

Summary
Endoplasmic reticulum (ER) stress has been implicated in the pathogenesis of viral hepatitis, insulin resistance, hepatosteatosis and non-alcoholic steatohepatitis (NASH), disorders that increase risk of hepatocellular carcinoma (HCC). To determine whether and how ER stress contributes to obesity-driven hepatic tumorigenesis we fed wild type (WT) and MUP-uPA mice, in which hepatocyte ER stress is induced by plasminogen activator expression, with high fat diet. Although both strains were equally insulin resistant, the MUP-uPA mice exhibited more liver damage, immune infiltration and increased lipogenesis, and as a result displayed classical NASH signs and developed typical steatohepatitic HCC. Both NASH and HCC development were dependent on TNF produced by inflammatory macrophages that accumulate in the MUP-uPA liver in response to hepatocyte ER stress.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and a leading cause of cancer deaths. More than 90% of HCC develops in the context of chronic liver disease, with HBV or HCV infections being the main causes. However, 30-40% of Western HCC patients do not exhibit viral infections (El-Serag, 2011). Most of these patients are obese with manifestations of the metabolic syndrome, and suffer from NASH, a severe form of non-alcoholic fatty liver disease (NAFLD) (Cohen et al., 2011). Indeed, obesity increases male HCC risk by up to 4.5-fold (Calle et al., 2005) and also increases HCC risk in viral hepatitis (Chen et al., 2008). Because the prevalence of obesity has been increasing worldwide, its association with hepatocarcinogenesis has attracted much attention. In previous studies we have shown that feeding mice exposed to the hepatic carcinogen diethylnitrosamine (DEN) with high fat diet (HFD) strongly enhanced HCC development (Park et al., 2010). Although low grade liver inflammation associated with TNF and IL-6 expression contributes to obesity-promoted HCC development in this model, it should be noted that WT mice do not develop NASH, even after DEN administration and prolonged HFD feeding. It is therefore not clear whether the mechanism identified in DEN-treated mice has much bearing on NASH-driven human HCC (Toffanin et al., 2010).
In considering possible mechanisms through which obesity may promote HCC development we decided to study the potential contribution of ER stress, because obesity (Hotamisligil, 2010; Ozcan et al., 2006) and HBV/HCV infections (Malhi and Kaufman, 2011) result in liver ER stress, which promotes hepatosteatosis (Rutkowski et al., 2008). Furthermore, several ER stress markers are elevated in NASH-affected livers (Puri et al., 2008) and ER stress was suggested to cause ballooning degeneration of hepatocytes, a classical sign of NASH (Caldwell et al., 2010). To this end, we placed MUP-uPA mice, that express high amounts of urokinase plasminogen activator (uPA) specifically in hepatocytes and therefore undergo transient ER stress (Sandgren et al., 1991; Weglarz et al., 2000), and WT mice on HFD. Whereas WT mice developed simple steatosis and no HCC, MUP-uPA mice developed NASH-like disease that spontaneously progressed to HCC, whose development was dependent on TNF production by inflammatory liver macrophages and TNFR1-IKKβ signaling in hepatocytes. Our results suggest that NASH and progression to steatohepatitic HCC may be prevented or ameliorated by anti-TNF drugs.
Results
HFD induces NASH signs and spontaneous HCC in MUP-uPA mice
WT and MUP-uPA mice were placed on HFD (60% of calories are fat derived), starting at 6 weeks of age. Body weight and glucose intolerance did not differ between the two strains (Figure S1A,B). As reported (Weglarz et al., 2000), serum alanine aminotransferase (ALT) in MUP-uPA mice on normal chow diet (LFD) was markedly elevated at 5 weeks of age but rapidly declined, likely due to replacement of dying hepatocytes with new cells in which uPA expression is extinguished (Sandgren et al., 1991; Weglarz et al., 2000) (Figure S1C). However, HFD feeding maintained high serum ALT throughout the observation period (Figure 1A), even though it did not restore uPA expression (Figure S1C). By contrast, in WT mice HFD substantially elevated ALT only after 32 weeks, reaching a level similar to MUP-uPA mice at 40 weeks. Examination of liver histology revealed hepatocyte damage, evidenced by tissue clearing, in 5 weeks old MUP-uPA mice but this had almost disappeared at 24 weeks on LFD, except for mild inflammation and spotty necrosis (Figure 1B). As reported (Park et al., 2010), HFD-fed WT mice showed pronounced steatosis but little inflammation by 24 weeks (Figure 1B). At that time, HFD-fed MUP-uPA mice exhibited extensive immune infiltration into the liver and numerous ballooning hepatocytes, both of which are important diagnostic features of human NASH (Brunt, 2001). Furthermore, HFD-fed MUP-uPA mice showed pericellular and bridging fibrosis, resembling the pattern in human NASH (Figure 1C and S1D). This was accompanied by increased expression of type 1 collagen α1 mRNA (Figure S1E).
Figure 1. HFD-fed MUP-uPA mice display classical NASH signs.
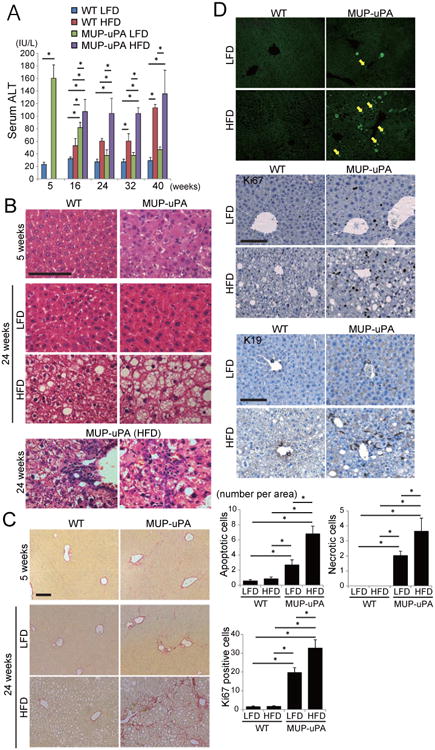
(A) Serum ALT in LFD- or HFD-fed WT and MUP-uPA mice was measured at indicated ages. HFD feeding was initiated at 6 weeks. Data are means ± S.D. (n = 3-5 per group). *p < 0.05. (B) H&E staining of liver sections from 5 weeks old mice on LFD and 24 weeks old mice kept on LFD or HFD (scale bar = 100 μm). Bottom two panels show infiltration of immune cells in HFD-fed MUP-uPA mouse livers (left, portal area; right, liver parenchyma). (C) Sirius red staining of liver sections described in B (scale bar = 100 μm). (D) TUNEL and IHC analyses of Ki67 and K19 in 24 weeks old mice that were kept on LFD or HFD (scale bar = 100 μm). Yellow and white arrows indicate apoptotic and necrotic cells, respectively. Bar graphs show numbers of apoptotic and necrotic cells and Ki67-positive cells per 200× field. Data are means ± S.D. (n = 5 per group). *p < 0.05. See also Figure S1.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining showed that both apoptotic (nuclear fragmentation) and necrotic (diffuse cytoplasmic staining) cell death were significantly increased in HFD-fed MUP-uPA livers and as a result, the numbers of Ki67-positive proliferating hepatocytes and K19-positive cells were also elevated (Figure 1D). Expression of cyclin D1 was also increased (Figure S1F). Thus, HFD-fed MUP-uPA mice exhibit continuous hepatocyte death and compensatory proliferation, a critical process in hepatocarcinogenesis (Maeda et al., 2005).
HFD-fed MUP-uPA mice developed small tumors on the liver surface by 32 weeks of age, and large tumors at 40 weeks (Figure 2A, S2A,B), when 78.6% (11/14 mice) of HFD-fed MUP-uPA mice had tumors larger than 2 mm, and 35.7% (5/14 mice) had tumors larger than 10 mm. Histologically, 30% of tumors larger than 2 mm were HCCs, similar to human steatohepatitic HCC, a histotype describing NASH-related HCC with ballooning cancer cells and inflammatory cell infiltration (Salomao et al., 2012), but some displayed a classical thick trabecular pattern, whereas the remaining 70% were either typical or steatohepatic adenomas (Figure 2B, S2C,D). Cancer cells were highly proliferative and frequently positive for α fetoprotein (AFP) with marked p62 aggregation (Figure S2E), a sign of impaired autophagy frequently observed in human HCC (Inami et al., 2011). Several oncogenic mediators, e.g. ERK, STAT3, and JNK, as well as cyclin D1, the liver oncogenes YAP and Myc, and the cancer stem cell markers EpCAM and CD44, were activated or upregulated (Figure S2F-H). By contrast, 30% LFD-fed MUP-uPA mice displayed a few tiny nodules in the liver even at 40 weeks of age, corresponding to simple hyperplasia (Figure S2C). Although 1 of 11 LFD-fed MUP-uPA mice developed a small 3 mm tumor, it was also classified as hyperplasia, which is not proliferative and AFP negative (Figure S2C, I). In WT mice, neither LFD nor HFD induced any liver tumors by 40 weeks. Of note, HFD-fed MUP-uPA mice showed microscopically visible foci of p62- and YAP-positive cells already at 24 weeks (Figure S2J). These foci may contain progenitors to the tumors detected at 32-40 weeks. Thus, HFD feeding of MUP-uPA mice induced complete NASH-like pathological features with continuous hepatocyte death and compensatory proliferation, and eventually led to spontaneous HCC and adenoma development that were not seen in LFD-maintained mice.
Figure 2. NASH to HCC progression in MUP-uPA mice.
