sequence polymorphism, as well as the highly polymorphic HLA region. A Q-Q plot of the raw p-values corresponding to the cis-associations tested before and after adjusting for somatic factors suggests systematic bias in the unadjusted data (λ = 1.29), which is eliminated after the adjustment (λ = 0.94∼1.01, Figure S1).
Significant associations were determined by applying a false-discovery rate (FDR) threshold of less than 0.1. As a result, from 8,107,200 unique SNP-gene pairs we identified 6,046 associations with raw P-values below 7.50×10-5. These associations mapped to a total of 6,145 SNP loci and 1,359 unique target genes (Table S2, related to Figure 1). The fraction of variation of the expression levels explained by the associated cis-acting SNP loci (R2) ranged from 18.5% to 73.2%.
689 (50.7%) of the target genes are regulated by a single cis-acting SNP locus (Figure S2A, related to Figure 1); the rest are regulated by multiple loci (2 to 83). After adjusting correlated loci by stepwise feature selection (Methods), 664 genes (48.9%) are explained by multiple SNP loci (median number 4), suggesting the existence of multiple independent eQTLs. 5,669 of the 5,893 cis-acting eQTL loci are associated with one target gene, suggesting the associations are highly locus specific (Figure S2B, related to Figure 1). Cis-associations typically occur within the 2 nearest transcripts (39.8%) of the eQTL; an additional 18.9% occur between 3 to 5 nearest transcripts (Figure S2C, related to Table 1).
Table 1.
Transcription factors mediating distant associations between risk loci for estrogen receptor positive breast cancer and gene expression.
| Region | Chr | Tested SNP | Physical Position | No of associated genes (P < 0.05) | P-value (Target genes P < 0.05) | TF within 1 mb | Distance between risk locus and TF (kb) |
|---|---|---|---|---|---|---|---|
| 6q25.1 | 6 | rs2046210 | 151990059 | 476 | 4.23×10-7 | ESR1 | 180 |
| 8q24.21 | 8 | rs418269 | 128415540 | 221 | 4.19×10-8 | MYC | 402 |
| 9q31.2 | 9 | rs471467 | 109927934 | 415 | 6.42×10-16 | KLF4 | 641 |
Determinants of transcript levels
Transcript levels of 8,568 genes (54.5% of the total genes tested) are significantly affected by the somatic copy number changes in the corresponding coding regions (FDR < 0.1). Among the 1,359 targets genes of the cis-acting SNP loci, 880 (64.8%) are also significantly associated with somatic copy number. We also identified 2,529 transcripts (16.1% of the total genes tested) that are affected by the methylation of CpG islands in the promoter region (FDR < 0.1). Among these genes, 210 are also target genes of eQTLs (15.5%, Figure 1A). Of the 15,732 genes tested, the cis-acting SNP loci account for 1.2% of the total variance of the expression. Somatic copy number alterations account for 7.3% and methylation status account for 3.3% of variation in gene expression (Figure 1B).
eQTL analysis of ER-negative breast cancer
To compare the genetic determinants of gene expression in different subtypes of breast cancer, we performed a cis-eQTL analysis for 48 ER-negative breast cancer samples. Based on the model described above, we identified 380 significant cis-associations (FDR < 0.1), mapping to 380 SNP loci and 179 target genes (Table S3). Notably, only 43 target genes (24.0%) from ER-positive tumors are represented in the significant associations in ER-negative tumors.
eQTL analysis of breast cancer risk loci
Cis- analysis
We focused on 15 breast cancer loci previously reported by genome-wide association studies (P < 5×10-8, Table S4, related to Table 1)(Hindorff LA). We identified three significant cis-associations mapping to risk loci at 2q35 (IGFBP5), 5q11 (C5orf35) and 16q12 (TOX3)(FDR<0.1, Table S5).
