

Abstract
Alcohol and amine oxidations are common reactions in laboratory and industrial synthesis of organic molecules. Aerobic oxidation methods have long been sought for these transformations, but few practical methods exist that offer advantages over traditional oxidation methods. Recently developed homogeneous Cu/TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidinyl-N-oxyl) and related catalyst systems appear to fill this void. The reactions exhibit high levels of chemoselectivity and broad functional-group tolerance, and they often operate efficiently at room temperature with ambient air as the oxidant. These advances, together with their historical context and recent applications, are highlighted in this minireview.
Keywords: aerobic oxidation, catalysis, nitroxyl radicals, alcohols, amines, copper
1. Introduction
Aldehydes and ketones are ubiquitous intermediates in the synthesis of pharmaceuticals, agrochemicals, and fine chemicals, and they are often prepared via alcohol oxidation. The target molecules for these applications commonly contain diverse functional groups, including amines, sulfur-containing groups, alkenes and heterocycles. Therefore, effective alcohol oxidation reactions must exhibit one or more types of “chemoselectivity”. Examples include the following: A) oxidation of a primary alcohol to an aldehyde without overoxidation to the carboxylic acid, B) selective oxidation of an alcohol in the presence of other oxidizable and/or potentially inhibitory functional groups, and C) oxidation of one alcohol in preference to another within a diol or polyol (Scheme 1). Numerous stoichiometric reagents and catalytic oxidation methods have been developed to meet these requirements,[1] but there continues to be widespread interest in methods capable of using O2 as an oxidant.[2] Aerobic oxidation methods, however, typically do not match the synthetic scope and utility of conventional alcohol oxidations, and they are rarely used in laboratory- or process-scale syntheses of complex molecules.
Scheme 1.

Chemoselectivity challenges encountered in alcohol oxidation reactions.
In this minireview, we survey Cu/TEMPO (TEMPO = 2,2,6,6-tetramethyl-1-piperidinyl-N-oxyl) and related Cu/nitroxyl catalyst systems (Scheme 2), which have emerged as some of the most efficient catalysts available for aerobic alcohol oxidation.[3] They are often capable of using ambient air as the oxidant, they are compatible with both activated (allylic, benzylic, propargylic) and unactivated (aliphatic) alcohols, and their chemoselectivity and functional-group compatibility rival traditional alcohol oxidation methods. Recent mechanistic studies have provided the basis for significant expansion of the efficiency and scope of these reactions with sterically less hindered, bicyclic nitroxyl cocatalysts, such as ABNO (ABNO = 9-azabicyclo[3.3.1]nonane-N-oxyl; cf. Scheme 2). These methods offer numerous advantages over earlier Cu-catalyzed aerobic alcohol oxidation methods that lack nitroxyl cocatalysts.[4] Similar Cu/nitroxyl catalyst systems have also been identified for aerobic oxidation of amines (Scheme 2B), with applications to the preparation of imines, nitriles and unsaturated heterocycles. In addition, other variants of these reactions have been developed, including oxidations of diols to lactones and oxidative coupling of alcohols and amines. Collectively, these methods represent some of the most versatile and accessible aerobic oxidation methods currently available to synthetic chemists.
Scheme 2.

Aerobic alcohol and amine oxidation reactions mediated by Cu/nitroxyl catalyst systems.
2. Copper/TEMPO-Catalyzed Aerobic Oxidation of Primary Alcohols
2.1 Nitroxyl Catalysts in Alcohol Oxidation
TEMPO is a well known, commercially available, stable nitroxyl radical.[5] One-electron oxidation of TEMPO affords an oxoammonium species (Scheme 3) that may be isolated and used as a stoichiometric oxidant.[ 6 ] Oxoammonium-mediated alcohol oxidation results in two-electron reduction of the oxoammonium species to afford a hydroxylamine (or hydroxylammonium species, depending on the solution pH). Numerous methods have been developed to use catalytic quantities of TEMPO in combination with an inexpensive stoichiometric oxidant, such as sodium hypochlorite (NaOCl), bromine, or PhI(OAc)2 (Scheme 4). One common protocol (the “Anelli oxidation”) features catalytic TEMPO (or a TEMPO derivative) and bromide in a buffered organic/aqueous biphasic mixture, with NaOCl as the stoichiometric oxidant.[7] Recent work by Iwabuchi[8 ] and others[ 9] has shown that bicyclic nitroxyl catalysts, such as ABNO, ketoABNO, AZADO, 1-methyl-AZADO, and 5-fluoro-AZADO (AZADO = 2-azaadamantane-N-oxyl) (Scheme 6), can significantly enhance the efficiency and scope of these reactions. Alcohol oxidations using TEMPO and related catalysts are widely used in laboratory and industrial syntheses, and they are the focus of a recent comprehensive Organic Reactions article by Bobbitt, Brückner, and Merbouh.[10]
Scheme 3.
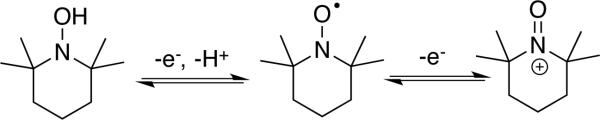
Different TEMPO oxidation/protonation states.
Scheme 4.
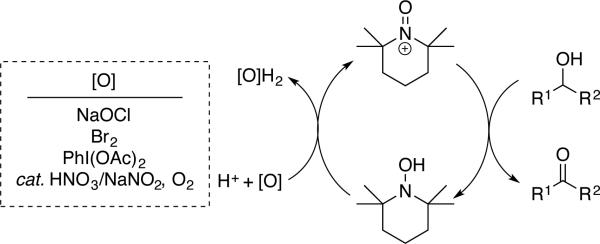
Hydroxylamine/oxoammonium mechanism for aerobic alcohol oxidation with diverse terminal oxidants.
Scheme 6.
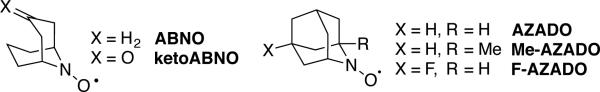
Bicyclic nitroxyl derivatives that significantly expand the efficiency and scope of nitroxyl-catalyzed alcohol oxidation reactions.
Considerable effort has been made to identify cocatalysts for regeneration of the oxoammonium species with O2 as the terminal oxidant. An important development in this area is the identification of NaNO2, HNO3, and other NOx sources that participate in a dioxygen-coupled NO/NO2 redox cycle and enable transition metal-free aerobic oxidation of alcohols. Such methods were first developed for TEMPO-based alcohol oxidation;[11] however, the bicyclic nitroxyl catalysts (cf. Scheme 6) again show significant advantages in these reactions.[12]
Aerobic alcohol oxidation with nitroxyl catalysts has also been achieved by using transition metal salts (e.g. Mn,[13] Fe,[14] Co,[15] and Ce[16]), polyoxometallates,[17] or metalloenzymes (laccase)[18] as cocatalysts.[19] Among these, the reactions with Cu cocatalysts have been the most extensively studied and exhibit particularly broad scope and synthetic utility. Mechanistic studies reveal that the reaction does not proceed via the classical hydroxylamine/oxoammonium cycle shown in Scheme 4, but involves a cooperative pathway with one-electron redox chemistry at Cu and TEMPO. These synthetic and mechanistic developments are the focus of the discussion below.
2.2 Cu/Nitroxyl Catalyst Systems for Aerobic Oxidation of Primary Alcohols
The use of Cu/nitroxyl catalysts for aerobic alcohol oxidation was first reported in 1966, when Brackman and Gaasbeek showed that di-tert-butylnitroxide significantly promotes the oxidation of methanol to formaldehyde by phenanthroline/CuII complexes in basic methanol solutions.[20] This work was missed by synthetic chemists until it was highlighted in 2004 review on nitroxyl radicals by Sheldon and Arends.[2b] The first exploration of the synthetic scope of Cu/nitroxyl-catalyzed aerobic alcohol oxidation was reported by Semmelhack in 1984.[21] Catalytic CuCl and TEMPO in DMF mediates the oxidation of numerous primary activated alcohols (i.e., allylic and benzylic) (Scheme 7). Aliphatic alcohols are much less reactive and stoichiometric quantities of copper and TEMPO were required to oxidize these substrates. The enhanced reactivity of activated alcohols is evident in many of the other Cu/TEMPO catalyst systems that have been reported in recent years, many of which probe the role of different copper sources, ligands, and/or reaction conditions.[22] Systems that use ionic liquids,[23] fluorous biphasic solvents,[24] and supported reaction components (e.g., polymer-supported nitroxyls,[ 25 ] ancillary ligands,[ 26 ] and metal-organic frameworks[27]) have also been reported.
Scheme 7.