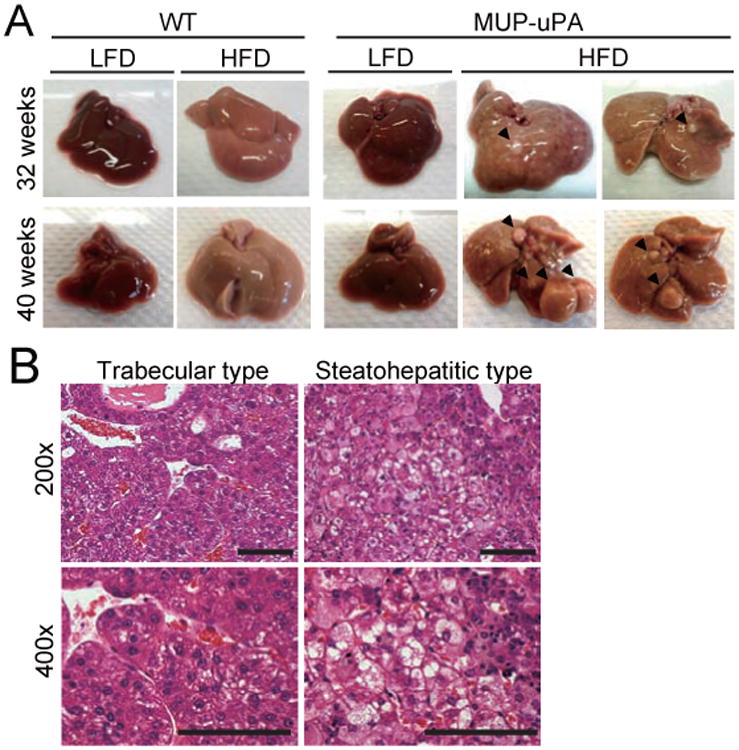
(A) Representative images of livers from 32 and 40 weeks old mice that were kept on LFD or HFD. (B) Representative H&E staining of tumor sections from 40 weeks old HFD-fed MUP-uPA mice. Left two panels show trabecular HCC and right two panels show steatohepatitic HCC (scale bar = 100 μm). See also Figure S2.
ER stress enhances lipogenesis and aggravates steatohepatitis
Although the mechanism responsible for hepatocyte death in young MUP-uPA mice is not entirely clear, their hepatocytes are ER stressed (Sandgren et al., 1991). Indeed, several ER stress markers, including CHOP, GRP78, spliced XBP1 (sXBP1), phosphorylated (p) eIF2α, p-IRE1α and p-JNK, were elevated in 5 weeks old MUP-uPA mice compared with WT (Figure 3A and S3A). While most markers declined in 16 weeks old MUP-uPA mice, paralleling the decline in uPA expression, HFD-fed MUP-uPA mice maintained strong eIF2α and JNK phosphorylation and CHOP expression, (Figure 3B, S3A). In WT mice, HFD feeding induced only a slight elevation in p-eIF2α and CHOP mRNA with no effect on CHOP protein (Figure 3B). Immunohistochemistry (IHC) confirmed nuclear CHOP in hepatocytes of 5 weeks old MUP-uPA mice, which was sustained at 16 weeks of age only in HFD-fed MUP-uPA mice (Figure 3C). TRB3 and DR5, two molecules capable of inducing cell death, were highly upregulated in MUP-uPA mice, especially after HFD feeding (Figure S3B). After 24 weeks of HFD, expression of the DR5 ligand TRAIL was also elevated in the MUP-uPA liver. Electron microscopy revealed distended and dilated ER in HFD-fed MUP-uPA mice (Figure S3C). Thus, whereas ER stress appears to be induced by uPA expression in 5 weeks old MUP-uPA mice, it declines due to transgene extinction. However, feeding these mice with HFD re-kindles the stress response and induces several cell death mediators that are not expressed in HFD-fed WT mice. To determine whether ER stress can cause ballooning degeneration and hepatocyte death, we injected HFD-fed WT mice with the protein glycosylation inhibitor and ER stress elicitor tunicamycin. This treatment led to rapid (36 hrs) induction of ballooning degeneration, hepatocyte apoptosis and ALT release only in HFD-fed mice (Figures S3D-F). The white appearance of the liver from HFD-fed mice treated with tunicamycin suggested that the liver had become more steatotic.
Figure 3. ER stress enhances lipogenesis and promotes steatohepatitis.
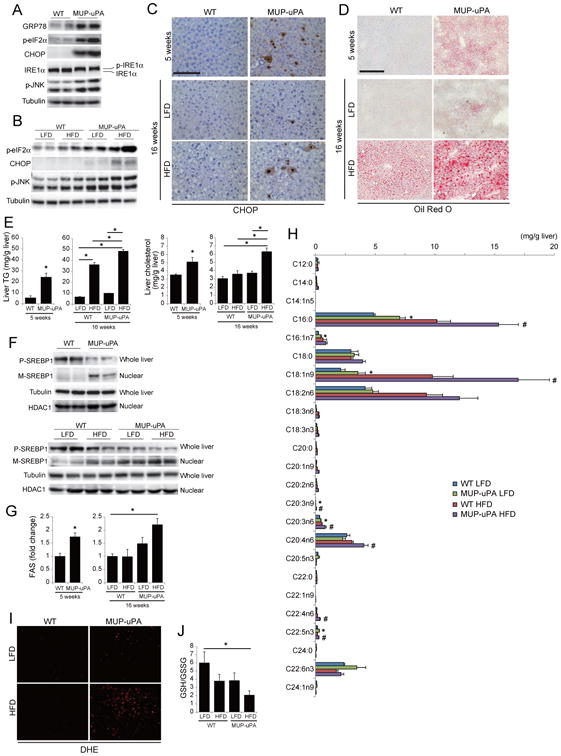
(A, B) IB analysis of ER stress markers in livers of 5 weeks old WT and MUP-uPA mice (A) and 16 weeks old WT and MUP-uPA mice kept on LFD or HFD (B). (C) IHC analysis of CHOP in livers of 5 weeks old mice on LFD and 16 weeks old mice kept on LFD or HFD (scale bar = 100 μm). (D) Oil Red O staining of mouse livers described in C (scale bar = 100 μm). (E) TG and cholesterol content of mouse livers described in C. (F) IB analysis of unprocessed precursor SREBP1 (P-SREBP1) in whole liver extract and mature SREBP1 (M-SREBP1) in liver nuclei of mice described in A (upper panels) and B (lower panels). (G) Real time PCR analysis of liver FAS mRNA. (H) Hepatic FA composition in 16 weeks old mice kept on LFD or HFD, analyzed by gas-chromatography. *p < 0.05, compared with LFD-fed WT mice. #p < 0.05, compared with HFD-fed WT mice. (I,J) ROS accumulation in 16 weeks old mice that were kept on LFD or HFD. Images of DHE staining (scale bar = 100 μm) (I) and GSH:GSSG ratio (J) are shown. All bar graphs represent means ± S.D. (n = 3 per group). *p < 0.05. See also Figure S3.
The above results are consistent with the ability of ER stress to cause liver steatosis (Rutkowski et al., 2008). Indeed, Oil Red O (ORO) staining showed mild spontaneous lipid accumulation in 5 weeks old MUP-uPA mice, that was diminished by 16 weeks of age under LFD (Figure 3D). However, HFD feeding induced more extensive lipid accumulation in MUP-uPA than in WT mice. Liver triglycerides (TG) and cholesterol were also elevated (Figure 3E). Decreased liver lipid export due to suppression of apoB expression/secretion and increased lipogenesis were suggested to be involved in ER stress-induced steatosis (Ota et al., 2008; Qiu et al., 2011; Rutkowski et al., 2008). Since apoB carries TG and cholesterol from the liver elsewhere, we examined serum TG and cholesterol and liver apoB mRNA. There were no differences in apoB mRNA between the four groups (Figure S3G) and serum TG and total cholesterol were similarly elevated in HFD-fed WT and MUP-uPA mice (Figure S3H), suggesting that liver lipid export was not fully impaired in MUP-uPA mice. Next, we examined mRNAs of lipogenic regulators. Although SREBP2 mRNA was slightly increased and PPARα and c/EBPα mRNAs were slightly decreased in 5 weeks old MUP-uPA mice compared with WT mice, these trends were not seen in 16 weeks old mice (Figure S3I). Expression of PPARγ was decreased in 16 weeks old MUP-uPA mice, but not in 5 weeks old mice. Therefore, enhanced lipogenesis in MUP-uPA mice could not be explained by differential expression of these molecules. However, among lipogenic regulators, SREBP1 is controlled not only by synthesis but also by cleavage and subsequent nuclear translocation (Goldstein et al., 2006), which are stimulated by ER stress (Kammoun et al., 2009). Indeed, SREBP1 precursor abundance was decreased in 5 weeks old MUP-uPA livers and mature, nuclear SREBP1, was elevated (Figure 3F). HFD feeding further accelerated SREBP1 processing in MUP-uPA mice, but also induced some SREBP1 processing in WT mice. mRNA expression of the SREBP1 target fatty acid synthase (FAS), was increased in 5 weeks old and HFD-fed MUP-uPA mice (Figure 3G). Consistent with elevated lipogenesis, gas-chromatography determination of hepatic FA composition revealed a significant increase in C16:0 palmitic acid (PA) and longer chain FA, in MUP-uPA mice compared with WT mice, which was further enhanced by HFD feeding (Figure 3H). Excess lipid accumulation leads to oxidative stress due to mitochondrial H2O2 production, which can induce cell death (Anderson et al., 2009). Accordingly, HFD-fed MUP-uPA mice displayed strong dihydroethidium (DHE) staining of hepatocytes and a decrease in liver GSH:GSSG ratio (Figure 3I, J). Oxidative stress in HFD-fed MUP-uPA mice may contribute to CHOP expression, JNK activation, lipotoxic hepatocyte death and oncogenic mutations.