Trans- analysis
We next hypothesized that the 15 GWAS-identified risk loci are cis-acting eQTLs of transcription factors (TFs), which in turn influence multiple downstream targets. In such cases, the activity level of a given TF may be more accurately reflected in the expression levels of its target genes (Essaghir et al., 2010; Wolfer et al., 2010). It is then possible to examine the set of eQTL-associated genes and then work “backwards” to evaluate if a TF binding motif is enriched in the target genes (Figure 2). Thus, each risk locus will be associated with a set of target genes. For each set of target genes, the putative enhancer regions, as defined by ENCODE generated DNaseI hypersensitivity (DHS) data from the MCF-7 cell line (a breast cancer cell line), were analysed for TF DNA binding motif enrichment (Methods). This analysis revealed 3 risk loci for which the target genes are significantly enriched for 3 particular TF motifs (6q25/ESR1; 9q31/KLF4, 8q24/MYC) (Table 1). Moreover, as mandated by our analysis, the particular TF whose motif is over-represented in the target genes is physically located nearby the risk locus. Of note, six of the 15 risk loci have a TF located nearby, but the TF motif is not enriched in the target genes (Table S6, related to Figure 2).
Figure 2.

Schematic of the hypothesis that risk alleles are cis-eQTLs of transcription factors. (A) A risk locus (blue triangle) cis-regulates a transcription factor (TF; red explosion). (B) the messenger RNA of the TF is translated into its active form and (C) binds to the target genes. (D) These target genes are associated with the risk allele, but not the TF because the TF is itself tightly regulated. (E) DNA sequences within DNaseI hypersensitive sites (yellow peak) are evaluated for TF binding motif enrichment. Please see also Table 1, Table S5.
The ESR1 enrichment is of particular significance as ESR1 encodes the estrogen receptor (ER), the defining TF of this class of breast cancer. To further evaluate the ER binding sites as defined by in vivo occupancy, we used the ER cistrome previously defined by chromatin immunoprecipitation in MCF-7 cells (Carroll et al., 2006). The regions within the 6q24 gene set computationally predicted to bind ER by the presence of an ER DNA binding motif are significantly enriched for in vivo ER binding sites defined by chromatin immunoprecipitation (P <1×10-6).
Of note, the ESR1, KLF4, and MYC TFs were not significantly associated with genotypic status. Since TF expression levels tend to be under tight regulatory control and are present at low cellular concentrations (Vaquerizas et al., 2009), TFs may be difficult to detect using an eQTL-based methodology.
A complementary method of identifying cis- regulated genes is allelic imbalance (AI). AI for a given gene can be measured when an individual is heterozygous for a transcribed polymorphism. Significant deviations in from a 1:1 ratio in RNA levels indicate genetic and/or epigenetic cis- influences on a gene (Babak et al., 2010; Campino et al., 2008; Ge et al., 2009; Heap et al., 2010). Recently released TCGA RNA-sequencing data enables evaluation of AI in tumor samples (Methods). The expectation is that individuals who are heterozygous for the functional regulatory polymorphism (or any variant in linkage disequilibrium with it) will display AI in the target gene while homozygous individuals will demonstrate less AI (Forton et al., 2007; Lefebvre et al., 2012)(Figure S3, related to Figure 2 and 3).
Figure 3.

Allelic imbalance (AI) of the ESR1, MYC, and KLF4 transcription factors by breast cancer risk genotypic status. Allelic specific expression measures of three TFs were derived from RNA-sequencing of 177 TCGA breast cancer samples. The association between ESR1, MYC and KLF4 and the corresponding risk loci of (A) 6q25 (rs2046210) (B) 8q24 (rs418269) and (C) 9q31 (rs471467) were evaluated using the F-test. Please see also Figure S3, Figure S4C.
Breast cancer TCGA RNA-seq data from 177 individuals were evaluated for the relationship between genotypic status at the risk alleles and AI (Table S7, related to Figure 3, Table S4). After removing cases with somatic copy number alterations, our results demonstrated that the 6q25 and 8q24 risk loci are significantly associated with allelic specific expression of ESR1 and MYC, respectively (P = 0.017 for rs2046210/ESR1 and P = 0.035 for rs418269/MYC; Figures 3A, B). As expected, the AI is greatest in individuals heterozygous for the risk variant. No significant association was observed for KLF4, however the power was limited due to fewer informative variants in the transcribed region (Figure 3C).
To validate the computational results, we sought experimental evidence by chromosome conformation capture (3C) to evaluate physical interactions between the risk loci and the promoters of the TFs. Since the MYC locus has already been shown to interact with the 8q24 risk locus (Ahmadiyeh et al., 2010; Wright et al., 2010; Yochum et al., 2010), we tested for interactions between the ESR1 and KLF4 promoters and the 6q25 and 9q31 risk regions, respectively (Methods). In estradiol stimulated MCF-7 cells, we observed sequence verified interaction products for 6q25 and 9q31 at distances of 189 kb and 644 kb, respectively (Figure 4).
