

Summary
Background
Posttraumatic stress disorder (PTSD) is associated with a 2–4 fold increased risk of developing Type 2 diabetes mellitus. However, detailed assessments of glucose metabolism and insulin secretion in a study designed to minimize confounders are lacking. Furthermore, few studies examine potential mechanisms involved. We analyzed data from a case-control study of medically healthy, medication-free adults to determine whether individuals with PTSD had abnormal glucose or insulin response to oral glucose tolerance test (OGTT) compared to controls. Secondarily, we assessed potential mediators such as sleep, cortisol and adiponectin.
Methods
Data was analyzed from 92 age and gender-matched subjects (44 PTSD, 48 controls). Chronic PTSD was diagnosed using the Structured Clinical Interview for DSM-IV and Clinician Administered PTSD Scale. Subjects underwent 75-gram OGTT, actigraphy and sleep diary (to quantify sleep duration), polysomnography (to assess slow wave sleep [SWS] and delta power), and overnight blood sampling (for cortisol and adiponectin).
Results
At baseline, individuals with PTSD had mildly increased insulin levels (by 19%, compared to controls, p=0.048) that was mediated primarily by weight. In response to OGTT, the PTSD group had higher levels of insulin at 120 min (by 44%, p=0.03) and insulin AUC (by 43%, p=0.015) compared to controls, after adjusting for confounders. Glucose levels were similar in the two groups. Although self-reported sleep duration, SWS, and delta power differed between PTSD subjects and controls, they did not mediate the effects of PTSD status on insulin response.
Conclusion
In this case-control study, individuals with PTSD had a hyperinsulinemic response to oral glucose challenge compared to controls, suggestive of insulin resistance.
Keywords: Oral glucose tolerance test, hyperinsulinemia, insulin resistance, glucose metabolism, sleep, posttraumatic stress disorder, adipocytokines, diabetes mellitus
Introduction
Posttraumatic stress disorder (PTSD) affects over 10 million Americans yearly, with an estimated lifetime prevalence of almost 7% (Kessler et al., 2005). Individuals with PTSD are at increased risk for numerous medical illnesses, including diabetes mellitus (DM), cardiovascular disease, and rheumatoid arthritis (Boscarino, 2004; Weisberg et al., 2002; O’Toole et al., 2007; Bedi et al., 2007). Epidemiological studies have shown that PTSD is associated with a 2–4 fold increased risk of Type 2 diabetes, compared to individuals without PTSD (Lukaschek et al., 2013; Agyemang et al., 2012; Boyko et al., 2010; Goodwin et al., 2005; Pietrzak et al., 2011; Trief et al., 2006). These studies have been conducted in a wide range of samples, including asylum seekers in Europe, veterans in the US, and population based samples of middle-aged and elderly individuals in Germany (Lukaschek et al., 2013; Agyemang et al., 2012; Boyko et al., 2010). Even after controlling for the presence of other psychiatric illnesses (such as depression), most studies have found an association between PTSD and DM (Lukaschek et al., 2013; Agyemang et al., 2012; Boyko et al., 2010, Trief et al., 2006). In fact, to our knowledge, only two studies have found no association between PTSD and abnormal glucose metabolism. One of these did not find an association between PTSD and DM, but did find a link between trauma exposure and diabetes in men (Norman et al., 2006). The other cross-sectional analysis found a numerical increase in self-reported history of DM in subjects with PTSD, but this did not reach statistical significance (Spitzer et al., 2009).
Insulin resistance is a key step in the development of DM and is associated with a variety of adverse health outcomes (Reaven, 1995; Reaven, 2003). It is defined as a state in which a given concentration of insulin produces a less than normal biological response (Ciaraldi, 2009). For a given concentration of insulin, glucose levels will be higher in individuals with insulin resistance, compared to those with normal insulin sensitivity. In lieu of using methods where exogenous insulin is infused and insulin levels standardized, tests that are easier (such as the oral glucose tolerance test [OGTT], and fasting glucose and insulin levels) can be used (Matsuda and DeFronzo, 1999).
Numerous studies have found an association between PTSD and DM, but none of these have closely examined the underlying physiological disturbances in metabolism. For example, compared to healthy individuals, do patients with PTSD have higher glucose levels, or higher insulin levels, or both? Furthermore, very few studies have adequately accounted for confounders. Patients with PTSD are more likely to have other chronic illnesses (such as hypertension and atherosclerosis), take medications, smoke and weigh more than those without PTSD (Pagoto et al., 2012; Walczewska et al., 2011; Kalman et al., 2005), factors that may affect glucose metabolism. The present analysis examines data from a case-control study of young, medically healthy individuals who were free of medications, providing an excellent opportunity to elucidate mechanisms involved early in the disease process and without potentially confounding comorbidities. Both individuals with PTSD and age and gender-matched controls underwent detailed metabolic testing (including OGTT), and hormonal and sleep assessments. The primary goal of this analysis was to determine whether, in a medically healthy group of participants, glucose metabolism and insulin secretion were abnormal in individuals with PTSD compared to controls.
Secondarily, we were interested in identifying factors that may mediate the effects of PTSD on glucose and insulin metabolism. We chose mediators that are either well established to be altered with insulin resistance (such as adiponectin), or that are known to be altered in PTSD and also have established effects on insulin and glucose metabolism (such as cortisol and parameters of sleep). Adiponectin is an adipocytokine secreted by adipose tissue; compared to other secreted adipocytokines (such as resistin, visfatin, retinol binding protein-4, tumor necrosis factor-α, and numerous interleukins), adiponectin has been extensively studied and is the most consistently associated with changes in insulin sensitivity (Tadowaki et al., 2003: Okamoto et al., 2006; Pajvani et al., 2003). Animal studies have shown that decreases in adiponectin levels parallels decreases in insulin sensitivity and precedes the development of Type 2 diabetes (Hu et al., 1996; Hotta et al., 2001; Kadowaki et al., 2006). Furthermore, both prospective and longitudinal studies in humans have shown that lower adiponectin levels are associated with both insulin resistance and with a higher incidence of diabetes (Yatagai et al., 2003; Yamamoto et al., 2004; Lindsay et al., 2002; Snehalatha et al., 2003; Spranger et al, 2003). Despite this wealth of studies, there are no published studies assessing adiponectin levels in patients with PTSD. Only one small study in a rodent model of PTSD (using chronic variable stress) has evaluated the role of adiponectin; they found that adiponectin levels were lower in PTSD animals compared to controls, and that these animals had impaired glucose metabolism as well (Castaneda et al., 2011). Our goal was to determine whether adiponectin levels were different in PTSD group, compared to controls, and whether this mediated the effects of PTSD status on glucose metabolism. The second potential mediator we assessed was cortisol. Elevated circulating levels of cortisol cause insulin resistance, impaired glucose tolerance and diabetes (Dineen et al., 1993; other refs). Furthermore, it has long been hypothesized that circulating cortisol levels are abnormal in patients with PTSD, compared with healthy controls. Published studies have been variable, with some groups finding decreased circulating cortisol levels, while others have found either no change or increased levels (Pittman et al., 1990; Rasmusson et al., 2001; Yehuda et al., 1990). However, to our knowledge, there are no studies assessing the role of cortisol in the impaired glucose metabolism or development of diabetes observed in PTSD. Lastly, we assessed sleep duration (measured by actigraphy and diary), slow wave sleep (SWS, quantified by visual analysis), and delta power (by quantitative analysis) as possible mediators. A substantial body of research has demonstrated that short sleep duration is associated with abnormal glucose metabolism and the development of diabetes. Furthermore, experimental studies have shown that decreased SWS adversely affect glucose tolerance in healthy adults (Spiegel et al., 1999; Tasali et al., 2008). However, there are no studies assessing the role of sleep duration or SWS in impairing glucose metabolism and insulin response in patients with PTSD. In this case-control study of young, medically healthy subjects, we examined how sleep duration, SWS, and specific hormones (adiponectin and cortisol) may mediate the effects of PTSD on glucose metabolism.