Aerobic oxidation of activated alcohols by CuCl/TEMPO in DMF reported by Semmelhack.[21]
In 2000, Knochel and coworkers described fluorous biphasic reaction conditions, in which CuBr·Me2S and a perfuoroalkyl-substituted bipyridine ligand could promote aerobic alcohol oxidation (Scheme 8).[24a] This catalyst system was the first to show broad utility with aliphatic alcohols. Some aliphatic alcohol reactivity was also observed with a Cu/salen catalyst system reported by Punniyamurthy, albeit under relatively forcing conditions (100 °C, 7 mol % TEMPO, 21–25 h).[28] In 2003, Sheldon and coworkers described a catalyst system consisting of CuBr2/bpy/TEMPO/KOtBu (5 mol % each; bpy = 2,2’-bipyridine) in acetonitrile/H2O as the solvent that was capable of promoting the oxidation of benzylic and allylic alcohols at room temperature with ambient air as the source of O2 in ≤ 5 h. [29] Selective oxidation of 1-octanol to 1-octanal was successful, but required a higher temperature and a longer reaction time (40 °C, 24 h).
Scheme 8.
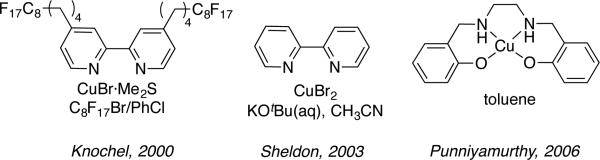
Ligand, Cu source and solvent used in early Cu/TEMPO catalyst systems capable of oxidizing aliphatic alcohols.[24, 29, 28]
The groups of Koskinen (Scheme 9)[30] and Stahl (Scheme 10) [31] subsequently developed two related catalyst systems that exhibit broad synthetic utility for the oxidation of primary alcohols to aldehydes, including aliphatic substrates. The catalyst system developed by Kumpulainen and Koskinen features Cu(OTf)2/bpy/TEMPO, two heterocyclic bases, N-methylimidazole (NMI) and 1,8-diazabicycloundec-7-ene (DBU), and 3Å molecular sieves. The oxidation of a number of aliphatic alcohols was achieved at room temperature within 1–5 h under an atmosphere of pure O2 (Scheme 9). Oxidation of Boc-protected valinol resulted in substantial epimerization of the stereocenter adjacent to the aldehyde probably arising from the presence of DBU, which is a relatively strong organic base (pKa DBU-H+ = 24 in MeCN). Nevertheless, the reaction tolerates alkenes, ethers, esters, and protected amines and alcohols. Variations of this catalyst system, differing in the identity of the organic base (among NMI, DBU, and 4-(N,N-dimethylamino)pyridine (DMAP)), showed excellent activity in the aerobic oxidation of allylic and benzylic alcohols.
Scheme 9.

Aerobic oxidation of aliphatic alcohols with bpy/CuII/NMI/DBU/TEMPO reported by Koskinen (GC yields in parentheses). NMI = N-methylimidazole, DBU = 1,8-diazabicycloundec-7-ene.[30]
Scheme 10.

Aerobic oxidation of aliphatic alcohols with bpy/CuI/NMI/TEMPO reported by Stahl.[31]
Hoover and Stahl noticed significant improvement in catalytic activity upon using a copper(I) source, rather than copper(II). CuI salts with a non-coordinating anion (e.g., CuIOTf) were particularly effective, and the CuOTf/bpy/TEMPO/NMI catalyst system was effective in the oxidation of benzylic, allylic, propargylic and aliphatic alcohols under ambient air.[31,32] Most of the reactions were performed at room temperature, although selected aliphatic alcohols required heating to 50 °C to achieve complete conversion. The operational simplicity of these conditions is complemented by a very broad substrate scope (Scheme 10). Numerous common functional groups, including aryl halides, anilines, nitrogen and sulfur heterocyles, and sulfides, are tolerated. NMI is a considerably weaker base than DBU (pKa NMI-H+ = 14.3 in MeCN), and the use of NMI as the sole organic base probably provides the basis for selective oxidation of (Z)-allylic alcohols without cis-trans isomerization of the base-sensitive enal and retention of enantioselectivity in the oxidation of N-Boc-prolinol. Like most other Cu/TEMPO catalyst systems, secondary alcohols showed poor reactivity; however, this feature was exploited to achieve selective oxidation of several diols containing both primary and secondary alcohols, resulting in near-exclusive formation of the aldehyde products.
2.3 Cu/Nitroxyl-Catalyzed Aerobic Oxidation of Primary Alcohols in Tandem or Sequential One-Pot Reactions
The mild reaction conditions associated with Cu/TEMPO-catalyzed alcohol oxidation make these reactions well suited for tandem or sequential reactions with an aldehyde intermediate. One implementation of this concept is the lactonization of diols. As an extension of selective oxidation of diols, Hoover and Stahl showed that a number of 1,5-diols undergo lactonization. The reaction selectivity is controlled by the reactivity of the respective alcohols, including primary vs. secondary, benzylic vs. aliphatic, and primary alcohols in different steric environments (Scheme 11).[31] The strong steric sensitivity of the reaction is highlighted by the last example in Scheme 11, in which the lactone arises from selective oxidation of a 1° aliphatic alcohol, rather than an electronically activated 2° benzylic alcohol.
Scheme 11.

Cu/TEMPO-catalyzed aerobic lactonization of diols.[31]
Cu/TEMPO-mediated lactonization of polyols has been implemented in the synthesis of more complex molecules (Scheme 12). Nonappa and Maitra prepared the steroidal lactone 1 by selective oxidation of the primary alcohol in the presence of several unprotected secondary alcohols.[33] This reaction employed Semmel-hack-type conditions (0.4 equiv CuCl/TEMPO),[21] but it seems well suited for implementation with one of the more recent aerobic catalyst systems that show good reactivity with aliphatic alcohols, as in Scheme 11. In a related application, the terpenoid precursor 2 was prepared by Cu/TEMPO-mediated oxidation of the allylic primary alcohol; in situ cyclization with the proximal secondary alcohol and further oxidation afforded the desired lactone.[34]
Scheme 12.

Synthetic applications of Cu/TEMPO-catalyzed aerobic lactonization reactions.[33,34]
Jang et al. showed that Cu/TEMPO-catalyzed oxidation of allylic alcohols could be performed in sequence with enantioselective organocatalytic Michael additions, without isolation of the intermediate aldehyde (Scheme 13).[35] Christmann and coworkers prepared (E)-α,β-unsaturated aldehydes selectively from a mixture of (Z)- and (E)-allylic alcohols with a catalyst system consisting of CuOTf/MeObpy/TEMPO/DMAP (MeObpy = 4,4’-dimethoxy-2,2’-bipyridine). DMAP was found to be effective as an alternative to NMI as a cocatalyst and was sufficiently basic to promote cis-trans isomerization of the enal product. This reactions was also implemented in a one-pot alcohol oxidation/enantioselective organocatalalytic Diels-Alder sequence (Scheme 14).[36]
Scheme 13.

Alcohol oxidation/conjugate addition, developed by Jang.[41]
Scheme 14.
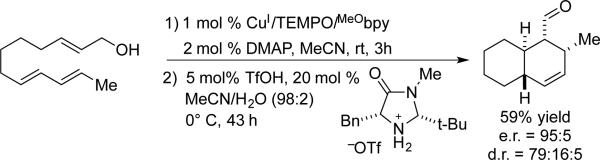
One-pot allylic alcohol oxidation/Diels-Alder cyclization demonstrated by Christmann.[36]
Masson and Jhu showed that alcohols could be used, rather than aldehydes, in Passerini three-component coupling reactions by performing in situ alcohol oxidation with a Cu/TEMPO catalyst system.[37] The groups of Porco[38] and Mehta[39] have used the Semmelhack catalyst system (cf. Scheme 7) to achieve selective oxidation of 1° allylic alcohols enroute to epoxyquinols and related molecules. One example, shown in Scheme 15, highlights the use of alcohol oxidation in tandem with [4 + 2] and [4 + 4] dimerization reactions to access epoxyquninoid dimers.[38b] Selective oxidation of the 1° allylic alcohol was accomplished without a need to protect the 2° allylic alcohol. Attempts to perform alcohol oxidation with MnO2 or Dess-Martin periodinane led to a mixture of alcohol oxidation products. Cu/TEMPO-based alcohol oxidation reactions have been used in numerous other syntheses of natural products and complex-molecule targets.[40]
Scheme 15.

Alcohol oxidation/6π-electrocyclization/cycloadditon sequence employed in the synthesis of epoxyquinoid molecules.[38b]
Tandem reactions initiated by Cu/nitroxyl-catalyzed oxidations of amines, as well as oxidative coupling reactions of alcohols and amines, will be discussed further below (see Sections 5.2 and 6, below). These applications, together with the reactions described in this section, highlight the significant potential utility of Cu/nitroxyl-based methods in organic chemical synthesis.
2.4 Toward Scalable Cu/TEMPO-Catalyzed Aerobic Alcohol Oxidation Reactions
The optimized conditions for Cu/TEMPO-catalyzed alcohol oxidation with ambient air at room temperature are very convenient for lab-scale reactions. The efficiency and synthetic scope of these reactions potentially also make them attractive for large-scale applications; however, the flash point of acetonitrile is well below room temperature (2 °C) and large volumes of acetonitrile under air or O2 represent a significant safety hazard. One way this issue can be addressed is to operate the reactions outside the O2/CH3CN flammability limits.[41] Root and Stahl recently reported a scalable application of this concept using a continuous flow reactor developed by Johnson and coworkers at Eli Lilly.[42,43] The reactions were performed by passing solutions of the catalyst and alcohol with dilute oxygen gas (9% O2 in N2; 35 bar total pressure) through a stainless steel tube reactor maintained at 100 °C. Near-quantitative yields of aldehydes could be obtained with reactor residence times as low as 5 min with activated alcohols, and benzyl alcohol oxidation was demonstrated on 100 g scale. Aliphatic alcohols reacted more slowly (as expected, cf. ref. 31) and the reaction temperature had to be lowered to 60 °C to minimize overoxidation of the aldehydes to carboxylic acids under the reaction conditions (carboxylic acids inhibit catalytic turnover, resulting in incomplete conversion of the starting alcohol). Near-quantitative yields of the aliphatic aldehydes could be obtained with 30-45 min residence times. These results are significantly better than analogous reactions with a homogeneous Pd catalyst reported earlier using the same reactor.[43] A Pd(OAc)2/pyridine catalyst used in the latter application required residence times from 2.5–4.5 h, was not compatible with primary aliphatic alcohols (over oxidation to the acid led to catalyst poisoning), and exhibited very limited functional group tolerance.
3. Mechanism of Copper/TEMPO-Catalyzed Aerobic Oxidation of Primary Alcohols
The mechanism of Cu/TEMPO-catalyzed alcohol oxidation reactions has both fundamental and practical implications. The involvement of three open-shell reagents (CuII, TEMPO, and O2) in a two-electron oxidation of a closed-shell organic molecule raises fundamental questions about the electronic coupling between individual reaction partners and the nature of redox steps involving these species. The origin of electronic effects that favor oxidation of activated alcohols over aliphatic alcohols, and steric effects that favor 1° over 2° alcohol oxidation, have important practical implications for the development of new catalysts. Insights into some of these issues have been gained, but a number of questions remain unanswered and results from different studies remain to be reconciled.
In their 1966 study of methanol oxidation by (phen)CuII/di-tertbutylnitroxyl, Brackman and Gaasbeek proposed that the nitroxyl abstracts a hydrogen atom from a CuII-coordinated methanol molecule (Scheme 17A).[20a] Loss of the H atom was proposed to occur in concert with reduction of CuII to CuI and formation of the formaldehyde. Inhibition of the reaction by NaOCH3 was interpreted as support for reaction of a methanol ligand, rather than methoxide, and evidence against a hydride-transfer pathway.
Scheme 17.