Figure 4.
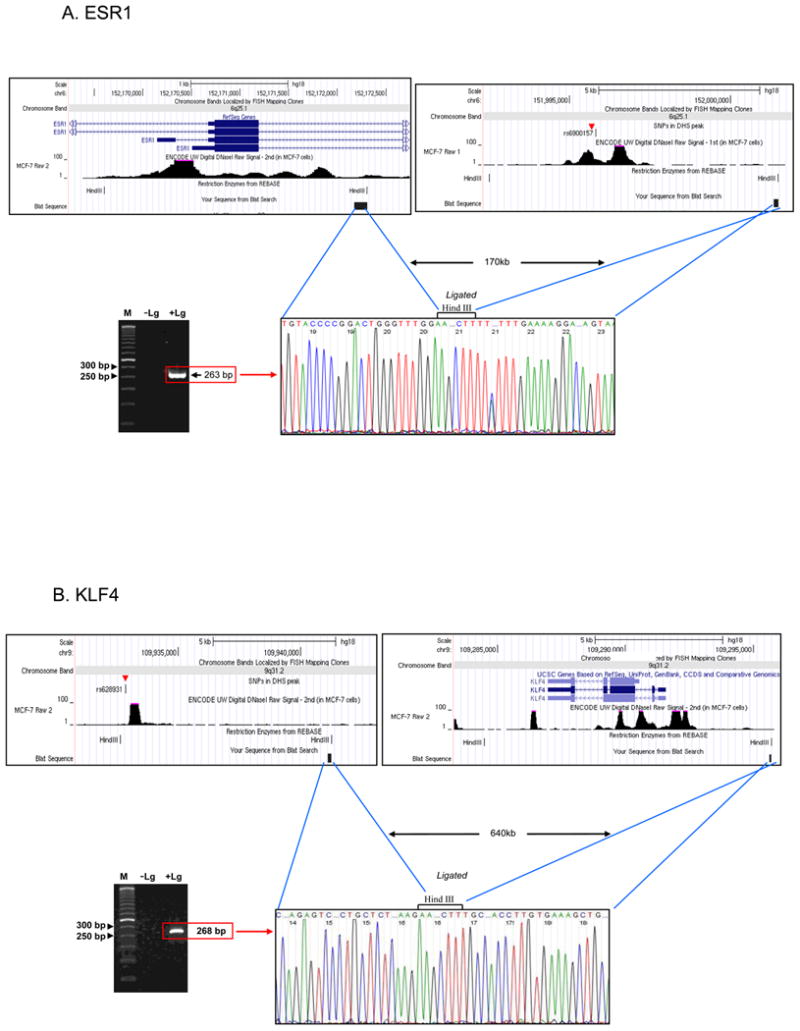
Chromosome conformation capture (3C) demonstrates physical interactions between the 6q25 risk locus and the ESR1 promoter and the 9q21 risk locus and the KLF4 promoter. (A) The top panel shows screenshots of the restriction fragments used for the 6q25/ESR1 interaction. These fragments are separated by approximately 170 kb. The lower left panel shows the gel image of the ligation band. The 263 base pair band is visualized only in the sample with ligase (+Lg) and no band is seen in the negative control sample without ligase (-Lg). The lower right panel demonstrates sequence verification of the +Lg band, confirming this interaction. (B) The 9q21 risk locus and KLF4 physically interact over a distance of 640 kb. Please see also Figure 3, Table 1.
Discussion
Most of our knowledge of the genetics of gene expression has been derived through studies conducted in cell lines and normal primary tissues. Understanding the germline contribution to gene expression levels in tumors is confounded by the acquisition of complex somatic and epigenetic alterations, as well as a dearth of datasets measuring this information on a common set of samples. Although eQTL studies using tumors have been reported, the effects of somatic genetic and epigenetic factors have not been addressed (Grisanzio et al., 2012; Kristensen et al., 2006; Pomerantz et al., 2010). Our method provides a practical solution to account for the multiple factors that determine gene expression levels. Moreover, the method is easily expandable to accommodate other factors, such as somatically acquired single nucleotide variants, that may influence gene expression and can be applied to the growing availability of genome wide tumor datasets.