Methods and Materials
Participants and Study Design
A total of 93 subjects were enrolled in this case-control study with a 2x2 design (PTSD/control x male/female), in which subjects were matched for age and gender. Subjects were recruited from the San Francisco Veterans Affairs Medical Center (SFVAMC) and the greater San Francisco Bay Area; ads and flyers were distributed in the community and in relevant local clinics (for the PTSD group). All participants were medically healthy and free of medications. They were screened with complete blood count, chemistry, liver function tests, thyroid function tests, and urine toxicology screen. Premenopausal females (as determined by medical screening interview) underwent urine pregnancy test and were studied in the follicular phase of the menstrual cycle (day 0–10).
Inclusion criteria for both groups was 20–50 years of age and body mass index (BMI) <30 kg/m2 (O’Donovan et al., 2011; Richards et al., 2013). Exclusion criteria included: history of traumatic brain injury; presence of neurologic disorders or systemic illness; sleep apnea; pregnancy; alcohol abuse in the previous 2 years, substance abuse in the previous 1 year; any history of psychiatric disorder with psychotic features, bipolar disorder or compulsive disorders; and use of any psychiatric, anticonvulsant, antihypertensive, steroidal, lipid-lowering, or any other prescription medications or estrogen replacement therapy. Control subjects were excluded if they had a lifetime history of major depressive disorder, panic disorder, or PTSD. Potential subjects were assessed for their habitual sleep habits by wearing a wrist actigraph for 7 days (details below), and keeping a 10-day sleep diary. Subjects were admitted to the Clinical Research Center (CRC) at the University of California, San Francisco (UCSF) for 4 days and 3 nights, and underwent polysomnography (PSG) on all 3 nights (night 1=adaptation, nights 2–3=pre- and post-metyrapone administration). While the primary study was designed to determine the effects of metyrapone on sleep architecture and pituitary hormones, the data used in this current analysis were all collected prior to the administration of metyrapone. Of the 93 enrolled participants, we have analyzable data with respect to glucose and insulin levels in 92 (44 with chronic PTSD, 48 control subjects). One subject dropped out prior to undergoing the glucose tolerance test. The study protocol was approved by the Committees on Human Research at UCSF and at SFVAMC. Written informed consent was obtained from all subjects.
Psychiatric Evaluation
PTSD and other psychiatric disorders were assessed through administration of the Structured Clinical Interview for DSM-IV by clinically trained interviewers (First et al., 1996). Lifetime and current PTSD were further assessed with the Clinician Administered PTSD Scale (CAPS) (Blake et al., 1995). Chronic PTSD of over 3 months was defined as satisfaction of DSM-IV criteria or by CAPS scores >40. This CAPS score was chosen as the criterion based on the definition of Weathers et al. (2001), who defined a score of >40 as moderate PTSD. Control subjects were negative for lifetime PTSD by DSM-IV criteria and had CAPS scores< 20. All diagnoses were made by clinically trained interviewers and were confirmed at weekly case consensus meetings by Ph.D. level psychologists.
Data Acquisition
Demographics
Self-report questionnaires were used to gather demographic data.
CRC Admission
Upon admission, subjects were gowned and measured for height and weight; BMI was calculated as weight in kilograms divided by height in meters squared. Subjects were not allowed to nap during their stay, and were permitted only one cup of caffeinated coffee each morning. A catheter was inserted into the antecubital vein on Day 1 for subsequent blood sampling. On Day 2, subject underwent the OGTT in the morning, and a dual energy X-ray absorptiometry (DEXA) scan in the afternoon. PSG was performed on all three nights of admission.
Actigraphy
Sleep-wake schedules were monitored at home using a wrist actigraph (Octagonal Basic Motionlogger-L, Ambulatory Monitoring, Inc.) worn continuously for 7 days. Actigraph data was available in 87 subjects (43 controls and 44 PTSD); six subjects did not have data due to equipment malfunction. The actigraph was worn in the PTSD group for a mean of 6.73 nights (SD 0.87) and for 6.52 nights (SD 1.32) in the control group (p=NS). Data was collected in 3 modes and are reported here based on the zero-crossing mode. Action W-2 software (version 7) was used to analyze the data with the Cole-Kripke algorithm. Actigraphic sleep duration was assessed in the secondary analysis as a potential mediator.
Sleep Diary
Subjects maintained a daily sleep diary for 10 consecutive days, coinciding with the actigraph. Data from the diary was used to score the actigraph and to determine subjective sleep duration. Subjective sleep duration was analyzed as a potential mediator in our analyses.
PSG
Subjects underwent 3 nights of ambulatory PSG (Nihon Kohden Trackit Ambulatory Recording System, Foothill Ranch, CA, USA) during the CRC admission. The recording montage consisted of C3/A2, C4/A1, O1/A2 and O2/A1 electroencephalograms, bilateral electroculograms, a bipolar submental electromyogram, bilateral anterior tibialis electromyograms and electrocardiograms (Richards et al., 2013; Rechtschaffen and Kales, 1968). Electrode impedance was set at <5000 ohms. Raw EEG signals were filtered, amplified, digitized at 256 Hz and recorded. Low and high frequency hardware filters on the recorder were single pole analogue filters with 3 db points at 0.5 and 100 Hz. Analyzable PSG data on Night 2 was available on 83 subjects (43 controls, 40 PTSD); data from 10 subjects were excluded due to poor quality EEG recordings. Of note, PSG data from night 1 (i.e., the night prior to the OGTT) was not used because it was the acclimatization night, and the “first night” phenomenon is well known to adversely affect sleep (Suetsugi et al., 2007; Toussaint et al., 1995).