Mechanistic pathways proposed for CuII/TEMPO-mediated alcohol oxidation.
In 1984, Semmelhack and coworkers proposed a two-electron oxoammonium/hydroxylamine redox cycle, mediated by CuI/II and O2. This mechanism resembles that of TEMPO-catalyzed alcohol oxidation with other terminal oxidants (cf. Scheme 4), but it was not studied in detail, and the Brackman/Gaasbeek results were not considered. Contel, Fish and coworkers studied a fluorous biphasic Cu/TEMPO system,[24c,d] and a loss of EPR signals during the reaction was interpreted in favor of an oxoammonium pathway, although β-hydride elimination from a Cu-alkoxide was also considered. Semmelhack supported their proposal by noting that the E1/2 of the CuCl2/CuCl redox couple is 370 mV higher than the TEMPO/TEMPO+ potential; however, the CuII/CuI potential is stongly solvent dependent. Whereas their catalytic reactions were performed in DMF, the electrochemical potentials were measure in acetonitrile. Stahl and coworkers recently showed that the CuII/CuI potential in DMF is lower than the TEMPO/TEMPO+ potential, providing evidence against an oxoammonium-based mechanism.[44]
Sheldon et al. revisited Semmelhack's Cu/TEMPO-catalyzed alcohol oxidation reaction in 2003.[ 45 ] They measured kinetic isotope effects (KIEs) via intramolecular competition with p-methoxybenzyl alcohol-d1 (i.e., ArCHDOH), and determined a Hammett correlation with para and meta-substituted benzyl alcohols. The results were compared to analogous data for alcohol oxidation mediated by TEMPO+Cl–, a Ru/TEMPO-based catalyst system believed to operate via an organometallic β-hydride elimination pathway,[19b] Galactose Oxidase (GOase),[46] and a small-molecule GOase mimic.[47] Differences between the Cu/TEMPO and the TEMPO+Cl– and Ru/TEMPO systems provided evidence against oxoammonium and organometallic alcohol oxidation pathways, while quantitative similarities between the KIEs and Hammett ρ values for Cu/TEMPO, GOase and the GOase mimic led the authors to conclude that these systems mediate alcohol oxidation by a similar mechanism. GOase features a CuII center with coordinated tyrosyl radical ligand (Scheme 18), and the proposed alcohol oxidation mechanism involves H-atom transfer from an alkoxide ligand to the oxyl radical.[46] Sheldon and coworkers suggested that Cu/TEMPO acts as a GOase mimic, in which TEMPO coordinates to CuII as an η2 ligand prior to abstracting a hydrogen atom from a bound alkoxide (Figure 17B), and this mechanism has also been proposed by Sheldon et al. for (bpy)Cu/TEMPO catalyst systems.[29]
Scheme 18.
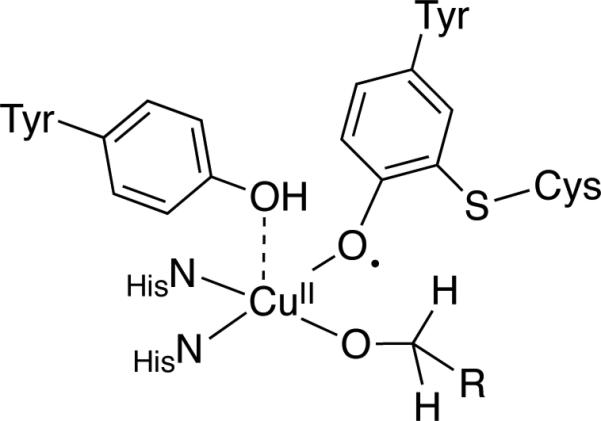
Structural representation of the Galactose Oxidase active site with the galactose substrate present as an alkoxide ligand.
Cu-TEMPO complexes have been crystallographically characterized, with examples including both η1- and η2-coordinated TEMPO (cf. Scheme 19).[48] These complexes are EPR silent, suggest strong coupling between the CuII and nitroxyl unpaired electrons. Structures of this type have been investigated by DFT computational methods to probe the nature of hydrogen transfer to a coordinated nitroxyl.[49] Both radical (H-atom) and hydride transfer pathways have been proposed, and mechanisms involving H-transfer to the nitrogen and oxygen atom of a coordinated nitroxyl have been considered. In the most compelling study to date, Baerends and coworkers identified a low-energy pathway for intramolecular hydride transfer from a Cu-alkoxide to an η1-O-coordinated nitroxyl ligand (cf. Scheme 17C).[49d] These studies employed a model piperidyl-N-oxyl that lacks the steric effects of TEMPO, and the results remain to be reconciled with experimental data, such as the first-order dependence of the reaction on [TEMPO] (see below).
Scheme 19.
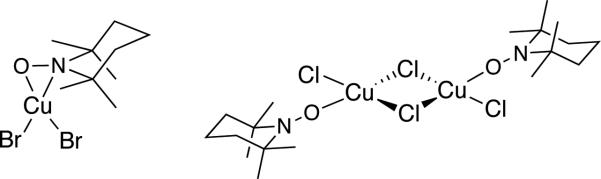
Examples of crystallographically characterized Cu-TEMPO complexes.
Koskinen[30] and Stahl[50] have carried out kinetic studies of their respective (bpy)Cu/TEMPO-catalyzed alcohol oxidation reactions, and Stahl also reported an independent comparative kinetic and mechanistic study of the catalytic reactions reported by Semmelhack, Sheldon, Koskinen and Stahl (cf. Schemes 7-10).[44] Several observations demonstrate clearly that (bpy)Cu/TEMPO-catalyzed alcohol oxidation does not involve an oxoammonium pathway: (1) different KIEs are observed for (bpy)Cu/TEMPO and TEMPO+ mediated alcohol oxidation (similar to Sheldon's observations with the Semmelhack system), (2) the CuII/CuI E1/2 is too low under the reaction conditions to oxidized TEMPOH or TEMPO to the oxoammonium species, and (3) TEMPO+-mediated alcohol oxidation was shown to be kinetically incompetent to account for the fast reaction rates observed with the (bpy)Cu/TEMPO catalysts.[44,50]
The mechanistic study of Stahl and coworkers provides the basis for the simplified catalytic mechanism in Scheme 20 (see original report for a more elaborate discussion).[50] Aerobic oxidation of CuI and TEMPOH affords a CuII–OH species and TEMPO (steps i and ii). This sequence explains why a strong base, such as KOtBu or DBU, is not required when a CuI salt is used as the catalyst source (cf. Scheme 10): the base (Ln CuII –OH) is generated upon reduction of O2. As noted above, this feature has beneficial implications in the oxidation of base-sensitive substrates. Alcohol oxidation proceeds via pre-equilibrium formation of a CuII–alkoxide species (step iii) followed by hydrogen-atom transfer to TEMPO (step iv). Details of the H-atom transfer step could not be discerned from these studies, but a first-order dependence on [TEMPO] was observed in reactions of aliphatic alcohols and no evidence was obtained for an interaction between TEMPO and CuII under the reaction conditions.[50,51] These observations are consistent with a bimolecular H-atom transfer step, similar to that proposed by Brackman and Gaasbeek (Scheme 17A); however, mechanisms involving rate-limiting coordination of TEMPO to CuII followed by rapid H-atom transfer cannot be excluded on the basis of available evidence.
Scheme 20.
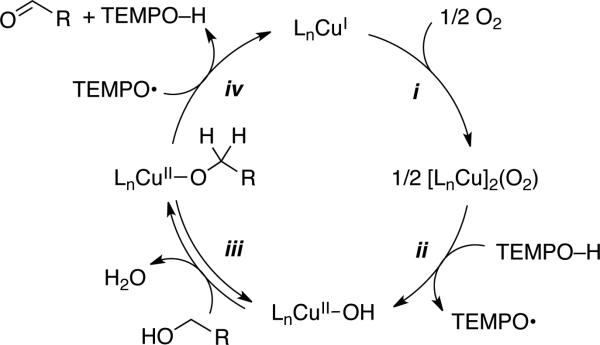
Mechanism of (bpy)Cu/TEMPO-catalyzed alcohol oxidation deduced by Stahl and coworkers.[50,52]
The latter studies provide important insights into the origin of reactivity difference between activated and aliphatic alcohols.[50] Benzylic alcohols exhibit faster rates and exhibit no kinetic dependence on [alcohol] or [TEMPO]. A rate dependence on [Cu] and [O2] suggests that aerobic oxidation of the Cu catalyst (step i, Scheme 20) is the turnover-limiting step of the reaction. In contrast, aliphatic alcohols react more slowly and the rate exhibits a saturation dependence on [alcohol] and a first-order dependence on [TEMPO]. These observations had a direct impact on the development of a new Cu/nitroxyl catalyst systems, described in the next section.[52]
4. Improved Aerobic Alcohol Oxidation with Bicyclic Nitroxyls
Stahl and coworkers expanded on the mechanistic studies above by testing different nitroxyl derivatives, with the hope of achieving faster rates and/or broader substrate scope with aliphatic alcohols.[52] They examined TEMPO analogs with different redox potentials and sterically less hindered bicylic nitroxyls, ABNO, ketoABNO and AZADO, as cocatalysts in combination with CuOTf/bpy/NMI. Reactions with benzylic and aliphatic primary and secondary alcohols reveal the dramatic effect of replacing TEMPO with ABNO under similar, but independently optimized, conditions (Scheme 21). Specifically, the Cu/TEMPO conditions show significantly different rates for different classes of alcohols, with the following trends: 1° benzylic > 1° aliphatic/2° benzylic >> 2° aliphatic/sterically hindered 2° benzylic. In contrast, The Cu/ABNO catalyst systems exhibited nearly identical rates with all classes of alcohols. The observations suggest that replacement of TEMPO with ABNO significantly increases the alcohol oxidation step (step iv, Scheme 20) and the rate is controlled by an alcohol-independent step, such as aerobic oxidation of CuI (step i, Scheme 20). This conclusion is suppported qualitatively by the beneficial effect of an electron-rich bpy ligand (MeObpy) and the ability to lower the ABNO loading from 5 to 1 mol % without affecting the rate. The optimized Cu/ABNO catalyst system show excellent reativity with a broad range of activated and aliphatic primary and secondary alcohols, including those bearing diverse functional groups and stereocenters adjacent to the product aldehyde group (Scheme 22). The good reactivity with secondary alcohols undoubtedly reflects the smaller steric profile of the bicyclic ABNO structure relative to TEMPO, although the precise origin of the steric effect remains to be resolved.
Scheme 21.