The IRE1α-XBP1 pathway has been reported to regulate hepatic lipid metabolism via XBP1-mediated induction of lipogenic enzymes and regulated IRE1-dependent mRNA decay (RIDD) (Lee et al., 2008; So et al., 2012). Although expression of the XBP1 target gene ERdj4 was upregulated in 5 weeks old and HFD-fed MUP-uPA mice, there were no differences in expression of DGAT2, a lipogenic enzyme regulated by XBP1 but not by SREBP1 (Figure S3J). RIDD-mediated downregulation of Angptl3 and Ces1 mRNAs can induce hypolipidemia and hepatosteatosis due to decreased lipid secretion from the liver. Although expression of Angptl3 mRNA was decreased in 5 weeks old MUP-uPA mice (Figure S3J), there were no differences in serum TG and total cholesterol levels between 5 weeks old WT and MUP-uPA mice and Angptl3 mRNA expression recovered in 16 weeks old MUP-uPA mice (Figure S3H, J). These results suggest that the IRE1α-XBP1 pathway does not play a major role in NASH development in MUP-uPA mice.
Chemical chaperons and GRP78 attenuate lipotoxicity and lipogenesis in MUP-uPA mice
To examine whether ER stress enhances lipotoxicity in MUP-uPA hepatocytes, we incubated WT and MUP-uPA hepatocytes with PA. After 24 hrs, lipotoxic cell death was seen in both WT and MUP-uPA hepatocytes, but was more extensive in the latter (Figure 4A). PA increased CHOP expression and SREBP1 maturation in WT hepatocytes, but these effects were more pronounced in MUP-uPA hepatocytes, which expressed both proteins prior to PA addition (Figure 4B). To examine the contribution of ER stress to these phenomena, we treated hepatocytes with the chemical chaperons 4-phenylbutyrate (4-PBA) and tauro-ursodeoxycholic acid (TUDCA), which reduce ER stress (Ozcan et al., 2006). Both compounds attenuated PA-induced cell death, but their pro-survival effect was more pronounced in MUP-uPA hepatocytes (Figure 4A). CHOP induction and SREBP1 maturation upon PA treatment were also reduced by 4-PBA (Figure 4B). Overexpression of the ER protein chaperon GRP78 in MUP-uPA hepatocytes also inhibited SREBP1 maturation and PA-induced cell death (Figure 4C, D), further supporting the role of ER stress in both phenomena. PA treatment activated JNK, but consistent with previous results that ER stress has only a partial role in JNK activation by PA (Holzer et al., 2011), the effect was restricted to WT hepatocytes and PBA treatment only partially reduced JNK phosphorylation (Figure 4B). Nonetheless, the JNK inhibitor D-JNKi protected both cell types from PA induced death (Figure 4E).
Figure 4. Chemical chaperons attenuate lipotoxicity and liver damage in MUP-uPA mice.
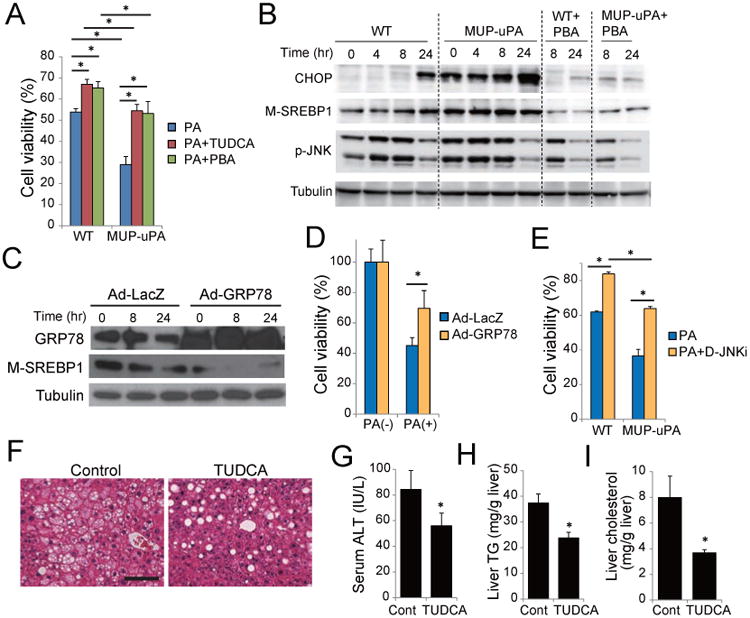
(A) Primary hepatocytes from WT and MUP-uPA mice were incubated with 300 μM PA for 24 hr with or without 500 μM TUDCA or 1 mM 4-PBA. Cell viability was assessed using Cell Counting Kit-8 assay. Data are means ± S.D. of triplicate wells. *p < 0.05. (B) Primary hepatocytes from WT and MUP-uPA mice were incubated with 200 μM PA with or without 4-PBA as above. CHOP expression, SREBP1 maturation, and JNK phosphorylation were assessed by IB. (C-D) Effect of GRP78 overexpression. MUP-uPA hepatocytes were infected with adenoviruses encoding LacZ or GRP78 and then incubated with PA. SREBP1 maturation (C) and cell viability (D) were assessed as above. (E) Hepatocytes from WT and MUP-uPA mice were incubated with 300 μM PA for 24 hr with or without 10 μM D-JNKi, and cell viability was assessed. Data are means ± S.D. of triplicate wells. *p < 0.05. (F-I) Effect of TUDCA on NASH in HFD-fed MUP-uPA mice. 16 weeks old HFD-fed MUP-uPA mice were i.p. injected with TUDCA (250 mg/kg) or vehicle, and after 4 weeks of daily treatment, liver histology (scale bar = 100 μm) (F), serum ALT (G), liver TG (H) and liver cholesterol (I) were evaluated. Bar graphs are means ± S.D. (n = 5 per group). *p < 0.05. See also Figure S4.
We examined the effect of TUDCA on NASH development. We initiated daily i.p. injections of TUDCA (250 mg/kg) or phosphate-buffered saline (PBS; vehicle control) to HFD-fed MUP-uPA mice at 16 weeks of age. After 4 weeks, hepatosteatosis and hepatocyte ballooning were attenuated (Figure 4F), and serum ALT and hepatic TG and cholesterol were significantly reduced (Figure 4G-I). Hepatocyte death and ROS accumulation were also suppressed (Figure S4A,B). TUDCA treatment also inhibited CHOP expression and SREBP1 maturation in livers of HFD-fed MUP-uPA mice (Figure S4C). We also found that in vivo overexpression of GRP78 using an adenovirus vector attenuated hepatic steatosis in HFD-fed MUP-uPA mice (Figure S4D,E). However, due to enhanced adenovirus toxicity in MUP-uPA mice we could not assess the effect on NASH and HCC development. Nonetheless, the results suggest that increased lipotoxicity caused by a positively reinforced cycle of ER stress, oxidative stress, and lipogenesis aggravates fatty liver disease in HFD-fed MUP-uPA mice.
Given the pronounced expression of CHOP in MUP-uPA mice and its postulated role in apoptosis (Malhi and Kaufman, 2011) we crossed MUP-uPA mice to ChopΔhep mice in which CHOP was deleted in hepatocytes. Despite efficient CHOP ablation, there was no reduction in liver damage, JNK and eIF2αphosphorylation or GRP78 expression in young ChopΔhep/MUP-uPA mice (Figure S4F,G). Correspondingly, CHOP ablation did not inhibit HCC development (Figure S4H). In fact, CHOP ablation increased tumor multiplicity without affecting tumor size, ER stress markers or NASH severity (Figure S4I-K), results that stand in marked contrast to the protective effect of whole body Chop ablation in DEN-induced hepatocarcinogenesis (DeZwaan-McCabe et al., 2013). CHOP was strongly expressed in some tumors and preneoplastic lesions of HFD-fed MUP-uPA mice but not in ChopΔhep/MUP-uPA mice, and the number of TUNEL-positive cells tended to be reduced in the tumor tissues of ChopΔhep/MUP-uPA mice (Figure S4L-M), suggesting that hepatocyte CHOP is not positively involved in NASH progression and HCC development, similar to what was observed in whole body Chop-/- mice on methionine-choline-deficient (MCD) diet (Soon et al., 2010). Nonetheless, CHOP may play a tumor suppressive role by inducing apoptosis of initiated hepatocytes.
TNF from liver macrophages promotes lipogenesis, NASH and HCC development
Next, we examined involvement of inflammatory cytokines in hepatosteatosis and steatohepatitis. In 24 weeks old mice, TNF and IL-1β, but not IL-6, mRNAs were elevated in HFD-fed MUP-uPA livers (Figure 5A). TNF production was confirmed by ELISA (Figure 5B) and immunofluorescence (IF) analysis localized it to F4/80-positive macrophages, whose number was elevated in HFD-fed MUP-uPA mouse livers (Figure 5C). The increase in macrophage infiltration and TNF expression was inhibited by TUDCA treatment (Figure S5A,B), suggesting it is stimulated, in part, by hepatocyte ER and oxidative stress.
Figure 5. TNFR1 signaling promotes tumor growth.