Pass Plus (Delta Software, University City, MO, USA) was used for visual scoring and quantitative analysis of PSG data. Visual scoring was conducted by a highly experienced registered PSG technician using the criteria of Rechtschaffen and Kales (1968). SWS was assessed as a potential mediator in this current analysis. Sleep activity in the delta band was analyzed from the C3 electrode by power spectral analysis (PSA). PSA was conducted on all epochs of sleep. As previously described (Richards et al., 2013), Pass Plus was used to perform Fast-fourier transformation analysis on 4.0-s Welch tapered windows with 2-s overlap, yielding 15 windows per 30-s epoch. Power spectra for delta (1–4 Hz) was used in this current analysis to determine whether delta energy was a potential mediator of the relationship between PTSD status and glucose tolerance.
DEXA
Total and regional fat mass and lean mass were measured by DEXA (Lunar DPX, Madison WI), with regional analysis as previously described (Lo et al., 1998). Six subjects did not complete the DEXA scan, thus we have analyzable data in 87 subjects (43 controls, 44 PTSD). Truncal fat percentage and total body fat percentage were assessed as covariates in our primary analyses, and were used instead of BMI in the sequentially adjusted analyses. Because the results did not alter significantly, we report the results with BMI.
Assays
On night 2 of the CRC admission, blood sampling was performed every 30 minutes for 8 hours while subjects slept through an indwelling catheter in the antecubital vein. Samples were processed in real time and stored at −70 °C. Plasma cortisol was measured at the Brigham Research Assay Core, using the Access Chemiluminescent Immunoassay (Beckman Coulter, Fullerton, CA). The intra-assay variation was 4.4–6.7% and the inter assay variation 6.4–7.9%. The integrated cortisol AUC was determined using the trapezoidal method; missing values were either imputed (for first or last time-points that were missing) with subject-specific mean values or interpolated (for any other missing time-points). Cortisol levels at some time points were not available due to technical issues with the catheters and blood draws, or because subjects woke up before 8 hours had passed. Eight subjects (4 PTSD, 4 control) did not have any overnight blood draws for assessment of cortisol, and one additional subject (with PTSD) did not have cortisol drawn at awakening. Of the 84 subjects with cortisol measured, 2 subjects had fewer than 3 measurements, and thus, cortisol AUC was not calculated. Morning (AM) cortisol level upon awakening and overnight cortisol AUC were assessed as potential mediators of the relationship between PTSD status and glucose tolerance. Total and high-molecular weight (HMW) adiponectin were assayed on serum samples by the Brigham Research Assay Core by ELISA (ALPCO Diagnostics Inc, Salem, NH). The intra-assay variation for total adiponectin and HMW adiponectin was 5.0–5.4% and 3.3–5.0%, respectively; the precision was 6% for both.
OGTT
OGTT was performed on the morning of Day 2 after a 10-hour overnight fast. Subjects were given 75-g dextrose in 250 cc of water, and blood samples were taken at baseline and 30, 60 and 120 min for measurement of glucose and insulin levels. Glucose was measured by the hexokinase method (Roche Applied Sciences, Indianapolis, IN) on the Cobas C501, at Washington University Core Laboratory. Insulin measurement was performed by EMD Millipore Corporation (St. Charles, MO, USA) by radioimmunoassay. Area under the curve (AUC) for glucose and insulin were determined using the trapezoidal method, and HOMA-IR was calculated (Matthews et al., 1985).
Statistical Analysis
Baseline characteristics were summarized using mean and standard deviation (SD) for continuous variables and counts and percentages for categorical data. These characteristics were compared between the PTSD and control groups using two-sample t-test for continuous data and chi square tests for categorical data.
For the primary analysis, we determined whether glucose and insulin parameters (i.e., HOMA-IR, glucose 0 min, glucose 30 min, glucose 60 min, glucose 120 min, glucose AUC, insulin 0 min, insulin 30 min, insulin 60 min, insulin 120 min, insulin AUC) were significantly different between the PTSD and control group using simple linear regression. Next, potential confounders were entered sequentially into the multivariable regression models, to assess their incremental effects. The covariates assessed are: age (as quartiles), gender, smoking (TOB), BMI. Multivariate models using truncal body fat percentage and total body fat percentage (instead of BMI) were also assessed. Because the associations between OGTT outcomes and PTSD were similar when adjusted for any of the three measure of body composition, we report only the models with BMI. All outcome variables had right-skewed distributions and were log-transformed for regression analyses. Beta coefficients were then back-transformed to show percent differences between the PTSD and control group. For all multivariate regression models, t-tests of whether the beta coefficient differed from zero were carried out, and the corresponding p-values were calculated. Interactions between gender and PTSD were tested for all outcomes. There was a statistically significant interaction (p=0.01) between gender and PTSD status with respect to glucose 0 min, but not with respect to the other outcome variables.
The secondary aim of this current study was to determine whether pre-specified variables known to be associated with glucose metabolism mediated the effects of PTSD status on the glucose and insulin parameters. The variables assessed were: actigraphic sleep duration, self-reported sleep duration (by diary), SWS (minutes and as a percentage of total sleep time), total and HMW adiponectin, and cortisol (AM cortisol and cortisol AUC). Sleep duration by PSG was not assessed because we were interested in sleep duration estimated over multiple days, rather than a single day which may less reliably measure typical sleep duration due to transient confounders (Suetsugi et al., 2007; Toussaint et al., 1995). All skewed variables were log transformed for statistical analyses; data presented in the tables were back-transformed to allow for ease of understanding. We performed mediation analysis by the classic regression approach (i.e., product method) (Baron and Kenny, 1986). We first determined whether the potential mediator was associated with the predictor variable. The variables that were associated with PTSD were then analyzed to determine association with the outcome variables. If the potential mediator fulfilled both criteria, mediation analysis was conducted to determine whether the indirect effect of PTSD on OGTT outcomes (via the mediator), adjusting for age, gender, smoking and BMI, was statistically significant. Significance of a mediation effect was determined using the boostrapping method (Preacher and Hayes, 2004); specifically, bias-corrected bootstrapped 95% confidence intervals (CIs) were derived for all direct and indirect effects and those effects with 95% CIs that excluded zero were considered statistically significant (p<0.05). All analyses were performed using SAS statistical software version 9.3 (SAS Institute, Cary, NC). A p-value < 0.05 was considered to be significant for all statistical analyses.