The overlap of eQTL-target genes between ER-positive and ER-negative samples is approximately 25%. Although the literature continues to emerge on this topic, this degree of eQTL sharing is what is typically observed between different tissue types (Dimas et al., 2009; Nica et al., 2010). The data are consistent with these tumors arising from different cell types.
The breast cancer risk SNP analysis provides a working model of the biology for non-protein-coding risk alleles. Both cis- based and trans- based analyses were performed. In the cis- analysis, we observed 3 transcripts associated with 3 risk loci at an FDR<0.1 (2q35/IGFBP5, 5q11/C5orf35, and 16q12/TOX3). Interestingly, the association with TOX3 (in the same direction) has been previously observed further demonstrating the utility of publicly available data (Riaz et al., 2012). IGFBP5 is a strong candidate gene for mediating the effect of the 2q35 risk locus. In breast cancer cell lines, IGFBP5 overexpression affected cell cycle regulation and apoptosis (Butt et al., 2003). In transgenic mice, IGFBP5 expression interferes with mammary epithelial development (Tonner et al., 2002). IGFBP5 expression levels have also been shown to be a marker of poor outcome in patients with breast cancer (Becker et al., 2012).
Our analysis does not support the previously described association between the 10q risk locus and FGFR2 levels (P = 0.537). Prior studies have yielded inconsistent results with 3 studies demonstrating an association and 2 that have not (Huijts et al., 2011; Martin et al., 2011; Meyer et al., 2008; Riaz et al., 2012; Sun et al., 2010). Variability between the studies may be due to different sample sizes, different tissue types (normal, tumor), and/or measurement of different isoforms. The largest study was performed on 1,401 breast tumor samples and the results showed no association. eQTL studies performed in different cell types and across the full spectrum of expressed isoforms will help to further clarify these observations.
The trans- analysis for the breast cancer risk loci support the assertion that some risk alleles act through TFs that, in turn, influence their target genes. The data demonstrate that target genes of a TF can be used as a surrogate readout for TF activity as has been shown in a recent study (Small et al., 2011). For example, 8q24 associated target genes are enriched for the MYC motif providing an additional layer of support that MYC is a common upstream TF. In addition to MYC, we discovered that the 6q25 and 9q31 risk loci act through the ESR1 and KLF4 TFs. Since the TFs themselves were not associated with genotypic status using an eQTL-based method, we turned to measuring allelic expression of the TFs.
AI is an intuitively advantageous method of detecting cis- regulatory effects. Theoretically, correlating AI with genotypic status should be more sensitive than eQTL based analyses because the comparison is made within an individual (using the other chromosome as a control) thereby removing sources of environmental noise (Fogarty et al., 2010). By applying AI to the TFs and correlating the AI with genotypic status at the risk loci, we demonstrated that MYC and ESR1 are significantly imbalanced in individuals that are heterozygous for the risk locus. A prior report also demonstrated allelic imbalance in MYC expression levels in a colon cancer cell line heterozygous for the 8q24 colon cancer risk allele (Wright et al., 2010). KLF4 did not reveal allelic imbalance most likely due to the low prevalence of informative markers in the KLF4 transcript. The AI method requires appreciable numbers of heterozygous (informative) sites in expressed transcripts. Nevertheless, KLF4 is a strong candidate gene. In prior studies, knockdown of KLF4 in MCF-7 cells affects proliferation, migration, and invasion (Akaogi et al., 2009; Yu et al., 2011). In addition, KLF4 has been shown to play a potent oncogenic role in mammary tumorigenesis by maintaining stem cell-like features (Yu et al., 2011). In agreement with the importance of KLF4 in driving stem cell features is the seminal observation that KLF4 is 1 of 4 TFs required for generating induced pluripotent stem cells (Takahashi et al., 2007).
eQTL-based analyses represent one strategy to detect how risk alleles exert their effects on gene expression. False negative results can be attributed to biologic and technical reasons. For example a variant may influence expression only at a certain developmental timepoint or in a non-cell autonomous fashion. In addition, most of the eQTL analyses are based on measures of steady-state expression levels, how ever a variant could be influencing the non-steady-state aspect of expression, such as rate of transcription. A negative eQTL result may also arise from technical limitations, which can occur due to a lack of power to detect a subtle, but biologically important change in transcript level. Testing associations with other transcribed elements, such as transcript isoforms as well as small and long non-coding RNAs will also be informative. Moreover, it will be interesting to compare eQTL-target gene results in tumors with results derived from normal tissues. The ability to systematically test many of these hypotheses in the near future will be possible given the proliferation of deeply characterized datasets (e.g., GTEx –http://commonfund.nih.gov/GTEx/;MuTHER -http://www.muther.ac.uk/).