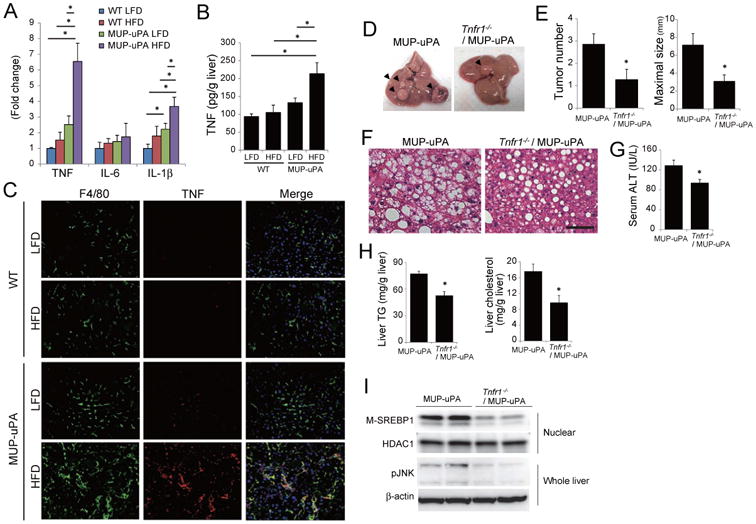
(A) Relative inflammatory cytokine mRNA in livers of 24 weeks old mice kept on LFD or HFD determined by real-time Q-PCR. Data are means ± S.D. (LFD-fed WT, n = 3; others, n =5 per group). *p < 0.05. (B) TNF protein in livers from A was measured by ELISA. Means ± S.D. *p < 0.05. (C) Double IF analysis of F4/80 (green) and TNF (red) of liver sections from A (scale bar = 100 um). Nuclei were labeled with DAPI (blue). (D-H) Effect of TNFR1 ablation on NASH and tumorigenesis in HFD-fed MUP-uPA mice. MUP-uPA and Tnfr1-/-/MUP-uPA mice were fed HFD from 6 to 40 weeks of age. Representative images of livers (D), tumor numbers and maximal sizes (E), H&E staining of non-tumor areas (scale bar = 100 μm) (F), and serum ALT (G) are shown. Bar graphs represent means ± S.E.M. (MUP-uPA, n = 14; Tnfr1-/-/MUP-uPA, n = 11). *p < 0.05. (H) TG and cholesterol content in non-tumor tissue of HFD-fed MUP-uPA and Tnfr1-/-/MUP-uPA mouse livers. Bar graphs represent means ± S.D. (n = 7 per group). *p < 0.05. (I) IB analyses showing effects of TNFR1 ablation on SREBP1 maturation and JNK phosphorylation in non-tumor tissue of HFD-fed MUP-uPA mice. See also Figure S5.
To investigate the role of TNF in NASH progression and HCC development, we generated TNF receptor 1 (TNFR1)-deficient MUP-uPA (Tnfr1-/-/MUP-uPA) mice. At 5 and 40 weeks of age, there were no differences in liver injury and body weights between MUP-uPA and Tnfr1-/-/MUP-uPA mice (Figure S5C-F). We placed these mice on HFD from 6 to 40 weeks of age, and assessed liver histology and tumorigenesis. Body weight gain at 40 weeks of age was similar between MUP-uPA and Tnfr1-/-/MUP-uPA mice (Figure S5F), but tumor development was substantially reduced upon TNFR1 ablation (Figure 5D,E). Importantly, hepatocyte ballooning, ALT release, liver TG and cholesterol, as well as SREBP1 and JNK activation, were reduced in Tnfr1-/-/MUP-uPA mice (Figure 5F-I). Therefore, TNFR1 signaling perpetuates NASH pathogenesis and HCC progression.
TNFR1 signaling directly promotes tumor growth
To determine whether TNFR1 signaling promotes HCC development by acting within HCC progenitor cells (HcPC), whose isolation we recently described (He et al., 2013), we transplanted HcPC from DEN-treated WT or Tnfr1-/- mice into MUP-uPA mice, that were placed on LFD or HFD (Figure 6A). Expression of the HcPC marker CD44 was comparable between WT or Tnfr1-/- HcPC (Figure S6A). After 5 months, non-transplanted MUP-uPA mice did not have any tumors larger than 2 mm, even after HFD feeding, whereas HcPC-transplanted mice developed multiple HCC nodules (Figure 6B). HFD feeding did not affect tumor number (which is determined by the number of transplanted HcPC), but significantly increased tumor size in mice transplanted with WT HcPC, but not in mice receiving Tnfr1-/- HcPC (Figure 6B,C). Thus, although TNFR1 signaling is dispensable for HcPC induction by DEN it strongly stimulates tumor growth in a cell autonomous manner. Control experiments confirmed that TNFR1 was deleted in HCC cells but not in non-tumor liver tissues (Figure S6B). In addition, there were no differences in NASH-like pathology and TNF expression in the background liver harboring either WT or Tnfr1-/- HcPC (Figure S6C-G). To further investigate the role of TNF signaling in these effects, we treated WT HcPCs-transplanted MUP-uPA mice with the TNF antagonist etanercept under HFD feeding. Etanercept treatment significantly suppressed HCC growth (Figure S6H). We also transplanted WT HcPC into Tnfr1-/-/MUP-uPA hosts and found that HFD still led to increased tumor size, albeit to a lower extent than the 2-fold effect seen in MUP-uPA hosts (Figure S6I).
Figure 6. TNFR1 signaling promotes tumor growth.
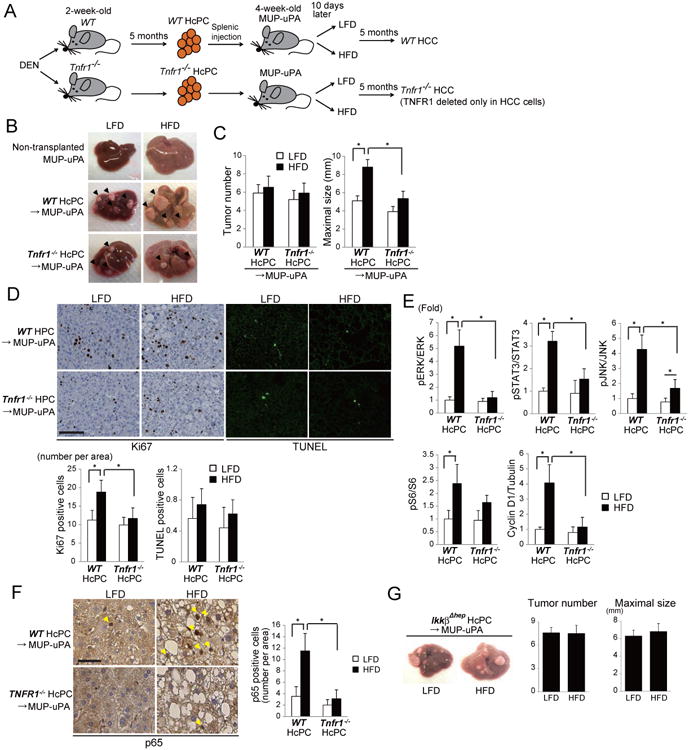
(A) HcPC isolation from DEN-treated WT and Tnfr1-/- mice and transplantation into MUP-uPA mice. HcPC-transplanted MUP-uPA mice were divided into two groups that were fed with either LFD or HFD, and 5 months later tumorigenesis was assessed. (B) Representative images of non-transplanted and HcPC-transplanted MUP-uPA mouse livers. (C) Tumor numbers and maximal sizes. Results are means ± S.E.M. (n = 10-11 per group). *p < 0.05. (D) Ki67 IHC and TUNEL staining of tumor areas in livers from C (scale bar = 100 μm). Bars represent numbers of apoptotic and necrotic cells and Ki67-positive cells per field. Results are means ± S.D. (n = 6 per group). *p < 0.05. (E) Tumor tissues from WT or Tnfr1-/- HcPC-transplanted MUP-uPA mice kept on LFD or HFD were IB analyzed for phosphorylation of ERK, STAT3, JNK, and S6, and expression of cyclin D1. Data were quantified using Image J software and are presented as means ± S.D. (n = 5-6 per group). *p < 0.05. (F) Activation of NF-κB analyzed by p65/RelA IHC in tumor tissues from MUP-uPA transplanted with WT or Tnfr1-/- HcPC and kept on LFD or HFD (scale bar = 25 μm). Bars show numbers of nuclear p65 positive cancer cells per 200× field. Data are means ± S.D. (n = 6 per group). *p < 0.05. (G) Effect of IKKβ ablation on HFD-stimulated HcPC progression. HcPCs isolated from DEN-injected liver-specific IkkβΔhep were transplanted into MUP-uPA mice as in A and the HcPC-transplanted mice were kept on LFD or HFD. 5 months later, tumor multiplicity and maximal sizes were determined. Data are means ± S.E.M. (n = 10 per group). See also Figure S6.
We assessed cell proliferation and apoptosis in HcPC-derived tumors. While no significant effects on apoptosis were observed, HFD enhanced cell proliferation in tumors formed by WT HcPC and this effect was diminished upon TNFR1 ablation (Figure 6D). Cyclin D1 expression and phosphorylation of ERK, STAT3, JNK, and S6 were enhanced by HFD feeding in WT HcPCs derived tumors (Figure 6E and S6J). Apart from S6 phosphorylation, these responses were abolished upon TNFR1 ablation. TNFR1 in HcPC was also required for NF-κB activation in tumors that developed in HFD-fed MUP-uPA mice (Figure 6F) and IKKβ ablation in HcPC prevented HFD-enhanced tumor growth (Figure 6G). Thus the TNF-TNFR1-IKKβ-NF-κB pathway is an important mediator of HCC growth in HFD-fed mice.
