

Abstract
Previously we have developed and statistically validated Quantitative Structure Property Relationship (QSPR) models that correlate drugs’ structural, physical and chemical properties as well as experimental conditions with the relative efficiency of remote loading of drugs into liposomes (Cern et al, Journal of Controlled Release, 160(2012) 14–157). Herein, these models have been used to virtually screen a large drug database to identify novel candidate molecules for liposomal drug delivery. Computational hits were considered for experimental validation based on their predicted remote loading efficiency as well as additional considerations such as availability, recommended dose and relevance to the disease. Three compounds were selected for experimental testing which were confirmed to be correctly classified by our previously reported QSPR models developed with Iterative Stochastic Elimination (ISE) and k-nearest neighbors (kNN) approaches. In addition, 10 new molecules with known liposome remote loading efficiency that were not used in QSPR model development were identified in the published literature and employed as an additional model validation set. The external accuracy of the models was found to be as high as 82% or 92%, depending on the model. This study presents the first successful application of QSPR models for the computer-model-driven design of liposomal drugs.
Keywords: Liposomes; Remote loading; QSPR; Virtual screening; Iterative Stochastic Elimination, k-nearest neighbors
1. Introduction
Liposomes present an effective drug delivery system (DDS): there are already more than 10 FDA-approved liposomal drugs [1,2] and many more preparations are in clinical trials [1]. A drug should fulfill several crucial conditions in order to become a successful liposomal product: (1) The concentration of the drug in the liposomes which is described as the drug to lipid (D/L) mole ratio should be high enough for administering a therapeutic dose. (2) The liposomal drug product should be stable at least at refrigerator temperature (4°C). Product stability includes all parameters related to chemical and physical stability, which includes minimal drug leakage, stability at higher temperature will be an advantage but not a must (3) Drug release in blood in vivo should occur slowly, enabling distribution of most of the liposomal drug to the target site.
For the purpose of parenteral administration, nano-size liposomes are mainly used [3,4]. Nano-size (<100 nm) liposomes are important because they enable passive targeting by the enhanced permeability and retention (EPR) effect. In addition, the nano-volume confers the liposomes with unique properties of highly efficient and stable drug loading as well as a controlled release profile. However, due to the very small internal volume, sufficient passive drug entrapment cannot usually be achieved [5]. The approach of remote loading was developed to overcome this obstacle and to achieve high drug concentrations in nano-liposomes [6–8]. This approach uses an ion gradient as the driving force for getting drugs into preformed liposomes to enable potentially high loading efficiency and good stability of the liposomal drug. Remote loading applies only to molecules that can accumulate in the internal aqueous phase of the liposome due to an ion or pH transmembrane gradient. Suitable candidates are amphipathic weak acids or weak bases which are defined by their logD at pH 7 in the range of −2.5 to 2. Amphipathic weak bases should have a pKa ≤11 and weak acids should have pKa >3 [8]. Drug molecules that are too hydrophobic associate mainly with the lipid bilayer and will not be good candidates for remote loading [5]. On the other hand, molecules which are not amphipathic enough will not be remote loaded, as they will not be able to diffuse across the liposome lipid bilayer. Basic or acidic drug molecules suitable for remote loading can achieve equilibrium between the neutral, uncharged state, when a molecule can easily diffuse across the liposome’s membrane, and a charged state, which in most cases prevents transport through the membrane. It is important to note that the success of this nanochemical engine is also due to the very small trapped aqueous volume of nanoliposomes, which supports faster and higher accumulation and intraliposome precipitation of a drug-counterion salt in crystalline or non-crystalline form. Amphipathic weak acids and bases can be effectively remote loaded to liposomes. However, the D/L ratios that will be loaded may be too low for administrating the therapeutic dose. For many drugs the therapeutic dose is relatively high (e.g. doxorubicin 50 mg/m2, [9]) that requires high D/L ratios in the formulation. In addition the formulation should maintain D/L ratio during storage (namely minimal drug leakage); while in vivo the release of drugs from the liposomes in the circulation should be low while in the diseased tissue it should not be too slow neither too fast [5].
Liposomal formulation development requires considerable time and effort calling for the development of computational modeling approach capable of predicting whether a drug is a good candidate for this DDS. To this end, recently we have begun to explore the utility of Quantitative Structure Property Relationships (QSPR) modeling as a computational tool to identify and prioritize drugs suitable for remote loading that satisfies the first and crucial condition for a good remote loading molecule, i.e., a high intra-liposomal drug concentration.