While the TCGA was primarily envisioned as a somatically oriented database, we utilized both germline and somatic information to reveal how the data can be used to shed light on risk alleles discovered through GWAS. Our analyses highlight the power and promise of using publicly available datasets to derive biologic insights in unanticipated ways.
Experimental Procedures
Data sets
The breast cancer data set was downloaded from “The Cancer Genome Atlas” data portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp). The germline genotypes are measured from blood-derived DNA samples and the expression profile, the somatic copy number and methylation are measured and inferred from matched tumor samples (Table S1, related to Figure 1).
Verification of ancestry
We selected 250,160 SNP loci with allele frequencies above 0.05 from the SNP profiles of 407 breast cancer samples (the subjects) and 415 HapMap cell lines (the controls). Then we combined the profiles of the 259,001 SNPs from the breast cancer samples and the HapMap cell lines. The combined SNP profiles were run on the EIGENSTRAT program and the top two principal components (PC1 and PC2) were retrieved. From the 273 samples that co-clustered with HapMap CEU controls, we further selected 171 cases with high ESR1 expression levels (above 1) and 48 cases with low ESR1 expression (below 0) with both segmented copy number and methylation measures available.
The somatic copy number and methylation of transcripts
To determine the copy number changes for a given transcript, we retrieved the segmented copy number scores of the tumor sample and the paired-normal control from the level 3 TCGA data, which were both inferred from the Affymetrix SNP 6.0 platform. Then for each transcript, we calculated the averages of the segmented copy number scores of the genetic interval between the transcription start and end sites as gene-based somatic copy number measure (Figure S4A, related to Figure 1).
The CpG methylation measure of each sample was profiled using the HumanMethylation27K array by Illumina, which assays 27,578 CpG sites per sample. These 27,578 sites represent 14,475 consensus coding sequences across the genome, providing approximately 2 CpG assays per gene, with increased targeting of cancer related genes. Then the CpG methylation status was determined by discretization CpG methylation measure with cut-off values of 0.2, 0.4 and 0.6 (Figure S4B, related to Figure 1).
Association analyses
In the cis- or local eQTL analysis we evaluated the associations between the genotype at a given SNP locus and transcripts located within a 2-Mb interval (1-Mb range up and down stream). We excluded 90,238 SNP loci with minor allele frequency < 0.05 and 396 genes with low expression levels - absent calls in more than 90% of the samples. The expression profile of each gene was first adjusted for somatic copy number effects and CpG methylation using a multivariate linear model. The p-value corresponds to the regression coefficient based on the residual expression level and germline genotype.
We performed cis-eQTL analysis between 816,362 SNP loci (22 autosomes and X chromosome) and corresponding mRNA transcripts in ER-positive breast cancer and ER-negative breast cancer respectively. To control for the false predictions, we excluded genes with absent call in over 90% of the samples; and genes of which the probes sets are affected by one or multiple known SNP loci. We also excluded the HLA locus given the high levels of polymorphism in this region. The Benjamini-Hochberg method was used to correct the raw P-values and a significant association was based on a threshold of false discovery rate (FDR) of 0.1.
We assessed 15 previously reported breast cancer risk loci for the regulatory potential in gene expression of breast cancer cells (Table S4, related to Figure 2). Any variant that achieved P < 5×10-8 was selected. To adjust for the effects of somatic factors, we performed multivariate linear regression of the tumor gene expression on somatic copy number and methylation and used the scaled residuals as adjusted measures of gene expression. Then a second regression of the adjusted gene expression on the germline genotypes of each of the 15 risk loci was performed.
The significance of the association between a given risk locus and a gene is given by the test p-value corresponding to the regression coefficient of the genotype. We called significant associations based on a p-value threshold of 0.05. The genes associated with a risk locus at a given significant level are then defined as the target genes of the locus.