TNFR1 signaling promotes tumor-associated inflammation
Some of the signaling effectors that are activated in HFD-fed mice are not directly regulated by TNFR1. We postulated that autocrine or paracrine signaling may mediate some of the observed responses and analyzed tumors generated by WT and Tnfr1-/- HcPC more closely. In HFD-fed mice, both WT and Tnfr1-/-HCCs were composed of steatotic cells, but immune infiltration was less extensive in Tnfr1-/- HCCs (Figure 7A). Real-time PCR and IHC analysis indicated that macrophage and B cell markers were significantly increased by HFD in WT but not in Tnfr1-/- HCCs (Figure 7B, C). In addition, mRNAs for numerous inflammatory cytokines, chemokines, and growth factors were upregulated by HFD in WT but not in Tnfr1-/- HCCs (Figure S7). IF analysis confirmed that expression of CCL7, which attracts macrophages and monocytes, was increased by HFD in WT but not in Tnfr1-/- HCCs (Figure 7D). Thus, TNFR1 signaling in HCC cells promotes tumor-associated inflammation, which can account for ERK and STAT3 activation in malignant cells.
Figure 7. TNFR1 signaling in cancer cells promotes tumor-elicited inflammation.
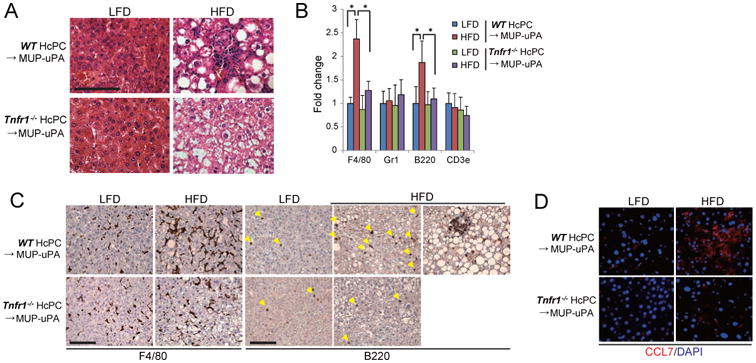
(A) H&E analysis of tumors from WT or Tnfr1-/- HcPC in MUP-uPA mice that were kept on LFD or HFD (scale bar = 100 μm). (B) Real time PCR determination of immune cell marker mRNAs in tumor tissues. Data are means ± S.D. (n = 5 per group). *p < 0.05. (C) IHC analysis of F4/80- and B220-positive cells in tumor tissues from A (scale bar = 100 μm). (D) IF analysis of CCL7 expression in tumor tissues (scale bar = 25 μm). See also Figure S7.
Discussion
ER stress and the unfolded protein response (UPR) are upregulated in many cancers and may be associated with drug resistance and adaptation to the transformed state (Wang et al., 2010). Elevated ER stress was also detected in precancerous conditions that precede HCC development, including HBV and HCV infections (Malhi and Kaufman, 2011) and NASH (Farrell et al., 2012; Tilg and Moschen, 2010). However, until recently, it has not been examined whether the ER stress response, which contributes to insulin resistance and hepatic steatosis (Hotamisligil, 2010) stimulates HCC development. Our results indicate that although transient ER stress in MUP-uPA mice that are kept on LFD does not trigger hepatocarcinogenesis, when combined with hypernutrition it ellicits a more sustained stress response that also includes extensive oxidative stress. This response leads to spontaneous NASH development and progression to HCC whose features closely resemble steatohepatitic HCC in NASH patients. Our studies suggest several potential mechanisms related to ER stress and HFD feeding that cooperate to induce HCC development. First, by stimulating hepatosteatosis (lipid droplet accumulation), HFD sustains a modest degree of ER stress in MUP-uPA mice, which otherwise would be switched off upon extinction of uPA expression. Second, ER stress promotes SREBP1 activation, enhancing lipogenesis and increasing the degree of hepatic steatosis beyond what is achieved by HFD alone. Third, ER stress and steatosis increase ROS production in hepatocytes to cause oxidative stress and its sequelae, which include genomic instability, oncogenic mutations and/or gene copy number changes. Fourth, ER and oxidative stress increase the sensitivity of hepatocytes to lipotoxic death, thereby releasing inflammatory mediators that attract and activate monocytes/macrophages. Fifth, TNF and other mediators produced by activated inflammatory macrophages stimulate compensatory hepatocyte proliferation and expand HCC progenitors. TNF further reinforces the inflammatory microenvironment and induces expression of chemokines (CCL2, CCL7 and CXCL13) and growth factors/cytokines (IL-1β, IL-6, TNF itself, lymphotoxin and HGF) both by HcPC and surrounding cells. The concerted action of these factors contributes to the development of NASH-like pathology and NASH to HCC progression. Mutually reinforcing ER stress and hepatosteatosis (Malhi and Kaufman, 2011) are needed to set this pathogenic cascade in motion.
The requirement for two hits (hepatosteatosis and ER stress) for induction of HCC development in MUP-uPA mice resembles what has been proposed to drive NASH development, a pre-HCC condition, in humans (Day and James, 1998; Tilg and Moschen, 2010). Although simple steatosis (“not-NASH”) is an extremely common disorder, affecting nearly 30% of the US population, only 10%-20% of these patients develop NASH. In the absence of known genetic factors it was proposed that NASH development depends on multiple secondary hits, which may include microbiota related factors, food additives, dysbiosis, IL-6 and TNF from adipose tissue, mitochondrial dysfunction and oxidative or ER stress (Farrell et al., 2012; Tilg and Moschen, 2010). Although these are considered secondary hits, they may act as pre-existing risk factors prior to hepatosteatosis caused by HFD. Nonetheless, in humans, unlike MUP-uPA mice, it has been extremely difficult to detect the presence of such risk factors as they do not lead to overt liver damage (elevated ALT) prior to development of a steatotic liver due to hypernutrition. Given its presence in other pre-HCC conditions (Malhi and Kaufman, 2011), we focused our study on the role of ER stress. Remarkably, feeding HFD to MUP-uPA mice resulted in steatohepatitis that closely resembled human NASH and two of the main pathological features, ballooning degeneration and hepatocyte death, were also rapidly induced by administration of tunicamycin to HFD-fed mice. By itself, short term tunicamycin did not damage the liver, but due to toxicity that may be associated with long term use, we did not examine if tunicamycin induces NASH and HCC in HFD-fed WT mice. Notably, NASH-like disease in MUP-uPA mice is associated with the same metabolic alterations linked to NASH in humans, and is not accompanied by weight loss, as seen in other NASH models that are based on feeding mice with toxic diets that induce liver damage (Farrell et al., 2012). Furthermore, the HFD-fed MUP-uPA mouse is currently the only model for studying obesity-induced HCC development that does not rely on administration of liver toxins or carcinogens. The major NASH-promoting effects of ER stress in this system are increased lipogenesis, oxidative stress and susceptibility to lipotoxic cell death. ER stress contributes to SREBP activation, thereby stimulating lipogenesis (Kammoun et al., 2009). ER and oxidative stress also upregulate several cell death mediators including TRB3 and DR5, but the exact mechanisms through which ER stress promotes cell death remain controversial (Xu et al., 2005) and our results indicate that in normal hepatocytes it is CHOP-independent. Although ER stress causes insulin resistance (Hotamisligil, 2010; Ozcan et al., 2006) and insulin resistance was proposed to contribute to HCC development, our results suggest that insulin resistance has no obvious role in HCC development because it is not higher in MUP-uPA mice than in HFD-fed WT mice.
A consequence of ER stress and lipotoxic hepatocyte death that contributes to HCC development is induction of TNF-dependent steatohepatitis. In addition to amplifying liver inflammation and shaping the inflammatory microenvironment nearby HcPC clusters, TNF contributes to hepatosteatosis and liver damage. Although TNFR1 engagement can trigger apoptosis, it is not responsible for ER stress-induced death in lean MUP-uPA mice and its contribution to liver damage in HFD-fed mice is proportional to its effect on lipogenesis and may be indirect. TNF, however, directly stimulates HCC growth through NF-κB activation. However, additional downstream TNFR1 effectors, such as JNK (Sakurai et al., 2008), may also contribute to HCC growth as well as hepatocyte death. TNF expression is also elevated in human NASH and anti-TNF therapy may reduce NASH activity (Schramm et al., 2008).
Although HFD feeding to MUP-uPA mice results in upregulation of multiple cytokines and growth factors including several that stimulate HCC development, namely HGF and lymphotoxin (Haybaeck et al., 2009), anti-TNF therapy inhibited the obesity-enhanced progression of HcPC to HCC and TNFR1 ablation almost completely blocked HCC development. We therefore suggest that anti-TNF drugs, perhaps in combination with improved intrahepatic delivery of chemical chaperons, such as TUDCA, should be evaluated for inhibition of NASH to HCC progression and treatment of steatohepatitic HCC along with more conventional chemotherapy.
Experimental Procedures
Animals
MUP-uPA mice were kindly provided by E.P Sandgren, University of Wisconsin-Madison (Weglarz et al., 2000). Liver-specific IkkβΔhep mice were described (Maeda et al., 2005). ChopΔhep mice were generated by crossing Alb-Cre mice with ChopF/F mice, that were developed by R.J.K together with Ira Tabas with the support of NIH grants DKD88227, DKD42394 and HLD52173. Tnfr1-/- mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animal studies were performed in accordance with NIH guidelines for the use and care of live animals and approved by University of California, San Diego (UCSD) Institutional Animal Care and Use Committee, S00218. All mouse lines were either on a pure C57BL/6 genetic background or crossed into it for at least 10 generations. Studies were conducted on male mice maintained in filter-topped cages on autoclaved water and regular chow diet (LFD, composed of 12% fat, 23% protein, 65% carbohydrates based on caloric content) or HFD (composed of 59% fat, 15% protein, 26% carbohydrates based on caloric content; BioServ) according to UCSD and NIH Guidelines.