The first study to establish a correlation between drug structural properties and experimental conditions with remote liposome loading efficiency employed a decision tree method [8]; this model was built using data from the Barenholz laboratory. Additional data was generated recently for a larger set of drug molecules, and the enlarged data set was employed to develop novel QSPR models for in silico prediction of drug suitability for remote loading [10]. In formulating the objectives of the QSPR studies we took into account that the requirement of being amphipathic weak acids or bases is insufficient since some of such molecules could achieve high loading efficiency only at low D/L ratios, whereas other such compounds can be efficiently loaded at higher D/L ratios. This important observation suggests that other structural features, as well as experimental conditions, influence the D/L ratio that affords high loading efficiency. Thus, the feasibility for a drug to achieve high loading efficiency at relatively high initial D/L ratio used in the preparation was chosen as the target property to be predicted by QSPR models, because this ratio determines whether a therapeutically effective dose could be administered using liposomal drug formulations. Taking into consideration that the amount of lipids allowed to be administered to a patient per day is limited, and that the D/L determines the final drug concentration in the product, and as a result the infusion volume, the D/L ratio in the preparation determines the maximum dose that can be administered. Therefore, from a therapeutic standpoint we are ultimately interested in achieving high loading efficiency at relatively high initial D/L ratios. In our studies, the threshold of this initial D/L ratio was chosen to be 0.3, i.e., drugs that were capable of achieving high loading efficiency (greater than 70%) at D/L ratio of 0.3 or above were considered as “positive” candidates, whereas those that showed low loading efficiency above this threshold (even if they had high loading efficiency at D/L ratio below this threshold) were regarded as “negative” candidates.
The QSPR models to predict the initial D/L ratio were built using structural descriptors of drug molecules combined with experimental conditions, forming a joint set of compound characteristics as described in our previous publication [10]. Several machine learning techniques were employed to generate both the binary classification models (predicting “high” vs. “low” loading efficiency at D/L ratio ≥ 0.3) and continuous models (predicting the D/L ratio that enables high loading efficiency) ; these models were statistically validated using the 5-fold external validation technique discussed in detail previously [10].
In the present study we have used these previously developed QSPR models [10] for virtual screening of the Comprehensive Medicinal Chemistry (CMC) database containing in excess of 8000 molecules. Two of the earlier classification models were considered suitable for virtual screening for candidate compound selection; these models predicting D/L ratio from drug structural descriptors and experimental conditions were built using methods of Iterative Stochastic Elimination (ISE) and k-Nearest Neighbors (kNN), as discussed in our previous publication [10]. Three molecules emerging from virtual screening were prioritized for a proof-of-concept experimental validation in this study. Two of the selected drugs were predicted to be “positives” (i.e., predicted by both ISE and kNN models to have high loading efficiency at D/L ≥0.3) and one drug was predicted as “negative” (predicted by both models to have low loading efficiency at D/L ≥0.3). Each of the tested molecules was loaded to PEGylated liposomes by remote loading and analyzed for its loading efficiency at several D/L ratios using HPLC methods. The experiments showed that all three compounds were indeed correctly classified by the models. Furthermore, we have identified in the recently published literature [11–20] the remote loadings data for 10 additional molecules that were not employed for model development in our previous study [10]. Nine of the drugs were reported as negatives and one was positive (using the same criteria as above). These 10 molecules were employed for additional validations of our models and the overall prediction accuracy for the external data set including both the three molecules tested by us and those reported in the literature was 92.3% and 81.8% for models built with ISE and kNN, respectively.
The vast amount of new data reported during 2011–2012 emphasizes the importance of the remote loading method in the field of liposomal drug delivery. Additionally, finding only one drug with positive remote loading out of the 10 in recent literature emphasizes the difficulty of finding good candidates for remote loading into liposomes. Our studies confirm that QSPR models developed by our team are applicable for virtual database screening to identify candidates with high drug liposomal concentration achieved by the remote loading method. The models also predict correctly those molecules that are not good candidates for remote loading, i.e., the “true negatives”. These observations underscore the power of the QSPR modeling method both to identify true positives and to reduce the number of false positives, saving experimental time, cost, and efforts.
2. Materials and methods
2.1. Computational methods
2.1.1. Database to select candidate drugs for virtual screening
The CMC database includes more than 8000 molecules that are used or have been studied as medicinal agents in humans, pharmacological agents or biologically active compounds. This database was analyzed using the liposome remote loading models generated previously and described in Section 2.1.2. The models employed both structural/property descriptors (typical for any cheminformatics investigation) as well as experimental conditions (regarded effectively as additional experimental “descriptors” of compounds) for building models. The experimental conditions entered for all molecules included the liposome composition of hydrogenated soy phospholipid (HSPC): cholesterol: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (mPEG DSPE) 55:40:5 mole ratio, loading duration of 10 min at 65°C (above the Tm of the selected phospholipid) and 200 mM concentration of buffer used for gradient creation. Structural/property descriptors for each molecule were calculated using Molecular Operating Environment (MOE) software [21].
2.1.2. QSPR models
Two types of QSPR models to predict loading efficiency were built [10]: binary classification (category) and continuous models. Binary classification models were built using Iterative Stochastic Elimination (ISE), k-Nearest Neighbors (kNN) and Support Vector Machines (SVM). Continuous models were built using kNN and Support Vector Regression (SVR) (for additional details see ref [10]).