Rate comparison of five different alcohols with CuI/TEMPO and CuI/ABNO alcohol oxidation systems. Data adapted from ref. [52]. See Schemes 10 and 22 for more details of reaction conditions.
Scheme 22.

A Cu/ABNO catalyst system effective with diverse primary and secondary alcohols under ambient conditions, reported by Stahl.[52]
Iwabuchi and coworkers recently reported a complementary study with a Cu/AZADO catalyst system, focused on chemoselective oxidation of alcohols within substrates bearing unprotected primary, secondary and tertiary amine substituents (Scheme 23).[53] The Cu/AZADO method was shown to be superior to several conventional alcohol oxidation reagents and methods (e.g., pyridinium chlorochromate, Swern, Dess-Martin periodinane, and tetrapropylammonium perruthenate) for a selection of substrates of this type. The results highlight the distinct reactivity of Cu/nitroxyl catalyst systems relative to oxoammonium reagents and catalytic methods. The latter are often incompatible with unprotected amines. The chemoselectivity of the reaction may be rationalized by the higher acidity of the O–H of the alcohol relative to the N–H of the amines, which promotes formation of a Cu-alkoxide (cf. Scheme 20), which is activated toward hydrogen-atom transfer to the nitroxyl. The methods in Scheme 22 and 23 are among the most efficient and general alcohol oxidation methods reported to date, even when considering non-aerobic methods, and they provide a powerful complement to the Cu/TEMPO-based methods, which show high selectivity for oxidation of 1° alcohols (cf. Schemes 9 and 10).
Scheme 23.

A Cu/AZADO catalyst system for selective oxidation of diverse amine-containing alcohol substrates under ambient conditions, reported by Iwabuchi.[53]
5. Copper/Nitroxyl-Catalyzed Aerobic Oxidation of Amines
The reactions outlined in the previous section show that alcohols can be oxidized in the presence of amines. Meanwhile, several groups have independently developed methods for Cu/nitroxylcatalyzed oxidation of amines to imines and nitriles (Scheme 24), as well as unsaturated nitrogen heterocycles (see below). In some cases, analogous Cu-catalyzed aerobic oxidation reactions (i.e., in the absence of nitroxyls) have been reported,[54] but the use of a nitroxyl cocatalyst enables faster rates, milder reaction conditions, and broader substrate scope.
Scheme 24.
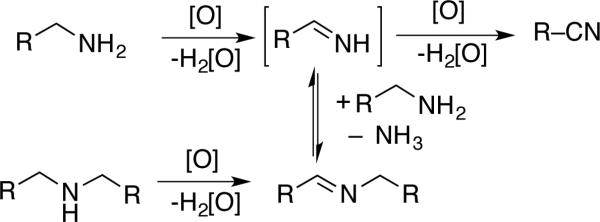
Amine oxidation pathways and products.
5.1 Oxidation of Primary and Secondary Amines to Form Imines, Nitriles, and Unsaturated Heterocycles
In 2012, the groups of Kerton[55] and Kanai[56] independently reported methods for Cu/nitroxyl-catalyzed aerobic oxidation of primary and secondary amines. In the Kerton report,[55] reactions were mostly limited to sterically unhindered, primary benzylic amines and they led to homocoupled imines via condensation of an amine substrate with the initially formed primary imine. In the presence of electron-rich and/or sterically unhindered anilines, selective cross-coupled imine products could be obtained (Scheme 25), and electron-rich anilines underwent homocoupling to the corresponding azo compounds in the absence of benzyl amine.
Scheme 25.
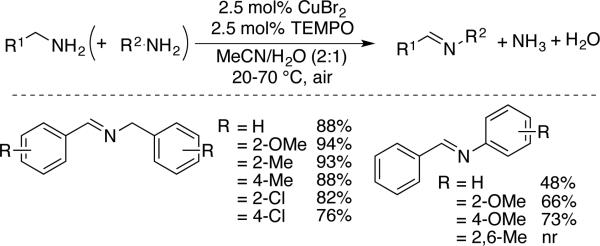
Cu/TEMPO-catalyzed oxidation of 1° benzylic amines resulting in imine formation, developed by Kerton.[55]
Kanai and coworkers developed a different catalyst system that uses ketoABNO, rather than TEMPO, as the cocatalyst.[56] They achieved similar homocoupling reactions of primary benzylic amines, including α-methylbenzylamine, which was not effective under the Kerton conditions. The smaller steric profile of ketoABNO relative to TEMPO probably accounts for the broader scope of this catalyst system. For example, this catalyst system achieved aerobic oxidation of secondary amines to the corresponding imines (Scheme 26). While the reactivity favors for benzylic substrates, a number of other substrates were effective, such as para-methoxyphenyl (PMP)-substituted aliphatic amines.
Scheme 26.

Cu/ketoABNO-catalyzed oxidation of 2° amines, developed by Kanai.[56]
Kim and Stahl reported a catalyst system that enables selective oxidation of benzylic and aliphatic primary amines to nitriles, with minimal formation of the imine homocoupling product (Scheme 27; cf. Scheme 24).[57] ABNO was used as the nitroxyl cocatalyst, and the reaction proceeds efficiently at room temperature with a variety of allylic, benzylic, and aliphatic substrates (Scheme 27). The similarity between this catalyst system and that reported by Kanai for imine formation is noteworthy, and shows how minor changes in reaction conditions and/or ligand architecture can alter the product selectivity. Use of acetonitrile as the solvent was found to be an important factor in order to achieve selective nitrile formation.
Scheme 27.

Cu/ABNO catalyzed oxidation of primary amines to nitriles, reported by Stahl.[57]
5.2 Cu/Nitroxyl-Catalyzed Aerobic Oxidation of Amines in Tandem or Sequential One-Pot Reactions
Copper/nitoxyl-catalyzed amine oxidation reactions have been incorporated within tandem reaction sequences. Han and coworkers showed that 2-aminobenzyl amines and alcohols could undergo in situ condensation with aldehydes, followed by oxidation of the secondary amine(s) to afford the corresponding quinazoline and 4H-3,1-benzoxazine heterocycles (Scheme 28).[58] The catalyst system was adapted from an alcohol oxidation catalyst developed earlier by Sekar and coworkers.[22h] The second oxidation step involved in the conversion of dihydroquinazoline to quinazoline is fast, and it may not require catalyst.
Scheme 28.

Condensation/amine oxidation sequence for the synthesis of quinazolines and 4H-3,1-benzoxazines, developed by Han.[58]
Kanai and coworkers showed that imines arising from Cu/ketoABNO-catalyzed oxidation of secondary amines are effective substrates for subsequent, in situ reactions.[56] Demonstrated examples include sequential Grignard addition and tandem Friedel-Crafts addition, aza-Diels-Alder cycloaddition, and Strecker reaction (Scheme 29). Perhaps the most noteworthy demonstration involves tandem amine oxidation/nitroalkane addition with a Cu catalyst system employing a chiral bisoxazoline ligand, (–)-Ph-Box (Scheme 30). The resulting product is obtained in excellent diastereo- and enantioselectivity. Collectively, these reactions represent an impressive class of “cross-dehydrogenative coupling” reactions.[59]
Scheme 29.

Tandem amine oxidation/addition reactions developed by Kanai.[56]
Scheme 30.
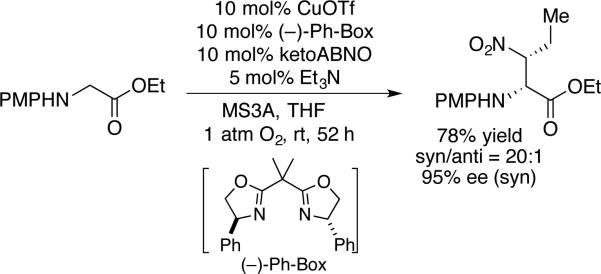
Amine oxidation followed by enantioselective aza-Henry reaction employing a single chiral CuI complex.
6. Oxidative Coupling Reactions of Alcohols and Amines
Alcohols are typically more readily available starting materials than amines. Recent studies by a number of groups have shown that Cu/nitroxyl catalyst systems enable efficient formation of imines, nitriles, and nitrogen heterocycles via aerobic oxidative coupling of alcohols and amines. Such reactions exploit the apparent chemoselectivity of Cu/nitroxyl for alcohol over amine oxidation (cf., Section 4, above).
Xu and coworkers reported an efficent method for preparation of imines from primary alcohols and amines at room temperature under ambient air (Scheme 31).[60] Benzyl, cinnamyl and phenylpropargyl alcohols were especially effective coupling partners with a range of aliphatic and benzylic amine and aniline derivatives. Two aliphatic alcohols were shown to react effectively, although isolation of the resulting imine complicated yield determination. Ideally, labile the products of this type would not need to be isolated, and future work focused on in situ transformations of the products could significantly enhance the synthetic utility. 2-Pyridylmethanol is a challenging substrate for Cu/TEMPO alcohol oxidation to the aldehyde (cf. Scheme 10),[31] but it undergoes efficient oxidative coupling under the reaction conditions of Scheme 31. This observation may suggest that the amine substrate minimized the inhibitory effect of the potentially chelating pyridyl substituent.
Scheme 31.