HcPC isolation and transplantation
DEN (Sigma) was i.p. injected into male mice (25 mg/kg) on postnatal day 14. After 5 months, HcPCs were isolated as described and transplanted into 4 weeks old MUP-uPA (He et al., 2013).
Primary hepatocytes and macrophage cultures
Primary hepatocytes were isolated (He et al., 2013) and cultured in William's E medium with 10% FBS on collagen-coated plates. PA (Sigma) was dissolved in ethanol at 50°C and then diluted in bovine serum albumin-containing RPMI-1640 medium that was applied to primary hepatocytes at a final concentration of 200 (to analyze signal transduction) or 300 (to analyze cell death) μM.
Hepatic lipid profile
Hepatic lipids were extracted with chloroform/methanol (2:1 v/v), and TG and total cholesterol contents were measured with Triglyceride Reagent Set (Pointe Scientific) and Cholesterol E (Wako), respectively. FA composition was analyzed by gas-chromatography at SRL Inc., Tokyo, Japan.
Biochemical analyses and reagents
Immunoblotting and real-time Q-PCR were described (Maeda et al., 2005). Antibodies used were against: phospho-ERK, ERK1/2, phospho STAT3, STAT3, phospho-JNK, JNK1/2, phospho-S6, S6, p65, cyclinD1, YAP (all from Cell Signaling); K19, GRP78, SREBP1, CHOP, CCL7, TNFR1 (all from Santa Cruz Biotechnology); phospho-eIF2α (Upstate); tubulin (Sigma); F4/80 (Molecular Probes); Ki67 (Gene Tex); AFP (Biocare Medical); TNF (R&D Systems); B220 (BD Pharmingen). TUDCA and 4-PBA were from Calbiochem and Sigma, respectively. GSH:GSSG ratio was analyzed using GSSG/GSH Quantification Kit (Dojindo).
Histology
Livers were fixed in 10% neutral-buffered formalin or 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E), Sirius Red, and processed for IHC. For frozen block preparation, tissue was embedded in Tissue-Tek OCT compound (Sakura Finetek). IHC and IF analyses were described (He et al., 2013). Stained areas were quantitated with Image J software. Slides were incubated with primary antibodies, followed by secondary antibodies labeled with Alexa488 or Alexa594 (Molecular Probes). TUNEL staining was performed using an Apoalert DNA Fragmentation Assay kit (Clontech). Accumulation of superoxide anions was examined by DHE staining (Sakurai et al., 2008). Tissue sample preparation and EM analysis were described (Lee et al., 2012).
Infection of recombinant adenovirus
Primary hepatocytes were infected with recombinant adenovirus encoding β-galactosidase (LacZ) and GRP78 at a titer of 50 plaque-forming unit/cell 4 hours after isolation.
Statistical analyses
Statistical analyses were performed using Student's t-test or one-way analysis of variance followed by the Tukey–Kramer test for multiple comparisons. A p value < 0.05 indicated statistical significance.
Supplementary Material
Figure S1 (Related to Figure 1). Effects of HFD on MUP-uPA mice. (A) Body weights of LFD- or HFD-fed WT and MUP-uPA mice. HFD was started at 6 weeks of age. Data are means ± S.D. (n = 6-8 per group). *p < 0.05. (B) Glucose tolerance tests of 24 weeks old WT and MUP-uPA mice that were kept on LFD or HFD. Blood glucose was measured at the indicated time points after i.p. injection of 0.8 g/kg (right graph) or 2.0 g/kg (left graph) glucose. (n = 4-5 per group). (C) Expression of the uPA transgene was examined by real-time PCR. Results are presented as means ± S.D. (n = 3 per group). (D) Comparison of liver histology between HFD-fed MUP-uPA mouse (24 weeks old) and human NASH by H&E and Sirius red staining (scale bar = 100 μm). Bottom panel shows high magnification image of Sirius red staining of HFD-fed MUP-uPA mouse liver (scale bar = 100 μm). Sirius red positive areas in livers from 24 weeks old WT and MUP-uPA mice that were kept on LFD or HFD were quantified with Image J software and shown as bar graphs. Data are means ± S.D. (n = 4 per group). *p < 0.05. (E) Relative mRNA amounts of type 1 collagen α1 were examined by real-time Q-PCR. Data are presented as means ± S.D. (LFD-fed WT, n = 3; others, n =5 per group). *p < 0.05. (F) Immunoblot evaluation of cyclin D1 in livers of 24 weeks old mice kept on LFD or HFD.
Figure S2 (Related to Figure 2). Characteristics of liver tumors in HFD-fed MUP-uPA mice. (A) The average numbers of liver tumors in LFD- or HFD-fed MUP-uPA mice at 32 and 40 weeks of age. (B) Frequencies of liver adenoma and HCC in LFD- or HFD-fed MUP-uPA mice at 32 and 40 weeks of age. (C) Representative H&E staining of tumor sections from 40 weeks old LFD- or HFD-fed MUP-uPA mice. Upper four panels show adenomas from HFD-fed MUP-uPA mice and lower two panels show hyperplastic nodule from LFD-fed MUP-uPA mice (scale bar = 100 μm). (D) Representative images of H&E stained human steatohepatitic HCC (scale bar = 100 μm). (E) IHC analysis of the indicated antigens in non-tumor (NT) and tumor areas of 40 weeks old HFD-fed MUP-uPA livers (scale bar = 100 μm). (F) IHC of YAP and EpCAM in tumor and non-tumor (NT) areas of HFD-fed MUP-uPA mouse livers (scale bar = 100 μm). (G) Relative CD44 and Myc mRNAs in tumor and NT areas of HFD-fed MUP-uPA mouse livers. Data are means ± S.D. (NT, n = 3; Tumor, n = 5). *p < 0.05. (H) IB analysis of the indicated proteins in liver tumors and non-tumor liver tissue (NT) from 40 weeks old HFD-fed MUP-uPA mice. (I) IHC of Ki67 and AFP in hyperplastic lesion from 40 weeks old LFD-fed MUP-uPA livers (scale bar = 100 μm). (J) IHC of p62 and YAP in liver premalignant foci of 24 weeks old HFD-fed MUP-uPA mice (scale bar = 100 μm).
Figure S3 (Related to Figure 3). ER stress is sustained by HFD in MUP-uPA mice. (A) Relative mRNA amounts of ER stress markers in livers of 5 weeks old WT and MUP-uPA mice and 16 weeks old WT and MUP-uPA mice that were kept on LFD or HFD. (B) Relative mRNA amounts of downstream targets for ER stress signaling in livers of 16 weeks old WT and MUP-uPA mice kept on LFD or HFD and TRAIL mRNA at 24 weeks. (C) Electron micrographs showing the ER in hepatocytes of HFD-fed WT and MUP-uPA mice. Arrows indicate dilated ER (scale bar = 1 μm). (D-F) LFD or HFD-fed 20 weeks old WT mice were intraperitoneally injected with 1.25 mg/kg tunicamycin (TM) or 150 mM dextrose (vehicle). Representative images of livers and H&E (D) and TUNEL (E) staining of liver sections prepared 36 hrs later (scale bar = 100 μm). (F) Serum ALT in LFD- or HFD-fed 20 weeks old WT mice at 36 hrs after injection of tunicamycin or vehicle. Data are means ± S.D. (n = 3-4 per group). (G) Relative expression of apoB mRNA in mouse livers described in A. (H) Serum TG and total cholesterol concentrations in 5 weeks and 16 weeks old WT and MUP-uPA mice kept on LFD or HFD. (I) Relative expression of lipogenic regulators in mouse livers described in A (left graph, 5 weeks old; right graph, 16 weeks old). (J) Relative expression of genes regulated by the IRE1α-XBP1 pathway in mouse livers described in A (left graph, 5 weeks old; right graph, 16 weeks old). All bar graphs represent means ± S.D. (n = 3 per group). *p < 0.05.
Figure S4 (Related to Figure 4). Effects of TUDCA and GRP78 overexpression and CHOP ablation on NASH and HCC development. (A,B) Cell death and ROS accumulation in livers of TUDCA- or vehicle-treated HFD-fed MUP-uPA mice were examined by TUNEL (A) and DHE staining (B), respectively (scale bar = 100 μm). (C) Effects of TUDCA treatment on ER stress and SREBP1 activation in HFD-fed MUP-uPA mouse livers. CHOP protein expression in whole liver and mature SREBP1 in the nuclear fraction were examined by IB analysis. Shown are three individual livers per condition. (D,E) Effects of GRP78 overexpression. HFD-fed MUP-uPA mice were intravenously injected with 1×109 pfu of Ad-LacZ or Ad-GRP78. After 6 days the mice (n=6 per group) were sacrificed and hepatic steatosis was analyzed by H&E staining (scale bar = 100 μm) (D) and GRP78 expression was determined by IB analysis (E). (F) Serum ALT in 5 weeks old ChopΔhep/MUP-uPA and ChopF/F/MUP-uPA mice. Data are means ± S.D. (n = 3 per group). (G) IB analysis of ER stress markers in livers of 5 weeks old ChopΔhep/MUP-uPA and ChopF/F/MUP-uPA mice. (H-K) Effect of hepatocyte CHOP ablation on tumor development and severity of NASH in HFD-fed MUP-uPA mice at 40 weeks of age. (H) Tumor numbers and maximal sizes are shown. Results are means ± S.E.M. (ChopF/F/MUP-uPA, n = 17; ChopΔhep/MUP-uPA, n = 11). *p < 0.05. Serum ALT (I), expression of indicated proteins in non-tumor tissue (J), and H&E staining of non-tumor areas (scale bar = 100 μm) (K) are shown. Data are presented as means ± S.D. (L) IHC analysis of CHOP expression in preneoplastic foci (24 weeks old) and liver tumors (40 weeks old) of HFD-fed ChopF/F/MUP-uPA and ChopΔhep/MUP-uPA mice (scale bar = 100 μm). (M) TUNEL staining of non-tumor (NT) and tumor areas of liver sections from 40 weeks old HFD-fed ChopF/F/MUP-uPA and ChopΔhep/MUP-uPA mice (scale bar = 100 μm). Bar graphs show numbers of TUNEL positive cells per 200× field. Data are means ± S.D. (n = 5-6 per group).