The binary classification models defined molecules as being “positive” when high remote loading efficiency (≥70%) was achieved at D/L ratio ≥0.3. “Negatives” were those that showed low loading efficiency above this threshold (even if they had high loading efficiency at D/L ratio below this threshold). It should be stated that for both ISE and kNN, binary classification models were built such that each compound was predicted either as “negative” (class “0” for kNN or class “−1” for ISE) or “positive” (class 1 for both ISE and kNN). Furthermore, both methods generate an ensemble of binary classification models where individual models could disagree in the assigned category for a compound. In both cases, the molecule category is calculated by averaging predictions made by contributing models such that the predicted category could be a value between “−1” and “+1” for ISE or between “0” and “1” for kNN. For this reason, a quantitative “index” of remote loading efficiency for each molecule is produced, that enables relative scoring and prioritization of compounds. For more information regarding these computational methods see Supplementary Information. As reported previously [10], the accuracy of models estimated by the means of 5-fold external cross-validation was as high as 93% for both ISE and kNN. This high accuracy enabled application of these models for virtual screening of external databases such as CMC.
2.2. Experimental materials
Mupirocin (Teva) was a gift from Foamix Ltd (Israel); pravastatin sodium was a gift from CCSB (Taiwan); piroxicam and Dowex 1X8–200 were obtained from Sigma Aldrich; HSPC, mPEG DSPE and cholesterol were obtained from Lipoid, Ludwigshafen, Germany. The solvents used for analysis were HPLC grade. All other chemicals were commercial products of reagent grade.
2.3. Experimental methods
2.3.1. Liposomes preparation
Liposomes were prepared using the calcium acetate gradient method [7]. Lipids in a mole ratio of 55:40:5 HSPC: cholesterol: mPEG DSPE were mechanically hydrated by 200 mM calcium acetate, pH 5.5, at a weight ratio of 1:9. A 65°C calcium acetate solution was used. Liposomal dispersion was downsized by stepwise extrusion by the Northern Lipids, (Burnaby, BC, Canada) extruder using polycarbonate filters starting with 3 times extrusion through a 400 nm pore size membrane, then 3 times through a 100 nm pore size membrane, and finally 10 times through a 50 nm pore size membrane. Liposomes were then dialyzed using a Cellu Sep regenerated cellulose membrane (Membrane Filtration Products, Inc. USA), against a 10% sucrose solution. Remote loading was performed by incubating at 65°C for 10 min a solution of the drug with the above liposome dispersion at a volumetric ratio of 1:1. Liposomes used for loading were freshly prepared and used within one week. Phospholipid concentration in liposome dispersion was in the range of 42–70 mM. Mupirocin was remote loaded from propylene glycol in 200 mM sodium phosphate buffer pH 6.3. Pravastatin was remote loaded from 10% sucrose solution, and piroxicam was remote loaded from polyethylene glycol 400 in 200 mM sodium phosphate buffer pH 6.3.
2.3.2. Analytical methods
2.3.2.1. Determination of phospholipid concentration
Phospholipid concentration was determined as organic phosphorus by the modified Bartlett method [22].
2.3.2.2. Determination of drug concentration
Drug concentrations were quantified using HPLC/UV methods (HPLC system, Hewlett-Packard Series II 1090). The column used for all three drugs tested was Waters, XBridge C18 5µm 4.6×150 column. The chromatographic conditions were based on published methods [23–25]. Total (free plus liposomal) drug concentration was determined by HPLC assay of the liposomal dispersion diluted with methanol. Liposomal drug concentration was determined after removing the free drug by mixing the dispersion with Dowex 1X8–200 (Sigma Aldrich) anion exchanger, which binds the free drug [26–28].
2.3.3. Drug to lipid (D/L) mole ratio
D/L ratio in the incubation was determined as the mole ratio of drug to phospholipid.
2.3.4. Loading efficiency
Loading efficiency was determined by the following equation:
2.3.5. Particle size measurements
Particle size was determined using the well established dynamic light scattering method, performed with Zetasizer Nano Series ZEN3600F (Malvern Instruments, Malvern, UK). Mean diameter was based on the volume mean. (For more details see [29].)
3. Results and Discussion
3.1. Virtual screening and candidate testing
3.1.1. Virtual screening of the CMC database
We employed models developed with both categorical and continuous approaches as described previously [10] for virtual screening of the CMC database (see Section. 2.1.2.). Each of these approaches by default generates an ensemble of models where each individual contributing model predicts a D/L value (for continuous models) or category (for category models) for each compound in the CMC database. The individual ISE model classified each molecule as either “−1” or “1”. The individual kNN and SVM models classified each molecule as“0” or “1” representing negative or positive classification, respectively. Individual models may differ in the predicted category for any given compound, so when these predictions are averaged across the entire ensemble of models, each compound receives a category score anywhere between “0” and “1” for kNN and SVM models or a weighted average between “−1” and “+1” for ISE (defined as ISE index). The higher is the concordance between categories predicted by individual models; the closer is the averaged predicted value to either of the extreme values. In this study, a score above 0.5 for kNN and SVM or above “0” for ISE was regarded as a positive hit since in this case more than 50% of the models scored the molecule as positive. (For more details regarding the computational models used see Supplementary Information.) In contrast to categorical approaches, the continuous kNN and SVR models predict the specific D/L ratio for each molecule (averaged over the ensemble of models), i.e., these models afford a quantitative estimate of loading efficiency.
