

Abstract
Objective
We evaluated the efficacy of the potent antioxidant C3 to salvage nigrostriatal neuronal function after MPTP exposure in nonhuman primates. C3 is a first-in-class functionalized water-soluble fullerene which reduces oxygen radical species associated with neurodegeneration in in vitro studies. However, C3 has not been evaluated as a neuroprotective agent in a Parkinson model in vivo.
Methods
Macaque fascicularis monkeys were used in a double-blind, placebo-controlled study design. MPTP-lesioned primates were given systemic C3 (n = 8) or placebo (n = 7) for two months starting one week after MPTP. Outcomes included in vivo behavioral measures of motor parkinsonism using a validated non-human primate rating scale, kinematic analyses of peak upper extremity velocity, PET imaging of 6-[18F]fluorodopa (FD, reflects dopa decarboxylase) and [11C]dihydrotetrabenazine (DTBZ; reflects vesicular monoamine transporter type 2), as well as ex vivo quantification of striatal dopamine (DA) and stereologic counts of tyrosine hydroxylase (TH) immunostained neurons in substantia nigra.
Results
After two months, C3 treated monkeys had significantly improved parkinsonian motor ratings, greater striatal FD and DTBZ uptake, and higher striatal dopamine levels. None of the C3 treated animals developed any toxicity.
Interpretation
Systemic treatment with C3 reduced striatal injury and improved motor function despite administration after the MPTP injury process had begun. These data strongly support further development of C3 as a promising therapeutic agent for PD.
Keywords: C3; fullerene; neuroprotection; drug therapy; 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP); positron emission tomography (PET); dopamine; Parkinson disease
Parkinson disease (PD) is a progressive neurological disorder characterized by tremor, bradykinesia, rigidity, masked facies, gait abnormalities, postural instability and multiple non-motor problems. While the exact etiology of PD remains to be established, animal models and human genetic studies have implicated altered mitochondrial metabolism, chronic activation of inflammatory pathways, oxidation of dopamine to toxic metabolites, and impaired growth factor (Powers et al., 2008; Shulman et al., 2011). Current therapies for PD are suboptimal, and there is an unmet need for development of new therapeutic options for this condition.
Since mitochondrial and inflammatory processes in PD lead to oxidative injury, a promising approach to new drug design involves compounds with antioxidant qualities. The fullerene compound C3 (Figure 1), is a promising drug for PD since it has potent anti-inflammatory properties in several disease models (Dugan et al., 1997; Kaplan SS, 1999; Lotharius et al., 1999; Ali et al., 2004; Behrens et al., 2007; Behrens et al., 2008) and high aqueous solubility. In this study, we evaluate the in vivo efficacy of parenteral C3 as a neuroprotectant using a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) primate model of PD.
Figure 1. Chemical structure of C3.
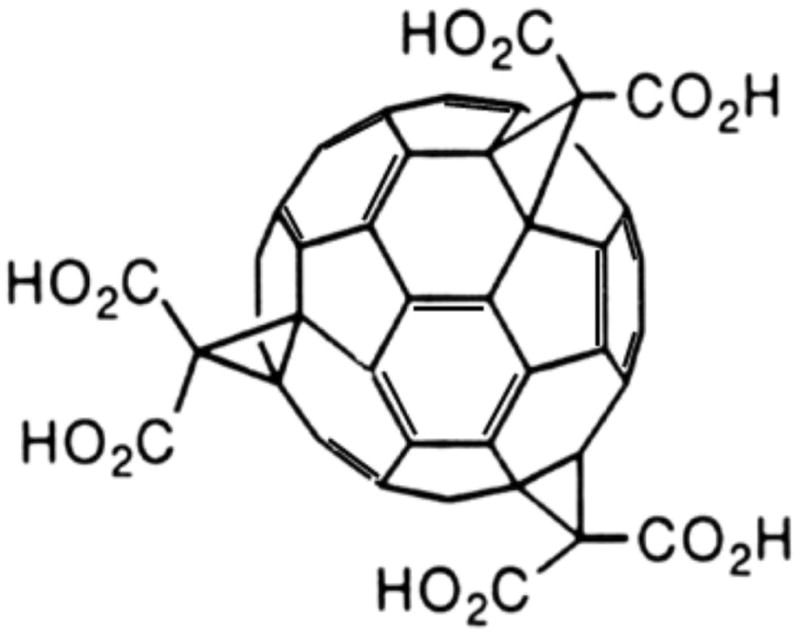
Molecular structure showing the three malonic acid groups of C3 attached to the fullerene C60 sphere through cyclopropane carbons at the e, e, e, positions of C60, giving the molecule C3 symmetry. The C3 symmetry gives the compound dipolar character, which enhances distribution across neuronal cell membranes.
The MPTP-lesioned non-human primate provides an excellent platform for testing symptomatic treatments and protective interventions that target processes believed to contribute to the PD pathology. One major difference between the primate-MPTP model and human pathology is the absence of Lewy body formation and the potential mechanism of cell death in human PD, which remains uncertain. However, because of the reliability and consistency of the model, its close similarity to the motor manifestations of PD, and its high predictive value for symptomatic therapies, the primate-MPTP model is quite reasonable for preclinical evaluation of PD drugs (Duty and Jenner, 2011). We chose unilateral intracarotid administration of MPTP to produce hemiparkinsonism thereby permitting the animal to care for itself and allowing the contralateral side of the brain to act as an internal control (Tabbal et al., 2006; Tabbal et al., 2012).
A key feature of our study is that we administered the test agent after induction of nigrostriatal injury by MPTP, and thus demonstrate effects that are representative of the post-injury neuroprotective properties of C3 rather than direct blockade of toxicity of MPTP itself.