Results
Characteristics of study participants
Subjects with PTSD (n=44) and control subjects (n=48) were similar in terms of age, sex and smoking status (Table 1). The control group included more Caucasians than the PTSD group. Subjects with PTSD had a mean CAPS score of 55 (SD=16); details of their trauma and other co-morbid conditions are summarized in Table 1. Subjects with PTSD had a higher BMI and greater total body fat than control subjects, but similar amounts of truncal fat. When stratified by gender, there was no significant difference in the BMI of females with PTSD compared to control subjects. However, males in the PTSD group had a significantly higher BMI than control subjects.
Table 1.
Baseline Characteristics
| PTSD (N=44) | Control (N=48) | p-value* | |
|---|---|---|---|
| Gender, N (%) | |||
| Female (n=48) | 22 (50%) | 26 (54%) | 0.69 |
| Male (n=45) | 22 (50%) | 23 (46%) | |
| Age (years), Mean (SD) | |||
| All subjects | 30.5 (6.6) | 30.5(8.2) | 0.97 |
| Female | 30.2 (6.8) | 30.5 (8.0) | 0.90 |
| Male | 30.9 (6.5) | 30.5 (8.8) | 0.86 |
| Race, N (%) | |||
| Caucasian | 23 (52%) | 36 (75%) | 0.02 |
| Non-Caucasian | 21 (48%) | 12 (25%) | |
| Smoking, N (%) | |||
| Yes | 11 (25%) | 9 (19%) | 0.47 |
| Prior EtOH Abuse | |||
| Yes | 15 (34%) | 1 (2%) | <0.001 |
| Prior Drug Abuse | |||
| Yes | 9 (21%) | 1 (2%) | <0.001 |
| Veteran Status | |||
| Civilian | 34 (77%) | 48 (100%) | <0.001 |
| Veteran | 10 (23%) | 0 (0%) | |
| BMI (kg/m2), Mean (SD) | |||
| All subjects | 26.9 (4.6) | 24.4 (3.8) | 0.01 |
| Female | 24.6 (3.9) | 25.2 (4.1) | 0.65 |
| Male | 29.3 (4.2) | 23.6 (3.3) | <0.001 |
| Total Body Fat (%), Mean (SD) | |||
| All subjects | 30.8 (8.4) | 26.6 (10.8) | 0.05 |
| Female | 33.1 (7.9) | 33.4 (8.1) | 0.89 |
| Male | 28.5 (8.4) | 19.2 (8.0) | 0.001 |
| Truncal Fat (%), Mean (SD) | |||
| All subjects | 53.3 (7.1) | 51.8 (6.1) | 0.41 |
| Female | 48.0 (5.6) | 48.9 (4.2) | 0.56 |
| Male | 58.0 (4.4) | 55.1 (6.4) | 0.09 |
| Trauma history | |||
| Sexual Trauma-Adult | 6 (14%) | 0 (0%) | ----- |
| Physical Trauma-Adult | 12 (27%) | 1 (2%) | ----- |
| Childhood Trauma** | 21 (47%) | 4 (8%) | ----- |
| Major Depressive Disorder | |||
| Current | 8 (18%) | 0 (0%) | ----- |
| Past | 25 (57%) | 0 (0%) | ----- |
| None | 11 (25%) | 0 (0%) | ----- |
P-values are calculated from chi-square test for categorical variables and Student’s t-tests for continuous variables.
Childhood trauma occurring at or before age 14.
-----Not Applicable
Glucose Tolerance Test
Unadjusted analyses of data from the glucose tolerance test showed little difference in glucose levels, either at baseline or in response to glucose administration (Figure 1). Specifically, glucose values from the OGTT in subjects with PTSD versus controls (mean±SE) were as follows: glucose 0 min 93.7±2.2 vs. 89.0±1.3 mg/dL (p=0.07), glucose 30 min 141.8±5.5 vs. 140.3±4.2 mg/dL (p=0.83), glucose 60 min 128.9±7.4 vs. 127.3±4.9 mg/dL (p=0.86), glucose 120 min 107.7±5.6 vs. 106.5±4.4 mg/dL (p=0.89), and glucose AUC 244±10.3 vs. 237±7.3 mg/dL (p=0.48). However, HOMA-IR, insulin level at all time points (0 min, 30 min, 60 min, 120 min) and insulin AUC were significantly higher in subjects with PTSD compared to controls (Figure 1). Specifically, subjects with PTSD had HOMA-IR that was 26% higher than control subjects (mean±SE: 2.66±0.19 versus 2.10±0.13, p=0.02). Likewise, in subjects with PTSD compared to controls, insulin 0 min was 19% higher (11.3±0.8 vs. 9.5±0.5 mIU/ml, p=0.03), insulin 30 min was 33% higher (74.9±7.3 vs. 55.9±5.0 mIU/ml, p=0.02), insulin 60 min was 51% higher (68.2±9.1 vs. 44.9±4.3 mIU/ml, p=0.02), insulin 120 min was 47% higher (48.6±8.1 vs. 33.0±3.7 mIU/ml, p=0.04) and insulin AUC was 55% higher (115.8±12.0 vs. 74.8±6.0, p=0.005). Of note, unadjusted analysis did not show any significant association between major depressive disorder (MDD) and measures of glucose or insulin (p=0.18).
Figure 1. Glucose and insulin responses to OGTT in PTSD+ and control subjects.
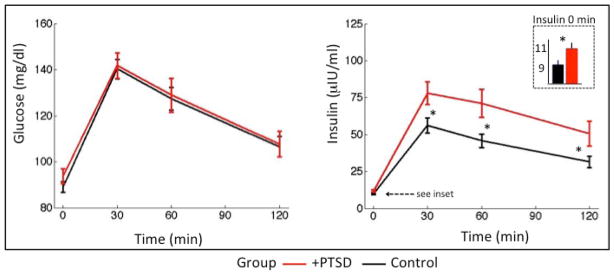
Mean (±SE) values of glucose and insulin in response to 75-g OGTT. Mean glucose levels were not significantly different in individuals with PTSD, compared to controls. Mean insulin level was significantly higher in the PTSD group both at baseline (see inset) and at each time point. *P-value <0.05; see text for exact values. P-value was calculated by Student’s t-test on log-transformed glucose and insulin values.
Regression models were performed with sequential adjustment for confounders. In the minimally adjusted model (Table 2, Model 1), subjects with PTSD continued to have a significantly higher HOMA-IR, insulin 0 min, insulin 120 min and insulin AUC compared to control subjects. After accounting for age, gender and smoking, the effect of PTSD compared to controls remained similar for HOMA-IR and insulin 0 min, but increased slightly for insulin 120 min and insulin AUC (Table 2, Model 2). When BMI was included in the fully adjusted model (Table 2, Model 3), the effect of PTSD on HOMA-IR and insulin 0 min levels decreased substantially; however, the association of PTSD with insulin 120 min and insulin AUC was only slightly reduced. Insulin 120 min was 44% higher (p=0.03) and insulin AUC was 43% higher (p=0.015) in the PTSD group compared to controls. Results were similar with insulin 30 min and insulin 60 min values (data not shown).