When applied to virtual screening of the CMC database, both SVM and SVR models identified no positive hits: the highest scoring compounds had values of 0.16 and 0.21, respectively. This could be a result of the high specificity [10] of the models developed with SVM and SVR, limiting their ability to identify high-loading candidates. The predicted D/L ratio resulting from application of kNN continuous models showed very high correlation (R2 of 0.99) with the kNN category score generated by the category models (Figure 1). Therefore, the two categorical methods, ISE and kNN, were chosen for virtual screening, and the molecules in the external library were ranked by their scores resulting from averaged multiple predictions by kNN or ISE ensembles of models.
Figure 1.

Correlation between kNN continuous prediction and kNN category score.
ISE identified 431 molecules (5% of the total data set of 8658 molecules) as good candidates (ISE index >0) and the kNN categorical model identified 1394 (16%) of the CMC molecules as positive hits. Figure 2 shows the relationship between ISE index and kNN category score. Note that both of these reflect the level of concordance between multiple models contributing to the ensemble, which can be interpreted as the confidence level in assigning positive or negative category to CMC molecules; thus, one should not expect any correlation between ISE index and kNN category score. Figure 2 illustrates that both methods partially agree in assigning categories to molecules, i.e., in the detection of drugs receiving ISE score below “0” and kNN score below 0.5 or ISE score above “0” and kNN score above 0.5. 197 molecules (2.3% of the CMC database) were predicted by both models to be good candidates for remote loading (red dotted quadrant). Most drugs were predicted by both models not to be good candidates (black dotted quadrant).
Figure 2.

The relationship between ISE index and kNN category score. Hits identified by both models as positives are found in the red dotted quadrant. Negative hits are found in the black dotted quadrant. The red squares indicate molecules tested in this study and green squares are molecules found in the literature; all these molecules comprised an additional external validation set to test our models.
The required condition for remote loading of molecules, i.e., to be amphipathic weak acid or base, was not set a priori and thus it is interesting to find that those two methods identified different numbers of drugs that were weak acids or bases according to calculated pKa values using ACDLab software [30]. It should not be surprising that virtual screening identified a large fraction of molecules that were not weak acids or bases since this feature is common to all compounds in the training set. Therefore it cannot be used as a descriptor but rather as a general filter following the QSPR modeling. (By default, only structural features or properties that vary within a data set should be employed as molecular descriptors in QSPR.) Among the top 10% molecules indexed with ISE (43 molecules), 77% were found to be weak acids or bases. Among the top 10% molecules as scored by kNN (139 molecules), 25% were weak acids or bases. These findings suggest that ISE has additional strength in predicting compound suitability for remote loading to liposomes.
It should be emphasized that liposome loading experiments are tedious and time- consuming, whereas QSPR models allow very rapid examination of molecules in silico. To examine the correspondence between the computational predictions and experimental data, as a proof of concept we tested three molecules experimentally, and as well, used results from recent literature for further validation of our models. In addition to the QSPR scores and filtering by logD and pKa values, the following features served to make a decision as to which molecules to pick for experimental assessment:
Availability of the drug. Testing the formulation requires a large amount of drugs (estimated to be ~1g), so the cost must be taken into account. In addition, drug availability was also derived from drug status; marketed drugs or drugs in clinical studies are easier to obtain.
Liposomes may not be the best DDS for all “positives” from both ISE and kNN. Therefore we have considered only those drugs for which loading into liposomes and being delivered as liposomal DDS may suggest therapeutic advantages.
The disease for which the drug is used should benefit substantially from having an enhanced permeability (EP) or enhanced permeability and retention (EPR) effect that leads to passive targeting to the disease site, such as in the case of most cancers and diseases having an inflammatory component.
The dose required is in the range of 150–200 mg per single administration. The dose limitation takes into consideration two aspects derived from the D/L of the product: the maximal approved lipid administration per patient and the infusion volume. Assuming that the HSPC concentration in the preparation will be similar to Doxil® liposomes (12 mM) [9], a D/L ratio of 0.3 will result in a drug concentration in the product of 3.6 mM, which for a drug of MW 500 would be 1.8 mg/ml. Limiting the infusion volume to 100 ml will thus limit the drug dose to 180 mg. Dose limitation was therefore set to 150–200 mg per single administration.
The overall workflow applied following the in silico screening for the purpose of candidate selection is summarized herein. CMC database classifies each molecule according to its pharmacological group. The 197 molecules classified by both models as positive hits were evaluated by their pharmacological group to examine whether the disease for which the drug is used could benefit from the effect of EPR (for cancer) or EP (for diseases involving inflammation). Pharmacological groups that may benefit from this enhanced permeability effect are anticancer, anti-inflammatory and antibiotic drugs. Suitable drugs were then screened and filtered by their pKa and logD properties. Only molecules that meet the criteria of being amphipathic weak acids or bases were then searched for their stage in drug development. Molecules that were marketed or in clinical studies were further considered (molecules in earlier stages of drug development are usually not available in large quantity). Other advantages in liposomal administration were examined for these molecules, such as pharmacokinetic and toxicologic properties of the drug. For example, drugs having short plasma half-life may result in increased circulation time by PEGylated liposomal administration and hence their efficacy may improve. The passive targeting may also result in a decrease in drug toxicity, which for the free (non-liposomal) drug may in many cases limit either the dose per treatment and dose per administration (exemplified by amphothericin B and AmbiSome®) or in total dose per treatment protocol (exemplified by doxorubicin and Doxil®). Remaining molecules were evaluated for their dose as molecules requiring very high doses are not suitable for liposomal delivery. Following the additional screening process, 25 molecules were found to be suitable for testing, and the two molecules predicted to be positive hits were selected based on their commercial availability. The same criteria were applied to select the negative hit compound. The consideration of other parameters such as stability during storage and in vivo release was reserved for future studies.