METHODS
Experimental Design
A generalized schematic of the experimental design is shown in Figure 2. The study begins with baseline behavioral (motor parkinsonism rating scales and kinematics) and PET ([11C]dihydrotetrabenazine [DTBZ] and 6-[18F]fluorodopa [FD]) measures on each animal. The animal is then treated with internal carotid artery injection of the neurotoxin MPTP, and a 7-day interval ensues during which nigrostriatal injury develops. At the end of the week-long fulmination interval, behavioral measures demonstrate the expected neurotoxicity from MPTP, and the animals are randomized into treatment and control arms that involve parenteral infusion of either C3 or placebo for a total of 8 weeks. During the treatment phase, behavioral measures (parkinsonism ratings and kinematics) are performed repeatedly to monitor the condition of the animals. At the end of the treatment phase (i.e., 9 weeks after MPTP injection, and 8 weeks after starting C3 or placebo administration), terminal PET measures (DTBZ and FD) are made. The animal is then euthanized, and the brain removed for in vitro measures of striatal dopamine (DA) and stereologic measures of tyrosine hydroxylase (TH) immunostained neurons in substantia nigra (SN). Note that the infusions begin seven days after administration of MPTP, so any differences observed between measures of the treatment and control arms can be attributed to the neuroprotective effects of the infusions, rather than due to simple blockade of the initial tissue insult from neurotoxin MPTP.
Figure 2. Schematic of Study Design.
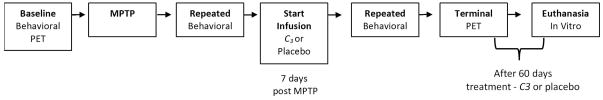
Illustration of the major aspects of the study. Baseline measures (behavioral, PET) precede treatment with intracarotid MPTP. One week after MPTP administration, each animal is assessed to verify nigrostriatal damage, and randomized to either a treatment (C3) or placebo infusion for 8 weeks. Repeated behavioral measures are made during the 8 weeks of infusion. At the end of the infusion interval, terminal PET measures are done, the animal euthanized and in vitro measures (DA, TH) are performed. Note that the process for the treatment and control arms are identical, and that both infusions begin well after (7 days) administration of neurotoxin.
Ethics Statement on Use of Animals
Experiments were conducted on 15 male Macaca fascicularis monkeys: seven controls and eight treated with C3 (mean ages 7.6 ± 2.2 and 8.1 ± 2.3, respectively) (Table 1). We used the minimum number of animals necessary for adequate statistical power, and in accordance with the recommendations of the Weatherall report “The use of non-human primates in research,” and took all steps to ameliorate suffering in our studies. Guidelines prescribed by the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animal were followed, and this work had the approval the Institutional Animal Care and Use Committee (IACUC) at Washington University in St. Louis. All animals were housed individually; maintained in facilities with 12-hour dark and light cycles; provided access to food and water ad libitum; and were equally engaged with a variety of psychologically-enriching tasks, such as watching movies or playing with appropriate toys.
Table 1. Subject Characteristics.
Fifteen male Macaque fascicularis monkeys were enrolled. There were no differences in age (p = 0.69) or weight (p=0.95) between treatment groups at baseline (just prior to MPTP injection).
| Treatment Group | Age range (years) | Age (mean, SD) | Weight (kg) | n |
|---|---|---|---|---|
| Placebo-treated | 5.1 – 12.0 | 7.6 ± 2.2 | 7.14 ± 1.18 | 7 |
| C3-treated | 5.8 – 13.0 | 8.1 ± 2.3 | 7.10 ± 1.22 | 8 |
Blinding of Study
Treatment randomization preceded acquisition of monkeys to avoid potential bias introduced by the size or health of an animal. For each primate, a pair of 2 mL mini-osmotic pumps (Alzet, model 2ML4, infusion rate 2.5 μl/hr) containing either C3 (200 mg/ml), delivering 3 mg/kg/day), or placebo solution (color-matched with Durkee red food coloring in 0.9% Sodium Chloride for Injection, USP) was shipped to Washington University from the University of California in San Diego labeled with the animal’s identification code but no other information. Behavioral measures, PET measures, and other analyses were carried out by investigators blinded to treatment group (i.e., C3 or placebo). After all data had been collected and compiled at the completion of the study, an investigator not involved in data acquisition or statistical analyses divided the data from the main spreadsheet into two spreadsheets based on treatment. This information was then sent to a blinded biostatistician who had not been involved with any of the study previously, and who was unaware of the identity of either treatment. No data were excluded for any animal.
MPTP Administration
Animals were fasted overnight, with free access to water until 2 hrs prior to MPTP injection. Anesthesia was induced with ketamine (10 mg/kg i.m.), followed by endotracheal intubation and maintenance of anesthesia using inhaled isoflurane (3%). MPTP (Sigma, St. Louis, MO) in 0.9% Sodium Chloride Injection, USP (0.1 mg/mL) at a dose of 0.25 mg/kg was infused over 30 minutes not faster than 1 mL/min into the right internal carotid artery (Tabbal et al., 2012). Location of the infusion catheter was confirmed with angiograms before and after MPTP infusion. Each animal was allowed to recover with constant observation until it was able to drink and care for itself independently. Full biohazard/neurotoxin precautions for MPTP were taken prior to, during, and after the injection, and all excretions for the next 24 hours were isolated and disposed of appropriately. Proper safety procedures were followed for handling MPTP and all contaminated tissues and waste products (Tian et al., 2012). Intracarotid MPTP injection as performed here has been shown to produce stable hemi-parkinsonism lasting more than 1.5 years contralateral to the lesioned side of the brain (Tabbal et al., 2006).