Table 2.
Sequential multivariate linear regression analysis of OGTT results
| Model 1 | Model 2 | Model 3 | ||||
|---|---|---|---|---|---|---|
| Outcome Variable | PTSD vs. Control % Difference (95% CI) | p-value | PTSD vs. Control % Difference (95% CI) | p-value | PTSD vs. Control % Difference (95% CI) | p-value |
| HOMA-IR | 27% (5%–52%) | 0.013 | 28% (6%–53%) | 0.01 | 12% (−5%–33%) | 0.18 |
| Glucose (mg/dl) t=0 min | 4.5% (−0.4%–9.6%) | 0.07 | 4% (−0.5%–9%) | 0.08 | 2% (−3%–7%) | 0.38 |
| Glucose (mg/dl) t=120 min | 0.3% (−11%–13%) | 0.96 | 0.1 (−11%–13%) | 0.99 | −0.8% (−12%–12%) | 0.88 |
| Glucose AUC | 2% (−8%–12%) | 0.75 | 1.3% (−8%–11%) | 0.78 | −1%(−10%–9.0%) | 0.83 |
| Insulin (μIU/ml) t=0 min | 20% (1.2%–42%) | 0.036 | 21% (2%–43%) | 0.03 | 9% (7%–28%) | 0.29 |
| Insulin (μIU/ml) t=120 min | 53% (10%–113%) | 0.01 | 57% (13%–118%) | 0.007 | 44% (3%–102%) | 0.03 |
| Insulin AUC | 58% (17%–114%) | 0.004 | 64% (23%–118%) | 0.001 | 43% (7%–90%) | 0.015 |
Model 1: adjusted for age, gender
Model 2: adjusted for age, gender, TOB
Model 43: adjusted for age, gender, TOB, BMI
P-value <0.05 indicates statistical significance.
Regression models were then stratified by gender (Table 3) as part of exploratory analyses assessing the role of gender-based differences in BMI (in subjects with PTSD vs. controls). In females with PTSD, insulin levels at 120 min were 46% higher (p=0.02) than in female controls, after adjusting for confounders. Insulin AUC in women with PTSD was also higher than controls by an average of 29% (p=0.04). Males with PTSD had higher insulin 120 min and insulin AUC levels than controls, but this did not reach statistical significance.
Table 3.
Fully adjusted linear regression model of OGTT results, stratified by gender
| PTSD vs Control, % difference (95% CI) | ||||
|---|---|---|---|---|
| Variable | Women (n=48) | p-value | Men (n=45) | p-value |
| HOMA-IR | 9.5% (−9.2%–32%) | 0.34 | 11% (−20%–54%) | 0.51 |
| Glucose (mg/dl) t=0 min | −2.2% (−7%–3.4%) | 0.39 | 8.7% (−0.6%–19%) | 0.06 |
| Glucose (mg/dl) t=120 min | −6.4% (−19%–8.7%) | 0.38 | 11% (−12%–39%) | 0.37 |
| Glucose AUC | −0.87% (−13%–13%) | 0.89 | 2.8% (−11%–21%) | 0.73 |
| Insulin (μIU/ml) t=0 min | 12% (−5.3%–33%) | 0.18 | −0.5% (−28%–38%) | 0.97 |
| Insulin (μIU/ml) t=120 min | 46% (6.1%–102%) | 0.02 | 34% (−36–178%) | 0.42 |
| Insulin AUC | 29% (0.8%–66%) | 0.04 | 40% (−24%–159%) | 0.27 |
Model adjusted for: age, TOB, BMI.
P-value<0.05 indicates statistical significance.
Potential mediators of the relationship between PTSD and glucose tolerance
Potential mediators that were examined are summarized in Table 4. There was no difference between the PTSD group and the control group with respect to adiponectin levels, AM cortisol, cortisol AUC or actigraphic sleep duration. Self-reported sleep time (by diary) was significantly lower in subjects with PTSD compared to controls, as were SWS minutes, SWS percentage and total log of delta power. We subsequently assessed whether these variables were associated with the outcome variables insulin 120 min and insulin AUC, and found that only mean sleep duration by diary was significantly associated with insulin 120 min (for every 10 min increase in mean diary sleep time, log insulin 120 min decreased by 3.4%, p=0.008). Mediation analysis indicated that the indirect effect of PTSD via sleep duration by diary was significant (natural indirect effect=0.14, 95% CI 0.02–0.34) and the proportion mediated was 0.40. However, diary sleep duration was not associated with insulin AUC.
Table 4.
Association between PTSD status and potential mediators
| Variable | N | Control Median (min-max) | PTSD Median (min-max) | P-value* |
|---|---|---|---|---|
| HMW Adiponectin (μg/ml) | 89 | 3.93 (0.96–14.6) | 3.36 (0.02–13.3) | 0.51 |
| Total Adiponectin (μg/ml) | 89 | 8.99 (2.35–29.6) | 6.88 (1.92–21.3) | 0.24 |
| AM Cortisol (ug/dl) | 83 | 13.8 (5.12–24.9) | 14.2 (7.7–26.9) | 0.60 |
| Cortisol AUC | 82 | 186 (105–300) | 195 (47.7–464) | 0.20 |
| Actigraphic sleep duration (minutes) | 86 | 446 (285–537) | 420 (178–562) | 0.14 |
| Diary-report of sleep duration (minutes) | 92 | 438 (330–606) | 399 (148–519) | <0.001 |
| SWS (minutes) | 82 | 58.3 (0.5–120) | 30.3 (0.0–130) | 0.008 |
| SWS (%) | 82 | 13.7 (0.1–28.4) | 8.48 (0.0–29.4) | 0.013 |
| Total log of delta power† | 80 | 2180 (0.0–3100) | 1830 (725–3010) | 0.046 |
P-values are calculated from Mann-Whitney test
Units of energy [absolute spectral power over the duration of non-rapid eye movement sleep] are μV2s per 100. Raw energy values must be multiplied by 100 to reflect actual energy and are shown as presented by Pass Plus software to enhance readability.