Oxidative coupling primary alcohols with amines to generate imines, developed by Xu.[60]
The oxidative coupling of alcohols (or aldehydes) and ammonia provides an efficient route to nitriles. In 1966, Gaasbeek et al. noted that (phen)Cu/di-tert-butylnitroxl converts methanol and ammonia to HCN,[20] but they did not explore this reactivity with other alcohols. In 1989, Capdevielle and coworkers reported a Cucatalyzed aerobic oxidation method for conversion of aromatic aldehydes and ammonia to nitriles, in the absence of a nitroxyl cocatalyst.[61,62] In 2013, the groups of Tao,[63] Huang,[64] Stahl[57] and Muldoon[65] independently reported catalytic methods for the conversion of primary alcohols to nitriles, by employing Cu/TEMPO catalyst systems in the presence of aqueous ammonia. The method reported by Huang and coworkers is the most versatile, enabling conversion of both activated and aliphatic alcohols to nitriles (Scheme 32). Like other Cu/nitroxyl oxidation methods, it shows broad functional group compatibility and often may be carried out at room temperature. Aromatic nitriles are especially versatile intermediates, and several benzonitrile derivatives were converted further, without isolation, into an aryl tetrazole, imidazoline, oxazoline, thiazoline, and triazolopyridine by traditional protocols.[64]
Scheme 32.

Preparation of aliphatic, aromatic and allylic nitriles from 1° alcohols, developed by Huang.[64]
The method of Tao employed a different Cu source and solvent (Cu(NO3)2 and DMSO) at 80 °C, and converted a wide range of benzylic and allylic alcohols and aldehydes to the corresponding nitriles. Stahl used conditions similar to those of Tao, but reported only a limited number of benzylic alcohols. Muldoon observed that NaOH accelerated the reactions and identified conditions for conversion of aryl aldehydes to nitriles with air as the source of O2. At elevated pressures of dilute O2 (8% O2 in N2), it was possible to lower the catalyst loading to 1 mol %.[65]
Intra- and intrermolecular oxidative coupling reactions of alcohols and amines can be used to prepare unsaturated heterocycles. Muldoon and coworkers reported one of the first applications of this concept in the oxidation of hydroxylalkyl-substituted anilines, leading to the formation of indoles and quinolines (Scheme 33).[66] An indole bearing an unprotected 2° alcohol substituent could be prepared, highlighting the low reativity of 2° alcohols under Cu/TEMPO oxidation conditions (cf. Schemes 10 and 11).
Scheme 33.

Alcohol oxidation/amine condensation to afford indoles and quinolines, reported by Muldoon.[66]
Facile oxidation of benzylic alcohols to benzaldehydes provided the basis for additional oxidative coupling routes to aromatic heterocycles (Schemes 34 and 35). The reaction of phenylene diamine with aromatic alcohols was developed by Lu and coworkers under solvent-free conditions with CuCl, bpy, and TEMPO to afford 2-arylbenzimidazoles (Scheme 12).[67] The method was also adapted for the preparation of benzoxazoles and benzothiazole products. And, finally, Wu and coworkers prepared quinazolines via a three-component oxidative coupling of 2-aminobenzyl alcohol, aryl aldehyde and ammonium chloride reagents (Scheme 35).[68] The reaction proceeds via formation of the benzaldehyde followed by condensation of the three components and oxidation of the heterocyclic ring.
Scheme 34.

Synthesis of benzimidazoles, benzoxazoles and benzothiazoles via oxidative coupling of aromatic alcohols with ortho-amino anilines, phenols, and thiophenols, reported by Lu.[64]
Scheme 35.

Three-component synthesis of quinazolines from 2-aminobenzyl alcohols, an aryl aldehyde and ammonium chloride, reported by Wu.[68]
7. Summary and Outlook
The development of aerobic oxidation reactions for chemical synthesis has been a long-standing goal in organic chemistry, and practical methods, suitable for widespread adoption by laboratory researchers have been elusive. Cu/TEMPO and related catalyst systems described here exhibit a level of practicality that could result in widespread adoption by laboratory researchers. Their scope, selectivity, efficiency and synthetic predictability rival or exceed that of many conventional oxidation methods commonly used in synthetic organic chemistry. The use of ambient air as the oxidant, room temperature reaction conditions, and common organic solvents, such as acetonitrile, add a level of convenience that removes key barriers associated with lab-scale use of aerobic oxidation reactions. Furthermore, the fast rates and readily available reagents make these reactions ideal targets for large-scale applications. Future work focused on the identification of lower-cost catalyst components and new catalysts effective at lower catalyst loading will further enhance large-scale opportunities.
The intimate coupling of CuI/CuII and TEMPOH/TEMPO redox processes illuminates a mechanistic paradigm similar to Galactose Oxidase whereby two one-electron redox partners mediate efficient two-electron oxidation of an organic molecule. Such pathways represent a powerful strategy for the use of first-row transition metals as alternatives to commonly used noble-metal catalysts. Nevertheless, several aspects of the mechanism remain unresolved (cf. Section 3), and future studies will be needed to gain further insights that can guide the development of new catalyst systems.
The synthetic scope and utility of these catalyst systems has only begun to be realized. The majority of the work thus far has focused simple dehydrogenation reactions, such as the oxidation of alcohols to carbonyl compounds and amines to imine and nitriles. The reactions highlighted in Sections 2.3, 5.2 and 6 demonstrate that that these catalyst systems are readily implemented in tandem and sequential, one-pot transformations that provide efficient routes to substituted heterocycles and other molecules from simple precursors. Reactions of this type hold significant promise for use in medicinal chemistry and applications where rapid access to diverse structure types is sought. Further discovery and creative implementation of multi-component/tandem reactions employing catalyst systems of the type described here are inevitable and will be facilitated by the development of improved catalyst systems.
Scheme 5.
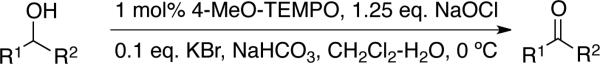
Commonly used TEMPO/bleach “Anelli” oxidation conditions for conversion of alcohols to carbonyl compounds.
Scheme 16.