Preparation & Insertion of Mini-osmotic Drug Infusion Pumps
Sterile C3 powder was prepared as previously described (Dugan et al., 1997; Behrens et al., 2007; Behrens et al., 2008), and formulated as a pH 7.4 stock solution just prior to loading of pumps. Sterile Alzet mini-osmotic pumps (model 2ML4 to deliver 2.5 μL/hr for 30 days) from Durect (CA) were loaded with C3 (200 mg/ml) or color-matched placebo, both dissolved in 0.9% Sodium Chloride for Injection, USP. At pH 7, C3 is soluble up to 400 mg/ml, so the target concentration was easily achieved (Dugan and Hardt, unpublished). At 37° C, and ambient light, C3 is stable for > 40 days, and this stability is enhanced when maintained as a concentrated solution (as in the Alzet pumps for this study) (Dugan, Hardt et al, manuscript in preparation). After preparation of C3 and loading of the pumps at the University of California in San Diego, each pair of pumps were placed in a sterile 50 mL plastic tube, and shipped overnight on wet ice to Washington University. After receipt, 0.9% Sodium Chloride Injection, USP was added to the tube to prime the pumps, which were inserted subcutaneously (first 4 animals) or intraperitoneally (remaining monkeys) the next day, while the animal was anesthetized. Each monkey had two pumps inserted at a time, and the total dose of C3 from both pumps was 3 mg/kg/day. This dose was selected based upon preliminary studies of tolerability and efficacy in mice and rats, which showed no long-term toxicity with 2 years of systemic treatment at 1mg/kg/day (Ali et al., 2006; Quick et al., 2008) or 3 mg/kg per day(Dugan et al., 1998), and the half-life of C3, which is ~ 8 hours in plasma (mice, monkeys). In mice, the lethal dose-50 (LD50) for a single bolus of C3 is 50 mg/kg, and for chronic infusion as in the current study, >30 mg/kg/day (Dugan, Hardt et al, manuscript in preparation); no LD50 studies were performed in monkeys, but blood tests for hematologic, renal and hepatic toxicity showed no difference in values between placebo and C3-treated animals. The placebo solution was color-matched to assure that the surgical team was blind to the treatment. After the first month of treatment, pumps were replaced for delivery of a second month of drug infusion.
Behavioral Measures
Nonhuman Primate Parkinsonism Rating Scale
We rated motor parkinsonism using a validated rating scale developed for nonhuman primates (Tabbal et al., 2012). Each animal was videotaped at the same time of day (early morning) several times before MPTP treatment for baseline behavioral ratings, then several times per week after MPTP until euthanasia. Animals were trained using positive conditioning only to follow a behavioral protocol that consisted of a) walking back and forth 3 times in a straight corridor for 15 feet, b) walking in a circle 6 times in a room (3 clockwise and 3 counterclockwise turns) and c) reaching 10 times with each hand at full arm length for a food item held by the trainer. Each behavioral session was video recorded, and the same observer blinded to the treatment group rated each video (Tabbal et al., 2012). Briefly, ratings included a 0 to 3 scale (0 = unaffected, 1 = mildly affected, 2 = moderately affected and 3 = severely affected) for parkinsonian features (bradykinesia, tremor and flexed posturing). This yielded a composite parkinsonism score ranging from 0 to 18 per side. We used for the analysis the parkinsonism score at various times up to 2 months after MPTP just prior to euthanasia. During these studies, animals were always able to care for themselves despite not receiving any anti-parkinsonism drugs. One C3 treated animal did not have final behavioral ratings included since the video was accidentally destroyed. All other data from this animal, including kinematic data were included.
Kinematic Analysis of Upper Extremity Motor Velocity
To assess the ability of treatment to maintain normal upper limb motor speed, animals were analyzed using a video-based kinematics assessment as previously described (Wenger et al., 1999). The primates had baseline ratings before MPTP, after MPTP, and then were repeatedly rated during the 60 days of infusion with either C3 or placebo. Monkeys were trained to sit in a primate chair and to reach for and eat bits of fruit as behavior reinforcement. Each animal had a training period of 2–3 months with five 30-minute sessions per week. Within a few weeks they became accustomed to the trainer and the transfers from home cage to training chair. Once an animal learned the task, the kinematics of the movement were measured using reflective markers placed on the lateral aspect of the index finger, wrist, elbow, and shoulder to aid in visualization and timing. Animals were videotaped in the sagittal plane during a minimum of 12 reaches to an apple slice target. The kinematic data were recorded at 60 frames per second using a motion analysis system from Peak Performance Technologies (Motus 2000). A time code was generated and recorded with the video information. The video images were digitized and the joint markers tracked to permit measurement of the movement. The reaching task was performed and videotaped in baseline sessions every other day for 5 sessions total, then continued every other day beginning the day after MPTP injection for the next 4 weeks and then twice a week for the following 9 weeks until euthanasia.
The kinematic data were analyzed off-line with the Peak Performance Motus 2000 motion analysis system and custom-written software to obtain velocity and acceleration information from the reaching task. The movement trajectories of the entire limb and of individual joints were quantified and analyzed with respect to joint angle relationships and variability from trial to trial in both the maintained posture and reaching phases of the task. All analyses were performed by investigators blind to treatment group.