Figure S5 (Related to Figure 5). Characteristics of Tnfr1-/-/MUP-uPA mice. (A, B) Effect of 4 weeks TUDCA treatment on HFD-fed MUP-uPA mouse liver. IHC of F4/80 (scale bar = 100 μm) (A) and TNF mRNA expression (B). Data are means ± S.D. (n = 5 per group). *p < 0.05. (C) H&E staining of liver sections from 5 weeks old MUP-uPA and Tnfr1-/-/MUP-uPA mice (scale bar = 100 μm). (D) Serum ALT in 5 weeks old MUP-uPA and Tnfr1-/-/MUP-uPA mice. Data are presented as means ± S.D. (n = 3 per group). (E) Serum ALT in 40 weeks old LFD-fed MUP-uPA and Tnfr1-/-/MUP-uPA mice. Data are presented as means ± S.D. (n = 5 per group). (F) Body weights of 40 weeks old LFD- or HFD-fed MUP-uPA and Tnfr1-/-/MUP-uPA mice (LFD group, MUP-uPA, n = 11, Tnfr1-/-/MUP-uPA, n = 10; HFD group, MUP-uPA, n = 14; Tnfr1-/-/MUP-uPA, n = 11). Results are shown as means ± S.D.
Figure S6 (Related to Figure 6). Analyses of HcPC transplanted MUP-uPA mice. (A) Relative mRNA amounts of CD44 in non-HcPC and HcPC isolated from DEN-treated WT and Tnfr1-/- mice. Data are means ± S.D. (n = 3 per group). (B) Confirmation of TNFR1 deletion in cancer cells but not in non-tumor liver tissues. Left panel shows that the deleted Tnfr1 allele is detected only in tumor tissues of Tnfr1-/-HcPC-transplanted MUP-uPA mice by genomic DNA PCR. Right panel shows an immunoblot of TNFR1 protein expression. (C-F) No differences in the severity of NASH and TNF expression in the background liver of MUP-uPA mice transplanted with either WT and Tnfr1-/- HcPC. Body weight (C), serum ALT (D), expression of TNF mRNA in non-tumor tissues (E), liver steatosis (F) and fibrosis (G) in MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD are shown (scale bar = 100 μm). Data are expressed as means ± S.D. (n = 10-11 per group). *p < 0.05. (H) Effect of the TNF antagonist etanercept on HFD-induced acceleration of tumor growth. WT HcPC-transplanted MUP-uPA mice were kept on HFD for 5 months as shown in Figure 6A. 5 mg/kg etanercept or vehicle control was i.p. injected three times a week during the last 7 weeks. Representative images of livers and maximal tumor sizes are shown. Data are expressed as means ± S.E.M. (control, n = 10; etanercept, n = 12). (I) HcPC were isolated from DEN- WT mice and transplanted into Tnfr1-/-/MUP-uPA mice. HcPC-transplanted Tnfr1-/-/MUP-uPA mice were divided into two groups that were fed with either LFD or HFD, and 5 months later tumorigenesis was assessed. Representative images of livers and maximal tumor sizes are shown. Data are expressed as means ± S.E.M. (n = 10 per group). (J) Non-tumor (NT) and tumor tissues from MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD were IB analyzed for phosphorylation of ERK, STAT3, JNK, and S6, and expression of cyclin D1.
Figure S7 (Related to Figure 7). Expression of inflammatory cytokines and chemokines in tumor tissues. Relative mRNA amounts of inflammatory cytokines and chemokines in HCC tissues from MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD were determined by real-time Q-PCR. (n = 5 per group). *p < 0.05. Results are shown as means ± S.D.
Significance.
ER stress is often observed in cancer, but its role in tumorigenesis has not been explored. ER stress aslso occurs in pre-malignant liver diseases, including NASH, which progress to HCC, a highly aggressive and common cancer. Our work demonstrates that when combined with hypernutrition, ER stress of liver parenchymal cells results in NASH-like disease that spontaneously progresses to HCC through an inflammatory mechanism dependent on TNF and IκB kinase signaling.
Highlights.
Creating mouse model for high fat diet (HFD)-induced NASH and HCC
ER stress increases NASH and HCC risk in response to HFD
NASH and HFD-driven HCC depend on TNF and IKKβ signaling
ER stress increases NASH and HCC risk independently of insulin resistance
Acknowledgments
We thank Drs. E.P. Sandgren for MUP-uPA, I. Tabas for ChopF/F mice and M. R. Mackey, G. A. Perkins and M. H. Ellisman for help for EM analysis. Research was supported by the Daiichi Sankyo Foundation of Life Science and Grant-in-Aid for Scientific Research (#25893042), and Astellas Foundation for Research on Metabolic Disorders (H.N.); a Global Grant Scholarship from The Rotary Foundation (A.U.); postdoctoral fellowship for Research Abroad, Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science, and the Uehara Memorial Foundation Fellowship (K.T.); California Institute for Regenerative Medicine Stem Cell Training Grant II (TG2-01154; J.F.B.); American Liver Foundation, (D.D.); Kanzawa Medical Research Foundation (H.O.) and grants from the NIH and Superfund Basic Research Program (CA155120-02; CA118165-06; ES010337-12). M.K. holds the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases and is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- Caldwell S, Ikura Y, Dias D, Isomoto K, Yabu A, Moskaluk C, Pramoonjago P, Simmons W, Scruggs H, Rosenbaum N, et al. Hepatocellular ballooning in NASH. J Hepatol. 2010;53:719–723. doi: 10.1016/j.jhep.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle EE, Teras LR, Thun MJ. Obesity and mortality. N Engl J Med. 2005;353:2197–2199. doi: 10.1056/NEJM200511173532020. [DOI] [PubMed] [Google Scholar]
- Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, Chen CJ. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135:111–121. doi: 10.1053/j.gastro.2008.03.073. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- DeZwaan-McCabe D, Riordan JD, Arensdorf AM, Icardi MS, Dupuy AJ, Rutkowski DT. The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 2013;9:e1003937. doi: 10.1371/journal.pgen.1003937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut and liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien PA, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, Seki E, Yost SE, Jepsen K, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–396. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012;16:311–321. doi: 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Qiu W, Zhang J, Dekker MJ, Wang H, Huang J, Brumell JH, Adeli K. Hepatic autophagy mediates endoplasmic reticulum stress-induced degradation of misfolded apolipoprotein B. Hepatology. 2011;53:1515–1525. doi: 10.1002/hep.24269. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomao M, Remotti H, Vaughan R, Siegel AB, Lefkowitch JH, Moreira RK. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum Pathol. 2012;43:737–746. doi: 10.1016/j.humpath.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–256. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- Schramm C, Schneider A, Marx A, Lohse AW. Adalimumab could suppress the activity of non alcoholic steatohepatitis (NASH) Zeitschrift fur Gastroenterologie. 2008;46:1369–1371. doi: 10.1055/s-2008-1027411. [DOI] [PubMed] [Google Scholar]
- So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, Lee AH. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell metabolism. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon RK, Jr, Yan JS, Grenert JP, Maher JJ. Stress signaling in the methionine-choline-deficient model of murine fatty liver disease. Gastroenterology. 2010;139:1730–1739. 1739–e1731. doi: 10.1053/j.gastro.2010.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- Toffanin S, Friedman SL, Llovet JM. Obesity, inflammatory signaling, and hepatocellular carcinoma-an enlarging link. Cancer Cell. 2010;17:115–117. doi: 10.1016/j.ccr.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang ZQ, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. American journal of translational research. 2010;2:65–74. [PMC free article] [PubMed] [Google Scholar]
- Weglarz TC, Degen JL, Sandgren EP. Hepatocyte transplantation into diseased mouse liver. Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am J Pathol. 2000;157:1963–1974. doi: 10.1016/S0002-9440(10)64835-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure 1). Effects of HFD on MUP-uPA mice. (A) Body weights of LFD- or HFD-fed WT and MUP-uPA mice. HFD was started at 6 weeks of age. Data are means ± S.D. (n = 6-8 per group). *p < 0.05. (B) Glucose tolerance tests of 24 weeks old WT and MUP-uPA mice that were kept on LFD or HFD. Blood glucose was measured at the indicated time points after i.p. injection of 0.8 g/kg (right graph) or 2.0 g/kg (left graph) glucose. (n = 4-5 per group). (C) Expression of the uPA transgene was examined by real-time PCR. Results are presented as means ± S.D. (n = 3 per group). (D) Comparison of liver histology between HFD-fed MUP-uPA mouse (24 weeks old) and human NASH by H&E and Sirius red staining (scale bar = 100 μm). Bottom panel shows high magnification image of Sirius red staining of HFD-fed MUP-uPA mouse liver (scale bar = 100 μm). Sirius red positive areas in livers from 24 weeks old WT and MUP-uPA mice that were kept on LFD or HFD were quantified with Image J software and shown as bar graphs. Data are means ± S.D. (n = 4 per group). *p < 0.05. (E) Relative mRNA amounts of type 1 collagen α1 were examined by real-time Q-PCR. Data are presented as means ± S.D. (LFD-fed WT, n = 3; others, n =5 per group). *p < 0.05. (F) Immunoblot evaluation of cyclin D1 in livers of 24 weeks old mice kept on LFD or HFD.