Discussion
In this case-control study of young, medically healthy, medication-free adults, we found that individuals with PTSD had an abnormal hyperinsulinemic response to the OGTT compared to control subjects suggesting the presence of insulin resistance. Specifically, the PTSD group had mild hyperinsulinemia at baseline that was mediated primarily by weight. With an oral glucose challenge, individuals with PTSD had much greater hyperinsulinemia that was only partially mediated by weight. After accounting for confounders (including BMI), insulin 120 min was 44% higher and insulin AUC was 43% higher in the PTSD group compared to the control group. Interestingly, the two groups did not differ in glucose levels, either at baseline or in response to OGTT. These results suggest the presence of insulin resistance with oral glucose challenge. Although a number of epidemiological studies have found an association between PTSD and diabetes as well as the metabolic syndrome (Lukaschek et al., 2013; Agyemang et al., 2012; Boyko et al., 2010; Goodwin et al., 2005; Pietrzak et al., 2011; Trief et al., 2006; Weiss et al., 2011; Heppner et al., 2009), factors such as medications and co-morbid health conditions may be significant confounders. A unique aspect of this current study is that the subjects are all free of medications, and do not have any chronic medical conditions.
We observed an interaction between gender and PTSD status with respect to our outcomes. Stratifying by gender showed that the observed impairment in insulin response to OGTT was present in both males and females. Importantly, BMI was not a confounding issue in females (since they were well matched) and we found a statistically significant difference in the insulin response (between PTSD and control subjects) in this group. This gender-specific finding is most likely due the difference in BMI observed in males versus females. However, the possibility of interaction between gender and PTSD status, as it relates to glucose and insulin metabolism cannot be ruled out, and would be an important topic to investigate in future studies.
The unique design of this study (i.e., case-control and 3-night admission to the CRC) also allowed for the detailed assessment of sleep and hormonal systems as possible mediators of the relationship between PTSD status and hyperinsulinemia. Numerous studies have shown that total sleep duration, as well as SWS duration, effect glucose metabolism in the general population (Spiegel et al., 1999; Tasali et al., 2008; Gottlieb et al., 2005). In this current study, we were able to assess sleep duration objectively (by actigraph) and subjectively (by sleep diary). While diary sleep time was significantly lower in subjects with PTSD compared to controls, it did not mediate the effects of PTSD status on insulin response in further analyses. In particular, sleep duration by diary did not mediate the effects of PTSD status on insulin AUC, which is an integrated measure of insulin response to the OGTT. Although diary sleep duration was found, statistically, to mediate the effects of PTSD status on insulin level at 120 minutes, this was an isolated finding and, given the results with respect to insulin AUC, is likely spurious. Actigraphic sleep duration was not significantly different between the two groups. The discrepancy between objectively measured and subjectively reported sleep duration has been noted in other studies (Lauderdale et al., 2008; Regestein et al., 2004; Rao et al., 2013), and suggests that reported sleep may be a marker for other social, physical or psychological issues. For example, individuals with more hyperarousal (as seen in PTSD) may report less sleep. Another possibility is that poor sleep quality is reflected in the self-reported sleep duration. In this current study, subjects with PTSD also had significantly lower SWS (both in minutes and percentage of total sleep) than controls; however, SWS did not mediate the effects of PTSD status on insulin response.
Lastly, we were able to closely examine levels of hormones (i.e., adiponectin) and patterns of hormone secretion (i.e., cortisol). A substantial body of literature has shown that adiponectin levels are positively associated with insulin sensitivity, in both humans and animal models (Oh et al., 2007; Yatagai et al., 2003; Yamamoto et al., 2004). Circulating adiponectin exists in a wide range of multimer complexes; the HMW isoform is believed to be more active in glucose metabolism than other isoforms (Kadowaki et al., 2006; Wang et al., 2008). In this current study, circulating levels of HMW adiponectin were not significantly different in the two groups. We were also able to closely study the pattern of cortisol secretion by obtaining blood every 30 minutes overnight. Studies in patients with PTSD have had widely differing results with respect to cortisol secretion (Pittman et al., 1990; Rasmusson et al., 2001; Yehuda et al., 1990). In the parameters of cortisol secretion we assessed (AM cortisol and cortisol AUC), we did not find any group differences. It is possible that other measures of cortisol secretion (such as 24-hr urine free cortisol or salivary cortisol, or a longer duration of blood sampling for cortisol) may have been useful to determine the mediating effects of cortisol in this population. Of note, we were interested in measuring circulating levels of cortisol rather than cortisol resistance (determined by monocyte glucocorticoid sensitivity, or other measures) for two reasons. First, our goal was to determine how circulating cortisol levels may mediate the effects of PTSD status on glucose metabolism; studies looking at the effects of elevated cortisol on glucose metabolism have quantified circulating cortisol levels, not other measures (Rizza et al., 1982)). Second, glucocorticoid sensitivity of monocytes determines how stress interacts with the immune system (Rohleder et al., 2001), which is not the focus of our current analyses.
The limitations of this study include the fairly small sample size (n=92 with OGTT data), as well as the differences between the PTSD and control group with respect to baseline characteristics. For example, BMI is higher in the males with PTSD than in male control, which could skew our results towards having an impaired insulin response in the PTSD group, compared to controls. We tried to address this limitation in two ways. First, because the females in this study were well matched for BMI, we performed gender-stratified analyses, which revealed that the association between PTSD status and impaired insulin response was just as strong in females as it was in males. Second, in multivariate analyses of the whole cohort, we adjusted our models for BMI and found the results to still be significant. The strengths of this study include the study design (2x2 case control, matched for age and gender), the opportunity to perform detailed assessments of sleep and hormonal secretion due to the 3-night CRC admission, and the robust control of the fasting condition prior to the OGTT. Further, the study was specifically designed to enroll medically healthy subjects who were medication-free; this allowed us to avoid significant confounders such as co-morbid illnesses and medications, which are present in most studies of PTSD.
In conclusion, we found that medically healthy, young individuals with PTSD have hyperinsulinemia in response to an oral glucose challenge compared to control subjects, despite no significant difference in glucose levels. These results suggest the presence of insulin resistance during oral glucose load in this population. However, definitive demonstration of insulin resistance requires the use of other techniques, such as the hyperinsulinemic-euglycemic clamp. Our results indicate that such studies are warranted in the future.
Acknowledgments
We thank the nurses and staff of the CRC for their assistance in performing the inpatient assessments.
Role of Funding Source: This research was supported by NIH grants R01MH073978 and R34MH077667, NIH GCRC grant UL1 RR024131 and grant funding from the Mental Illness Research and Education Clinical Center of the US Veterans Health Administration. The funding agencies had no role in the collection or analysis of the data or in the preparation of this manuscript.
Footnotes
Role of Contributors: Authors MNR and TCN designed the study. Authors MNR, AC, and EM undertook statistical analyses. Authors LR and AR performed analysis of sleep data. Authors MNR, AC, EM, SI, LT, AR, AO, CG and TCN all contributed to writing and editing the final draft of the manuscripts. All authors contributed to and have approved the final manuscript.