PET Measures
In addition to the behavioral measures, we also carried out PET imaging to assess striatal dopa decarboxylase using FD and vesicular monoamine transporter type 2 (VMAT2) using DTBZ. Both PET measures were done before MPTP injection to acquire baseline data. Repeat PET measures using both tracers were also done at the end of the two-month infusion with either C3 or placebo solution. To mimic the clinical situation in which PD patients have pre-existing dopaminergic neuronal injury, administration of C3 or placebo was initiated 7 days after MPTP. Note that unlike systemic administration of MPTP, which causes bilateral loss of dopamine neurons, the unilateral intracarotid MPTP infusion model used in these studies spares the contralateral side (Tabbal et al., 2012) This situation allowed the non-injured side to serve as an internal control for each individual animal.
PET Tracers
Carbon-11 and fluorine-18 for radiotracer syntheses were produced using the Washington University RDS-Eclipse cyclotron, and labeling substrates were from ABX Advanced Biochemical Compounds (Radeberg, Germany). DTBZ was prepared via N-[11C]methylation in ≥ 90% radiochemical purity and specific activity ≥ 1000 Ci/mmol. FD was prepared via electrophilic [18F]fluorination in ≥ 90% radiochemical purity and specific activity ≥ 100 mCi/mmol.
PET Imaging
Animals were imaged under anesthesia (Karimi et al., 2013) using the Siemens Microsystems (Knoxville, TN) MicroPET Focus 220 scanner. A transmission scan was collected for individual attenuation measurements at each imaging session. Afterwards, up to 10 mCi (10 nmole) of DTBZ) was infused i.v. over 60 seconds, followed by PET scans with three 1-minute frames, four 2-minute frames, three 3-minute frames and then eight 5-minute frames for the next 40 minutes (1 hour total). Three hours later, 3–10 mCi (33–100 μmole) of FD was infused i.v. over 60 seconds, and PET scans collected for 120 min (three 1-minute frames, four 2-minute frames, three 3-minute frames, and twenty 5-minute frames) (Karimi et al., 2013).
MRI
Magnetic resonance images permit identification of relevant volumes of interest (VOIs) from the PET images. A Siemens Magnetom Sonata 1.5 T system was used for these studies. At each imaging session, gradient and shim coil currents were adjusted to correct for spatial inhomogeneities using the scanner’s multi-axis-projection shim program. We collected a 3-D MP-RAGE sequence (TR = 2400 ms, TE = 3.93 ms, TI = 1100 ms, flip angle = 7, 256 x 256 x 192 matrix, 2 averages, acquisition time = 15:23) to produce high-resolution T1 images (0.8 mm isotropic voxels).
PET Image Analysis
All image analysis was done by investigators blinded to treatment status of the monkeys. PET image reconstruction considered randoms, scatter, attenuation and deadtime. Reconstructed resolution was < 2.0 mm full-width half-maximum for all 3 dimensions at the center of the field of view. Correction for movement of individual frames was unnecessary since the anesthetized animal’s head was immobile. The first baseline PET image for each animal acted as the target image, with subsequent PET images collected on subsequent days co-registered to it. Caudate, putamen and an occipital region (as a reference) were manually traced on the MP-RAGE for each animal. The MP-RAGE was co-registered to the target PET scan of the corresponding animal using a vector gradient method, and the traced VOIs were transformed into PET space using the same transformation matrix (Rowland et al., 2005; Brown et al., 2013). To reduce volume averaging with the neighboring regions, the MRI based striatal VOIs were trimmed to include only voxels with at least 90% of the maximal counts within that VOI. The reference region included a pair of hemi-cylinders in occipital cortex on either side of the midline, set back from midline and the posterior edges to avoid vascular activity and transformed to PET space as described above. After extracting the tissue activity curves, we calculated the influx constant KOCC for FD (Patlak and Blasberg, 1985) (data from 24–94 min), and the non-displaceable binding potential (BPND) for DTBZ (Logan et al., 1996) (15–60 min), as previously described (Karimi et al., 2013).
In Vitro Measures
Euthanasia & Sample Preparation
Animals were euthanized with intravenous pentobarbital (100 mg/kg; Somnasol Euthanasia, Butler Schein Animal Health, Dublin, OH), 2 months after initiation of osmotic pump infusions, which corresponds to 9 weeks after MPTP injection. Brain tissue was handled as described previously (Tabbal et al, 2012). The brain was removed within 10 minutes, and the hemispheres and midbrain were separated. The hemispheres were sliced in the coronal plane. Standard punch biopsies were collected from caudate and putamen bilaterally, frozen quickly on dry ice snow and stored in a −80° C freezer until dopamine assay. The midbrain was fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline solution (PBS).
Analysis of Striatal Dopamine Levels
Striatal dopamine was measured in the tissue extracts using high performance liquid chromatography with electrochemical detection (HPLC-EC) (Karimi et al., 2006; Tian et al., 2012). The analytical data were expressed as ng/g of brain tissue. The concentration of dopamine in the lesioned caudate, putamen, and striatum was divided by the concentration in the non-lesioned side to normalize for differences across animals.
Cell Counts of Dopaminergic TH Immunostaining Nigral Neurons
The brains were sliced and immunostained with TH using 3, 3′-diaminobenzidine solution amplification. Unbiased stereological counts of nigrostriatal neurons were done on immunostained midbrain slices, focusing on the substantia nigra pars compacta (SNpc) as previously described (Tabbal et al., 2012).
Statistics Analysis
All data from the above studies were analyzed by repeated measures ANOVA followed by Tukey’s post-hoc test by SigmaStat statistical software, with significance set at p < 0.05. For in vivo PET measures as well as in vitro quantification of striatal dopamine concentration and TH-positive cell counts, the ratio of the lesioned side/unlesioned side for each animal was used for statistical analysis to control for inter-individual differences. Statistical analyses with the parkinsonian rating scales were done with the non-parametric Mann-Whitney U (rank sum) test. Data for the various study results are presented as the mean ± SEM for all the data in each group. In the statistical analysis, no data were excluded from any subject or any test.