Motif Analysis
For each risk locus, we selected the target genes based on significant associations (P < 0.05). Then for each of the target genes, we selected 1-50kb regions on both sides of the transcription start site as putative enhancer regions. We next retrieved all the sites overlapping the DNaseI hypersensitivity (DHS) peaks in the ENCODE MCF-7 DNaseI-seq data generated by the Stamatoyannopoulos group (Birney et al., 2007) (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwDnase/). Thus for each risk locus, we obtained a set of DNA sequences from the putative enhancer regions of the target genes. To control for directional regulatory effects, we considered up-regulated and down-regulated target genes separately. Then we identified the binding motifs of known transcription factors (TFs) from databases embedded in cistrome, including Transfac, JASPAR, UniPROBE (pbm) and hPDI (Liu et al., 2011) which are significantly overrepresented in these sites (P <1×10-6). We further mandated that the TF whose motif was enriched in the target genes to be located within 1 MB range of the corresponding risk locus. If a transcription factor satisfies these criteria, we considered it as a candidate for empirical 3C validation.
To verify that the ESR1 binding site is enriched in the 476 target genes of 6q25 risk locus, we randomly sampled the entire genome 1 million times, each time we pick up 476 genes and counted the the occurence of ESR1 binding site in the putative enhancer regions (1kb to 50kb) according to ChIPseq study. Then we compare the number of ESR1 binding site found in the radom gene sets to that of the 476 6q25 target genes to compute a simulated P value for enrichment.
Determination of allelic imbalance (AI) and correlation with risk allele status
177 breast cancer samples with Caucasian ancestry were selected from TCGA database using EIGENSTRAT. The corresponding RNA-sequencing datasets are available in BAM format from “Cancer Genomics Hub” (CGHub)(https://cghub.ucsc.edu/). We marked all the exonic SNP loci (marker SNP, Table S6, related to Figure 2) mapping to the three TF genes (ESR1, MYC and KLF4) based on NCBI dbSNP build 135. Then for each sample j, we retrieved all the RNA-sequencing reads mapped to the marker SNP locus i and counted for the occurrence of reference (Aij) and alternative (Bij) alleles.
We excluded all SNP loci with low coverage (less than 15x), homozygosity (only one allele present in the mapped reads) and somatic copy number changes (copy number measure 1.5-2.5). For each individual and for a given transcribed SNP, we calculated a measure of allelic imbalance μ given by (3):
| (3) |
Marker SNP loci with extreme major allele fraction (above 0.8) are excluded as many of these loci are falsely called homozygous by sequencing errors. When multiple SNP loci were found for agene in sample j, the average allelic imbalance across all of the marker SNP loci was used to represent that gene.
For each of the three breast cancer risk loci and corresponding TF, we assessed the association between μij and the heterozygosity (AB vs. AA and BB) using F-test p values (Figure S3, related to Figure 3).
In order to address the reference bias introduced by aligning sequencing reads to the reference genome, we also examined the distribution of the reference allele fraction for all the exonic SNP loci that have been used to determine AI status, which showed no bias towards the reference allele (Figure S4C, related to Figure 3).
Chromosome Conformation Capture (3C) and PCR
MCF7 cells were purchased from ATCC and grown in estrogen-depleted media for 3 days and treated with 10 nM of 17β-estradiol for 45min as performed in (Shang and Brown, 2002). A 3C library was then prepared as previously described (Pomerantz et al., 2009). Briefly, after hormone treatment, MCF7 cells were trypsinized, fixed with 1% formaldehyde and, lysed with 3C lysis buffer (10 mM TrisHCl pH 8, 10 mM NaCl, 0.2% Nonidet P-40). The pelleted cell nuclei were resuspended in restriction butter with SDS (final concentration 0.1%) and incubated for 10 min at 65°C. After then, TritonX-100 (final concentration 1%) and HindIII 160 U per 1 × 106 cells were added and incubated for 24 hr at 37°C on a rotating platform. The digested samples were added to the 3C ligation mix buffer with T4 DNA ligase and incubated for 24 hr at 16 °C. The ligated samples were decrosslinked at 65 °C with proteinase K for overnight and were Phenol/Chloroform extracted, EtOH precipitated and desalted. Target primers were designed against HindIII digested fragments within or close to DHS around SNPs in the region of risk loci and, anchor primers were designed against the fragments cut by HindIII in the TF promoters (primers available upon request). PCR was performed using Taq polymerase (Qiagen), 3C library and the oligonucleotides listed in Table S3. PCR conditions; 5 min at 94°C, (20 s at 94°C, 20 s at 57, and 30 s at 72°C) ×42 cycles, and 10 min at 72°C for extension. 7ul of PCR samples were loaded on the 1.7% agarose gel. The PCR products were then gel purified and sequenced. Since spurious interactions are typically undetectable beyond 150 kilobases, the ESR1 and KLF4 ligation fragments represent bona fide interactions.