Two “positive” hits and one “negative” hit (by both models) were selected for testing. The positive hits were pravastatin and mupirocin, and the negative hit was piroxicam. The red squares in Figure 2 represent the three molecules selected for testing. Table 1 shows the chemical structure of the selected drugs and their calculated pKa and log D at pH 7.0. These parameters are the parameters defining a molecule as being an amphipathic weak acid or base [8]. All three molecules selected are amphipathic weak acids (Table 1) and thus are suitable for remote loading. However, piroxicam was identified by these models to be a negative hit, i.e., to have low loading efficiency at D/L ratio ≥0.3. Detailed discussion of the selected molecules follows.
Table 1.
Chemical structure of the selected molecules and their calculated pKa and logD at pH 7.0
| Chemical structure | pKa | LogD at pH 7.0 |
|
|---|---|---|---|
| Pravastatin | 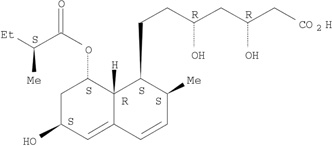 |
4.31 | −0.54 |
| Mupirocin | 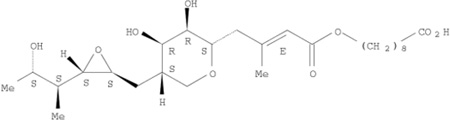 |
4.78 | 0.02 |
| Piroxicam | 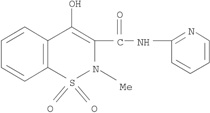 |
4.50 | −1.94 |
Pravastatin is used orally to lower cholesterol. However, pre-clinical studies suggest that statins may be effective anticancer agents, but the required doses are 100–500-fold higher than those needed to lower cholesterol levels. Liposomal pravastatin [31] was found to inhibit tumor growth by targeting cancer-related inflammation in mice, the D/L ratio was low (0.02) by a passive loading method [31]. Our in silico models suggest that the D/L ratio affording high loading efficinecy may be significantly increased (≥ 0.3) by remote loading. A hypothesis that we did validate for pravastatin in this study (see section 3.1.2. below).
Mupirocin (pseudomonic acid) is a relatively new antibiotic. When absorbed into the blood stream or administered parenterally, mupirocin is rapidly degraded to form the inactive monic acid. Therapy with mupirocin is therefore confined to topical application [32]. Toxicological studies were carried out as described in the Bactroban product monograph [33] and showed that mupirocin may be safely administered systemically even at high doses. Nano-liposomes may protect mupirocin from degradation and passively target the drug to the infected tissue by the inflammation related enhanced permeability (EP) effect [34,35].
Piroxicam, a non-steroidal anti-inflammatory drug (NSAID) was predicted to be “negative” and was selected to test this prediction. Piroxicam was chosen according to the four criteria (section 3.1.1.) and it was predicted to have low loading efficiency at D/L ratio of ≥0.3. Our predictions for piroxicam were experimentally validated indeed (vide infra).
3.1.2. Candidate testing
All three selected drugs were amphipathic weak acids (Table 1) and were therefore remote loaded using a calcium acetate transmembrane gradient [7]; the results of these experiments are shown in Table 2. Pravastatin and mupirocin showed high loading efficiency (93–100%) at all D/L ratios tested (0.2–0.5). Thus, both drugs were confirmed to be true positives. Piroxicam had medium loading efficiency (35–54%) at all D/L ratios tested; thus, it was confirmed to be a true negative.
Table 2.
Percent loading efficiency as a function of D/L ratio for 3 selected drugs
| Pravastatin | Mupirocin | Piroxicam | |||
|---|---|---|---|---|---|
| D/L ratio | % Loading efficiency |
D/L ratio |
% Loading efficiency |
D/L ratio | % Loading efficiency |
| 0.20 | 100 | 0.20 | 100 | 0.05 | 35 |
| 0.30 | 100 | 0.30 | 100 | 0.14 | 51 |
| 0.40 | 93 | 0.40 | 100 | 0.25 | 45 |
| 0.50 | 100 | 0.48 | 95 | 0.42 | 54 |
| Model score: | ISE: 0.41 kNN: 0.93 |
Model score: | ISE: 0.14 kNN: 0.83 |
Model score: | ISE: −0.07 kNN: 0.34 |
3.2. Model validation using recently published data
We have collected data for remote loading of 10 additional drugs from recent publications [11–20]. None of these molecules was used to build our QSPR models; they are marked as green squares in Figure 2. Table 3 summarizes the data related to these 10 drugs. Their chemical structures and calculated pKa and logD are shown in Table 1 in the Supplementary Information. The experimental data on their loading efficiency are found in Table 2 of the Supplementary Information.
Table 3.