RESULTS
Behavioral Measures
Parkinsonism Ratings
Monkeys were rated throughout the study. Figure 3 shows the mean parkinsonism motor ratings for the C3-treated and control animals as a function of time. Note that early after MPTP administration, the scores for the two arms of the study are not significantly different. However, as infusion times increase, the two curves first statistically differ (Mann-Whitney Wilcoxon, p = 0.026) beginning 40 days after the start of the C3 infusion with declining parkinsonism ratings in the treated animals but no drop in these ratings in the control animals. The final ratings at the end of the 2-month treatment period are shown in Figure 4. Data are the mean ± SEM for all animals in the treatment (n = 7) and control (n = 7) groups. C3-treated animals had significantly lower parkinsonian scores than placebo-treated animals, indicating less bradykinesia, flexed posturing and tremor when the animals received C3 infusion (p = 0.007, Mann-Whitney-Wilcoxon Rank Sum test).
Figure 3. Dynamic functional assessment of Parkinson symptoms for C3-treated and control animals.

Animals were rated using an 18-point system which assesses the severity of parkinsonian symptoms (0= no symptoms; 18= most severely affected). Data are the mean and standard deviation of scores for all animals in each arm. Note the similarity of scores shortly after MPTP treatment, but prior to starting infusion of C3 or placebo, and the progressively different scores attained with longer infusions of C3. Scores for the two groups of animals diverged significantly beginning 30 days after the start of C3 infusion (p=0.026 by Mann-Whitney-Wilcoxon). Filled circles, C3-treated animals; open circles, control animals.
Figure 4. Parkinsonian motor scores in the primates at the end of the study.

Data represent the mean ± SEM at the end of the 8-week infusions. At the end of 2 months of treatment, C3-treated monkeys had a significantly lower rating compared to controls (Mann-Whitney-Wilcoxon rank sum test: U=4.5, n1 = n2 = 7, p = 0.007).
Kinematic scores of upper extremity peak velocity
The upper extremity peak velocity was compared to the baseline velocity of the same limb following the same stimulus prior to MPTP lesioning. Results are presented as the mean percent decrease in velocity from baseline, with a smaller decrease indicating preservation of function (less bradykinesia) (Figure 5). Preservation of peak upper extremity reaching in the C3-treated monkeys was not significantly different from control animals, but showed a strong trend (p = 0.078) by ANOVA).
Figure 5. Kinematic scores of upper extremity peak velocity.

Kinematic data are analyzed as the ratio of the peak movement velocity for the affected (right) upper extremity in animals at the end of the 8-week treatment compared to that of the same limb prior to MPTP lesioning. Results are presented as the mean percent decrease in velocity from baseline, with a smaller decrease indicating preservation of function. Data represent the means ± SEM at the end of the 8-week infusions; n = 8 for C3 and n = 7 for placebo. Compared to controls, C3-treated monkeys appeared to retain upper extremity movement speed (less bradykinesia), but this did not quite reach statistical significance (p = 0.078) by ANOVA t test).
PET Measures
PET Imaging: FD and DTBZ
Figure 6A illustrates FD and DTBZ striatal uptake at baseline and 2 months after MPTP in two control monkeys, and Figure 6B shows the similar PET images for two C3 treated monkeys.
Figure 6. PET imaging of DTBZ and FD.
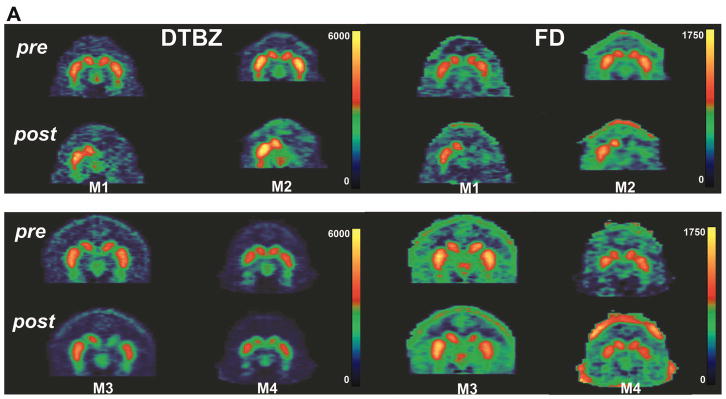
Representative coronal PET images are shown from two control primates (A; monkeys M1 and M2) and two C3-treated monkeys (B; monkeys M3 and M4) just prior to intracarotid MPTP (top panels) and at the end of two months of treatment with placebo or C3 (bottom panels). Images of DTBZ (left panels) and FD (right panels) are shown. Note the bilateral uptake of each tracer pre-MPTP, showing the tear-drop-shaped substantia nigra bilaterally for all four animals. However, at the end of the study, there is significantly less uptake of both tracers on the lesioned side in placebo-treated animals compared to C3-treated monkeys, or conversely, there is preservation of DTBZ and FD in C3-treated animals.
Figure 7 presents the quantified PET measures taken from all animals in the C3-treated and placebo-infused arm before and after 2 months of infusion. Data are the means ± SEM for all animals; C3, n = 8; placebo, n = 7. Compared to placebo, infusion of C3 provides substantial preservation of striatal DTBZ BPND (Figure 7A) and striatal FD Kocc (Figure 7B) compared to controls.
Figure 7. Summary of PET measures after completion of C3 or placebo infusion.