Figure S2 (Related to Figure 2). Characteristics of liver tumors in HFD-fed MUP-uPA mice. (A) The average numbers of liver tumors in LFD- or HFD-fed MUP-uPA mice at 32 and 40 weeks of age. (B) Frequencies of liver adenoma and HCC in LFD- or HFD-fed MUP-uPA mice at 32 and 40 weeks of age. (C) Representative H&E staining of tumor sections from 40 weeks old LFD- or HFD-fed MUP-uPA mice. Upper four panels show adenomas from HFD-fed MUP-uPA mice and lower two panels show hyperplastic nodule from LFD-fed MUP-uPA mice (scale bar = 100 μm). (D) Representative images of H&E stained human steatohepatitic HCC (scale bar = 100 μm). (E) IHC analysis of the indicated antigens in non-tumor (NT) and tumor areas of 40 weeks old HFD-fed MUP-uPA livers (scale bar = 100 μm). (F) IHC of YAP and EpCAM in tumor and non-tumor (NT) areas of HFD-fed MUP-uPA mouse livers (scale bar = 100 μm). (G) Relative CD44 and Myc mRNAs in tumor and NT areas of HFD-fed MUP-uPA mouse livers. Data are means ± S.D. (NT, n = 3; Tumor, n = 5). *p < 0.05. (H) IB analysis of the indicated proteins in liver tumors and non-tumor liver tissue (NT) from 40 weeks old HFD-fed MUP-uPA mice. (I) IHC of Ki67 and AFP in hyperplastic lesion from 40 weeks old LFD-fed MUP-uPA livers (scale bar = 100 μm). (J) IHC of p62 and YAP in liver premalignant foci of 24 weeks old HFD-fed MUP-uPA mice (scale bar = 100 μm).
Figure S3 (Related to Figure 3). ER stress is sustained by HFD in MUP-uPA mice. (A) Relative mRNA amounts of ER stress markers in livers of 5 weeks old WT and MUP-uPA mice and 16 weeks old WT and MUP-uPA mice that were kept on LFD or HFD. (B) Relative mRNA amounts of downstream targets for ER stress signaling in livers of 16 weeks old WT and MUP-uPA mice kept on LFD or HFD and TRAIL mRNA at 24 weeks. (C) Electron micrographs showing the ER in hepatocytes of HFD-fed WT and MUP-uPA mice. Arrows indicate dilated ER (scale bar = 1 μm). (D-F) LFD or HFD-fed 20 weeks old WT mice were intraperitoneally injected with 1.25 mg/kg tunicamycin (TM) or 150 mM dextrose (vehicle). Representative images of livers and H&E (D) and TUNEL (E) staining of liver sections prepared 36 hrs later (scale bar = 100 μm). (F) Serum ALT in LFD- or HFD-fed 20 weeks old WT mice at 36 hrs after injection of tunicamycin or vehicle. Data are means ± S.D. (n = 3-4 per group). (G) Relative expression of apoB mRNA in mouse livers described in A. (H) Serum TG and total cholesterol concentrations in 5 weeks and 16 weeks old WT and MUP-uPA mice kept on LFD or HFD. (I) Relative expression of lipogenic regulators in mouse livers described in A (left graph, 5 weeks old; right graph, 16 weeks old). (J) Relative expression of genes regulated by the IRE1α-XBP1 pathway in mouse livers described in A (left graph, 5 weeks old; right graph, 16 weeks old). All bar graphs represent means ± S.D. (n = 3 per group). *p < 0.05.
Figure S4 (Related to Figure 4). Effects of TUDCA and GRP78 overexpression and CHOP ablation on NASH and HCC development. (A,B) Cell death and ROS accumulation in livers of TUDCA- or vehicle-treated HFD-fed MUP-uPA mice were examined by TUNEL (A) and DHE staining (B), respectively (scale bar = 100 μm). (C) Effects of TUDCA treatment on ER stress and SREBP1 activation in HFD-fed MUP-uPA mouse livers. CHOP protein expression in whole liver and mature SREBP1 in the nuclear fraction were examined by IB analysis. Shown are three individual livers per condition. (D,E) Effects of GRP78 overexpression. HFD-fed MUP-uPA mice were intravenously injected with 1×109 pfu of Ad-LacZ or Ad-GRP78. After 6 days the mice (n=6 per group) were sacrificed and hepatic steatosis was analyzed by H&E staining (scale bar = 100 μm) (D) and GRP78 expression was determined by IB analysis (E). (F) Serum ALT in 5 weeks old ChopΔhep/MUP-uPA and ChopF/F/MUP-uPA mice. Data are means ± S.D. (n = 3 per group). (G) IB analysis of ER stress markers in livers of 5 weeks old ChopΔhep/MUP-uPA and ChopF/F/MUP-uPA mice. (H-K) Effect of hepatocyte CHOP ablation on tumor development and severity of NASH in HFD-fed MUP-uPA mice at 40 weeks of age. (H) Tumor numbers and maximal sizes are shown. Results are means ± S.E.M. (ChopF/F/MUP-uPA, n = 17; ChopΔhep/MUP-uPA, n = 11). *p < 0.05. Serum ALT (I), expression of indicated proteins in non-tumor tissue (J), and H&E staining of non-tumor areas (scale bar = 100 μm) (K) are shown. Data are presented as means ± S.D. (L) IHC analysis of CHOP expression in preneoplastic foci (24 weeks old) and liver tumors (40 weeks old) of HFD-fed ChopF/F/MUP-uPA and ChopΔhep/MUP-uPA mice (scale bar = 100 μm). (M) TUNEL staining of non-tumor (NT) and tumor areas of liver sections from 40 weeks old HFD-fed ChopF/F/MUP-uPA and ChopΔhep/MUP-uPA mice (scale bar = 100 μm). Bar graphs show numbers of TUNEL positive cells per 200× field. Data are means ± S.D. (n = 5-6 per group).
Figure S5 (Related to Figure 5). Characteristics of Tnfr1-/-/MUP-uPA mice. (A, B) Effect of 4 weeks TUDCA treatment on HFD-fed MUP-uPA mouse liver. IHC of F4/80 (scale bar = 100 μm) (A) and TNF mRNA expression (B). Data are means ± S.D. (n = 5 per group). *p < 0.05. (C) H&E staining of liver sections from 5 weeks old MUP-uPA and Tnfr1-/-/MUP-uPA mice (scale bar = 100 μm). (D) Serum ALT in 5 weeks old MUP-uPA and Tnfr1-/-/MUP-uPA mice. Data are presented as means ± S.D. (n = 3 per group). (E) Serum ALT in 40 weeks old LFD-fed MUP-uPA and Tnfr1-/-/MUP-uPA mice. Data are presented as means ± S.D. (n = 5 per group). (F) Body weights of 40 weeks old LFD- or HFD-fed MUP-uPA and Tnfr1-/-/MUP-uPA mice (LFD group, MUP-uPA, n = 11, Tnfr1-/-/MUP-uPA, n = 10; HFD group, MUP-uPA, n = 14; Tnfr1-/-/MUP-uPA, n = 11). Results are shown as means ± S.D.
Figure S6 (Related to Figure 6). Analyses of HcPC transplanted MUP-uPA mice. (A) Relative mRNA amounts of CD44 in non-HcPC and HcPC isolated from DEN-treated WT and Tnfr1-/- mice. Data are means ± S.D. (n = 3 per group). (B) Confirmation of TNFR1 deletion in cancer cells but not in non-tumor liver tissues. Left panel shows that the deleted Tnfr1 allele is detected only in tumor tissues of Tnfr1-/-HcPC-transplanted MUP-uPA mice by genomic DNA PCR. Right panel shows an immunoblot of TNFR1 protein expression. (C-F) No differences in the severity of NASH and TNF expression in the background liver of MUP-uPA mice transplanted with either WT and Tnfr1-/- HcPC. Body weight (C), serum ALT (D), expression of TNF mRNA in non-tumor tissues (E), liver steatosis (F) and fibrosis (G) in MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD are shown (scale bar = 100 μm). Data are expressed as means ± S.D. (n = 10-11 per group). *p < 0.05. (H) Effect of the TNF antagonist etanercept on HFD-induced acceleration of tumor growth. WT HcPC-transplanted MUP-uPA mice were kept on HFD for 5 months as shown in Figure 6A. 5 mg/kg etanercept or vehicle control was i.p. injected three times a week during the last 7 weeks. Representative images of livers and maximal tumor sizes are shown. Data are expressed as means ± S.E.M. (control, n = 10; etanercept, n = 12). (I) HcPC were isolated from DEN- WT mice and transplanted into Tnfr1-/-/MUP-uPA mice. HcPC-transplanted Tnfr1-/-/MUP-uPA mice were divided into two groups that were fed with either LFD or HFD, and 5 months later tumorigenesis was assessed. Representative images of livers and maximal tumor sizes are shown. Data are expressed as means ± S.E.M. (n = 10 per group). (J) Non-tumor (NT) and tumor tissues from MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD were IB analyzed for phosphorylation of ERK, STAT3, JNK, and S6, and expression of cyclin D1.
Figure S7 (Related to Figure 7). Expression of inflammatory cytokines and chemokines in tumor tissues. Relative mRNA amounts of inflammatory cytokines and chemokines in HCC tissues from MUP-uPA mice transplanted with either WT or Tnfr1-/- HcPC and kept on LFD or HFD were determined by real-time Q-PCR. (n = 5 per group). *p < 0.05. Results are shown as means ± S.D.