Continuous flow method for Cu/TEMPO-catalyzed aerobic alcohol oxidation.[42]
Acknowledgments
We thank Kelsey Miles, Janelle Steves and Dr. Jinho Kim for editorial assistance and helpful feedback on this manuscript. We are grateful for a number of organizations that provided financial support of our work on Cu/nitroxyl-catalyzed aerobic oxidation. The work was initiated with support from the Camille and Henry Dreyfus Foundation (Environmental Chemistry Postdoctoral Fellowship for Dr. Jessica Hoover), the National Institutes of Health (RC1-GM091161 and NIGMS T32 GM008505), and the American Chemical Society Green Chemistry Institute - Pharmaceutical Roundtable. Recent synthetic studies in our lab have been supported by a consortium of pharmaceutical companies (Eli Lilly, Pfizer, and Merck) and mechanistic studies by the Department of Energy (DE-FG02-05ER15690).
Biography
 Bradford L. Ryland received his B.A. from Columbia University in 2008, where he did research in the group of Prof. Jack Norton. He carried out his graduate work in the lab of in Prof. Shannon Stahl, studying the mechanism of homogeneous copper-catalyzed aerobic oxidation reactions. He obtained his PhD in October 2013 and is currently employed at AkzoNobel in Brewster, NY (USA).
Bradford L. Ryland received his B.A. from Columbia University in 2008, where he did research in the group of Prof. Jack Norton. He carried out his graduate work in the lab of in Prof. Shannon Stahl, studying the mechanism of homogeneous copper-catalyzed aerobic oxidation reactions. He obtained his PhD in October 2013 and is currently employed at AkzoNobel in Brewster, NY (USA).
 Shannon Stahl is a Professor of Chemistry at the University of Wisconsin-Madison, where his research group specializes in the characterization and development of aerobic oxidation reactions. He obtained his B.S. in 1992 from the University of Illinois at Urbana-Champaign, and his PhD in 1997 from Caltech, where he worked with Prof. John Bercaw. He was a postdoctoral fellow with Prof. Stephen Lippard at MIT from 1997-1999.
Shannon Stahl is a Professor of Chemistry at the University of Wisconsin-Madison, where his research group specializes in the characterization and development of aerobic oxidation reactions. He obtained his B.S. in 1992 from the University of Illinois at Urbana-Champaign, and his PhD in 1997 from Caltech, where he worked with Prof. John Bercaw. He was a postdoctoral fellow with Prof. Stephen Lippard at MIT from 1997-1999.
References
- 1.a Tojo G, Fernández M. In: Oxidation of Alcohols to Aldehydes and Ketones. Tojo G, editor. Springer; New York: 2010. [Google Scholar]; b Tojo G, Fernández M. In: Oxidation of Primary Alcohols to Carboxylic Acids. Tojo G, editor. Springer; New York: 2010. [Google Scholar]
- 2.For reviews of aerobic alcohol oxidation, see Arends IWCE, Sheldon RA. In: Modern Oxidation Methods. Bäckvall J-E, editor. Wiley-VCH Verlag Gmb & Co.; Weinheim: 2004. pp. 83–118. Mallat T, Baiker A. Chem. Rev. 2004;104:3037–3058. doi: 10.1021/cr0200116. Markó IE, Giles PR, Tsukazaki M, Chellé-Regnaut I, Gautier A, Dumeunier R, Philippart F, Doda K, Mutonkole J-L, Brown SM, Urch CJ. Adv. Inorg. Chem. 2004;56:211–240. Zhan B-Z, Thompson A. Tetrahedron. 2004;60:2917–2935. Schultz MJ, Sigman MS. Tetrahedron. 2006;62:8227–8241. Matsumoto T, Ueno M, Wang N, Kobayashi S. Chem.–Asian J. 2008;3:196–214. doi: 10.1002/asia.200700359. Parmeggiani C, Cardona F. Green Chem. 2012;14:547–564.
- 3.For a recent comprehensive review of copper-catalyzed aerobic oxidation reactions in organic chemistry, see: Allen SE, Walvoord RR, Padilla-Salinas R, Kozlowski MC. Chem. Rev. 2013;113:6234–6458. doi: 10.1021/cr300527g.
- 4.For leading references, see: Jallabert C, Lapinte C, Riviere H. J. Mol. Catal. 1982;14:75–86. Liu X, Qiu A, Sawyer DT. J. Am. Chem. Soc. 1993;115:3239–3243. Capdevielle P, Sparfel D, Baranne-Lafont J, Cuong NK, Maumy M. J. Chem. Res. (S) 1993:10–11.
- 5.For reviews, including those discussing applications of nitroxyls in catalytic alcohol oxidation, see Rozantsev EG, Sholle VD. Synthesis. 1971:401–414. Sheldon RA, Arends IWCE. Adv. Synth. Catal. 2004;346:1051–1071. Vogler T, Studer A. Synthesis. 2008:1979–1993. Karoui H, Moigne FL, Ouari O, Tordo P. In: Stable Radicals. Hicks RG, editor. John Wiley & Sons, Ltd; 2010. pp. 173–229. Tebben L, Studer A. Angew. Chem. 2011;123:5138–5174. Angew. Chem. Int. Ed. 2011;50:5034–5068. doi: 10.1002/anie.201002547. Wertz S, Studer A. Green Chem. 2013;15:3116–3134. Cao Q, Dornan LM, Rogan L, Hughes NL, Muldoon MJ. Chem. Commun. 2014;50:4524–4543. doi: 10.1039/c3cc47081d.
- 6.For leading references, see Yamaguchi M, Takata T, Endo T. Tetrahedron Lett. 1988;29:5671–5672. Yamaguchi M, Miyazawa T, Takata T, Endo T. Pure Appl. Chem. 1990;2:217–222. de Nooy AEJ, Besemer AC, van Bekkum H. Synthesis. 1996:1153–1174. Bobbitt JM. J. Org. Chem. 1998;63:9367–9374. Merbouh N, Bobbitt JM, Brückner C. J. Org. Chem. 2004;69:5116–5119. doi: 10.1021/jo049461j.
- 7.Anelli PL, Biffi C, Montanari F, Quici S. J. Org. Chem. 1987;52:2559–2562. [Google Scholar]
- 8.a Shibuya M, Tomizawa M, Suzuki I, Iwabuchi Y. J. Am. Chem. Soc. 2006;128:8412–8413. doi: 10.1021/ja0620336. [DOI] [PubMed] [Google Scholar]; b Tomizawa M, Shibuya M, Iwabuchi Y. Org. Lett. 2009;11:1829–1831. doi: 10.1021/ol900441f. [DOI] [PubMed] [Google Scholar]; c Shibuya M, Tomizawa M, Sasano Y, Iwabuchi Y. J. Org. Chem. 2009;74:4619–4622. doi: 10.1021/jo900486w. [DOI] [PubMed] [Google Scholar]; d Hayashi M, Shibuya M, Iwabuchi Y. J. Org. Chem. 2012;77:3005–3009. doi: 10.1021/jo300088b. [DOI] [PubMed] [Google Scholar]; e Shibuya M, Doi R. s., Shibuta T, Uesugi S.-i., Iwabuchi Y. Org. Lett. 2012;14:5006–5009. doi: 10.1021/ol3021429. [DOI] [PubMed] [Google Scholar]
- 9.See the following and ref. 12: Graetz B, Rychnovsky S, Leu W-H, Farmer P, Lin R. Tetrahedron: Asymmetry. 2005;16:3584–3598. Demizu Y, Shiigi H, Oda T, Matsumura Y, Onomura O. Tetrahedron Lett. 2008;49:48–52.
- 10.Bobbitt JM, Brückner C, Merbouh N. Org. Reactions. 2010;74:103–424. [Google Scholar]
- 11.a Liu R, Liang X, Dong C, Hu, X. X. J. Am. Chem. Soc. 2004;126:4112–4113. doi: 10.1021/ja031765k. [DOI] [PubMed] [Google Scholar]; b Liu R, Dong C, Liang X, Wang X, Hu X. J. Org. Chem. 2005;70:729–731. doi: 10.1021/jo048369k. [DOI] [PubMed] [Google Scholar]; c Xie Y, Mo W, Xu D, Shen Z, Sun N, Hu B, Hu X. J. Org. Chem. 2007;72:4288–4291. doi: 10.1021/jo0705824. [DOI] [PubMed] [Google Scholar]; d Karimi B, Biglari A, Clark JH, Budarin V. Angew. Chem. 2007;119:7348–7351. doi: 10.1002/anie.200701918. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:7210–7213. doi: 10.1002/anie.200701918. [DOI] [PubMed] [Google Scholar]; e Wang X, Liu R, Jin Y, Liang X. Chem. Eur. J. 2008;14:2679–2685. doi: 10.1002/chem.200701818. [DOI] [PubMed] [Google Scholar]; f He X, Shen Z, Mo W, Sun N, Hu B, Hu X. Adv. Synth. Catal. 2009;351:89–92. [Google Scholar]; g Miao C-X, He L-N, Wang J-Q, Wang J-L. Adv. Synth. Catal. 2009;351:2209–2216. [Google Scholar]; h Miao C-X, He L, Wang J, Wu F. J. Org. Chem. 2010;75:257–260. doi: 10.1021/jo902292t. [DOI] [PubMed] [Google Scholar]; i Kuang Y, Rokubuichi H, Nabae Y, Hayakawa T, Kakimoto M-A. Adv. Synth. Catal. 2010;352:2635–2642. [Google Scholar]; j Qiu C, Jin L, Huang Z, Tang Z, Lei A, Shen Z, Sun N, Mo W, Hu B, Hu X. ChemCatChem. 2012;4:76–80. [Google Scholar]; k Rahimi A, Azarpira A, Kim H, Ralph J, Stahl SS. J. Am. Chem. Soc. 2013;135:6415–6418. doi: 10.1021/ja401793n. [DOI] [PubMed] [Google Scholar]; l Aellig C, Scholz D, Conrad S, Hermans I. Green Chem. 2013;15:1975–1980. [Google Scholar]