Quantification of (A) FD Kocc and (B) DTBZ BP at the end of the study. Data are the mean ± SEM of the nigral tracer uptake before MPTP, and at the end of the 2-month drug or placebo infusion. There was significant preservation of uptake for both tracers (FP Kocc p=0.038; DTBZ p=0.02) in the C3-treated group by repeated measures ANOVA, with Tukey’s post-hoc test. C3, n = 8; controls, n = 7.
In Vitro Measures
Striatal Dopamine Levels
The dopamine concentrations per mg of tissue for caudate (caud), putamen (put) and striatum (striat) are shown in Figure 8. Consistent with the PET measures, dopamine levels in all striatal tissues of the lesioned striata were significantly higher in the C3-treated animals compared to the controls (p < 0.001; 2-way ANOVA, Tukey’s post-hoc test).
Figure 8. Striatal dopamine levels.

Tissue dopamine concentration per mg wet weight tissue was determined by HPLC (a) at the completion of the study, following the 2-month drug treatment. The concentration of dopamine in the lesioned caudate (caud), putamen (put), and striatum (striat) was divided by the concentration in the non-lesioned side to control for differences in brain size or time to dissect the tissue. There was significant (p < 0.001) preservation of dopamine in all three regions in C3-treated animals compared to placebo-treated controls (2-way ANOVA, Tukey’s post-hoc test).
TH Immunoreactive Neurons in the SNpc
Qualitatively, we observed substantial loss of TH immunoreactivity in processes in the nigra, which was in many cases more dramatic than the loss of TH positive cell bodies (Figure 9A, Figure 9B, C). Images of TH stained slices containing the SN from two separates pairs of animals are shown in Figure 9B,C (and see insets) with the insets comparing fiber staining in the non-lesioned and lesioned sides. The SNpc on the lesioned side had greater than 50% decrease in the number of TH immunoreactive neurons, compared to the unlesioned side, in all animals (Figure 9D). A trend towards preservation of TH positive cells in C3 treated animals did not reach significance (Figure 9D), but the degree of nigral cell loss (about 50%) is close to that which is associated with nearly complete loss of striatal dopamine (Tabbal et al., 2012). Quantitative fiber density measurements were not performed because TH staining intensity was calibrated to allow reliable cell counts, not fiber counts.
Figure 9. Imaging of tyrosine hydroxylase (TH) immunoreactive neurons and processes in substantia nigra in placebo and C3 treated monkeys.

Brains were sliced and immunostained for TH using DAB amplification. TH is shown in brown, with some slices counterstained with cresyl violet. The corresponding left and right (MPTP lesioned) hemispheres from each animal are shown from two independent pairs of monkeys. (A) Comparison of a placebo and a C3 treated monkey showing substantial loss of TH immunoreactivity on the lesioned side with placebo, and less loss in the C3-treated animal. (B, C) different pair of monkeys showing similar loss of TH positive neurons (magnified in insets) in the C3 treated animal, which is more dramatic in the placebo-treated animals. (D) Quantification showing a nonsignificant trend (p = 0.078, by ANOVA) toward preservation of nigral TH cell bodies by stereological counts. Qualitatively, the degree of preservation of TH immunoreactivity in neuronal processes appears substantially greater than that in cell bodies.
DISCUSSION
Treatment with C3 reduced striatal injury and improved functional outcomes in a primate model of PD, even after the injury process had begun. These data strongly support further development of C3 as a therapeutic agent for PD.
C3 is a functionalized fullerene bearing three malonic acid groups (Fig. 1) that impart aqueous solubility to the C60 core, as well as amphiphilic (dipolar) character that improves intercalation into neuronal membranes to enhance neuroprotective effects. C3 harnesses the unique electronic properties of the C60 fullerene core to catalytically eliminate superoxide anion and other reactive oxygen species believed to play a role in excitotoxic neuronal death. C3 also exhibits anti-inflammatory properties (Kaplan SS, 1999; Behrens et al., 2007; Behrens et al., 2008), and reduces oxidative injury in several disease models (Dugan et al., 1997; Lotharius et al., 1999; Behrens et al., 2007), including MPTP and 6-hydroxydopamine-induced neuronal cell death (Lotharius et al., 1999). The compound localizes in mitochondria (Foley et al., 2002; Ali et al., 2004), and functionally replaces lost mitochondrial superoxide dismutase, Sod2 (MnSOD)(Ali et al., 2004) in Sod2−/− mice. Unlike other antioxidants, C3 does not require active recycling to maintain activity, and does not become a pro-oxidant. Rather, the fullerene demonstrates activity that is similar to the native superoxide dismutase (SOD) enzyme, allowing C3 to eliminate the reactive oxygen species, superoxide, by catalytic dismutation.
There are several special characteristics of C3 relevant for neuroprotection in PD that also may help attenuate MPTP injury. First of all, C3 scavenges injurious free radicals regardless of whether due to excitotoxic or neuroinflammatory origin. Rather than targeting a specific step of a neurochemical pathway, the scavenging mechanism of C3 can provide comprehensive benefit by neutralizing radicals generated from multiple simultaneous biochemical pathways. Second, C3 has a fullerene core that acts as a high capacity free-radical catalyst, resulting in a high stoichiometric ratio of radical to fullerene. Third, aqueous solubility designed onto the lipophilic core provides an important pharmaceutical advantage that enhances penetration of the blood brain barrier after parenteral administration. Taken together, these characteristics make C3 a highly favorable neuroprotective drug for testing in human PD.