Comparison between experimental category and the prediction by ISE index and kNN category score for 10 compounds found in literature
| Observed category | ISE indexa | kNN category scoreb |
|
|---|---|---|---|
| Sanguinarine [13] | Negative | −0.88 | 0.24 |
| Fluoxetine [37] | Negative | −0.61 | 0.14 |
| Edaravone [38] | Negative | −0.711 | 0.222 |
| Gefitinib [16] | Negative | −0.642 | 0.383 |
| Istaroxime [11] | Positive | −0.77 | 0.100 |
| 5-carboxyfluorescein [17] | Negative | −0.602 | 0.012 |
| Vancomycin [18] | Negative | −0.892 | NCc |
| AR-67[12] | Negative | −0.012 | 0.637 |
| Boanmycin (BAM) [19] | Negative | −0.896 | NCc |
| 3-deazaneplanocin A (DZNep) [20] | Negative | −0.622 | 0.241 |
ISE index below “0” implies negative category, and above “0” implies positive category.
kNN score below 0.5 implies negative category, and above 0.5 implies positive category.
NC- Not calculated for this model because it is outside the applicability domain: in Figure 1, kNN score for these molecules was assigned as 0.5.
The ISE model classified all 10 molecules as negatives; thus, it misclassified istaroxime [11], which was found to be positive. It should be noted that istaroxime was borderline in the experiment. Its loading efficiency at D/L ratio of 0.3 was found to be only 72–74%. The kNN model classified only 8 molecules of the 10 tested because two molecules were found to be outside of the kNN models’ applicability domain (applicability domain is a specially defined chemical similarity threshold that prevents making predictions for molecules that are chemically different from those in the training set used for model development— see [36] for additional discussion). Of the eight drugs predicted by kNN, one was false negative (istaroxime— also by ISE) and one was false positive (AR-67 [12]). Evaluating the drugs based on pKa and logD shows that only four molecules fit the requirements for being amphipathic weak acids or bases, as discussed above (logD at pH 7 in the range of −2.5 to 2 and pKa ≤11 for an amphipathic weak base or pKa >3 for an amphipathic weak acid): fluoxetine, istaroxime, 5-carboxyfluorescein and DZNep, while only one of them was experimentally positive and none of them was identified by the models as positives. These observations illustrate the advantages of using QSPR models as opposed to simple criteria based on logD and pKa ranges to achieve accurate selection of drugs for remote liposome loading.
4. Conclusions
Our study describes the first application of QSPR models to virtually screen drugs for their suitability for efficient remote loading into liposomes. Results obtained in this study confirmed the significant predictive power of the binary classification models built with ISE and kNN approaches. Based on the predictions from these models and applying other experimental considerations, three molecules were selected for experimental testing: two of those that were predicted to be “positives” (pravastatin and mupirocin) and one predicted to be “negative” (piroxicam). Indeed, all three compounds were found to be correctly classified by both models. Data for 10 other molecules published in recent literature were used for additional validation of QSPR models. The ISE method misclassified one compound (false negative) that was found experimentally to be borderline positive. kNN misclassified two instances (one false negative and one false positive), and two instances were outside of its applicability domain. The accuracy of models built with ISE and kNN was found to be 92.3% (12 correctly classified instances out of 13 predictions) and 81.8% (9 correctly classified instances out of 11 predictions), respectively.
The accuracy of QSPR models for a test set containing compounds that were not used for producing the model is exciting, especially given the diversity of their chemical structures, both for "positives” and “negatives”. In addition, each drug was also evaluated based on its pKa and logD at pH 7.0 for its suitability for remote loading (as these two parameter defined if the dug is an amphiphatic weak acid or base). It was found that both in cases where pKa and logD were similar for different molecules, as well as in cases in which they were very different, our models were able to identify both the best candidates for remote loading as well as those compounds that were not good candidates.
This study demonstrates the high utility of QSPR models for selecting new drug candidates for liposome remote loading. The QSPR model-driven virtual screening protocol was able to generate good candidates for this DDS: both of the compounds that were predicted to be positive by both models and selected for experimental validation in this proof-of-concept study were indeed confirmed experimentally to be positive. These results strongly support the notion that QSPR models may serve as a reliable and useful tool to identify candidates to achieve high intra-liposomes drug concentration and move them from in silico prediction to in vitro and in vivo studies.
One should always keep in mind that drug loading (predicted by the QSPR modeling) is indeed a major parameter for liposomal drug formulation development as it constitutes the initial crucial step. However, in order to make a more complete prediction of drug suitability to become a liposomal drug product other important parameters have to be considered. Good candidates can be determined by in silico predictions followed by the amphipathic acid or base filter (with the help of pKa and logD predictions). Other considerations include the disease for which the drug is intended and whether administration via liposomes may have a therapeutic advantage. The therapeutic dose should also be considered, as too high doses are not suitable for liposomal administration. Additional parameters, which were not considered in the present study, include storage stability and drug release at storage temperature (for stability) and at 37°C for efficacy. Only liposomes that release enough drug at the disease site, which is taken up and processed by cells have the chance to show an improved therapeutic efficacy. At the same time a too rapid release rate will not enable achieving the benefits of liposomal delivery [39]. We are currently studying the stability of loaded liposomes and their release profile; the latter property will also be examined by QSPR modeling.