- 12.See ref. 11i and the following: Shibuya M, Osada Y, Sasano Y, Tomizawa M, Iwabuchi Y. J. Am. Chem. Soc. 2011;133:6497–6500. doi: 10.1021/ja110940c. Lauber M, Stahl SS. ACS Catal. 2013;3:2612–2616.
- 13.Cecchetto A, Fontana F, Minisci F, Recupero F. Tetrahedron Lett. 2001;42:6651–6653. [Google Scholar]
- 14.a Wang N, Liu R, Chen J, Liang X. Chem. Commun. 2005:5322–5324. doi: 10.1039/b509167e. [DOI] [PubMed] [Google Scholar]; b Wang X, Liang X. Chin. J. Catal. 2008;29:935–939. [Google Scholar]; c Ma S, Liu J, Li S, Chen B, Cheng J, Kuang J, Liu Y, Wan B, Wang Y, Ye J, Yu Q, Yuan W, Yu S. Adv. Synth. Catal. 2011;353:1005–1017. [Google Scholar]; d Miao C-X, Wang J-Q, Yu B, Cheng W-G, Sun J, Chanfreau S, He L-N, Zhang S-J. Chem. Commun. 2011;47:2697–2699. doi: 10.1039/c0cc04644b. [DOI] [PubMed] [Google Scholar]; e Wang L, Li J, Lv Y, Zhao G, Gao S. Appl. Organomet. Chem. 2012;26:37–43. [Google Scholar]
- 15.a Jing Y, Jiang J, Yan B, Lu S, Jiao J, Xue H, Yang G, Zheng G. Adv. Synth. Catal. 2011;353:1146–1152. [Google Scholar]; b Guan Y, Zhou G, Yang W. Heterocyclic Commun. 2014;20:11–13. [Google Scholar]
- 16.Yan Y, Tong X, Wang K, Bai X. Catal. Commun. 2014;43:112–115. [Google Scholar]
- 17.Ben-Daniel R, Alsters P, Neumann R. J. Org. Chem. 2001;66:8650–8653. doi: 10.1021/jo0105843. [DOI] [PubMed] [Google Scholar]
- 18.a Fabbrini M, Galli C, Gentili P, Macchitella D. Tetrahedron Lett. 2001;42:7551–7553. [Google Scholar]; b d'Acunzo F, Galli C, Masci B. Eur. J. Biochem. 2002;269:5330–5335. doi: 10.1046/j.1432-1033.2002.03256.x. [DOI] [PubMed] [Google Scholar]; c d'Acunzo F, Baiocco P, Fabbrini M, Galli C, Gentili P. Eur. J. Org. Chem. 2002;2002:4195–4201. [Google Scholar]; d Arends IWCE, Li Y-X, Ausan R, Sheldon RA. Tetrahedron. 2006;62:6659–6665. [Google Scholar]; e Liebminger S, Siebenhofer M, Guebitz G. Bioresour. Technol. 2009;100:4541–4545. doi: 10.1016/j.biortech.2009.04.051. [DOI] [PubMed] [Google Scholar]; f Tromp SA, Matijošytė I, Sheldon RA, Arends IWCE, Mul G, Kreutzer MT, Moulijn JA, de Vries S. ChemCatChem. 2010;2:827–833. [Google Scholar]; g Díaz-Rodríguez A, Lavandera I, Kanbak-Aksu S, Sheldon RA, Gotor V, Gotor-Fernández V. Adv. Synth. Catal. 2012;354:3405–3408. [Google Scholar]
- 19.For other mechanistic roles of TEMPO in metal-catalyzed alcohol oxidation, see: ten Brink G-J, Arends IWCE, Sheldon RA. Science. 2000;287:1636–1639. doi: 10.1126/science.287.5458.1636. Dijksman A, González AM, i Payeras AM, Arends IWCE, Sheldon RA. J. Am. Chem. Soc. 2001;123:6826–6833. doi: 10.1021/ja0103804. Matsumoto T, Ueno M, Kobayashi J, Miyamura H, Mori Y, Kobayashi S. Adv. Synth. Catal. 2007;349:531–534.
- 20.a Brackmann W, Gaasbeek C. Rec. Trav. Chim. 1966;85:221–241. [Google Scholar]; b Brackmann W, Gaasbeek C. Rec. Trav. Chim. 1966;85:242–256. [Google Scholar]; c Brackmann W, Gaasbeek C. Rec. Trav. Chim. 1966;85:257–267. [Google Scholar]
- 21.Semmelhack MF, Schmid CR, Cortes DA, Chou CS. J. Am. Chem. Soc. 1984;106:3374–3376. [Google Scholar]
- 22.a Geißlmeir D, Jary WG, Falk H. Monatsh. Chem. 2005;136:1591–1599. [Google Scholar]; b Jiang N, Ragauskas AJ. J. Org. Chem. 2006;71:7087–7090. doi: 10.1021/jo060837y. [DOI] [PubMed] [Google Scholar]; c Striegler S. Tetrahedron. 2006;62:9109–9114. [Google Scholar]; d Jiang N, Vinci D, Liotta CL, Eckert CA, Ragauskas AJ. Ind. & Eng. Chem. Res. 2007;47:627–631. [Google Scholar]; e Guo Y, Zhao J, Xu J, Wang W, Tian F, Yang G, Song M. J. Nat. Gas Chem. 2007;16:210–212. [Google Scholar]; f Salinas Uber J, Vogels Y, van den Helder D, Mutikainen I, Turpeinen U, Fu WT, Roubeau O, Gamez P, Reedijk J. Eur. J. Inorg. Chem. 2007:4197–4206. [Google Scholar]; g Figiel PJ, Leskelä M, Repo T. Adv. Synth. Catal. 2007;349:1173–1179. [Google Scholar]; h Mannam S, Alamsetti SK, Sekar G. Adv. Synth. Catal. 2007;349:2253–2258. [Google Scholar]; i Gassama A, Hoffmann, N. N. Adv. Synth. Catal. 2008;350:35–39. [Google Scholar]; j Lu Z, Sánchez Costa J, Roubeau O, Mutikainen I, Turpeinen U, Teat SJ, Gamez P, Reedijk J. J. Dalton Trans. 2008:3567–3573. doi: 10.1039/b802109k. [DOI] [PubMed] [Google Scholar]; k Jiang N, Ragauskas AJ. ChemSusChem. 2008;1:823–825. doi: 10.1002/cssc.200800144. [DOI] [PubMed] [Google Scholar]; l Figiel PJ, Sibaouih A, Ahmad JU, Nieger M, Räisänen MT, Leskelä M, Repo T. Adv. Synth. Catal. 2009;351:2625–2632. [Google Scholar]; m Ahmad JU, Figiel PJ, Räisänen MT, Leskelä M, Repo T. Appl. Catal., A. 2009;371:17–21. [Google Scholar]; n Lu A, Ladrak T, Roubeau O, van der Toorn J, Teat SJ, Massera C, Gamez P, Reedijk J. Dalton Trans. 2009:3559–3570. doi: 10.1039/b820554j. [DOI] [PubMed] [Google Scholar]; o Figiel PJ, Kirillov AM, Karabach YY, Kopylovich MN, Pombeiro AJL. J. Mol. Catal. A: Chem. 2009;305:178–182. [Google Scholar]; p Gartshore CJ, Lupton DW. Adv. Synth. Catal. 2010;352:3321–3328. [Google Scholar]; q Hossain MM, Shyu S-G. Adv. Synth. Catal. 2010;352:3061–3068. [Google Scholar]; r Striegler S, Dunaway NA, Gichinga MG, Milton LK. Tetrahedron. 2010;66:7927–7932. [Google Scholar]; s Wang Q, Zhang Y, Zheng G, Tian Z, Yang G. Catal. Commun. 2011;14:92–95. [Google Scholar]; t Kopylovich MN, Karabach YY, Guedes da Silva MFC, Figiel PJ, Lasri J, Pombeiro AJL. Chem. Eur. J. 2012;18:899–914. doi: 10.1002/chem.201101688. [DOI] [PubMed] [Google Scholar]; u Gamba I, Mutikainen I, Bouwman E, Reedijk J, Bonnet S. Eur. J. Inorg. Chem. 2013;2013:115–123. [Google Scholar]; v Liu X, Xia Q, Zhang Y, Chen C, Chen W. J. Org. Chem. 2013;78:8531–8536. doi: 10.1021/jo401252d. [DOI] [PubMed] [Google Scholar]
- 23.a Ansari IA, Gree R. Org. Lett. 2002;4:1507–1509. doi: 10.1021/ol025721c. [DOI] [PubMed] [Google Scholar]; b Jiang N, Ragauskas AJ. Org. Lett. 2005;7:3689–3692. doi: 10.1021/ol051293+. [DOI] [PubMed] [Google Scholar]; c Lin L, Liuyan, J. J, Yunyang W. Catal. Commun. 2008;9:1379–1382. [Google Scholar]; d Lin L, Juanjuan M, Liuyan J, Yunyang W. J. Mol. Catal. A: Chem. 2008;291:1–4. [Google Scholar]; e Liu LL, Ma JJ, Xia JY, Li LD, Li C, Zhang XB, Gong JY, Tong ZW. Catal. Commun. 2011;12:323–326. [Google Scholar]
- 24.a Betzemeier B, Cavazzini M, Quici S, Knochel P. Tetrahedron Lett. 2000;41:4343–4346. [Google Scholar]; b Ragagnin G, Betzemeier B, Quici S, Knochel P. Tetrahedron. 2002;58:3985–3991. [Google Scholar]; c Contel M, Izuel C, Laguna M, Villuendas PR, Alonso PJ, Fish RH. Chem. Eur. J. 2003;9:4168–4178. doi: 10.1002/chem.200304771. [DOI] [PubMed] [Google Scholar]; d Contel M, Villuendas PR, Fernandez-Gallardo J, Alonso PJ, Vincent J-M, Fish RH. Inorg. Chem. 2005;44:9771–9778. doi: 10.1021/ic051220m. [DOI] [PubMed] [Google Scholar]
- 25.a Dijksman A, Arends IWCE, Sheldon RA. Synlett. 2001:102–104. [Google Scholar]; b Chung CWY, Toy PH. J. Comb. Chem. 2007;9:115–120. doi: 10.1021/cc060111f. [DOI] [PubMed] [Google Scholar]
- 26.a Herbert M, Montilla F, Galindo A. Dalton Trans. 2010;39:900–907. doi: 10.1039/b914788h. [DOI] [PubMed] [Google Scholar]; b Hu Z, Kerton FM. Appl. Catal., A. 2012:413–414. 332–339. [Google Scholar]; c Zhang G, Han X, Luan Y, Wang Y, Wen X, Xu L, Ding C, Gao J. RSC Adv. 2013;3:19255–19258. [Google Scholar]
- 27.a Kopylovich MN, Mahmudov KT, Haukka M, Figiel PJ, Mizar A, da Silva JAL, Pombeiro AJL. Eur. J. Inorg. Chem. 2011;2011:4175–4181. [Google Scholar]; b Dhakshinamoorthy A, Alvaro M, Garcia, H. H. ACS Catal. 2011;1:48–53. [Google Scholar]
- 28.Velusamy S, Srinivasan A, Punniyamurthy T. Tetrahedron Lett. 2006;47:923–926. [Google Scholar]
- 29.a Gamez P, Arends IWCE, Reedijk J, Sheldon RA. Chem. Commun. 2003:2414–2415. doi: 10.1039/b308668b. [DOI] [PubMed] [Google Scholar]; b Gamez P, Arends IWCE, Sheldon RA, Reedijk, J. J. Adv. Synth. Catal. 2004;346:805–811. [Google Scholar]