We hypothesized that systemically administered C3 one week after MPTP would attenuate nigrostriatal injury and behavioral deficits. To evaluate this hypothesis, we carried out a randomized, double-blind, placebo-controlled longitudinal study in MPTP-lesioned M. fascicularis monkeys and emphasized blinding of all investigators to treatment throughout the study and data analysis. Furthermore, we waited seven days after MPTP administration to start treatment with C3 or placebo. Thus, differences between the C3 and placebo groups can be attributed to neuroprotective effects of C3 and not to interference with the initial steps of MPTP-induced neurotoxicity. This experimental design provides additional confidence in the efficacy of C3 and likely success in humans with PD that already have begun the degenerative process.
The behavioral measures suggest a neuroprotective benefit of C3. Parkinsonian ratings were performed periodically throughout the study using a validated rating scale (Fig. 2). Both C3-treatment and placebo arms start with similar degrees of parkinsonism after MPTP (Fig. 3). However, animals treated with C3 began to improve 3–4 weeks after MPTP (first reaching a statistically significant difference between the two groups of animals at 40 days after start of C3; Fig. 3) and approach baseline measures by the end of the study, whereas, the animals in the placebo arm did not improve. The slow onset of behavioral effects of C3 may reflect the pharmacokinetics of the drug. Although steady state in the plasma compartment may have been reached before 30 days, partitioning of C3 across the BBB and distribution to target sites within the CNS is likely slower. After two months of treatment, the C3 treated animals had significantly less parkinsonism, but the faster quantified reaching velocity did not reach statistical significance. The parkinsonism rating scale integrates more motor behaviors than the reaching task and may be a more comprehensive measure of motor performance.
The PET measures of nigrostriatal terminal fields reflect two different components of presynaptic dopaminergic nigrostriatal neurons. FD reflects primarily decarboxylase activity and storage whereas DTBZ reflects the status of VMAT2 specific binding sites on presynaptic vesicles that store dopamine. As revealed in Figures 6 and 7, the striatal uptake of both of these radiotracers is significantly preserved in the C3 treated animals. Together with the behavioral measures, these PET measures of CNS changes are convincing evidence for a central mechanism involving nigrostriatal neurons whereby C3 provides its pharmacological effect.
The in vitro measures from brain tissue (Fig. 2) provide biochemical validation for the behavioral and in vivo PET measures. The in vitro measures of dopamine concentration within caudate and putamen show a significant increase for the C3-treatment group (Fig. 8).
In contrast, stereologic counts of nigral cell bodies were not statistically different between the C3 and control groups. In all animals, the lesioned SNpc had close to 50% decrease in in nigral cell counts compared to the non-lesioned side. This may have implications for the site of recovery or protection provided by C3. C3 could either preferentially have its effects in terminal fields or, alternatively, it could have a more general action on nigrostriatal neurons, with terminal field recovery just exceeding the flooring threshold that provides clinical benefit. Preferential action of C3 at terminal fields would be reasonable if this molecule is taken up at terminal fields where one might anticipate a higher concentration than at distant cell bodies. One might speculate that either longer time of treatment or possibly a higher dose could enhance recovery at the cell body level. Further investigations are needed to clarify these issues. We also observed qualitatively substantial greater loss of TH immunoreactive fibers in SNpc in placebo treated animals, which was more dramatic than the loss of TH positive cell bodies (Fig. 9). We could not perform quantitative fiber density measurements since TH staining intensity was calibrated to allow reliable cell body counts, and not optimized for fiber counts. Nevertheless, these findings provide a suggestion about the potential mechanisms of action, and targets, of C3.
Our results demonstrate that systemically administered C3 modified severity in the nonhuman primate-MPTP model of PD. Two caveats require consideration. The current study was relatively short, only two months of treatment. Whether treatment effects would persist or potentially improve with longer therapy remain unknown. Clearly the MPTP model provides an excellent system to screen pharmacologic interventions to ameliorate parkinsonian motor manifestations and some non-motor manifestations (Brown et al., 2012), but it is a more tenuous model for testing treatments aimed at the etiopathology of PD in humans. Nevertheless, testing interventions using the MPTP-induced injury model in nonhuman primates provides important clues for potential candidates for therapeutic trials in PD. However, only a properly controlled trial in human subjects with PD can determine clinical efficacy of C3. Fortunately, in these pre-clinical studies we found little evidence of toxicity for C3. Therefore the therapeutic doses used in this study suggest that this therapy should be well-tolerated in humans.
Acknowledgments
This work was supported by NIH RO1 NS039913, RO1NS05425, R01AG033679, R21AG030320, RO1 NS37688, RO1NS058714, NS075321, the Larry L. Hillblom Foundation, the Selma I. Hartke bequest, American Parkinson Disease Association (APDA) Center for Advanced PD Research at Washington University; Greater St. Louis Chapter of the APDA; McDonnell Center for Higher Brain Function; Barnes-Jewish Hospital Foundation (Elliot Stein Family Fund for PD Research & the Parkinson Disease Research Fund). We thank Darryl Craig, Christina Zukas and Terry Anderson for outstanding technical assistance.
Footnotes
The authors wish to dedicate this to the memory of Lester M. Perlmutter, who lived with parkinsonism for 14 years, but never succumbed. He was a constant source of inspiration for this study.