The vast amount of new data on liposomal delivery of drugs published in recent years demonstrates the importance of this DDS in the field of formulations. Additionally, the fact that only one drug out of the 10 found in recent literature is “positive” (although borderline) emphasizes the difficulty of finding good candidates for remote loading. To the best of our knowledge, the studies presented herein represent the first successful example of using QSPR modeling for evaluating drugs suitable for liposomal delivery. Thus, this pilot study suggests that computational models may be used effectively to accelerate the development of novel drug delivery systems.
Supplementary Material
Acknowledgments
We would like to thank the Barenholz lab members, with special thanks to Dr. Erez Koren, for their help in the experimental part of the work. AT appreciates the support from the NIH (grants GM66940 and GM 096967). The work was supported in part by the Barenholz Fund.
The authors would like to thank Sigmund Geller for editing this paper.
Contributor Information
Yechezkel Barenholz, Email: chezyb@ekmd.huji.ac.il.
Amiram Goldblum, Email: amiram@vms.huji.ac.il.
References
- 1.Chang H-I, Yeh M-K. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. International Journal of Nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1217. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenberg D, Barenholz Y. Liposomes: Preparation, Characterization and Preservation. Methods of Biochemical Analysis. 1988;33:337–462. doi: 10.1002/9780470110546.ch7. [DOI] [PubMed] [Google Scholar]
- 4.Barenholz Y, Crommelin DJA. Encyclopedia of Pharmaceutical Technology. New York: Marcel Dekker; 1994. Liposomes as pharmaceutical Dodage Forms; pp. 1–39. [Google Scholar]
- 5.Barenholz Y. Relevancy of drug loading to liposomal formulation therapeutic efficacy. J Liposome Res. 2003;13:1–8. doi: 10.1081/lpr-120017482. [DOI] [PubMed] [Google Scholar]
- 6.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochimica Et Biophysica Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 7.Clerc S, Barenholz Y. Loading of amphipathic weak acids into liposomes in response to transmembrane calcium acetate gradients. Biochimica Et Biophysica Acta. 1995;1240:257–265. doi: 10.1016/0005-2736(95)00214-6. [DOI] [PubMed] [Google Scholar]
- 8.Zucker D, Marcus D, Barenholz Y, Goldblum A. Liposome drugs’ loading efficiency: a working model based on loading conditions and drug's physicochemical properties. J Control Release. 2009;139:73–80. doi: 10.1016/j.jconrel.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 9.Doxil Product Information. 2013 [Google Scholar]
- 10.Cern A, Golbraikh A, Sedykh A, Tropsha A, Barenholz Y, Goldblum A. Quantitative structure - property relationship modeling of remote liposome loading of drugs. Journal of Controlled Release. 2012;160:147–157. doi: 10.1016/j.jconrel.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luciani P, Fevre M, Leroux J-C. Development and physico-chemical characterization of a liposomal formulation of istaroxime. European Journal of Pharmaceutics and Biopharmaceutics : Official Journal of Arbeitsgemeinschaft Für Pharmazeutische Verfahrenstechnik e.V. 2011;79:285–293. doi: 10.1016/j.ejpb.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Modi S, Xiang T-X, Anderson BD. Enhanced active liposomal loading of a poorly soluble ionizable drug using supersaturated drug solutions. Journal of Controlled Release : Official Journal of the Controlled Release Society. 2012;162:330–339. doi: 10.1016/j.jconrel.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Ke X, Bei JH, Zhang Y, Li J. In vitro and in vivo evaluation of sanguinarine liposomes prepared by a remote loading method with three different ammonium salts. Pharmazie. 2011;66 [PubMed] [Google Scholar]
- 14.Ong JC-L, Sun F, Chan E. Development of stealth liposome coencapsulating doxorubicin and fluoxetine. Journal of Liposome Research. 2011;21:261–271. doi: 10.3109/08982104.2010.545070. [DOI] [PubMed] [Google Scholar]
- 15.Hironaka K, Inokuchi Y, Fujisawa T, Shimazaki H, Akane M, Tozuka Y, et al. Edaravone-loaded liposomes for retinal protection against oxidative stress-induced retinal damage. European Journal of Pharmaceutics and Biopharmaceutics : Official Journal of Arbeitsgemeinschaft Für Pharmazeutische Verfahrenstechnik e.V. 2011;79:119–125. doi: 10.1016/j.ejpb.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Zhou X LR, Yung B, Huang Y, Li H, Hu X, Xiang G. Novel liposomal gefitinib (L-GEF) formulations. Anticancer Res. 2012;32(7):2919–2923. [PubMed] [Google Scholar]
- 17.Hironaka K, Fujisawa T, Sasaki H, Tozuka Y, Tsuruma K, Shimazawa M, et al. Fluorescence investigation of the retinal delivery of hydrophilic compounds via liposomal eyedrops. Biological & Pharmaceutical Bulletin. 2011;34:894–897. doi: 10.1248/bpb.34.894. [DOI] [PubMed] [Google Scholar]