- 30.Kumpulainen ETT, Koskinen AMP. Chem.–Eur. J. 2009;15:10901–10911. doi: 10.1002/chem.200901245. [DOI] [PubMed] [Google Scholar]
- 31.a Hoover JM, Stahl SS. J. Am. Chem. Soc. 2011;133:16901–16910. doi: 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hoover JM, Steves JE, Stahl SS. Nat. Protoc. 2012;7:1161–1167. doi: 10.1038/nprot.2012.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hoover JM, Stahl SS. Org. Synth. 2013;90:240–250. [Google Scholar]
- 32.This protocol has been adapted as an undergraduate teaching laboratory: Hill NJ, Hoover JM, Stahl SS. J. Chem. Ed. 2013;90:102–105.
- 33.Nonappa U. Maitra. Eur. J. Org. Chem. 2007:3331–3336. [Google Scholar]
- 34.Mehta G, Bhat BA, Kumara THS. Tetrahedron Lett. 2010;51:4069–4072. [Google Scholar]
- 35.Ho X-H, Oh H-J, Jang H-Y. Eur. J. Org. Chem. 2012:5655–5659. [Google Scholar]
- 36.Könning D, Hiller W, Chistmann M. Org. Lett. 2012;14:5258–5261. doi: 10.1021/ol302420k. [DOI] [PubMed] [Google Scholar]
- 37.Brioche J, Masson G, Zhu J. Org. Lett. 2010;12:1432–1435. doi: 10.1021/ol100012y. [DOI] [PubMed] [Google Scholar]
- 38.a Li C, Bardhan S, Pace EA, Liang M-C, Gilmore TD, Porco JA., Jr. Org. Lett. 2002;4:3267–3270. doi: 10.1021/ol026513n. [DOI] [PubMed] [Google Scholar]; b Li C, Porco JA. J. Org. Chem. 2005;70:6053–6065. doi: 10.1021/jo050897o. [DOI] [PubMed] [Google Scholar]; c Kleinke AS, Li C, Rabasso N, Porco JA., Jr. Org. Lett. 2006;8:2847–2850. doi: 10.1021/ol060954f. [DOI] [PubMed] [Google Scholar]
- 39.a Mehta G, Islam K. Org. Lett. 2004;6:807–811. doi: 10.1021/ol036521j. [DOI] [PubMed] [Google Scholar]; b Mehta G, Pan SC. Org. Lett. 2004;6:811–813. doi: 10.1021/ol0499699. [DOI] [PubMed] [Google Scholar]; c Mehta G, Roy S. Org. Lett. 2004;6:2389–2392. doi: 10.1021/ol0492288. [DOI] [PubMed] [Google Scholar]; d Mehta G, Pan SC. Org. Lett. 2004;6:3985–3988. doi: 10.1021/ol0483551. [DOI] [PubMed] [Google Scholar]; e Mehta G, Ramesh SS. Tetrahedron Lett. 2004;45:1985–1987. [Google Scholar]; f Mehta G, Islam K. Tetrahedron Lett. 2004;45:7683–7687. [Google Scholar]; g Mehta G, Pan SC. Tetrahedron Lett. 2005;46:3045–3048. [Google Scholar]; h Mehta G, Roy S. Tetrahedron Lett. 2008;49:1458–1460. [Google Scholar]; i Mehta G, Kumar YCS, Khan TB. Tetrahedron Lett. 2010;51:5112–5115. [Google Scholar]
- 40.For examples, see: Knaus GH, Paust J. Vol. 24. BASF; DE: Feb, p. 1987. DE Patent Application 3,705,785. Kobertz WR, Essigmann JM. J. Am. Chem. Soc. 1996;118:7101–7107. Ernst H, Klaus H. Vol. 12. Keil&Weinkauf; USA: Mar, p. 2002. US Patent Application 10,094,819. Kuwahara S, Imada S. Tetrahedron Lett. 2005;46:547–549. Fürstner A, Aïssa C, Chevrier C, Teplý F, Nevado C, Tremblay M. Angew. Chem. 2006;118:5964–5969. doi: 10.1002/anie.200601859. Angew. Chem. Int. Ed. 2006;45:5832–5837. doi: 10.1002/anie.200601859.
- 41.Brooks MR, Crowl DA, Loss Prev J. Process Ind. 2007;20:144–150. [Google Scholar]
- 42.Greene JF, Hoover JM, Mannel DS, Root TW, Stahl SS. Org. Process Res. Dev. 2013;17:1247–1251. [Google Scholar]
- 43.Ye X, Johnson MD, Diao T, Yates MH, Stahl SS. Green Chem. 2010;12:1180–1186. doi: 10.1039/c0gc00106f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoover JM, Ryland BL, Stahl SS. ACS Catal. 2013;3:2599–2605. doi: 10.1021/cs400689a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dijksman A, Arends IWCE, Sheldon RA. Org. Biomol. Chem. 2003;1:3232–3237. doi: 10.1039/b305941c. [DOI] [PubMed] [Google Scholar]
- 46.Whittaker JW. Chem. Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, DuBois JL, Hedman B, Hodgson KO, Stack TDP. Science. 1998;279:537–540. doi: 10.1126/science.279.5350.537. [DOI] [PubMed] [Google Scholar]
- 48.a Caneschi A, Grand A, Laugier J, Rey P, Subra R. J. Am. Chem. Soc. 1988;110:2307–2309. [Google Scholar]; b Laugier J, Latour JM, Caneschi A, Rey P. Inorg. Chem. 1991;30:4474–4477. [Google Scholar]; c Dickman MH, Doedens RJ. Inorg Chem. 1981;20:2677–2681. [Google Scholar]
- 49.a Michel C, Belanzoni P, Gamez P, Reedijk, J. J, Baerends EJ. Inorg. Chem. 2009;48:11909–11920. doi: 10.1021/ic902155m. [DOI] [PubMed] [Google Scholar]; b Cheng L, Wang J, Wang M, Wu Z. Dalton Trans. 2010;39:5377–5387. doi: 10.1039/b926098f. [DOI] [PubMed] [Google Scholar]; c Cheng L, Wang J, Wang M, Wu Z. Inorg. Chem. 2010;49:9392–9399. doi: 10.1021/ic100996b. [DOI] [PubMed] [Google Scholar]; d Belanzoni P, Michel C, Baerends EJ. Inorg. Chem. 2011;50:11896–11904. doi: 10.1021/ic200725k. [DOI] [PubMed] [Google Scholar]
- 50.Hoover JM, Ryland BL, Stahl SS. J. Am. Chem. Soc. 2013;135:2357–2367. doi: 10.1021/ja3117203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lack of interaction between CuII and TEMPO was also noted in a different catalyst system, see ref. 22u.
- 52.Steves JE, Stahl SS. J. Am. Chem. Soc. 2013;135:15742–15745. doi: 10.1021/ja409241h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasano Y, Nagasawa S, Yamazaki M, Shibuya M, Park J, Iwabuchi Y. Angew. Chem. 2014;126:1–6. doi: 10.1002/anie.201309634. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014;53:1–6. [Google Scholar]
- 54.For leading references, see: Capdevielle P, Lavigne A, Maumy M. Synthesis. 1989;1989:453–454. Maeda Y, Nishimura T, Uemura S. Bull. Chem. Soc. Jpn. 2003;76:2399–2403. Landge SM, Atanassova V, Thimmaiah M, Török B. Tetrahedron Lett. 2007;48:5161–5164. Grirrane A, Corma A, Garcia H. J. Catal. 2009;264:138–144. Zhang C, Jiao N. Angew. Chem. 2010;122:6310–6313. Angew. Chem. Int. Ed. 2010;49:6174–6177. doi: 10.1002/anie.201001651. Patila RD, Adimurthy S. Adv. Synth. Catal. 2011;353:1695–1700.
- 55.Hu Z, Kerton FM. Org. Biomol. Chem. 2012;10:1618–1624. doi: 10.1039/c2ob06670j. [DOI] [PubMed] [Google Scholar]
- 56.Sonobe T, Oisaki K, Kanai M. Chem. Sci. 2012;3:3249–3255. [Google Scholar]
- 57.Kim J, Stahl SS. ACS Catal. 2013;3:1652–1656. doi: 10.1021/cs400360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han B, Yang X-L, Wang C, Bai Y-W, C Pan T-, Chen X, Yu W. J. Org. Chem. 2012;77:1136–1142. doi: 10.1021/jo2020399. [DOI] [PubMed] [Google Scholar]
- 59.For an introduction to “cross-dehydrogenative coupling”, see: Li C-J. Acc. Chem. Res. 2008;42:335–344. doi: 10.1021/ar800164n.
- 60.Tian H, Yu X, Li Q, Wang J, Xu Q. Adv. Synth. Catal. 2012;354:2671–2677. [Google Scholar]
- 61.Capdevielle P, Lavigne A, Maumy M. Synthesis. 1989:451–452. [Google Scholar]
- 62.For other catalysts systems that promote the oxidative coupling of alcohols and ammonia to nitriles, see: Mori K, Yamaguchi K, Mizugaki T, Ebitani K, Kaneda K. Chem. Commun. 2001:461–462. Oishi T, Yamaguchi K, Mizuno N. Angew. Chem. 2009;121:6404–6406. doi: 10.1002/anie.200900418. Angew. Chem. Int. Ed. 2009;48:6286–6288. doi: 10.1002/anie.200900418. Oishi T, Yamaguchi K, Mizuno N. Top. Catal. 2010;53:479–486. Ishida T, Watanabe H, Takei T, Hamasaki A, Tokunaga M, Haruta M. Applied Catalysis A: General. 2012:425–426. 85–90. Taketoshi A, Beh XN, Kuwabara J, Koizumi T.-a., Kanbara T. Tetrahedron Lett. 2012;53:3573–3576.
- 63.Tao C, Liu F, Zhu Y, Liu W, Cao Z. Org. Biomol. Chem. 2013;11:3349–3354. doi: 10.1039/c3ob00002h. [DOI] [PubMed] [Google Scholar]
- 64.Yin W, Wang C, Huang Y. Org. Lett. 2013;15:1850–1853. doi: 10.1021/ol400459y. [DOI] [PubMed] [Google Scholar]
- 65.Dornan LM, Cao Q, Flanagan JCA, Cook MJ, Crawford MJ, Muldoon MJ. Chem. Commun. 2013:6030–6032. doi: 10.1039/c3cc42231c. [DOI] [PubMed] [Google Scholar]
- 66.Flanagan JCA, Dornan LM, McLaughlin MG, McCreanor NG, Cook MJ, Muldoon MJ. Green Chem. 2012;14:1281–1283. [Google Scholar]
- 67.Yu J, Xu J, Lu M. Appl. Organomet. Chem. 2013;27:606–610. [Google Scholar]
- 68.Chen Z, Chen J, Liu M, Ding J, Gao W, Huang X, Wu H. J. Org. Chem. 2013;78:11342–11348. doi: 10.1021/jo401908g. [DOI] [PubMed] [Google Scholar]