References
- Ali SS, Hardt JI, Quick KL, Kim-Han JS, Erlanger BF, Huang TT, Epstein CJ, Dugan LL. A biologically effective fullerene (C60) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–1202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Anderson DG, Mariappan SV, Buettner GR, Doorn JA. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem. 2011;286:26978–26986. doi: 10.1074/jbc.M111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28:13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons Is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Bezard E, Przedborski S. A tale on animal models of Parkinson’s disease. MovDisord. 2011;26:993–1002. doi: 10.1002/mds.23696. [DOI] [PubMed] [Google Scholar]
- Brown CA, Campbell MC, Karimi M, Tabbal SD, Loftin SK, Tian LL, Moerlein SM, Perlmutter JS. Dopamine pathway loss in nucleus accumbens and ventral tegmental area predicts apathetic behavior in MPTP-lesioned monkeys. Exp Neurol. 2012;236:190–197. doi: 10.1016/j.expneurol.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Karimi MK, Tian L, Flores H, Su Y, Tabbal SD, Loftin SK, Moerlein SM, Perlmutter JS. Validation of midbrain PET measures for nigrostriatal neurons in macaques. Ann Neurol. 2013 doi: 10.1002/ana.23939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, O’Malley K. Axon degeneration in Parkinson’s disease. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 2012;135:2058–2073. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Deleidi M, Gasser T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, Neunlist M, Derkinderen P. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–48. doi: 10.1016/j.nbd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Lovett E, Cuddihy S, Almli CR, Lin T-S, Choi DW. Carboxyfullerenes as neuroprotective antioxidants. In: Kreiglstein J, editor. Pharmacology of Cerebral Ischemia. New York: Academic Press; 1998. [Google Scholar]
- Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, Shen CK, Luh TY, Choi DW, Lin TS. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci U S A. 1997;94:9434–9439. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley S, Crowley C, Smaihi M, Bonfils C, Erlanger BF, Seta P, Larroque C. Cellular localization of a water-soluble fullerene derivative. Biochem Biophys Res Commun. 2002;294:116–119. doi: 10.1016/S0006-291X(02)00445-X. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Blesa J, Przedborski S. Animal models of Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S183–185. doi: 10.1016/S1353-8020(11)70057-8. [DOI] [PubMed] [Google Scholar]
- Kaplan SSGJ, Dugan LL, Gonzales E, Perez R, Park TS. Leukocyte dependent blood-brain barrier breakdown is attenuated by a novel carboxyfluerence derivative in a model of newborn hypoxic-ischemicencephalopathy in piglets. Abstract, Annual Meeting of Section of Pediatric Neurological Surgery, AANS and CNS..1999. [Google Scholar]
- Karimi M, Carl JL, Loftin S, Perlmutter JS. Modified high-performance liquid chromatography with electrochemical detection method for plasma measurement of levodopa, 3-O-methyldopa, dopamine, carbidopa and 3,4-dihydroxyphenyl acetic acid. Journal of chromatography B, Analyticaltechnologies in the biomedical and life sciences. 2006;836:120–123. doi: 10.1016/j.jchromb.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Karimi M, Tian L, Brown CA, Flores HP, Loftin SK, Videen TO, Moerlein SM, Perlmutter JS. Validation of nigrostriatal positron emission tomography measures: critical limits. Ann Neurol. 2013;73:390–396. doi: 10.1002/ana.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D, Kaufman E, Brundin L, Hall S, Surova Y, Hansson O. Non-motor symptoms in patients with Parkinson’s disease - correlations with inflammatory cytokines in serum. PLoS ONE. 2012;7:e47387. doi: 10.1371/journal.pone.0047387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Dugan LL, O’Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab. 1985;5:584–590. doi: 10.1038/jcbfm.1985.87. [DOI] [PubMed] [Google Scholar]
- Phani S, Loike JD, Przedborski S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S207–209. doi: 10.1016/S1353-8020(11)70064-5. [DOI] [PubMed] [Google Scholar]
- Powers WJ, Videen TO, Markham J, Black KJ, Golchin N, Perlmutter JS. Cerebral mitochondrial metabolism in early Parkinson’s disease. J Cereb Blood Flow Metab. 2008;28:1754–1760. doi: 10.1038/jcbfm.2008.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Djaldetti R, Liberatore G, Vila M, Vukosavic S, Almer G. The parkinsonian toxin MPTP: action and mechanism. Restorative neurology and neuroscience. 2000;16:135–142. [PubMed] [Google Scholar]
- Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. 2008;29:117–128 E. doi: 10.1016/j.neurobiolaging.2006.09.014. pub Oct. 2006. [DOI] [PubMed] [Google Scholar]
- Rowland DJ, Garbow JR, Laforest R, Snyder AZ. Registration of [18F]FDG microPET and small-animal MRI. Nucl Med Biol. 2005;32:567–572. doi: 10.1016/j.nucmedbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics and pathogenesis. Annual review of pathology. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- Tabbal SD, Mink JW, Antenor JA, Carl JL, Moerlein SM, Perlmutter JS. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience. 2006;141:1281–1287. doi: 10.1016/j.neuroscience.2006.04.072. [DOI] [PubMed] [Google Scholar]
- Tabbal SD, Tian L, Karimi M, Brown CA, Loftin SK, Perlmutter JS. Low nigrostriatal reserve for motor parkinsonism in nonhuman primates. Exp Neurol. 2012;237:355–362. doi: 10.1016/j.expneurol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Karimi M, Loftin SK, Brown CA, Xia H, Xu J, Mach RH, Perlmutter JS. No differential regulation of dopamine transporter (DAT) and vesicular monoamine transporter 2 (VMAT2) binding in a primate model of Parkinson disease. PLoS ONE. 2012;7:e31439. doi: 10.1371/journal.pone.0031439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, Effros RB, Chesselet MF. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. 2012;237:318–334. doi: 10.1016/j.expneurol.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger KK, Musch KL, Mink JW. Impaired reaching and grasping after focal inactivation of globus pallidus pars interna in the monkey. J Neurophysiol. 1999;82:2049–2060. doi: 10.1152/jn.1999.82.5.2049. [DOI] [PubMed] [Google Scholar]