- 18.Muppidi K, Pumerantz AS, Wang J, Betageri G. Development and stability studies of novel liposomal vancomycin formulations. ISRN Pharmaceutics. 2012;2012:636743. doi: 10.5402/2012/636743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Yoo SD, Li L, Fang L, Wen Z, Li T. Formulation and characterization of boanmycin-loaded liposomes prepared by pH gradient experimental design. Drug Delivery. 2012;19:90–101. doi: 10.3109/10717544.2011.649217. [DOI] [PubMed] [Google Scholar]
- 20.Sun F, Li J, Yu Q, Chan E. Loading 3-deazaneplanocin A into pegylated unilamellar liposomes by forming transient phenylboronic acid-drug complex and its pharmacokinetic features in Sprague-Dawley rats. European Journal of Pharmaceutics and Biopharmaceutics : Official Journal of Arbeitsgemeinschaft Für Pharmazeutische Verfahrenstechnik e.V. 2012;80:323–331. doi: 10.1016/j.ejpb.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 21.MOE. 2012.10. Molecular Operating Environment. 2012 [Google Scholar]
- 22.Shmeeda H, Even-Chen S, Honen R, Cohen R, Weintraub C, Barenholz Y. Enzymatic assays for quality control and pharmacokinetics of liposome formulations: comparison with nonenzymatic conventional methodologies. Methods in Enzymology. 2003;367:272–292. doi: 10.1016/S0076-6879(03)67017-5. [DOI] [PubMed] [Google Scholar]
- 23.Mupirocin official monograph. USP. 2012;35 [Google Scholar]
- 24.Abdallah OLAM. RP-HPLC Determination of Three Anti-Hyperlipidemic Drugs in Spiked Human Plasma and in Dosage Forms. Methodology. 2011;8:753–761. [Google Scholar]
- 25.Song HH, Choi KS, Kim C-W, Kwon YE. Pharmacokinetic Profiles of Two Branded Formulations of Piroxicam 20mg in Healthy Korean Volunteers by a Rapid Isocratic HPLC Method. Journal of Bioequivalence & Bioavailability. 2009;01:074–079. [Google Scholar]
- 26.Druckmann S, Gabizon a, Barenholz Y. Separation of liposome-associated doxorubicin from non-liposome-associated doxorubicin in human plasma: implications for pharmacokinetic studies. Biochimica Et Biophysica Acta. 1989;980:381–384. doi: 10.1016/0005-2736(89)90329-5. [DOI] [PubMed] [Google Scholar]
- 27.Amselem S, Gabizon A, Barenholz Y. Optimization and Upscaling of Doxorubicin-Containing Liposomes for clinical use. Journal of Pharmaceutical Science. 1990;79:1045–1052. doi: 10.1002/jps.2600791202. [DOI] [PubMed] [Google Scholar]
- 28.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, et al. Prolonged Circulation Time and Enhanced Accumulation in Malignant Exudates of Doxorubicin Encapsulated in Polyethylene-Glycol Coated Liposomes. Cancer Research. 1994;54:987–992. [PubMed] [Google Scholar]
- 29.Barenholz Y, Amselem S. Quality control assays in the development and clinical use of liposome-based formulations. In: Gregoriadis G, editor. Liposome Technology. 2nd Ed. Liposome Preparation and Related Techniques; 1993. pp. 527–616. [Google Scholar]
- 30.Advanced Chemistry Development Software V11.02. 2013 [Google Scholar]
- 31.Coimbra M, Banciu M, Fens MHaM, de Smet L, Cabaj M, Metselaar JM, et al. Liposomal pravastatin inhibits tumor growth by targeting cancer-related inflammation. Journal of Controlled Release : Official Journal of the Controlled Release Society. 2010;148:303–310. doi: 10.1016/j.jconrel.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 32.Pappa K. The clinical development of mupirocin. Journal of the American Academy of Dermatology. 1990;22:873–879. doi: 10.1016/0190-9622(90)70116-y. [DOI] [PubMed] [Google Scholar]
- 33.Product monograph Bactroban. 2001 [Google Scholar]
- 34.Azzopardi Ea, Ferguson EL, Thomas DW. The enhanced permeability retention effect: a new paradigm for drug targeting in infection. The Journal of Antimicrobial Chemotherapy. 2013;68:257–274. doi: 10.1093/jac/dks379. [DOI] [PubMed] [Google Scholar]
- 35.Huh AJ, Kwon YJ. Nanoantibiotics”: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. Journal of Controlled Release : Official Journal of the Controlled Release Society. 2011;156:128–145. doi: 10.1016/j.jconrel.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Tropsha A, Golbraikh A. Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr. Pharm. Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 37.Ong JC, Sun F, Chan E. Development of stealth liposome coencapsulating doxorubicin and fluoxetine. Journal of Liposome Research. 2011;21:261–271. doi: 10.3109/08982104.2010.545070. [DOI] [PubMed] [Google Scholar]
- 38.Hironaka K, Inokuchi Y, Fujisawa T, Shimazaki H, Akane M, Tozuka Y, et al. Edaravone-loaded liposomes for retinal protection against oxidative stress-induced retinal damage. European Journal of Pharmaceutics and Biopharmaceutics : Official Journal of Arbeitsgemeinschaft Für Pharmazeutische Verfahrenstechnik e.V. 2011;79:119–125. doi: 10.1016/j.ejpb.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. Journal of Controlled Release : Official Journal of the Controlled Release Society. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.