Supplementary Material
Highlights.
A novel method to identify eQTLs from tumors by adjusting for somatic alterations
Application of method to the cancer genome atlas (TCGA) breast cancer dataset
Discovery of candidate causal genes for 6 breast cancer risk loci
Acknowledgments
The results published here are in part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov. We thank Dr. Giovanni Parmigiani for his suggestions with respect to the analytic modeling. We thank Dr. Gad Getz for suggestions on data processing. MLF is a Howard Hughes Medical Institute Physician-Scientist Early Career Awardee (www.hhmi.org) and a recipient of a 2009 Claudia Adams Barr award (www.dana-farber.org). This work was supported by grants from the US National Institutes of Health (ELLIPSE/GAME-ON-U19 CA148537 to MLF and R01 CA131341 to TL and IP) the Mayer Foundation (to MLF), the H.L. Snyder Medical Foundation (to MLF), the Kohlberg Foundation, and the A. David Mazzone Awards Program (to MLF).
Footnotes
Additional information: The results published here are in whole or part based upon data generated by The Cancer Genome Atlas Pilot Project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov.
References
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaogi K, Nakajima Y, Ito I, Kawasaki S, Oie SH, Murayama A, Kimura K, Yanagisawa J. KLF4 suppresses estrogen-dependent breast cancer growth by inhibiting the transcriptional activity of ERalpha. Oncogene. 2009;28:2894–2902. doi: 10.1038/onc.2009.151. [DOI] [PubMed] [Google Scholar]
- Babak T, Garrett-Engele P, Armour CD, Raymond CK, Keller MP, Chen R, Rohl CA, Johnson JM, Attie AD, Fraser HB, et al. Genetic validation of whole-transcriptome sequencing for mapping expression affected by cis-regulatory variation. BMC Genomics. 2010;11:473. doi: 10.1186/1471-2164-11-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MA, Hou X, Harrington SC, Weroha SJ, Gonzalez SE, Jacob KA, Carboni JM, Gottardis MM, Haluska P. IGFBP ratio confers resistance to IGF targeting and correlates with increased invasion and poor outcome in breast tumors. Clin Cancer Res. 2012;18:1808–1817. doi: 10.1158/1078-0432.CCR-11-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AJ, Dickson KA, McDougall F, Baxter RC. Insulin-like growth factor-binding protein-5 inhibits the growth of human breast cancer cells in vitro and in vivo. J Biol Chem. 2003;278:29676–29685. doi: 10.1074/jbc.M301965200. [DOI] [PubMed] [Google Scholar]
- Campino S, Forton J, Raj S, Mohr B, Auburn S, Fry A, Mangano VD, Vandiedonck C, Richardson A, Rockett K, et al. Validating discovered Cis-acting regulatory genetic variants: application of an allele specific expression approach to HapMap populations. PLoS One. 2008;3:e4105. doi: 10.1371/journal.pone.0004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33:422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- Essaghir A, Toffalini F, Knoops L, Kallin A, van Helden J, Demoulin JB. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res. 2010;38:e120. doi: 10.1093/nar/gkq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MP, Xiao R, Prokunina-Olsson L, Scott LJ, Mohlke KL. Allelic expression imbalance at high-density lipoprotein cholesterol locus MMAB-MVK. Hum Mol Genet. 2010;19:1921–1929. doi: 10.1093/hmg/ddq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forton JT, Udalova IA, Campino S, Rockett KA, Hull J, Kwiatkowski DP. Localization of a long-range cis-regulatory element of IL13 by allelic transcript ratio mapping. Genome Res. 2007;17:82–87. doi: 10.1101/gr.5663007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman ML, Monteiro AN, Gayther SA, Coetzee GA, Risch A, Plass C, Casey G, De Biasi M, Carlson C, Duggan D, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet. 2011;43:513–518. doi: 10.1038/ng.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge B, Pokholok DK, Kwan T, Grundberg E, Morcos L, Verlaan DJ, Le J, Koka V, Lam KC, Gagne V, et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat Genet. 2009;41:1216–1222. doi: 10.1038/ng.473. [DOI] [PubMed] [Google Scholar]