Conflict of Interest and Financial Disclosures: Drs. Rao, Chau, Madden, Inslicht, Talbot, O’Donovan, Grunfeld and Ms. Ruoff reported no financial interests or potential conflicts of interest. Dr. Neylan reported receiving study medication from Actelion for a study funded by the Department of Defense and receiving study medication from Glaxo Smith Kline for a study funded by the Department of Veterans Affairs.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agyemang C, Goosen S, Anujuo K, Ogedegbe G. Relationship between posttraumatic stress disorder and diabetes among 105,180 asylum seekers in the Netherlands. Eur J Public Health. 2012;22:658–662. doi: 10.1093/eurpub/ckr138. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research:Conceptual, strategic and statistical considerations. Journal of personality and social psychology. 1986;51:1173. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Bedi US, Arora R. Cardiovascular manifestation of posttraumatic stress disorder. J Natl Med Assoc. 2007;99:642–649. [PMC free article] [PubMed] [Google Scholar]
- Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8:75–90. doi: 10.1007/BF02105408. [DOI] [PubMed] [Google Scholar]
- Boscarino JA. Posttraumatic stress disorder and physical illness: results from clinical and epidemiologic studies. Ann NY Acad Sci. 2004;1032:141–153. doi: 10.1196/annals.1314.011. [DOI] [PubMed] [Google Scholar]
- Boyko EJ, Jacobson IG, Smith B, Ryan MA, Hooper TI, Amoroso PJ, et al. Risk of diabetes in U.S. military service members in relation to combat deployment and mental health. Diabetes Care. 2010;33:1771–1777. doi: 10.2337/dc10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda TR, Nogueiras R, Muller TD, Krishna R, Grant E, et al. Decreased glucose tolerance and plasma adiponectin:resisin ratio in a mouse model of posttraumatic stress disorder. Diabetologia. 2011;54:900–909. doi: 10.1007/s00125-010-2019-y. [DOI] [PubMed] [Google Scholar]
- Ciaraldi T. Cellular mechanisms of insulin action. In: Poretsky L, editor. Principles of Diabetes Mellitus. 2. Springer; New York: 2009. [Google Scholar]
- Dinneen S, Alzaid A, Miles J, Rizza R. Metabolic effects of the nocturnal rise in cortisol on carbohydrate metabolism in normal humans. J Clin Invest. 1993;92:2283–90. doi: 10.1172/JCI116832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First M, Spitzer R, Williams J, Gibbon M. Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders-IV (DSM-IV) 4. New York: State Psychiatric Institute, Biometrics Research; 1996. [Google Scholar]
- Goodwin RD, Davidson JR. Self-reported diabetes and posttraumatic stress among adults in the community. Prev Med. 2005;40:570–574. doi: 10.1016/j.ypmed.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Gottlieb DJ, Punjabi NM, Newman AB, Resnick HE, Redline S, Baldwin CM, et al. Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch Intern Med. 2005;165:863–867. doi: 10.1001/archinte.165.8.863. [DOI] [PubMed] [Google Scholar]
- Heppner PS, Crawford EF, Haji UA, Afari N, Hauger RL, Dashevsky BA, et al. The association of posttraumatic stress disorder and metabolic syndrome: a study of increased health risk in veterans. BMC Med. 2009;7:1. doi: 10.1186/1741-7015-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen B, et al. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50:1126–1133. doi: 10.2337/diabetes.50.5.1126. [DOI] [PubMed] [Google Scholar]
- Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated gene in obesity. J Biol Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Dubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman D, Morissette SB, George TP. Co-Morbidity of smoking in patients with psychiatric and substance use disorders. Am J Addict. 2005;14:106–123. doi: 10.1080/10550490590924728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Walters EE. Prevalence, severity, and comorbidity of twelve-month DSM-IV disorders in the National Comorbidity Survey Replication (NCS-R) Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderdale DS, Knutson KL, Yan LL, Liu K, Rathouz PJ. Self-reported and measured sleep duration: how similar are they? Epidemiology. 2008;19:838–845. doi: 10.1097/EDE.0b013e318187a7b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RS, et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet. 2002;360:57–58. doi: 10.1016/S0140-6736(02)09335-2. [DOI] [PubMed] [Google Scholar]
- Lo JC, Mulligan K, Tai VW, Algren H, Schambelan M. “Buffalo hump” in men with HIV-1 infection. Lancet. 1998;351:867–870. doi: 10.1016/S0140-6736(97)11443-X. [DOI] [PubMed] [Google Scholar]
- Lukaschek K, Baumert J, Kruse J, Emeny RT, Lacruz ME, Huth C, et al. Relationship between posttraumatic stress disorder and Type 2 Diabetes in a population-based cross-sectional study with 2970 participants. J Psychosom Res. 2013;74:340–345. doi: 10.1016/j.jpsychores.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and B-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- Norman SB, Means-Christensen AJ, Craske MG, Sherbourne CD, Roy-Byrne PP, Stein MB. Associations between psychological trauma and physical illness in primary care. J Traum Stress. 2006;19:461–470. doi: 10.1002/jts.20129. [DOI] [PubMed] [Google Scholar]
- O’Donovan A, Epel E, Lin J, Wolkowitz O, Cohen B, Maguen S, et al. Childhood trauma associated with short leukocyte telomere length in posttraumatic stress disorder. Biol Psychiatry. 2011;70:465–471. doi: 10.1016/j.biopsych.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DK, Ciaraldi T, Henry RR. Adiponectin in health and disease. Diabetes Obes Metab. 2007;9:282–289. doi: 10.1111/j.1463-1326.2006.00610.x. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Kihara S, Funahashi T, Matsuzawa Y, Libby P. Adiponectin: a key adipocytokine in metabolic syndrome. Clin Sci. 2006;110:267–278. doi: 10.1042/CS20050182. [DOI] [PubMed] [Google Scholar]
- O’Toole BI, Catts SV. Trauma, PTSD and physical health: an epidemiological study of Australian Vietnam veterans. J Psychosom Res. 2007;64:33–40. doi: 10.1016/j.jpsychores.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Pagoto SL, Schneider KL, Bodenlos JS, Appelhans BM, Whited MC, Ma Y, et al. Association of posttraumatic stress disorder and obesity in a nationally representative sample. Obesity. 2012;20:200–205. doi: 10.1038/oby.2011.318. [DOI] [PubMed] [Google Scholar]
- Pajvani UB, Scherer PE. Adiponectin: Systemic contributor to insulin sensitivity. Current Diabetes Reports. 2003;3:207–213. doi: 10.1007/s11892-003-0065-2. [DOI] [PubMed] [Google Scholar]
- Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Medical comorbidity of full and partial posttraumatic stress disorder in US adults: results from Wave 2 of the national epidemiologic survey on alcohol and related conditions. Psychosom Med. 2011;73:697–707. doi: 10.1097/PSY.0b013e3182303775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK, Orr SP. Twenty-four hour urinary cortisol and catecholamine excretion in combat-related posttraumatic stress disorder. Biol Psychiatry. 1990;27:245–247. doi: 10.1016/0006-3223(90)90654-k. [DOI] [PubMed] [Google Scholar]
- Preacher KJ, Hayes AF. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behavior Research Methods, Instruments and Computers. 2004;36:717–731. doi: 10.3758/bf03206553. [DOI] [PubMed] [Google Scholar]
- Rao MN, Blackwell T, Redline S, Punjabi NM, Barrett-Connor E, et al. Association between sleep duration and 24-hour urine free cortisol in the MrOS Sleep Study. PLoS ONE. 2013;8:e75205. doi: 10.1371/journal.pone.0075205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson AM, Lipschitz DS, Wang S, Hu S, Vojvoda D, Bremner JD, et al. Increased pituitary and adrenal reactivity in premenopausal women with posttraumatic stress disorder. Biol Psychiatry. 2001;50:965–977. doi: 10.1016/s0006-3223(01)01264-1. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Pathophysiology of insulin resistance in human disease. Physiological Reviews. 1995;75:473–486. doi: 10.1152/physrev.1995.75.3.473. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Insulin resistance/compensatory hyperinsulinemia, hypertension, and cardiovascular disease. J Clin Endocrinol Metab. 2003;88:2399–2403. doi: 10.1210/jc.2003-030087. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales A, editors. A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. Washington DC: National Institutes of Health; 1968. NIH Publication 204. [DOI] [PubMed] [Google Scholar]
- Regestein QR, Friebely J, Shifren JL, Scharf MB, Wilita B, Carver J, et al. Self-reported sleep in postmenopausal women. Menopause. 2004;11:198–207. doi: 10.1097/01.gme.0000097741.18446.3e. [DOI] [PubMed] [Google Scholar]
- Richards A, Metzler TJ, Ruoff LM, Inslicht SS, Rao M, Talbot LS, et al. Sex differences in objective measures of sleep in posttraumatic stress disorder and healthy control subjects. J Sleep Res. 2013;22:679–687. doi: 10.1111/jsr.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza RA, Lawrence JM, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor defect of insulin action. J Clin Endocrinol Metab. 1982;54:131–138. doi: 10.1210/jcem-54-1-131. [DOI] [PubMed] [Google Scholar]
- Rohleder N, Schommer NC, Hellhammer DH, Engel R, Kirschbaum C. Sex differences in glucocorticoid sensitivity of proinflammatory cytokine production after psychosocial stress. Psychosomatic medicine. 2001;63:966–972. doi: 10.1097/00006842-200111000-00016. [DOI] [PubMed] [Google Scholar]
- Snehalatha C, et al. Plasma adiponectin is an independent predictor of type 2 diabetes in Asian Indians. Diabetes Care. 2003;26:3226–3229. doi: 10.2337/diacare.26.12.3226. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- Spitzer C, Barnow S, Volzke H, John U, Freyberger HJ, Grabe HJ. Trauma, posttraumatic stress disorder and physical illness: findings from the general population. Psychosom Med. 2009;71:1012–1017. doi: 10.1097/PSY.0b013e3181bc76b5. [DOI] [PubMed] [Google Scholar]
- Spranger J, Kroke A, Mohlig M, Bergmann MM, Ristow M, Boeing H, et al. Adiponectin and protection against type 2 diabetes mellitus. Lancet. 2003;361:226–228. doi: 10.1016/S0140-6736(03)12255-6. [DOI] [PubMed] [Google Scholar]
- Suetsugi M, Mizuki Y, Yamamoto K, Uchida S, Watanabe Y. The effect of placebo administration on the first-night effect in healthy young volunteers. Prog Neuropsychoparmacol Biol Psychiatry. 2007;31:839–847. doi: 10.1016/j.pnpbp.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Tasali E, Leproult R, Ehrmann DA, Van Cauter E. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci. 2008;105:1044–1049. doi: 10.1073/pnas.0706446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint M, Luthringer R, Schaltenbrand N, Carelli G, Lainey E, Jacqmin A, et al. First-night effect in normal subjects and psychiatric inpatients. Sleep. 1995;18:463–469. doi: 10.1093/sleep/18.6.463. [DOI] [PubMed] [Google Scholar]
- Trief PM, Ouimette P, Wade M, Shanahan P, Weinstock RS. Post-traumatic stress disorder and diabetes: co-morbidity and outcomes in a male veterans sample. J Behav Med. 2006;29:411–418. doi: 10.1007/s10865-006-9067-2. [DOI] [PubMed] [Google Scholar]
- Walczewska J, Rutkowski K, Wizner B, Cwynar M, Grodzicki T. Stiffness of large arteries and cardiovascular risk in patients with posttraumatic stress disorder. Eur Heart J. 2011;32:730–736. doi: 10.1093/eurheartj/ehq354. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lam KS, Yau MN, Xu A. Post-translational modifications of adiponectin: mechanisms and functional implications. Biochem J. 2008;409:623–633. doi: 10.1042/BJ20071492. [DOI] [PubMed] [Google Scholar]
- Weathers FW, Keane TM, Davidson JR. Clinician-adminstered PTSD scale: A review of the first ten years of research. Depress Anxiety. 2001;13:132–156. doi: 10.1002/da.1029. [DOI] [PubMed] [Google Scholar]
- Weisberg RB, Bruce SE, Machan JT, Kessler RC, Culpepper L, Keller MB. Nonpsychiatric illness among primary care patients with trauma histories and posttraumatic stress disorder. Psychiatr Serv. 2002;53:848–854. doi: 10.1176/appi.ps.53.7.848. [DOI] [PubMed] [Google Scholar]
- Weiss T, Skelton K, Phifer J, Jovanovic T, Gillespie CF, Smith A, et al. Posttraumatic stress disorder is a risk factor for metabolic syndrome in an impoverished urban population. Gen Hosp Psychiatry. 2011;33:135–142. doi: 10.1016/j.genhosppsych.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Hirose H, Saito I, Nishikai K, Saruta T. Adiponectin, an adipocyte-derived protein, predicts future insulin-resistance: two-year follow-up study in Japanese population. J Clin Endocrinol Metab. 2004;89:87–90. doi: 10.1210/jc.2003-031163. [DOI] [PubMed] [Google Scholar]
- Yatagai T, Nagasaka S, Taniguchi A, Fukushima M, Nakamura T, Kuroe A, et al. Hypoadiponectinemia is associated with visceral fat accumulation and insulin resistance in Japanese men with type 2 diabetes mellitus. Metabolism. 2003;52:1274–1278. doi: 10.1016/s0026-0495(03)00195-1. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL, Jr, Mason JW. Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis. 1990;178:366–369. doi: 10.1097/00005053-199006000-00004. [DOI] [PubMed] [Google Scholar]