- Grisanzio C, Werner L, Takeda D, Awoyemi BC, Pomerantz MM, Yamada H, Sooriakumaran P, Robinson BD, Leung R, Schinzel AC, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci U S A. 2012;109:11252–11257. doi: 10.1073/pnas.1200853109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap GA, Yang JH, Downes K, Healy BC, Hunt KA, Bockett N, Franke L, Dubois PC, Mein CA, Dobson RJ, et al. Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum Mol Genet. 2010;19:122–134. doi: 10.1093/hmg/ddp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, M J, E.B.I. Wise A, Junkins HA, Hall PN, Klemm AK, Manolio TA. A Catalog of Published Genome-Wide Association Studies [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijts PE, van Dongen M, de Goeij MC, van Moolenbroek AJ, Blanken F, Vreeswijk MP, de Kruijf EM, Mesker WE, van Zwet EW, Tollenaar RA, et al. Allele-specific regulation of FGFR2 expression is cell type-dependent and may increase breast cancer risk through a paracrine stimulus involving FGF10. Breast Cancer Res. 2011;13:R72. doi: 10.1186/bcr2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen VN, Edvardsen H, Tsalenko A, Nordgard SH, Sorlie T, Sharan R, Vailaya A, Ben-Dor A, Lonning PE, Lien S, et al. Genetic variation in putative regulatory loci controlling gene expression in breast cancer. Proc Natl Acad Sci U S A. 2006;103:7735–7740. doi: 10.1073/pnas.0601893103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JF, Vello E, Ge B, Montgomery SB, Dermitzakis ET, Pastinen T, Labuda D. Genotype-based test in mapping cis-regulatory variants from allele-specific expression data. PLoS One. 2012;7:e38667. doi: 10.1371/journal.pone.0038667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AJ, Grant A, Ashfield AM, Palmer CN, Baker L, Quinlan PR, Purdie CA, Thompson AM, Jordan LB, Berg JN. FGFR2 protein expression in breast cancer: nuclear localisation and correlation with patient genotype. BMC Res Notes. 2011;4:72. doi: 10.1186/1756-0500-4-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KB, Maia AT, O'Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Shrestha Y, Flavin RJ, Regan MM, Penney KL, Mucci LA, Stampfer MJ, Hunter DJ, Chanock SJ, Schafer EJ, et al. Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010;6:e1001204. doi: 10.1371/journal.pgen.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Riaz M, Berns EM, Sieuwerts AM, Ruigrok-Ritstier K, de Weerd V, Groenewoud A, Uitterlinden AG, Look MP, Klijn JG, Sleijfer S, et al. Correlation of breast cancer susceptibility loci with patient characteristics, metastasis-free survival, and mRNA expression of the nearest genes. Breast Cancer Res Treat. 2012;133:843–851. doi: 10.1007/s10549-011-1663-3. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, Hunt S, Kahl B, Antonarakis SE, Tavare S, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Olopade OI, Di Rienzo A. rs2981582 is associated with FGFR2 expression in normal breast. Cancer Genet Cytogenet. 2010;197:193–194. doi: 10.1016/j.cancergencyto.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tonner E, Barber MC, Allan GJ, Beattie J, Webster J, Whitelaw CB, Flint DJ. Insulin-like growth factor binding protein-5 (IGFBP-5) induces premature cell death in the mammary glands of transgenic mice. Development. 2002;129:4547–4557. doi: 10.1242/dev.129.19.4547. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Wolfer A, Wittner BS, Irimia D, Flavin RJ, Lupien M, Gunawardane RN, Meyer CA, Lightcap ES, Tamayo P, Mesirov JP, et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc Natl Acad Sci U S A. 2010;107:3698–3703. doi: 10.1073/pnas.0914203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JB, Brown SJ, Cole MD. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol Cell Biol. 2010;30:1411–1420. doi: 10.1128/MCB.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc Natl Acad Sci U S A. 2010;107:145–150. doi: 10.1073/pnas.0912294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.